
Novel spiro[fluorene-9,9′-xanthene]-based hole transport layers for red and green PHOLED devices with high efficiency and low efficiency roll-off†
Tao
Wang
a,
Ming
Shi
a,
Daqi
Fang
a,
Junpeng
He
a,
Meng
Zhang
b,
Siwei
Zhang
b,
Guodan
Wei
 *b and
Hong
Meng
*a
*b and
Hong
Meng
*a
aSchool of Advanced Materials, Peking University Shenzhen Graduate School, Peking University, Shenzhen, 518055, P. R. China
bTsinghua-Berkeley Shenzhen Institute (TBSI), Tsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen, 518055, P. R. China
First published on 25th January 2021
Abstract
Two N-phenyl-1-naphthylamine (PNA)/spiro[fluorene-9,9′-xanthene] (SFX) hybrid materials (DPNA-SFX and DOPNA-SFX) were successfully obtained by incorporating two PNA groups with a bridging phenyl ring to an SFX center. Both materials possess similar photophysical properties and an outstanding glass-transition temperature (Tg) beyond 150 °C surpassing that of most typical spirobifluorene and SFX materials. Both green and red phosphorescent organic light-emitting diodes (PHOLEDs) based on two materials achieved remarkable results. The DPNA-based green devices exhibited decent performance with the maximum current efficiency (CEmax), power efficiency (PEmax) and external efficiency (EQEmax) of 89.8 cd A−1, 94.2 lm W−1 and 24.7%, respectively. More importantly, the pure red PHOLEDs with DPNA-SFX as the hole-transporting material achieved the CEmax, PEmax and EQEmax of 41.1 cd A−1, 46.4 lm W−1 and 34.7%, respectively, which is comparable to those of the best red PHOLEDs. And the EQE at 1000 cd m−2 maintained 96% of its peak value, demonstrating an extremely low efficiency roll-off.
Introduction
Organic light-emitting diodes (OLEDs) have attracted extensive research interest both in academia and industry due to their superior merits such as wider viewing angle, lower energy consumption, light weight, excellent colour purity and mechanical flexibility. Within the past few years, OLEDs have been steadily well-developed and rapidly expanded in the mainstream display and solid-state lighting technology for consumer electronics. OLEDs are typically composed of multi-layered thin films sandwiched between two respective anode and cathode. Each organic layer is constructed for its own function, such as carrier injection layer (CIL), hole transporting layer (HTL), electron transporting layer (ETL), and emissive layer (EML), specifically designed for effective injection, the transport of the charge carriers and electron–hole balance in the emissive region.1–5,6 Therefore, OLED efficiency is predominantly determined by charge balance, the radiative decay of excitons and light outcoupling. Due to the relatively low intrinsic carrier concentrations of the transport layer, the current–voltage characteristic is injection-limited, and it requires balancing charge injection to improve the current performance of OLED devices. Useful and feasible HTL plays a critical role in improving electroluminescence efficiency since it directly relates the turn-on voltage of OLED, balancing hole transport and light-extraction efficiency.4,7–11In general, hole transport materials (HTMs) need to possess strong thermal stability with high glass transition temperature (Tg), high hole mobility, morphologically stable thin films and an appropriate HOMO level to ensure a low energy barrier for hole injection and a suitable LUMO level to block electron injection from the emissive layer. Traditionally, commonly used HTLs are N,N′-di(1-naphthyl)-N,N′-diphenylbenzidine (NPB), 4,4′-bis[N-(p-tolyl)-N-phenylamino]biphenyl (TPD), N,N′-di(biphenyl-4-yl)-N,N′-diphenyl-[1,1′-biphenyl]-4,4′-diamine (p-BPD) and 1-bis[4-[N,N-di(4-tolyl)amino]phenyl]-cyclohexane (TAPC), due to their high hole mobility, suitable HOMO/LUMO energy levels and ease of sublimation.12–16 It is quite apparent that triarylamine (TPA) and its derivatives with a nitrogen atom connected with aromatic rings are excellent building blocks because of their good electron donating properties.17–24 In addition, HTMs are expected to have good thermal stability and morphological durability, which are conducive to OLED fabrication and operation. Introducing a bulky and rigid moiety into a molecule is a common strategy to increase thermal stability.24,25 The utilization of a spirocyclic structure endows HTMs with an intrinsic nonplanar three-dimensional architecture, which results in reduced intermolecular interactions, restricted crystallization tendency and increased molecule rigidity along with higher Tg.26–30
Spiro[fluorene-9,9′-xanthene] (SFX) is a famous spiro-type platform for one-pot synthesis to design new organic semiconductors for optoelectronic devices.31–38 For example, Zhang et al. synthesized and characterized several dicarbazole-substituted SFX-based molecules as host materials for blue and green PHOLEDs.39 Two similar SFX derivatives (SFX-Cr1 and SFX-Cr2) with tetra-carbazole substitution were reported by Wang and co-workers. SFX-Cr2 possesses a respectable Tg of 130 °C and a high hole mobility (∼10−3 cm2 V−1 s−1). Green PHOLEDs with SFX-Cr2 as the HTL showed a high EQE of 16.3%.40 Several compounds with carbazole, diphenylamine, N,N′-di(4-methoxyphenyl)amino, and phenothiazine motifs have been demonstrated as compromising HTMs for high-efficiency PSCs by replacing the corresponding moiety in commercialized spiro-MeOTAD to the SFX core.41,42 Recently, the relationship between the thermal and redox properties of the HTMs and different modified positions of TPA groups as well as their further influence on PSC efficiency were also analyzed.43 Therefore, these reports have established solid progress in developing high-quality HTMs based on the SFX core for high-performance OLEDs.
However, poor thermal stability renders their low Tg, thus leading to the detriment of device efficiency and lifetime.44–47 It is essential to further develop new types of HTMs to improve the OLED performance. In this work, we have designed two new hole transporting materials, spiro[fluorene-9,9′-xanthene]-2,7-di-(N1,N3-di-1-naphthalenyl-N1,N3-diphenyl-1,3-benzenediamine) (DPNA-SFX) and spiro[fluorene-9,9′-xanthene]-2′,7′-di-(N1,N3-di-1-naphthalenyl-N1,N3-diphenyl-1,3-benzenediamine) (DOPNA-SFX), applying the same SFX core with bridge phenyl rings at different positions (fluorene or xanthene core), which are meta-substituted by two electron-donating arylamine groups, N-phenyl-1-naphthylamine (PNA). It was found that both HTMs show relatively high Tg beyond 150 °C, indicating dramatically enhanced thermal stability compared to NPB. The hole mobilities of DPNA-SFX and DOPNA-SFX are 7.6 × 10−5 cm2 V−1 s−1 and 6.8 × 10−5 cm2 V−1 s−1, respectively, both of which are superior to those of NPB. All the multilayer green and red PHOLEDs exhibit superb efficiency data with a low turn-on voltage when adopting new HTLs. The DPNA-based green OLED device represents the greatest green device with CE, PE and EQE peaks of 89.8 cd A−1, 94.2 lm W−1 and 24.7%. More significantly, the DPNA-based red device R1 displays outstanding performance with the CE, PE and EQE peaks of 41.1 cd A−1, 46.4 lm W−1 and 34.7%, which is comparable to the world record of red PHOLEDs. And the EQE of the device R1 at 1000 cd m−2 maintained 96% of its highest value, which shows an extremely low efficiency roll-off in pure red PHOLED devices.
Results and discussion
Synthesis and thermal properties
All the chemical structures and the synthetic processes of the target compounds are shown in Scheme 1. The main intermediates and the final products were synthesized using common methods published in the literature.40 All of the compounds were confirmed by 1H-NMR, 13C-NMR and high-resolution mass spectrometry (HR-MS).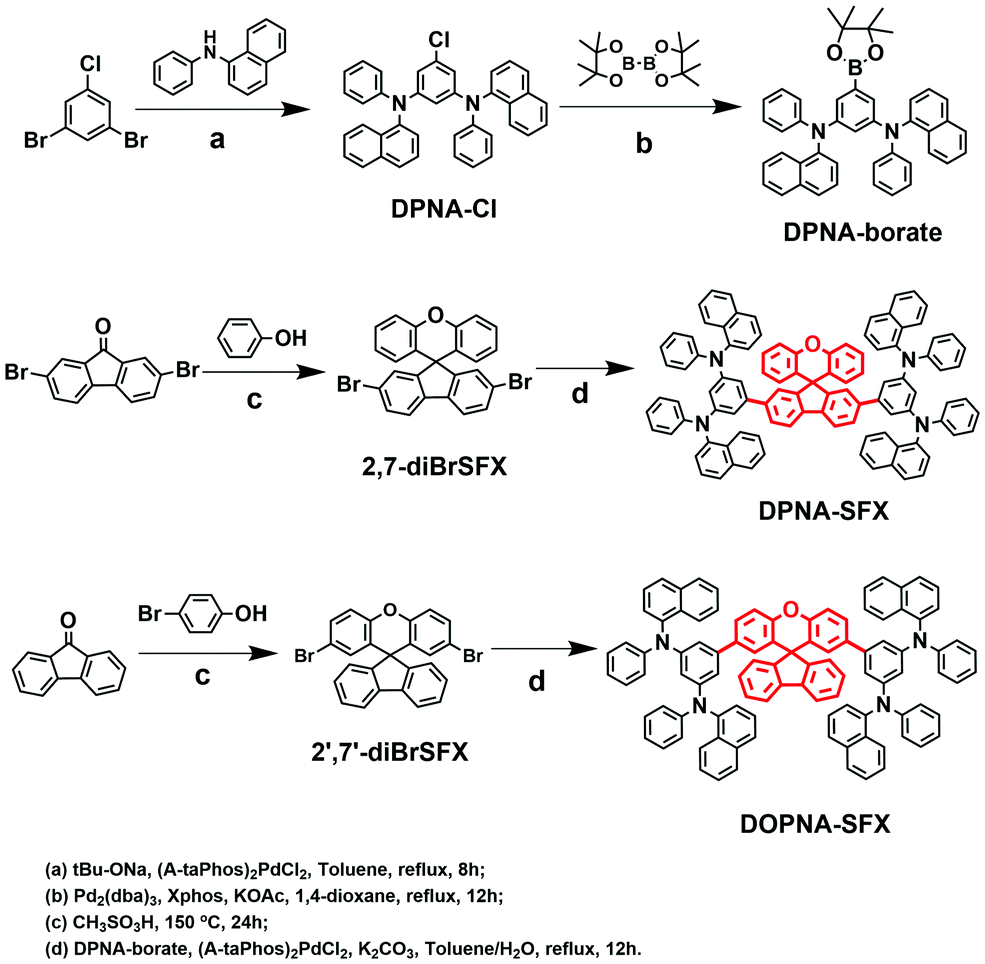 | ||
| Scheme 1 Synthetic routes of DPNA-SFX and DOPNA-SFX. | ||
To determine the thermal properties and morphological stabilities of the synthesized HTMs, differential scanning calorimetry (DSC) and thermogravimetric analysis (TgA) were measured under a nitrogen atmosphere, as depicted in Fig. S5 (see the ESI†). The decomposition temperature (Td) and glass transition temperature (Tg) values are summarized in Table 1. The Td values were found to be 543 °C and 535 °C for DPNA-SFX and DOPNA-SFX, respectively, indicating the excellent thermal stability of both compounds due to the rigidity of the spiro molecular structure. According to the DSC curves, a clear glass transition phenomenon can be observed with the Tg values of DPNA-SFX and DOPNA-SFX up to 151 °C and 153 °C, respectively, suggesting it is beneficial to form continuous thin films compared with the typical hole transporting material NPB with a Tg of 95 °C. Moreover, the PL spectra of thin films of three HTMs under different temperatures have been measured (see Fig. S6, ESI†). The PL spectra of DPNA-SFX and DOPNA-SFX remain consistent even under 300 °C; however, the PL intensity relative to that under room temperature of NPB has been largely decreased, which indicates the better thermal stability of our HTMs than NPB.
| Compound | T d /Tgb | λ abs | λ em,s /λem,fd | HOMO/LUMO (eV) | E e (eV) | E T (eV) | |
|---|---|---|---|---|---|---|---|
| Exp.e | Cal.f | ||||||
| a Decomposition temperature (5% weight loss). b Glass transition temperature. c Measured in degassed dichloromethane. d Measured in thin solid films on quartz. e HOMO was calculated by the formula: HOMO = E1/2,Fc − EOX − 4.8; LUMO was calculated by the sum of HOMO and optical band gap. f Calculated by the density functional theory level (B3LYP/6-311G**). g Estimated from the absorption edge in degassed dichloromethane. h Estimated from the phosphorescence spectra in degassed dichloromethane at 77 K. | |||||||
| DPNA-SFX | 543/151 | 308 | 447/429 | −5.23/−1.98 | −4.87/−1.37 | 3.25 | 2.45 |
| DOPNA-SFX | 535/153 | 297 | 435/420 | −5.23/−2.03 | −4.85/−1.45 | 3.20 | 2.44 |
Electrochemical properties and theoretical calculations
To investigate the electrochemical behaviour of DPNA-SFX and DOPNA-SFX, cyclic voltammetry (CV) measurements were conducted with Ag/AgCl as the reference electrode. The CV curves are displayed in Fig. S7 (see the ESI†) and the corresponding data are summarized in Table 1, indicating the same CV profile for DPNA-SFX and DOPNA-SFX with similar oxidation couples and no reduction process. Both DPNA-SFX and DOPNA-SFX show good stability of electrochemical oxidation. During the anodic scan in the DCM solution, two HTMs exhibit four quasi-reversible, four electron oxidation processes, which can be assigned to the oxidation of four 1-(N-phenylamino)naphthalene units. The HOMO energy levels are determined from the onset of the oxidation curve using the empirical formula, which is at the same value of −5.23 eV for both materials, deeper than the HOMO level of NPB (−5.12 eV), benefiting the promotion of hole injection. On the other hand, the LUMO energy level can be calculated using the HOMO energy level and band gap (Eg), which are thus −1.98 eV and −2.03 eV, respectively. To have a better understanding of the electronic structures of DPNA-SFX and DOPNA-SFX, density functional theory (DFT) calculations were conducted to simulate the frontier molecular orbital (FMO) levels and spatial distributions (Fig. 1(a)). The DFT results show that the HOMOs of both molecules are mainly localized at the two PNA moieties and the bridge phenyl ring, and the calculated HOMOs show no big difference which is in agreement with the experimental results. However, the LUMO of DPNA-SFX is distributed in the fluorene part of the SFX core and the bridge phenyl ring, while that of DOPNA-SFX is just located at the PNA moiety due to the breakdown of conjugation extension by the xanthene moiety.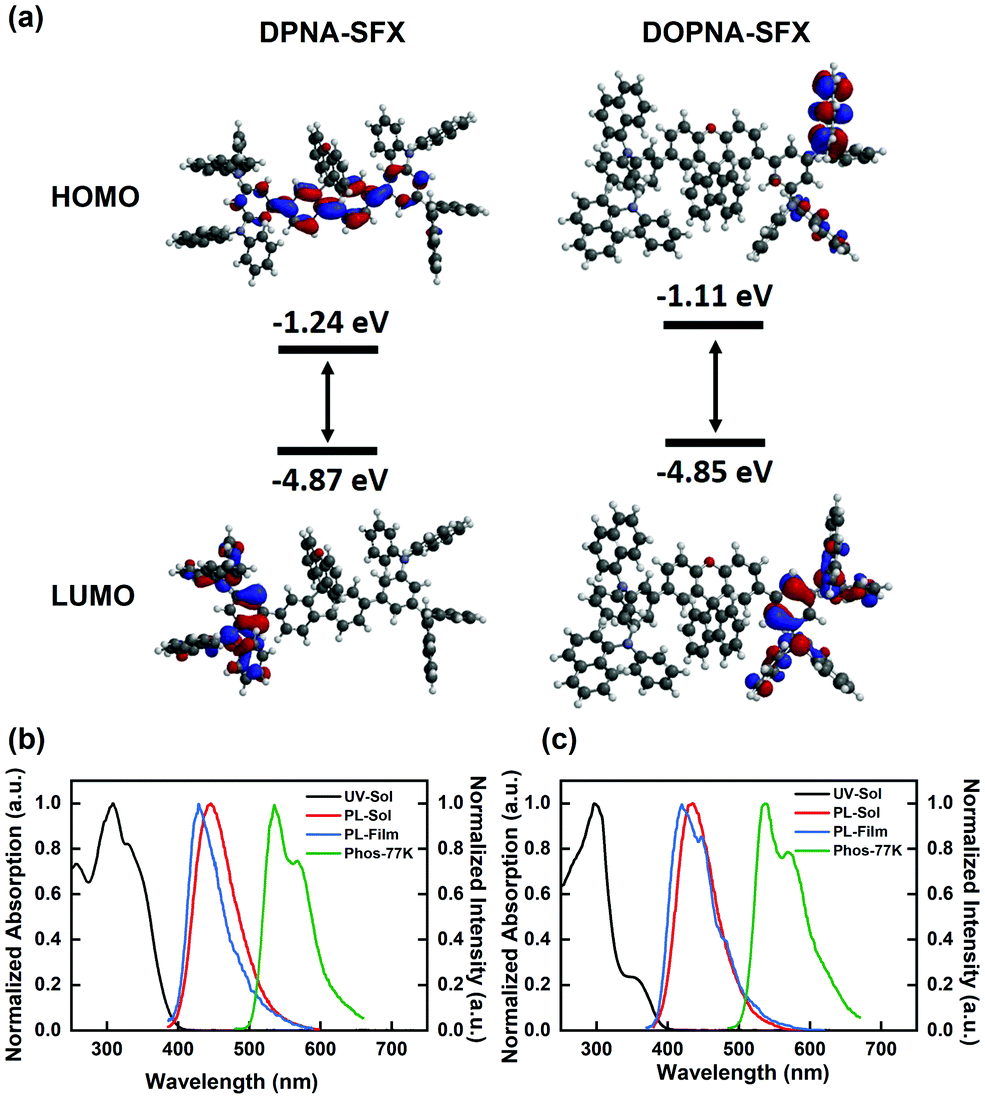 | ||
| Fig. 1 (a) HOMO and LUMO distributions of DPNA-SFX and DOPNA-SFX; (b and c) UV-Vis absorption (in DCM solution), PL spectra (in DCM solution and in film at room temperature, in DCM solution at 77 K) of (b) DPNA-SFX; and (c) DOPNA-SFX. | ||
Photophysical properties
To investigate the photophysical behaviours of the new HTMs, UV-Vis absorption spectra and photoluminescence (PL) spectra in dilute dichloromethane (DCM) solution and thin film, as well as phosphorescence spectra in DCM at 77 K were recorded respectively, and are shown in Fig. 1(b) and (c), and the summarized data are presented in Table 1. For both DPNA-SFX and DOPNA-SFX, the strongest absorption bands around 300 nm are attributed to localized transitions associated with the PNA core. In the UV-Vis spectra of DPNA-SFX, a shoulder absorption peak was observed at 320 nm due to the π–π* transition from the PNA moiety to the fluorene moiety. The longer wavelength band at 350 nm with weaker absorption of DOPNA-SFX could be assigned to the π–π* transition from the PNA moiety to the xanthene moiety, revealing the formation of the intramolecular charge transfer (ICT) state made by some degree of weaker spiro-conjugation effect. In addition, the band gaps (Eg) are estimated from the absorption edge, which are 3.25 eV and 3.20 eV for DPNA-SFX and DOPNA-SFX, respectively. Both compounds show blue emission in dichloromethane (DCM) and their maximum emission peaks were at 447 nm and 435 nm, respectively. The maximum wavelength of DPNA-SFX exhibits a 14 nm red-shift relative to DOPNA-SFX, which could be due to the extended conjugation of DPNA-SFX. The fluorescence spectra of two HTMs reveal a solvatochromic effect (see Fig. S8, ESI†), which indicates that DPNA-SFX and DOPNA-SFX are slightly sensitive to solvent polarity. To measure the triplet energy of the two HTMs, low temperature phosphorescence spectra (77 K, in DCM solution) were collected under a nitrogen atmosphere (shown in Fig. 1(b) and (c)), corresponding to T1 of 2.45 eV and 2.44 eV (Table 1), respectively, which are both higher than NPB (2.33 eV). Higher T1 guarantees better exciton confinement in terms of OLED device structure.Single carrier devices
Single carrier devices of DPNA-SFX and DOPNA-SFX were fabricated to study the corresponding hole-transporting properties, according to the architecture of ITO/HAT-CN (10 nm)/DPNA-SFX or DOPNA-SFX (100 nm)/HAT-CN (10 nm)/Al (100 nm), in which HAT-CN was used as the hole injection layer and electron blocking layer (inserted picture in Fig. 2(a)). No electroluminescence phenomenon was detected during measurement implying these are singlet-only devices. The J–V curves of the hole-only devices in Fig. 2(a) reveal that the current densities at the same voltage follow the sequence: DPNA-SFX > DOPNA-SFX > NPB. The difference between the curves could further indicate that, according to the HOMO energy level of HAT-CN and the above HTMs, a better charge injection from HAT-CN could be achieved in DPNA-SFX and DOPNA-SFX based devices than in the NPB-based one. | ||
| Fig. 2 (a) J–V curves of the hole-only devices and (b) comparative field dependence mobility of DPNA-SFX and DOPNA-SFX. | ||
Space charge limited current (SCLC) theory can be used to evaluate the carrier mobility of semiconductors when quasi-Ohmic contacts between the metal electrode and organic layer are well established, which gives the formula: J = 9ε0εrμE2/8L. Considering the electric-field-dependent charge mobility of organic semiconductors according to Poole–Frenkel mechanics (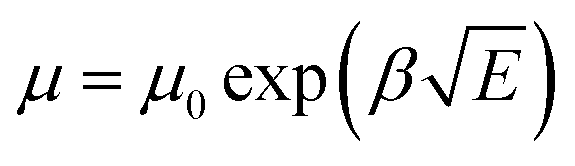 ), the expression can be written as
), the expression can be written as
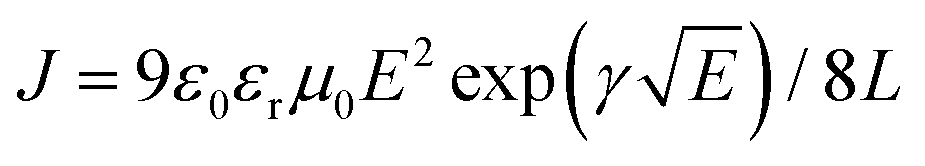
The value of μ0 and γ was determined from the J–V curves by the linearization of the equation above, which is 7.6 × 10−5 cm2 V−1 s−1, double helical C/Co@CNT nanocomposite with hierarchical structures for enhancing microwave absorption for DPNA-SFX, 6.3 × 10−5 cm2 V−1 s−1, 1.48 × 10−2 V−1/2 cm1/2 for DOPNA-SFX and 2.8 × 10−5 cm2 V−1 s−1, 1.38 × 10−2 V−1/2 cm1/2 for NPB. It can be clearly observed that the zero-field mobilities show the following sequence: DPNA-SFX > DOPNA-SFX > NPB, which indicates the better hole-transporting properties of the synthesized HTMs and the potential replacement for commercialized NPB. The hole mobility dependent on the electric field ascribed to the materials can be calculated with the two parameters, with the results depicted in Fig. 2(b). It is obviously shown that the hole mobilities increased rapidly at a high bias voltage. At the high electric field of 1.9 × 105 V cm−1, the hole mobilities of DPNA-SFX and DOPNA-SFX reach 2.75 × 10−3 cm2 V−1 s−1 and 2.1 × 10−3 cm2 V−1 s−1, respectively. High hole-transporting mobility with good hole injection ability renders DPNA-SFX and DOPNA-SFX as potential HTM candidates for application in OLEDs.
PHOLED performance
To evaluate the practical application for phosphorescent organic light-emitting diodes (PHOLEDs), green and red devices using the new HTMs were fabricated. Control devices were fabricated with the commonly-used HTL of N,N′-bis(naphthalen1-yl)-N,N′-bis(phenyl)-benzidine (NPB). The optimized PHOLED device structure is ITO/HAT-CN (5 nm)/HTM (30 nm)/Spiro-3-BFP (10 nm)/DMIC-TRZ: DMIC-CZ: emitter (40 nm)/ANT-BIZ (70 nm)/Liq (2.5 nm)/Al (100 nm) (the HTM is DPNA-SFX for device G1, R1; DOPNA-SFX for device G2, R2), with Ir(ppy)2(acac) ([bis(2-phenylpyridine)(acetylacetonato) iridium(III)]) and Ir(dmpibq)2(acac) (bis[2-(3,5-dimethylphenyl)-5-isobutylquinoline-C2,N′](acetylacetonato)iridium(III)) as the dopant for green and red emitting layers, respectively. In these devices, HAT-CN (dipyrazino[2,3-f: 2′,3′-h]quinoxaline-2,3,6,7,10,11-hexacarbonitrile) and Liq (8-quinolinolato lithium) were used as the hole-injection and electron-injection materials, respectively. Spiro-3-BFP (N-([1,1′-biphenyl]-2-yl)-N-(9-dimethyl-9H-fluoren-2-yl)-9,9′-spirobi[9H-fluoren]-4-amine) was used as the electron-blocking material to balance the electrons and holes for green and red devices. In the emitting layer, DMIC-TRZ (5-(3-(4,6-diphenyl-1,3,5-triazin-2-yl)phenyl)-7,7-dimethyl-5,7-dihydroindeno[2,1-b]carbazole) and DMIC-CZ (7,7-dimethyl-5-phenyl-2-(9-phenyl-9H-carzole-3-yl)-5,7-dihydroindeno[2,1-b]carbazole) served as the co-host system with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for both green and red PHOLEDs, and the doping concentrations of Ir(ppy)2(acac) and Ir(dmpibq)2(acac) were 5 wt% and 2.5 wt%, respectively. ANT-BIZ (1-(4-(10-([1,1′-biphenyl]-4-yl)anthracen-9-yl)phenyl)-2-ethyl-1H-benzo[d]-imidazole) was employed as the electron-transporting material. The device configuration and energy level diagram and the corresponding molecule structures are shown in Fig. 3(a) and (b) and Fig. S9 (ESI†). The device performance of the green and red PHOLED devices, including current density–voltage–luminance (J–V–L) curves, current efficiency (CE), power efficiency (PE) and external quantum efficiency (EQE) versus luminance (L) curves, as well as the EL spectra are shown in Fig. 3. All the key device parameters are listed in Table 2.
:1 for both green and red PHOLEDs, and the doping concentrations of Ir(ppy)2(acac) and Ir(dmpibq)2(acac) were 5 wt% and 2.5 wt%, respectively. ANT-BIZ (1-(4-(10-([1,1′-biphenyl]-4-yl)anthracen-9-yl)phenyl)-2-ethyl-1H-benzo[d]-imidazole) was employed as the electron-transporting material. The device configuration and energy level diagram and the corresponding molecule structures are shown in Fig. 3(a) and (b) and Fig. S9 (ESI†). The device performance of the green and red PHOLED devices, including current density–voltage–luminance (J–V–L) curves, current efficiency (CE), power efficiency (PE) and external quantum efficiency (EQE) versus luminance (L) curves, as well as the EL spectra are shown in Fig. 3. All the key device parameters are listed in Table 2.
 | ||
| Fig. 3 (a) Device architecture of PHOLEDs and (b) energy diagram of multilayer PHOLEDs. Device performances of green PHOLEDs: (c) EL spectra (4.0 V), (e) J–V–L curves, (f) CE–L–PE curves, and (g) EQE–L curves. | ||
| Devices | V on (V) | CEb (cd A−1) | PEc (lm W−1) | EQEd (%) | CIEe |
|---|---|---|---|---|---|
| Max/103/104 (cd m−2) | Max/103/104 (cd m−2) | Max/103/104 (cd m−2) | |||
|
a Turn-on voltage at 1 cd m−2.
b Current efficiency at maximum values/1000 cd m−2/10000 cd m−2.
c Power efficiency at maximum values/1000 cd m−2/10000 cd m−2.
d External quantum efficiency at maximum values/1000 cd m−2/10000 cd m−2.
e CIE coordinates at 1000 cd m−2.
|
|||||
| G1 | 2.4 | 89.8/88/72.2 | 94.2/90/50.4 | 24.7/24.2/19.9 | (0.32, 0.62) |
| G2 | 2.4 | 85.6/81.4/70.5 | 93.2/76.5/54.5 | 23.6/22.4/21.4 | (0.33, 0.62) |
| R1 | 2.2 | 41.1/39.5/31.6 | 46.4/29.6/15.5 | 34.7/33.3/13.1 | (0.66, 0.34) |
| R2 | 2.2 | 31.4/26.5/25.4 | 35.6/18.5/13.8 | 25.1/21.2/20.3 | (0.66, 0.34) |
As depicted in Fig. 3(c), both devices G1 and G2 emit green light with roughly identical emission peaks at 521 nm and 524 nm, respectively, which was ascribed to Ir(ppy)2(acac) by efficient energy transfer from co-hosts to the guest. The EL spectra remain unchanged over a wide range of operating voltage from 3 V to 7 V (see Fig. S10, ESI†). The corresponding CIE coordinates are (0.32, 0.62) and (0.33, 0.62), respectively. The two devices exhibited a comparatively low turn-on voltage of 2.4 V simultaneously (determined from Fig. 3(d)). It is apparently demonstrated that the device G1 has better device performance, with the maximum CE, PE and EQE of 89.8 cd A−1, 94.2 lm W−1 and 24.7%, respectively, compared with the corresponding data of device G2 (85.6 cd A−1, 93.2 lm W−1 and 23.6%). This is chiefly attributed to the higher hole mobility of DPNA-SFX and more harmonious charge recombination inside device G1. It is noteworthy that the green devices based on DPNA-SFX and DOPNA-SFX are both far better than the NPB-based PHOLEDs (68.5 cd A−1, 79.7 lm W−1 and 18.7%) shown in Fig. S12 and Table S1 (ESI†), which is presumably due to the imbalance of hole/electron injection and transport. The efficiency roll-off values of green PHOLEDs based on DPNA-SFX, DOPNA-SFX and NPB are 2%, 5% and 1%. As we can see, both green devices show a relatively low efficiency roll-off. Sure, the roll-off of the NPB-based device is the lowest one, and the EQE is far behind that of the others.
The device performance of the red PHOLED devices based on DPNA-SFX and DOPNA-SFX was also measured (see Fig. 4). As shown in Fig. 4(a), the devices R1 and R2 present saturated red emissions with the same maximum peak of 620 nm, with the CIE coordinates of (0.66, 0.34). The EL spectra remain unchanged over a wide range of operating voltage from 3 V to 7 V (see Fig. S11, ESI†). The J–V–L curve of the red devices indicates that the turn-on voltage of R1 and R2 is as low as 2.2 V. The DPNA-SFX based device possesses a superb device performance, with a CE of 41.1 cd A−1, a PE of 46.4 lm W−1, and an EQE of 34.7%, which completely outperforms that of the other one. The EQE of the DPNA-based red device is almost 40% higher than that of the NPB-based one. To the best of our knowledge, this excellent device performance is comparable with the best records of pure red PHOLEDs48–57 (some representative EQE values of red PHOLEDs are shown in Fig. 5). The efficiency roll-off values of red PHOLEDs based on DPNA-SFX, DOPNA-SFX and NPB are 4%, 16% and 30%, respectively. To note, the device R1 also shows better efficiency roll-off compared with that of R2. It retains an EQE of 33.3% even at 1000 cd m−2, which is also significantly superior compared with those of R2 and the NPB-based device. Therefore, the DPNA-SFX based red device possesses an extremely high EQE and low roll-off compared with DOPNA-SFX and NPB. It is worth mentioning that we have also tested the devices using an integrating sphere in order to exclude the possibility of anisotropic emission of the green and red emitters. Almost similar results were exhibited (shown in Fig. S17 and S18, ESI†), which indicates the impossibility of anisotropic emission of green and red emitters which might cause an overestimated performance.
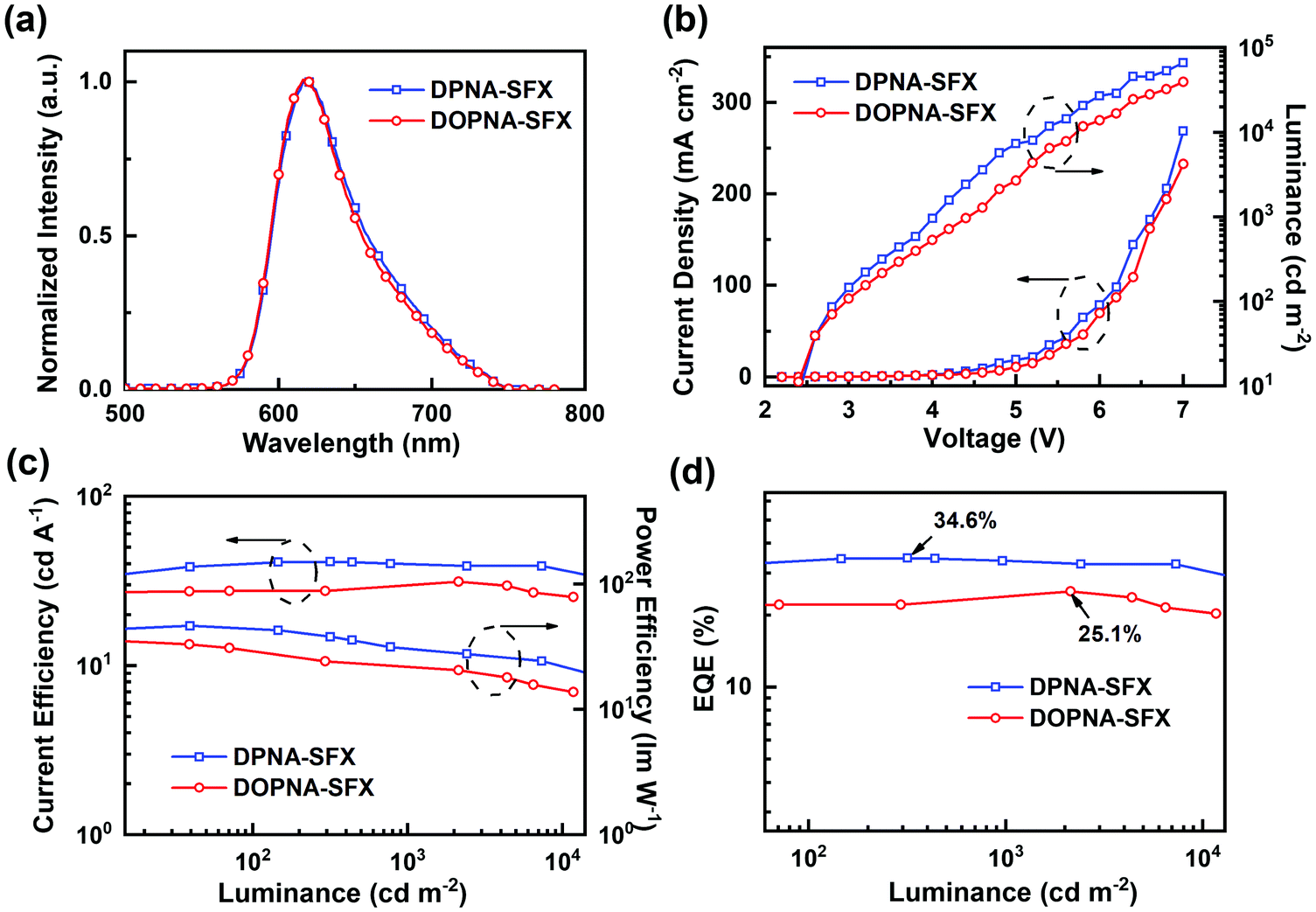 | ||
| Fig. 4 Device performance of red PHOLEDs: (a) EL spectra (4.0 V), (b) J–V–L curves, (c) CE–L–PE curves, and (d) EQE–L curves. | ||
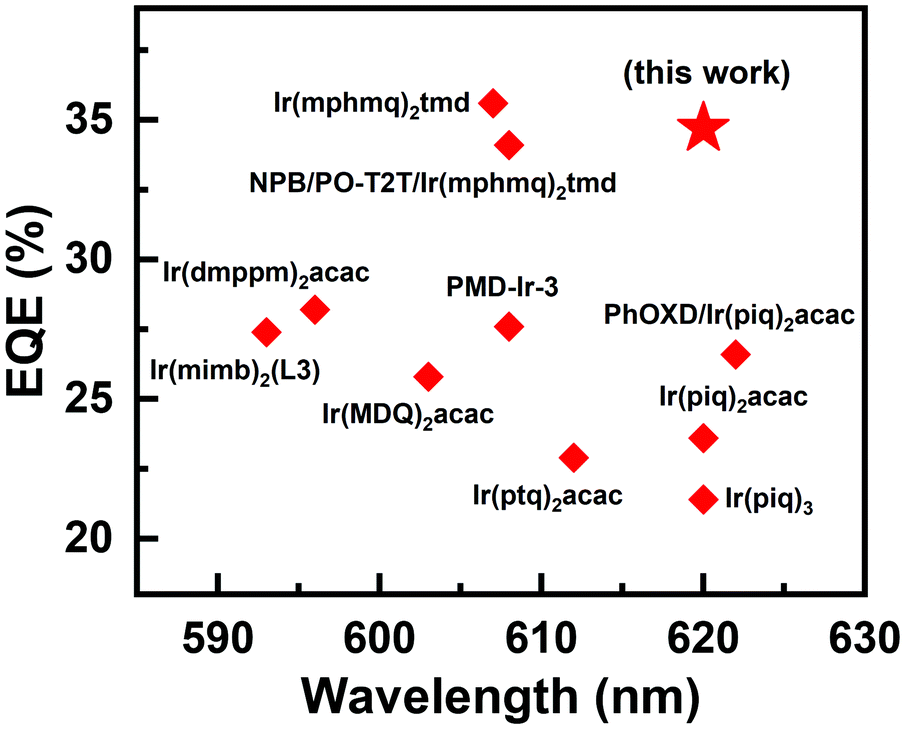 | ||
| Fig. 5 EQE versus EL peaks of red PHOLEDs based on several representative iridium complexes. | ||
In order to explore the possibility of high performance blue PHOLEDs, we fabricate blue devices based on the architecture of ITO/HAT-CN (5 nm)/HTM (40 nm)/TCTA (10 nm)/TCTA: TmPyPB:FIrpic (20 nm)/TmPyPB (30 nm)/Liq (2.5 nm)/Al (100 nm). The devices performance is shown in Fig. S13 (ESI†) and all the key parameters are listed in Table S2 (ESI†). Three kinds of devices possess a low turn-on voltage of 2.8 V. The DPNA-SFX based device exhibits the best CE, PE and EQE of 17.93 cd A−1, 14.87 lm W−1 and 8.70%, respectively. Comparing with the NPB-based device, the performance of the DPNA-SFX and DOPNA-SFX based devices is much better, demonstrating their potential to replace commercialized HTMs.
Besides high efficiency, a long lifetime of PHOLEDs is also essential and is affected by the charge balance. We have confirmed the better thermal stability of our HTMs compared with NPB, which should contribute to the operational stability of the corresponding PHOLEDs as well. The lifetime of red and green PHOLEDs with the best EQE was evaluated. The lifetime curves shown in Fig. 6 of the green and red PHOLEDs were measured at a high initial luminance of 50000 cd m−2 and 10000 cd m−2, respectively. The DPNA-SFX and DOPNA-SFX based red and green devices showed a longer lifetime than NPB, especially the DPNA-SFX based ones, indicating that the crucial role of the thermal stability of HTMs and much better charge balance render a longer lifetime for PHOLEDs.
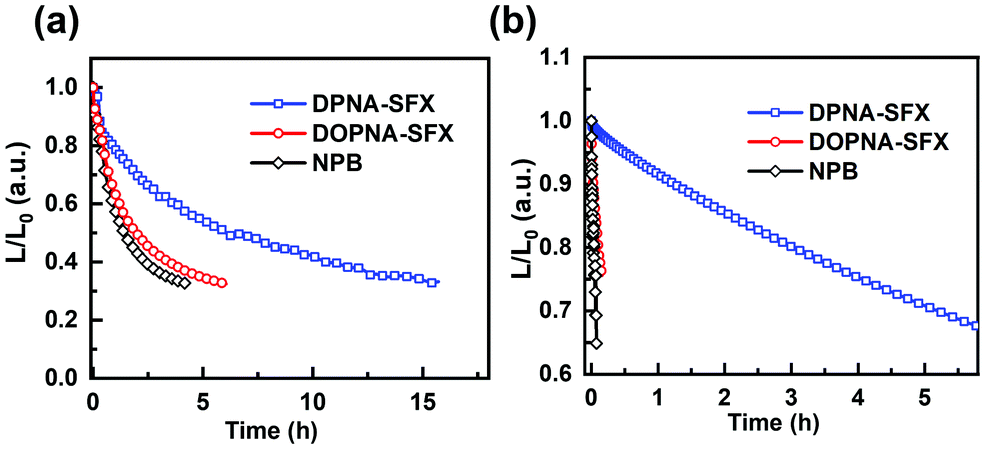 | ||
| Fig. 6 Normalized luminance as a function of operating time for (a) red and (b) green PHOLEDs with different HTMs. | ||
To reveal the reasons for superb PHOLED performance, electron-only devices based on ANT-BIZ have also been fabricated ITO/Liq (10 nm)/ANT-BIZ (100 nm)/Liq (10 nm)/Al (100 nm) (see Fig. S14, ESI†). The calculated electron mobility under zero electric field is 2.4 × 10−4 cm2 V−1 s−1, which is higher than the hole mobilities of the applied HTMs. Considering the sequences of hole-transporting properties of DPNA-SFX, DOPNA-SFX and NPB, the DPNA-based PHOLEDs exhibiting the best device performance could be attributed to more efficient charge injection and better charge transport balance, which guarantee good exciton confinement. It is also in accordance with the optimization results of the thickness of the ETL layers (Table S3, ESI†). In addition, we also fabricate two single carrier devices based on our PHOLED architecture without the emitting layers. The structures of the hole-only and electron-only devices are ITO/HAT-CN (5 nm)/HTM (30 nm)/Spiro-3-BFP (10 nm)/Al and ITO/ANT-BIZ (70 nm)/Liq (2.5 nm)/Al (100 nm) (see Fig. S15, ESI†). It is apparent that the mobility of three HTMs show the same sequence: DPNA-SFX > DOPNA-SFX > NPB. Similarly, the electron transporting properties of ANT-BIZ are better than those of HTMs, thus DPNA-SFX possesses the best performance because of better charge balance.
According to the device performance of green and red PHOLEDs, respectable high efficiency, low turn-on voltage and minor efficiency roll-off behavior can be achieved by the strategy of utilizing DPNA-SFX and DOPNA-SFX as HTMs.
Conclusions
In summary, two new materials, spiro[fluorene-9,9′-xanthene] (SFX) derivatives, spiro[fluorene-9,9′-xanthene]-2,7-di-(N1,N3-di-1-naphthalenyl-N1,N3-diphenyl-1,3-benzenediamine) (DPNA-SFX) and spiro[fluorene-9,9′-xanthene]-2′,7′-di-(N1,N3-di-1-naphthalenyl-N1,N3-diphenyl-1,3-benzenediamine) were designed, synthesized, and characterized completely. The green PHOLEDs using DPNA-SFX and DOPNA-SFX as the hole-transporting material exhibited maximum EQE values of 24.7% and 23.6%, respectively. Moreover, the red PHOLEDs with DPNA-SFX achieved the highest EQE of 34.7% and the EQE at 1000 cd m−2 maintained 96% of its peak value. To the best of our knowledge, these performances are among the highest efficiencies for pure red PHOLEDs. This research work suggests that the SFX-core based derivatives show great potential for application as good hole-transporting materials in PHOLED displays with high EQE and low turn-on voltage as well as low efficiency roll-off.Experimental section
General information
1H and 13C NMR spectra were acquired using a Bruker AVANCE 500 MHz NMR spectrometer. UV–Vis spectra were obtained using a PerkinElmer Lambda 750 spectrophotometer. Fluorescence and phosphorescence spectra at room temperature and 77 K were recorded using a Horiba Fluorolog-3 in chromatographically pure DCM. Density functional theory (DFT) calculations were performed using the 6-31g(d,p) basis set with Gaussian 09 package. Thermo-gravimetric analysis (TGA) and differential scanning calorimetry (DSC) were carried out using a METTLER TOLEDO TGA/DSC1 system with the samples under pure nitrogen at a heating rate of 10 °C min−1. A typical three-electrode configuration was applied in cyclic voltammetry (CV) measurements, with Ag/AgNO3 (0.1 M, CH3CN) as the reference electrode, glass carbon as the working electrode and platinum wire as the counter electrode.Synthetic details
:1). Yield 53%. 1H NMR (400 MHz, CDCl3) δ 7.87 (s, 1H), 7.85 (s, 2H), 7.83 (s, 1H), 7.72 (s, 1H), 7.70 (s, 1H), 7.50–7.45 (m, 2H), 7.40 (s, 1H), 7.38 (d, J = 0.7 Hz, 2H), 7.36 (s, 1H), 7.24 (d, J = 0.9 Hz, 1H), 7.22 (d, J = 1.0 Hz, 1H), 7.06 (d, J = 1.9 Hz, 1H), 7.05 (d, J = 1.2 Hz, 2H), 7.03 (s, 1H), 6.90 (s, 2H), 6.88 (s, 2H), 6.87 (s, 1H), 6.85 (d, J = 1.0 Hz, 1H), 6.44 (t, J = 2.0 Hz, 1H), 6.41 (d, J = 2.0 Hz, 2H).
MALDI-TOF-MS: m/z [M + H]+ calcd, 546.19; found, 547.51. Anal. Calcd for C38H27ClN2: C, 83.43; H, 4.97; Cl, 6.48; N, 5.12. Found: C, 83.41; H, 4.97; Cl, 6.47; N, 5.13.
N 1,N3-di-1-naphthalenyl-N1,N3-diphenyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-benzenediamine (DPNA-borate).
DPNA-Cl (9.84 g, 18 mmol) and bis(pinacolato)diboron (5.48 g, 21.6 mmol) were added to dry 1,4-dioxane (100 mL). Potassium acetate (5.30 g, 54 mmol), tris(dibenzylideneacetone)dipalladium (0.99 g, 1.08 mmol) and 2-dicyclohexylphosphino-2′,4′,6′-triisoprophylbiphenyl (1.03 g, 2.16 mmol) were added to the container quickly under a nitrogen atmosphere. The mixture was heated to reflux temperature and stirred overnight. After the reaction, the mixture was filtrated, washed with dichloromethane (100 mL), followed by evaporation. The residue was subjected to silica gel column chromatography (petroleum ether/dichloromethane = 4:1) to obtain 10.33 g of yellow solid. Yield 90%. 1H NMR (300 MHz, CDCl3) δ 7.84 (d, J = 8.6 Hz, 4H), 7.66 (d, J = 8.2 Hz, 2H), 7.47–7.40 (m, 2H), 7.39–7.27 (m, 4H), 7.22 (d, J = 0.9 Hz, 1H), 7.20 (d, J = 0.9 Hz, 1H), 7.11 (d, J = 2.2 Hz, 2H), 7.05–6.97 (m, 4H), 6.84–6.74 (m, 7H), 1.21 (s, 12H).
MALDI-TOF-MS: m/z [M + H]+ calcd, 638.31; found, 639.47. Anal. Calcd for C44H39BN2O2: C, 82.75; H, 6.16; B, 1.69; N, 4.39; O, 5.01. Found: C, 82.71; H, 6.18; B, 1.68; N, 4.38; O, 4.99.
2,7-dibromospiro[fluorene-9,9′-xanthene] (2,7-diBrSFX)
A 100 mL round flask was charged with 2,7-dibromofluorenone (14.58 g, 43.14 mmol), phenol (40.6 g, 431.4 mmol) and methane sulfonic acid (16.93 g, 172.6 mmol). The mixture was heated at 150 °C under a nitrogen atmosphere to carry out the reaction for 24 h. Then, the reaction mixture in the flask was slowly poured into water (50 mL) and extracted three times with 200 mL of dichloromethane. After this, the extracts were collected, dried over MgSO4 and evaporated; the residue was subjected to silica gel column chromatography (petroleum ether/dichloromethane = 4:1) to obtain 15.01 g of white solid. Yield 71%. 1H NMR (500 MHz, CDCl3) δ 7.63 (d, J = 8.1 Hz, 2H), 7.50 (dd, J = 8.1, 1.8 Hz, 2H), 7.27 (s, 2H), 7.24 (dd, J = 4.6, 1.0 Hz, 4H), 6.85–6.80 (m, 2H), 6.40–6.36 (m, 2H).
MALDI-TOF-MS: m/z [M + H]+ calcd, 487.94; found, 488.65. Anal. Calcd for C25H14Br2O: C, 61.24; H, 2.88; Br, 32.60; O, 3.26. Found: C, 61.19; H, 2.88; Br, 32.62; O, 3.27.
2′,7′-dibromospiro[fluorene-9,9′-xanthene] (2′,7′-diBrSFX)
A 100 mL round flask was charged with 9H-fluoren-9-one (2.85 g, 15.8 mmol), 4-bromophenol (16.4 g, 94.8 mmol) and methane sulfonic acid (6.2 g, 63.2 mmol). The mixture was heated at 150 °C under a nitrogen atmosphere to carry out the reaction for 24 h. Then, the reaction mixture in the flask was slowly poured into water (50 mL) and extracted three times with dichloromethane (200 mL). After this, the extracts were collected, dried over MgSO4 and evaporated; the residue was subjected to silica gel column chromatography (petroleum ether/dichloromethane = 4:1) to obtain 4.88 g of white solid. Yield 63%. 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 7.6 Hz, 2H), 7.42 (td, J = 7.5, 1.0 Hz, 2H), 7.30 (d, J = 2.4 Hz, 1H), 7.27 (t, J = 2.0 Hz, 2H), 7.25 (d, J = 1.0 Hz, 1H), 7.12 (dd, J = 15.1, 8.2 Hz, 4H), 6.47 (d, J = 2.4 Hz, 2H).
MALDI-TOF-MS: m/z [M + H]+ calcd, 487.94; found, 488.63. Anal. Calcd for C25H14Br2O: C, 61.24; H, 2.88; Br, 32.60; O, 3.26. Found: C, 61.21; H, 2.86; Br, 32.63; O, 3.28.
Spiro[fluorene-9,9′-xanthene]-2,7-di-(N1,N3-di-1-naphthalenyl-N1,N3-diphenyl-1,3-benzenediamine) (DPNA-SFX).
DPNA-borate (3.82 g, 6 mmol) and 2,7-diBrSFX (1.47 g, 3 mmol) were added into a 250 mL round flask and dissolved in dry toluene (80 mL) by magnetic stirring. Potassium carbonate (1.24 g, 9 mmol) was dissolved in 20 mL of water and poured into the round flask. Then, dichlorobis[di-tert-butyl(4-dimethylaminophenyl)phosphino]palladium(II) (0.064 g, 0.09 mmol) was added into the container quickly. The mixture was degassed under a nitrogen atmosphere for 20 min and heated to 110 °C. After overnight reaction, the mixture was filtered, poured into water and extracted with dichloromethane (200 mL). 2.78 g of white solid was obtained by silica gel column chromatography (petroleum ether/dichloromethane = 4:1). Yield 69%. 1H NMR (400 MHz, CDCl3) δ 7.83 (d, J = 8.6 Hz, 8H), 7.66 (d, J = 8.2 Hz, 4H), 7.54 (d, J = 7.9 Hz, 2H), 7.42 (ddd, J = 8.2, 6.9, 1.0 Hz, 4H), 7.36–7.27 (m, 8H), 7.17 (dddd, J = 14.9, 9.7, 7.8, 1.3 Hz, 10H), 7.05–6.98 (m, 8H), 6.96 (d, J = 1.3 Hz, 2H), 6.90–6.77 (m, 12H), 6.65–6.60 (m, 2H), 6.58 (dd, J = 3.6, 1.5 Hz, 6H), 6.25 (dd, J = 7.9, 1.2 Hz, 2H). 13C NMR (400 MHz, CDCl3) δ 155.23, 151.56, 149.16, 148.05, 143.25, 142.61, 141.34, 138.51, 135.27, 131.12, 128.95, 128.45, 128.02, 127.61, 127.04, 126.94, 126.48, 126.36, 126.25, 126.08, 125.03, 124.40, 124.26, 123.29, 121.78, 121.62, 119.96, 116.79, 114.17, 113.94, 54.46. MALDI-TOF-MS: m/z [M + H]+ calcd, 1352.54; found, 1353.55. Anal. Calcd for C101H64N4O: C, 89.68; H, 5.06; N, 4.14; O, 1.18. Found: C, 89.31; H, 5.05; N, 4.14; O, 1.17.
Spiro[fluorene-9,9′-xanthene]-2′,7′-di-(N1,N3-di-1-naphthalenyl-N1,N3-diphenyl-1,3-benzenediamine) (DOPNA-SFX)
DPNA-borate (4.7 g, 7 mmol) and 2,7-diBrSFX (1.72 g, 3.5 mmol) were added into a 250 mL round flask and dissolved in 80 mL of dry toluene by magnetic stirring. Potassium carbonate (1.45 g, 10.5 mmol) was dissolved in water (20 mL) and poured into the round flask. Then, dichlorobis[di-tert-butyl(4-dimethylaminophenyl)phosphino]palladium(II) (0.074 g, 0.11 mmol) was added into the container quickly. The mixture was degassed under a nitrogen atmosphere for 20 min and heated to 110 °C. After overnight reaction, the mixture was filtered, poured into water and extracted with dichloromethane (200 mL). 3.55 g of white solid was obtained by silica gel column chromatography (petroleum ether/dichloromethane = 4:1). Yield 75%. 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 8.2 Hz, 4H), 7.78 (d, J = 8.4 Hz, 4H), 7.66 (dd, J = 16.1, 7.9 Hz, 6H), 7.44 (dd, J = 11.1, 4.0 Hz, 4H), 7.31 (dd, J = 15.7, 7.8 Hz, 8H), 7.27 (d, J = 2.1 Hz, 1H), 7.25 (d, J = 1.8 Hz, 1H), 7.16 (d, J = 6.6 Hz, 4H), 7.07 (d, J = 1.8 Hz, 1H), 7.05–7.03 (m, 3H), 7.00–6.95 (m, 8H), 6.90 (t, J = 5.8 Hz, 4H), 6.80 (dd, J = 14.4, 7.4 Hz, 12H), 6.52 (t, J = 1.9 Hz, 2H), 6.34 (d, J = 2.0 Hz, 4H), 6.20 (d, J = 1.8 Hz, 2H). 13C NMR (400 MHz, CDCl3) δ 154.07, 151.17, 149.19, 148.03, 143.23, 141.76, 139.52, 136.11, 135.32, 131.25, 128.95, 128.44, 128.25, 127.92, 127.11, 126.73, 126.50, 126.36, 126.25, 126.07, 125.98, 125.39, 124.39, 121.78, 121.59, 119.82, 116.83, 113.65, 113.32, 54.49. MALDI-TOF-MS: m/z [M + H]+ calcd, 1352.54; found, 1353.53. Anal. Calcd for C101H64N4O: C, 89.62; H, 5.06; N, 4.14; O, 1.18. Found: C, 89.24; H, 5.03; N, 4.16; O, 1.16.
Device fabrication and measurements
All the materials except DPNA-SFX and DOPNA-SFX for multilayer PHOLED fabrication and ITO-coated glass substrates were purchased from commercial sources. The ITO-coated glass substrates were washed with acetone, deionized water and isopropanol in order and put into a UV-ozone environment for 20 min. The deposition process of all organic layers and metal electrode was performed under vacuum (∼8 × 10−7 Pa). The characterization of PHOLED performance including current density–voltage–luminance (J–V–L) curves, electroluminescence (EL) spectra and Commission Internationale de ĺEclairage (CIE) coordinates were carried out using a Keithley 2400 semiconductor characterization system with BM-7A luminance colorimeter and PR-788 photometer. Further measurements were also conducted using equipment with calibrated integrating spheres and with the device's substrate edges masked. The external quantum efficiency of all the fabricated devices were calculated according to electroluminescence performance and EL spectra.All the measurements except device lifetime measurements were conducted in ambient environment without further protection at room temperature. Device stability of PHOLEDs was evaluated by collecting time dependent luminance data at a constant current density and constant initial luminance. The lifetime measurements were carried out in a glovebox with encapsulation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Key-Area Research and Development Program of Guangdong Province (2019B010924003), Guangdong Basic and Applied Basic Research Foundation (2020B1515120030), Shenzhen Peacock Plan (KQTD2014062714543296), Shenzhen Engineering Research Center (Shenzhen development and reform commission [2018]1410), and Shenzhen Science and Technology Research Grant (JCYJ20180302153451987).References
- J. Kido, M. Kimura and K. Nagai, Science, 1995, 267, 1332 CrossRef CAS.
- S. Chen, L. Deng, J. Xie, L. Peng, L. Xie, Q. Fan and W. Huang, Adv. Mater., 2010, 22, 5227–5239 CrossRef CAS.
- Z. Wu and D. Ma, Mater. Sci. Eng., R, 2016, 107, 1–42 CrossRef.
- Z. Zheng, Q. Dong, L. Gou, J.-H. Su and J. Huang, J. Mater. Chem. C, 2014, 2, 9858–9865 RSC.
- S. Zhang, Q.-L. Xu, J.-C. Xia, Y.-M. Jing, Y.-X. Zheng and J.-L. Zuo, New J. Chem., 2015, 39, 7954–7960 RSC.
- T.-Y. Chu and O.-K. Song, Appl. Phys. Lett., 2007, 90 Search PubMed.
- R. Braveenth, H. W. Bae, I. J. Ko, W. Qiong, Q. P. B. Nguyen, P. G. S. Jayashantha, J. H. Kwon and K. Y. Chai, Org. Electron., 2017, 51, 463–470 CrossRef CAS.
- S. Zhang, L.-S. Xue, Y.-M. Jing, X. Liu, G.-Z. Lu, X. Liang, H.-Y. Li, Y.-X. Zheng and J.-L. Zuo, Dyes Pigm., 2015, 118, 1–8 CrossRef CAS.
- A. M. Thaengthong, S. Saengsuwan, S. Jungsuttiwong, T. Keawin, T. Sudyoadsuk and V. Promarak, Tetrahedron Lett., 2011, 52, 4749–4752 CrossRef CAS.
- S. Jhulki and J. N. Moorthy, J. Mater. Chem. C, 2018, 6, 8280–8325 RSC.
- J. Li, C. Ma, J. Tang, C.-S. Lee and S. Lee, Chem. Mater., 2005, 17, 615–619 CrossRef CAS.
- Z. Li, Z. Wu, W. Fu, D. Wang, P. Liu, B. Jiao, X. Lei, G. Zhou and Y. Hao, Electron. Mater. Lett., 2013, 9, 655–661 CrossRef CAS.
- R. Griniene, J. V. Grazulevicius, K. Y. Tseng, W. B. Wang, J. H. Jou and S. Grigalevicius, Synth. Met., 2011, 161, 2466–2470 CrossRef CAS.
- O. Usluer, S. Demic, D. A. M. Egbe, E. Birckner, C. Tozlu, A. Pivrikas, A. M. Ramil and N. S. Sariciftci, Adv. Funct. Mater., 2010, 20, 4152–4161 CrossRef CAS.
- H. Kanai, S. Ichinosawa and Y. Sato, Synth. Met., 1997, 91, 195–196 CrossRef CAS.
- J. Zhuang, W. Su, W. Li, Y. Zhou, Q. Shen and M. Zhou, Org. Electron., 2012, 13, 2210–2219 CrossRef CAS.
- B. Liu, J. Zhao, C. Luo, F. Lu, S. Tao and Q. Tong, J. Mater. Chem. C, 2016, 4, 2003–2010 RSC.
- Q. D. Liu, J. Lu, J. Ding, M. Day, Y. Tao, P. Barrios, J. Stupak, K. Chan, J. Li and Y. Chi, Adv. Funct. Mater., 2007, 17, 1028–1036 CrossRef CAS.
- X.-Y. Liu, Y.-J. Zhang, X. Fei, Q. Ran, M.-K. Fung and J. Fan, J. Mater. Chem. C, 2019, 7, 1370–1378 RSC.
- Q. P. Nguyen, S. J. Baek, M. J. Kim, N. Y. Shin, G. W. Kim, D. C. Choe, J. H. Kwon and K. Y. Chai, Molecules, 2014, 19, 14247–14256 CrossRef.
- T. L. Wu, S. Y. Liao, P. Y. Huang, Z. S. Hong, M. P. Huang, C. C. Lin, M. J. Cheng and C. H. Cheng, ACS Appl. Mater. Interfaces, 2019, 11, 19294–19300 CrossRef CAS.
- S.-h. Ye, L. Li, M. Zhang, Z. Zhou, M.-h. Quan, L.-F. Guo, Y. Wang, M. Yang, W.-Y. Lai and W. Huang, J. Mater. Chem. C, 2017, 5, 11937–11946 RSC.
- H. Fukagawa, T. Shimizu, H. Kawano, S. Yui, T. Shinnai, A. Iwai, K. Tsuchiya and T. Yamamoto, J. Phys. Chem. C, 2016, 120, 18748–18755 CrossRef CAS.
- J. Kwak, Y.-Y. Lyu, S. Noh, H. Lee, M. Park, B. Choi, K. Char and C. Lee, Thin Solid Films, 2012, 520, 7157–7163 CrossRef CAS.
- Z. Zhong, X. Wang, T. Guo, J. Cui, L. Ying, J. Peng and Y. Cao, Org. Electron., 2018, 53, 35–42 CrossRef CAS.
- X.-Y. Liu, X. Tang, Y. Zhao, D. Zhao, J. Fan and L.-S. Liao, J. Mater. Chem. C, 2018, 6, 1023–1030 RSC.
- M. Romain, C. Quinton, D. Tondelier, B. Geffroy, O. Jeannin, J. Rault-Berthelot and C. Poriel, J. Mater. Chem. C, 2016, 4, 1692–1703 RSC.
- M. Romain, D. Tondelier, B. Geffroy, A. Shirinskaya, O. Jeannin, J. Rault-Berthelot and C. Poriel, Chem. Commun., 2015, 51, 1313–1315 RSC.
- X.-D. Zhu, Y.-L. Zhang, Y. Yuan, Q. Zheng, Y.-J. Yu, Y. Li, Z.-Q. Jiang and L.-S. Liao, J. Mater. Chem. C, 2019, 7, 6714–6720 RSC.
- C. Poriel and J. Rault-Berthelot, J. Mater. Chem. C, 2017, 5, 3869–3897 RSC.
- L.-H. Xie, F. Liu, C. Tang, X.-Y. Hou, Y.-R. Hua, Q.-L. Fan and W. Huang, Org. Lett., 2006, 8, 2787–2790 CrossRef CAS.
- H. Li, K. Fu, A. Hagfeldt, M. Gratzel, S. G. Mhaisalkar and A. C. Grimsdale, Angew. Chem., 2014, 53, 4085–4088 CrossRef CAS.
- B.-Y. Ren, D.-K. Zhong, Y.-G. Sun, X.-H. Zhao, Q.-J. Zhang, Y. Liu, M. Jurow, M.-L. Sun, Z.-S. Zhang and Y. Zhao, Org. Electron., 2016, 36, 140–147 CrossRef CAS.
- M. Sun, R. Xu, L. Xie, Y. Wei and W. Huang, Chin. J. Chem., 2015, 33, 815–827 CrossRef CAS.
- D. P. Tabor, V. A. Chiykowski, P. Friederich, Y. Cao, D. J. Dvorak, C. P. Berlinguette and A. Aspuru-Guzik, Chem. Sci., 2019, 10, 8360–8366 Search PubMed.
- X.-H. Zhao, Y.-N. Xu, J.-X. Wu, G.-D. Zou, X.-W. Zhang, L.-H. Xie, J.-F. Zhao, C.-M. Han, X. Hui and J.-J. Zhang, Dyes Pigm., 2019, 168, 228–234 CrossRef CAS.
- L. Calio, S. Kazim, M. Gratzel and S. Ahmad, Angew. Chem., 2016, 55, 14522–14545 CrossRef CAS.
- H.-T. Cao, C.-S. Hong, D.-Q. Ye, L.-H. Liu, L.-H. Xie, S.-F. Chen, C. Sun, S.-S. Wang, H.-M. Zhang and W. Huang, J. Mol. Struct., 2019, 1196, 132–138 CrossRef CAS.
- H.-j. Jiang, J. Sun, K. Yuan and Q.-w. Zhang, Synth. Met., 2014, 197, 217–224 CrossRef CAS.
- X. Liang, K. Wang, R. Zhang, K. Li, X. Lu, K. Guo, H. Wang, Y. Miao, H. Xu and Z. Wang, Dyes Pigm., 2017, 139, 764–771 Search PubMed.
- M. Maciejczyk, A. Ivaturi and N. Robertson, J. Mater. Chem. A, 2016, 4, 4855–4863 RSC.
- B. Xu, D. Bi, Y. Hua, P. Liu, M. Cheng, M. Grätzel, L. Kloo, A. Hagfeldt and L. Sun, Energy Environ. Sci., 2016, 9, 873–877 RSC.
- V. A. Chiykowski, Y. Cao, H. Tan, D. P. Tabor, E. H. Sargent, A. Aspuru-Guzik and C. P. Berlinguette, Angew. Chem., Int. Ed., 2018, 57, 15529–15533 CrossRef CAS.
- Z. Jiang, T. Ye, C. Yang, D. Yang, M. Zhu, C. Zhong, J. Qin and D. Ma, Chem. Mater., 2011, 23, 771–777 CrossRef CAS.
- D.-H. Lee, Y.-P. Liu, K.-H. Lee, H. Chae and S. M. Cho, Org. Electron., 2010, 11, 427–433 CrossRef CAS.
- H. Sasabe, E. Gonmori, T. Chiba, Y.-J. Li, D. Tanaka, S.-J. Su, T. Takeda, Y.-J. Pu, K.-I. Nakayama and J. Kido, Chem. Mater., 2008, 20, 5951–5953 CrossRef CAS.
- J. Li, D. Liu, Y. Li, C.-S. Lee, H.-L. Kwong and S. Lee, Chem. Mater., 2005, 17, 1208–1212 CrossRef CAS.
- C.-H. Chen, L.-C. Hsu, P. Rajamalli, Y.-W. Chang, F.-I. Wu, C.-Y. Liao, M.-J. Chiu, P.-Y. Chou, M.-J. Huang, L.-K. Chu and C.-H. Cheng, J. Mater. Chem. C, 2014, 2, 6183–6191 RSC.
- L. S. Cui, Y. Liu, X. Y. Liu, Z. Q. Jiang and L. S. Liao, ACS Appl. Mater. Interfaces, 2015, 7, 11007–11014 CrossRef CAS.
- B. Jiang, X. Ning, S. Gong, N. Jiang, C. Zhong, Z.-H. Lu and C. Yang, J. Mater. Chem. C, 2017, 5, 10220–10224 RSC.
- B. Jiang, C. Zhao, X. Ning, C. Zhong, D. Ma and C. Yang, Adv. Opt. Mater., 2018, 6, 1800108 CrossRef.
- K. H. Kim, S. Lee, C. K. Moon, S. Y. Kim, Y. S. Park, J. H. Lee, J. Woo Lee, J. Huh, Y. You and J. J. Kim, Nat. Commun., 2014, 5, 4769 CrossRef CAS.
- C. Y. Kuei, W. L. Tsai, B. Tong, M. Jiao, W. K. Lee, Y. Chi, C. C. Wu, S. H. Liu, G. H. Lee and P. T. Chou, Adv. Mater., 2016, 28, 2795–2800 CrossRef CAS.
- J. H. Lee, H. Shin, J. M. Kim, K. H. Kim and J. J. Kim, ACS Appl. Mater. Interfaces, 2017, 9, 3277–3281 CrossRef CAS.
- X. Y. Liu, F. Liang, Y. Yuan, L. S. Cui, Z. Q. Jiang and L. S. Liao, Chem. Commun., 2016, 52, 8149–8151 RSC.
- C. H. Shih, P. Rajamalli, C. A. Wu, W. T. Hsieh and C. H. Cheng, ACS Appl. Mater. Interfaces, 2015, 7, 10466–10474 CrossRef CAS.
- C.-H. Shih, P. Rajamalli, C.-A. Wu, M.-J. Chiu, L.-K. Chu and C.-H. Cheng, J. Mater. Chem. C, 2015, 3, 1491–1496 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tc04676k |
| This journal is © The Royal Society of Chemistry 2021 |