
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand-assisted solid phase synthesis of mixed-halide perovskite nanocrystals for color-pure and efficient electroluminescence†
Simon F.
Solari
 a,
Sudhir
Kumar
a,
Jakub
Jagielski
a,
Nikolas M.
Kubo
a,
Frank
Krumeich
b and
Chih-Jen
Shih
*a
a,
Sudhir
Kumar
a,
Jakub
Jagielski
a,
Nikolas M.
Kubo
a,
Frank
Krumeich
b and
Chih-Jen
Shih
*a
aInstitute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zürich, Vladimir Prelog Weg 1, CH-8093 Zürich, Switzerland. E-mail: chih-jen.shih@chem.ethz.ch
bLaboratory of Inorganic Chemistry, Department of Chemistry and Applied Biosciences, ETH Zürich, Vladimir Prelog Weg 1, CH-8093 Zürich, Switzerland
First published on 25th March 2021
Abstract
Colloidal nanocrystals (NCs) of lead halide perovskites have generated considerable interest in the fabrication of optoelectronic devices, such as light emitting-diodes (LEDs), because of their tunable optical bandgap, narrow spectral width, and high defect tolerance. However, the inhomogeneous halide distribution within individual NCs remains a critical challenge in order to obtain color-stable electroluminescence in mixed-halide systems. Here, we demonstrate a new post-synthetic approach, ligand-assisted solid phase synthesis (LASPS), for the preparation of electroluminescent colloidal NCs of methylammonium (MA) lead halide perovskites, at room temperature. The slow reaction kinetics preserves the morphology, size, and shape in the resulting NCs whose emission covers the entire visible spectral region with photoluminescence (PL) quantum yields (QYs) of up to >90% and colloidal stability up to several months. The LEDs fabricated using the prepared mixed-halide NCs display narrowband electroluminescence (EL) ranging from 476 to 720 nm. The optimized red LEDs exhibit an external quantum efficiency, ηext, of up to 2.65%, with the CIE 1931 color coordinates of (0.705, 0.290), nearly identical to those of the red primary in the recommendation (rec.) 2020 standard (0.708, 0.292).
Colloidal nanocrystals (NCs) of organic–inorganic hybrid perovskites (OIHPs) have emerged as the most promising class of semiconductors for optoelectronic applications, including solar cells, lasers, and light-emitting diodes (LEDs).1–5 OIHPs have the general formula of ABX3, where A- and B-sites are cationic species and X represents halides.6 The emerging semiconductor system has demonstrated a wide range of tunability in optical and photophysical characteristics through composition engineering owing to its soft nature, which also leads to fast migration of anionic species and high defect tolerance.7 In addition to reduced dimensionality,8 the composition engineering, including halide and A- and B-site mixing, has shown its versatility to tune the optical bandgap of perovskite NCs.9–12 Indeed, although the quantum-confined OIHP NCs exhibit a high exciton binding energy without sacrificing the photoluminescence (PL) quantum yields (QYs), the large surface-to-volume ratio demands a large amount of capping ligands,13 which compromise the charge transport characteristics that are essential for obtaining high-efficiency electroluminescence (EL).14 In contrast, the three-dimensional (3D) OIHP NCs can be stabilized with a relatively small amount of ligands,15 and the emission wavelength can be readily tuned by composition engineering.9 Accordingly, we consider them to be better candidates for electroluminescent materials.
As far as halide mixing is concerned, there are two general approaches: (i) direct synthesis of mixed-halide perovskites via mixing different halide precursors,16 and (ii) post-synthetic anion exchange of OIHP and pure inorganic lead halide perovskites.17–21 The latter is analogous to the well-established surface modification strategies, such as ligand exchange, in colloidal quantum dot synthesis for enhancing stability and optoelectronic performance.20,22–27 Indeed, as the halide molecular orbitals strongly contribute to the valence band maximum (VBM) of the resulting perovskite compound, a variation in the halide composition would regulate the VBM and the optical bandgap.28 The first post-synthetic anion exchange in inorganic CsPbX3 was reported by Nedelcu et al.,17 by injecting a halide precursor solution at elevated temperatures. Similar strategy was thereafter used to synthesize mixed-halide perovskite NCs.9 Other approaches are also reported. For example, Parobek et al. demonstrated a photo-induced anion exchange of colloidal CsPbBr3 NCs in dihalomethane solvents; the wavelength and intensity of photo-excitation source could control the degree of halide mixing. However, only Br/Cl mixing could work.29 Yoon et al. reported a reversible post-synthetic anion exchange route using the haloalkane solvents with a strong nucleophile trioctylphosphine (TOP).30 The nucleophile TOP promotes halogen–carbon bond dissociation through the SN2 reaction. Guhrenz et al. also reported a solid state post-synthetic anion exchange reaction by mixing solid potassium halides (KX, where X = Cl, Br, or I) in an NC colloidal solution, followed by evaporating the solvents,31 but the reaction kinetics was considerably slow. Accordingly, the abovementioned methods often require complex synthetic protocols at elevated temperatures, which may hinder large-scale synthesis.
Here, we present a facile post-synthetic anion exchange method, ligand-assisted solid phase synthesis (LASPS), for the preparation of highly luminescent methylammonium lead halide (MAPbX3) NCs. The proposed synthetic protocol is entirely performed at room temperature and under ambient conditions. The synthesized NCs were directly used in light-emitting diode (LED) devices. We demonstrate their maximum external quantum efficiencies, ηext, ranging from 0.4% (476 nm) to 2.65% (656 nm), as compared to 3.5% based on parent MAPbBr3 NCs (524 nm). Finally, we further demonstrate an electric-field driven irreversible post-synthetic anion exchange technique for the OIHP NCs.
We synthesized the parent MAPbBr3 colloidal NCs dispersed in toluene using the ligand-assisted reprecipitation (LARP) method, followed by mixing with a solid powder of halide precursor and a small amount of ligands in a flask at room temperature (Fig. 1a; for details, see ESI†). Methylammonium halide (MAX, X = Cl or I) powder was used as the solid source to ensure that only anion exchange takes place. Two surfactants, oleylamine (OAM) and oleic acid (OLA), were used, identical to those in the parent MAPbBr3 NC preparation. Upon mixing the reactants, the solution color gradually turned either dark or colorless depending on the halide precursors used (ESI,† Fig. S1). Fig. 1b presents a schematic diagram describing the halide exchange process. Specifically, upon mixing of halide powder and surfactants, halide ions get gradually dissolved in the nonpolar toluene by forming reverse micelles.32 The formation of halide-containing micelles increases the chemical potential of the halide ions, thereby slowly replacing the bromide ions in the perovskite NC lattices. The protocol was successful in both MAPb(Br/I)3 and MAPb(Br/Cl)3 systems. Prior to device fabrication, the excess ligands and unreacted species in solution were removed by washing with anti-solvents. A number of anti-solvents, including methyl acetate (MeOAc), ethyl acetate (EtOAc), dimethyl sulfoxide (DMSO), propionitrile, acetonitrile (ACN), acetone, n-butanol, chloroform, and their mixtures, have been investigated to maximize the perovskite NC concentration. As reported in the literature, the anti-solvent washing often considerably compromises the yield.33 After extensive experimentation, we found that MeOAc is the most optimal anti-solvent for the purification of parent MAPbBr3 and MAPb(Br/Cl)3 NCs, and both MeOAc and n-butanol are highly effective anti-solvents for MAPb(Br/I)3 NCs.
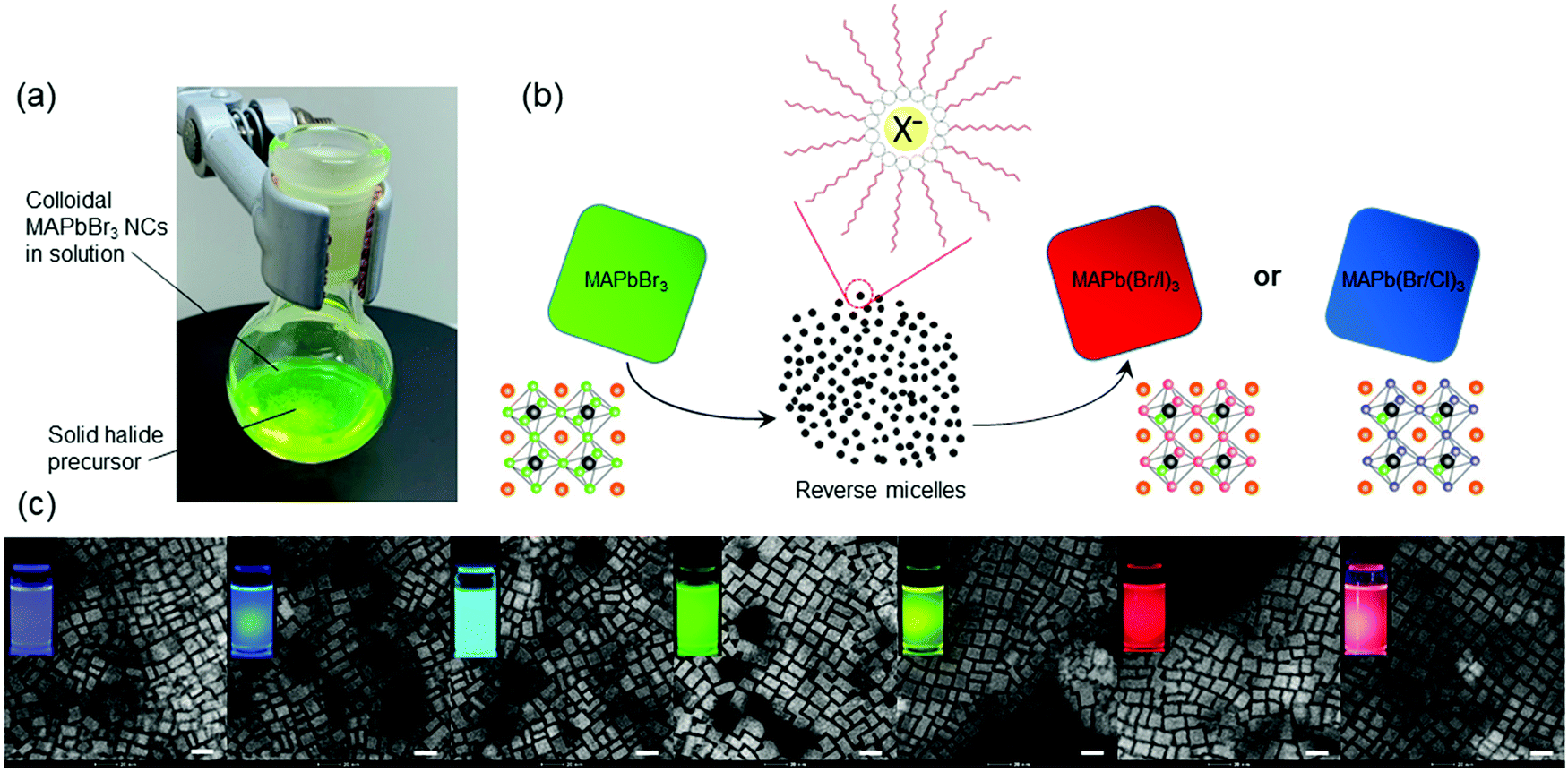 | ||
| Fig. 1 (a) Reaction of colloidal MAPbBr3 NCs with a solid halide precursor at the bottom of the flask. (b) Schematic diagram of the LASPS process showing the formation of reverse micelles upon solid mixing. Positively charged long-chain hydrocarbon ammonium ions attach to the halide ion forming reverse micelles. The crystal structures of MAPbBr3 and its halide exchanged mixtures demonstrate partial anion exchange. (c) Representative transmission electron micrographs and photographs under UV excitation for the synthesized MAPbX3 NC solutions (scale bar: 20 nm). | ||
After halide exchange, the majority of parent bromide ions were substituted by either chloride or iodide ions, as revealed by the energy dispersive X-ray spectroscopy (EDXS) (ESI,† Fig. S2–S8). We find that the long-chain hydrocarbon ligands OLA and OAM play a crucial role in the reaction system by partially dissolving the MAX salts, such that excess halides become accessible for anion exchange. The OAM ligands are protonated by OLA to yield positively charged ammonium, so that they can form reverse micelles by surrounding the halide ions. We notice that the halide exchange kinetics is relatively slow ranging from few minutes to several hours, hypothetically due to the low solubility of halide ions in toluene. More experiments were carried out to estimate the proper amount of halide powder used in LASPS (ESI,† Fig. S9 and S10).
Taking MAPb(Br/I)3 NCs as an example, we notice that the reaction reaches equilibrium within 1.5 hours; the emission wavelengths between 1.5 and 17 hours did not show any noticeable difference (ESI,† Fig. S11). Mixed-halide NCs can be obtained by either terminating the reaction before reaching equilibrium or by mixing fully exchanged halide NCs with the parent MAPbBr3 NCs via the internanocrystal exchange (ESI,† Table S1). We also noticed that anionic mixing between iodide and chloride did not occur because of a considerable difference between their ionic radii (220 and 181 pm, respectively).34 Luminescent mixed-halide OIHP NCs covering the entire visible spectrum were synthesized (Fig. 1c). Their transmission electron micrographs suggest that the NC morphology, size, and shape are preserved upon halide mixing, consistent with the directly synthesized MAPbX3 NCs.35,36
It is well-known that at room temperature, the crystalline phase of pure MAPbCl3 and MAPbBr3 is cubic but transforms to tetragonal in MAPbI3.35–39 The cubic phase in MAPbI3 can only become thermodynamically stable at temperatures greater than 330 K.40,41Fig. 2 compares the X-ray diffraction (XRD) patterns of parent MAPbBr3 and the mixed-halide MAPb(Br/Cl)3 and MAPb(Br/I)3 NC systems considered here. The parent MAPbBr3 is confirmed to be of the cubic phase, as revealed by the measured d-spacings of 5.82 and 4.12 Å for the (100) and (110) planes, respectively, consistent with the literature.38,39 Interestingly, the crystallographic features of MAPbBr3 and MAPb(Br/Cl)3 NCs are nearly identical. The lattice expansion and contraction upon iodide and chloride alloying are revealed by the degree of peak shifting. The d-spacing corresponding to the (100) peak in the cubic phase for fully reacted MAPb(Br/Cl)3 perovskites is 5.63 Å, which is similar to those reported for cubic MAPb(Br/Cl)3 alloys.42 Upon exchanging Br with I, the X-ray diffraction peaks are gradually shifted towards lower angles due to lattice expansion. We observe splitting of the (100) plane peak to 2θ of 14.39° and 14.82° in the fully exchanged MAPb(Br/I)3 system, which could be attributed to a transition between the cubic and tetragonal phase. Note that the pure MAPbBr3 has a cubic crystal structure, whereas pure MAPbI3 has a tetragonal crystal structure at room temperature.39 It has been reported that when x > 0.2 in the MAPb(BrxI1−x)3 NCs, the phase transition from tetragonal to a cubic would take place.43 Accordingly, we deduce that the I-rich MAPb(Br/I)3 NCs are composed of both crystal phases, which is completely different from the parent MAPbBr3 NCs.
 | ||
| Fig. 2 Powder X-ray diffraction (XRD) patterns of the synthesized MAPbX3 films. | ||
The absorption and photoluminescence (PL) spectra of parent MAPbBr3 and mixed-halide NC solutions are shown in Fig. 3a and b, and ESI,† Fig. S12. Their PL quantum yields, ηPL, are also shown in Fig. 3a. All samples show narrowband emission with the peak positions, λPL, covering the entire visible spectral region. The parent MAPbBr3 and the fully reacted MAPb(Br/I)3 and MAPb(Br/Cl)3 NCs exhibit ηPL values of 91% (λPL = 524 nm), 89% (λPL = 724 nm), and 35% (λPL = 415 nm), respectively, comparable to, or even higher than, those reported in the literature for the directly synthesized perovskite NCs.35,36,44 For pure MAPbBr3 and fully exchanged MAPb(Br/I)3 NCs, the ηPL can reach high values (Fig. 3a). Similar behavior to PL characteristics upon halide mixing was also observed for all-inorganic CsPbX3 NCs.45 The drop of ηPL and its later recovery in the case of MAPb(Br/I)3 mixtures can be attributed to halide segregation.46 The ηPL trend for MAPb(Br/Cl)3 halide mixtures can also be associated with the ion migration processes.47 Both parent MAPbBr3 and mixed-halide MAPb(Br/Cl)3 perovskite NCs (λPL = 490–524 nm) show a plateau with a constantly high ηPL of approximately 85%. For MAPbBr3−xClx halide mixtures, when x ≤ 0.45 (Br-rich), the ion migration in these mixed-halide perovskite materials seemed to be suppressed by Cl incorporation. This yielded an improvement of charge carrier transport in these materials.48 The suppression of ion migration within this PL emission range (λPL = 490–524 nm) may explain the outstanding ηPL. By further replacing Br with Cl in the mixed-halide MAPb(Br/Cl)3, the ηPL decreases continuously, which can be attributed to an increasing population of halide vacancies and a boost of the ion migration within the crystal lattice. These defects introduce energy levels in the bandgap that may act as traps for photogenerated charge carriers.49 We also find that the optimized anti-solvent washing protocol could get rid of excess ligands in the MAPb(Br/I)3 NCs with slight increase in ηPL, for example, from 79% in unwashed MAPb(Br/I)3 to 89% in the washed counterparts and further narrowing the emission bandwidth from 46 nm (unwashed) to 41 nm (washed) (ESI,† Fig. S13). Surprisingly, upon the anti-solvent washing treatment, the λPL of MAPb(Br/I)3 NCs also show a slight redshift from 721 nm to 724 nm, whereas the optical density also decreases. Nevertheless, for the partially reacted MAPb(Br/I)3 solutions, the anti-solvent washing would lead to a high degree of blueshift in the emission (ESI,† Fig. S14).50 To this end, in order to get rid of excess ligands for LEDs and prevent the PL emission from blueshifting during purification, partially anion-mixed MAPb(Br/I)3 NCs were prepared by using the internanocrystal exchange method. On the other hand, for MAPb(Br/Cl)3 samples, they are very robust upon anti-solvent washing. We observed a sudden drop of ηPL down to 11% in the yellow-emitting MAPb(Br/I)3 NCs. The low ηPL in the yellow-emitting MAPb(Br/I)3 is attributed to the large population of non-radiative trap states possibly because of halide segregation46,51 that often emerged due to the fast dynamics of halide species within the crystal lattice.7 Our yellow-emitting MAPb(Br/I)3 NCs are composed of a Br-rich environment within the crystal lattice as revealed by Energy-dispersive X-ray (EDX) spectroscopy (ESI,† Fig. S6). In particular, a number of reports have suggested that when 0.2 < x < 1 in MAPb(BrxI1−x)3 (Br-rich), the halide segregation becomes more pronounced.46,52 Indeed, upon decreasing the Br content, the halide segregation seems suppressed as reflected by an increase in ηPL. Recently, several trap passivation strategies have been exploited to mitigate halide segregation in perovskite materials such as KI, KBr, and iodide treatment.53–55
 | ||
| Fig. 3 Normalized (a) PL and (b) absorption spectra of the parent MAPbBr3 and mixed-halide perovskite NCs together with their ηPL. (c) Time-resolved photoluminescence (TRPL) responses of the synthesized NCs. (d) Representative transmission electron micrograph of 2D MAPbBr3 NPLs (scale bar: 50 nm). (e) Absorption (solid lines) and PL (dashed lines) of the LASPS synthesized mixed-halide perovskite NPLs. | ||
The colloidal stability for mixed-halide perovskite NCs is excellent, as reflected by the observation that high ηPL can retain over several months (ESI,† Table S2). The outstanding colloidal stability of the parent MAPbBr3 and mixed-halides MAPb(Br/Cl)3 and MAPb(Br/I)3 could be attributed to long-chain alkylamines such as oleylamine that regulate the kinetics of crystallization during the reactions. The oleic acids also prevent the NC aggregation, which results in the long-term colloidal stability of parent and exchanged halide NCs.15,56–58 We anticipate that the colloidal stability may be further improved by the post-synthetic treatment, which could passivate the trap-sites on the NC surfaces.59,60
The time-resolved photoluminescence (TRPL) responses of a number of representative samples are shown in Fig. 3c. The carrier lifetimes of MAPbBr3 and mixed-halide perovskite NCs are shown in ESI,† Table S3. In general, we observed the average excited state lifetime, τavg, between 12.4 ns for fully reacted MAPb(Br/Cl)3 to 40.4 ns for fully reacted MAPb(Br/I)3 NCs. The yellow-emitting MAPb(Br/I)3 NCs (λPL = 573 nm) exhibit a relatively short τavg of 23.5 ns as compared to the parent green-emitting MAPbBr3 NCs, τavg = 30.7 ns. This is possibly due to its significantly low ηPL = 11%, as a result of the non-radiative trap sites caused by the halide segregation processes.46
Finally, we demonstrated that the protocol also apply to the quantum-confined two-dimensional (2D) MAPbBr3 nanoplatelets (NPLs) (Fig. 3d), emitting at 455 nm. Fig. 3e presents the absorption and PL spectra of parent blue-emitting MAPbBr3 NPLs and their halide exchanged products. In particular, the sharp excitonic absorption features are preserved upon halide mixing.
The protocols established here, including LASPS and the anti-solvent washing, allow us to prepare mixed-halide perovskite NCs for EL devices. The LED devices were fabricated using the parent MAPbBr3 and the anion exchanged MAPb(Br/Cl)3 as well as MAPb(Br/I)3 NCs. Fig. 4a schematically shows the device architecture with the layering sequence of ITO/PEDOT:PSS/emissive layer/TPBi/LiF/Al. The energy diagram is shown in Fig. 4b. Five devices with EL emitting at 476 nm (blue), 524 nm (green), 584 nm (yellow), 656 nm (red), and 720 ± 4 nm (near-infrared, NIR) were fabricated (Fig. 4c). The EL emission bandwidths, the full width at half maximum (fwhm) range from 18 to 42 nm (Table 1). The Commission Internationale d'Eclairage (CIE) color coordinates for red, green, and blue devices yield a wide color gamut, covering 84% and 110% of the Rec. 2020 and NTSC standards in the 1931 CIE color space (ESI,† Fig. S15).61 We were not able to obtain efficient deep-blue devices; otherwise the Rec. 2020 gamut coverage can reach 95%, based on the measured λPL of our deep-blue-emitting MAPb(Br/Cl)3 NCs.
 | ||
| Fig. 4 EL characteristics using our synthesized anion exchanged MAPbX3 perovskite NCs. (a) Schematic device architecture. (b) Schematic energy level diagram based on the parent MAPbBr3 NCs. (c) EL spectra of the fabricated LEDs. Characterized (d) current density, (e) luminance, and (f) external quantum efficiency as a function of voltage. | ||
| Device | η ext (%) | λ EL (nm) | fwhm (nm) | CIEx,y coordinates |
|---|---|---|---|---|
| Blue | 0.40 | 476 | 18.0 | (0.125, 0.144) |
| Green | 3.50 | 524 | 22.4 | (0.150, 0.771) |
| Yellow | 0.48 | 584 | 35.0 | (0.476, 0.391) |
| Red | 2.65 | 656 | 40.0 | (0.705, 0.290) |
| NIR | 2.36 | 720 ± 4 | 42.4 | (0.644, 0.320) |
The current density–voltage–luminance (J–V–L) characteristics of our representative LEDs are shown in Fig. 4d and e. The green LED device based on parent MAPbBr3 NCs shows a turn-on voltage (Von) of 3 V with a luminance of 18.1 cd m−2. A maximum luminance of 1861 cd m−2 was realized at 5.0 V under a current density of 41.4 mA cm−2. The maximum external quantum efficiency, ηext, reaches 3.5% (Fig. 4f) corresponding to the maximum current efficiency, ηCE, of 14.6 cd A−1. The blue and yellow devices reached relatively low ηext values of 0.40 and 0.48%, respectively. The lower device performance of the blue LED device could be attributed to the deep lying valence band maximum (VBM) energy level, which is indeed the result of increasing the chloride content in the MAPbX3 NCs9 that creates a high energy barrier for hole injection.62 It has been suggested that all forms of lead halide perovskites have a considerable degree of electron traps; together with a high energy barrier for hole injection, the undesirable carrier imbalance would move the recombination zone closer to the hole transport layer (HTL)/emissive layer interface.62,63
For MAPb(Br/l)3 NCs, on the other hand, an expected low ηext and spectral stability of the yellow device is possibly due to its low ηPL. Indeed, ηext is substantially enhanced to 2.65% in the red device (Fig. 4f), following the increase of ηPL from 15 to 58%. All EL spectra are consistent with their PL. It is noteworthy that our red-emitting device exhibits color coordinates of (0.705, 0.290), very close to those for the Rec. 2020 standard (0.708, 0.292).64 The NIR device reaches ηext of 2.36% at 720 ± 4 nm with an fwhm of 42.4 nm. The EL spectra of devices remained unchanged at different driving voltages (ESI,† Fig. S16). The LED devices based on parent MAPbBr3 NCs and mixed-halide perovskite NCs, MAPb(Br/l)3 and MAPb(Br/Cl)3, show excellent EL spectral stability over the entire operating voltage range. The ultimate EL spectral stability in the LEDs could be attributed to highly emissive NCs with a reduced number of defects. This could be explained by the synthesis via LASPS and the efficient purification protocols for the mixed-halide perovskite NCs. Advanced photophysical characterization is required to understand the origin of the observed spectral stability of LEDs. We also examined the operational stability of the resulting green, blue, and red LEDs based on MAPbBr3, MAPb(Br/Cl)3, and MAPb(Br/I)3 NCs, respectively. Our encapsulated devices showed the LT50, defined as the time of the luminance to decay to 50% of the initial luminance, of 23, 0.5, and 6.2 minutes, respectively, in the green, blue, and red LED devices (ESI,† Fig. S17). Note that all devices were stressed at a high constant J = 20 mA cm−2.
We found that potassium iodide (KI) powder can also be used as the solid source in the anion exchange reactions of MAPbBr3 NCs (see ESI† for further details). In short, KI powder (1.125 mmol) was mixed with MAPbBr3 NCs for 2.5 hours, followed by centrifugation and MeOAc anti-solvent washing, yielding yellow-emitting solution (λPL = 580 nm) with an ηPL of 10%. The EL spectrum, however, tends to be unstable upon electrical excitation. Fig. 5a shows the evolution of the EL spectra as a function of driving voltage, which reveal the transformation from yellow to deep-red (702 nm) perovskite phase. It is clear that the deep-red-emitting phase that appears is not pure MAPbI3 NCs, but it is color-stable upon further electrical stress (Fig. 5b). The origin for the formation of this stable MAPb(Br/I)3 phase is under further investigation. We anticipate that the electric-field-driven, irreversible post-synthetic technique may open a new avenue for color-stable perovskite LEDs.
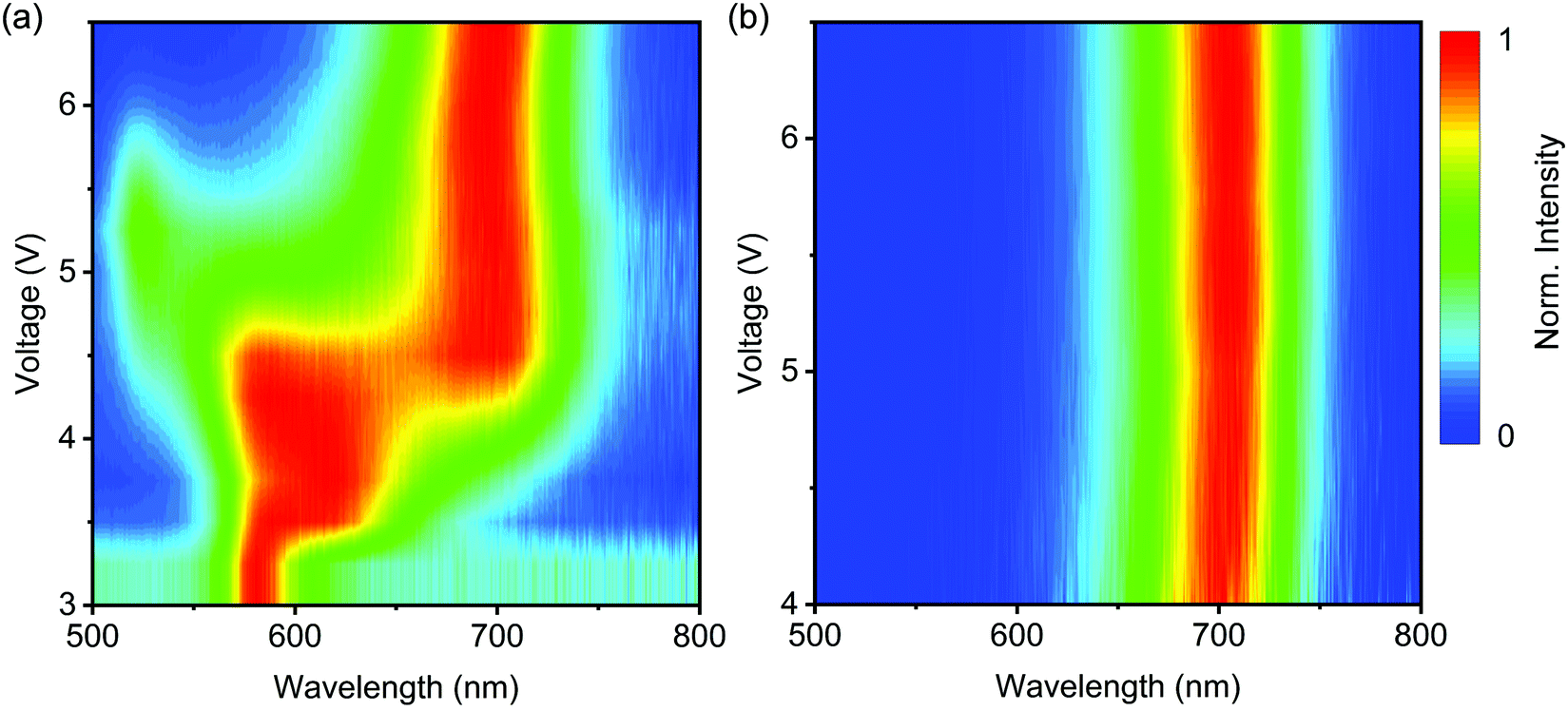 | ||
| Fig. 5 EL spectra of KI-exchanged MAPb(Br/I)3 under electrical stress. (a) The evolution of EL spectra as a function of driving voltage. (b) EL spectra of the color-stable mixed-halide MAPb(Br/I)3 NCs after applying a reverse bias of −4 V for 20 minutes. | ||
In conclusion, we present a facile synthetic route, ligand-assisted solid phase synthesis (LASPS), together with anti-solvent washing protocols for the preparation of electroluminescent colloidal NCs of mixed-halide lead perovskites. The synthesized MAPb(Br/Cl)3 and MAPb(Br/I)3 NCs exhibit narrowband PL covering the entire visible spectral region. The protocols preserve the morphology, size, and shape of the perovskite NCs throughout the attainable halide composition. We also prepared quantum-confined mixed-halide perovskite NPLs, which have been difficult to synthesize using direct synthesis methods. Decent LED external quantum efficiencies were achieved using the standard device architecture. In particular, the red device reached color coordinates nearly identical to the newly defined Rec. 2020 standard. Further optimization of synthetic protocols, solid sources, and ligands may allow us to obtain high-efficiency and color-stable EL enduring electrical stress based on mixed-halide perovskite NCs.
Conflicts of interest
The authors declare that they have no other competing interests.Acknowledgements
The authors are grateful for the financial support from the Swiss National Science Foundation (project number: 200021-178944), ETH research grant (ETH-33 18-2), and European Research Council (ERC) starting grant (N849229 – CQWLED). The authors are grateful to ScopeM (ETH Zürich) for access the electron microscopy facilities.References
- S. D. Stranks and H. J. Snaith, Nat. Nanotechnol., 2015, 10, 391 CrossRef CAS PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- B. R. Sutherland and E. H. Sargent, Nat. Photonics, 2016, 10, 295–302 CrossRef CAS.
- Z.-K. Tan, R. S. Moghaddam, M. L. Lai, P. Docampo, R. Higler, F. Deschler, M. Price, A. Sadhanala, L. M. Pazos, D. Credgington, F. Hanusch, T. Bein, H. J. Snaith and R. H. Friend, Nat. Nanotechnol., 2014, 9, 687 CrossRef CAS PubMed.
- Y.-H. Kim, C. Wolf, Y.-T. Kim, H. Cho, W. Kwon, S. Do, A. Sadhanala, C. G. Park, S.-W. Rhee, S. H. Im, R. H. Friend and T.-W. Lee, ACS Nano, 2017, 11, 6586–6593 CrossRef CAS PubMed.
- S. F. Solari, S. Kumar, J. Jagielski and C.-J. Shih, J. Soc. Inf. Disp., 2019, 27, 667–678 CrossRef CAS.
- Q. A. Akkerman, G. Rainò, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394–405 CrossRef CAS PubMed.
- J. Jagielski, S. F. Solari, L. Jordan, D. Scullion, B. Blülle, Y.-T. Li, F. Krumeich, Y.-C. Chiu, B. Ruhstaller, E. J. G. Santos and C.-J. Shih, Nat. Commun., 2020, 11, 387 CrossRef CAS PubMed.
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS PubMed.
- M. Hao, Y. Bai, S. Zeiske, L. Ren, J. Liu, Y. Yuan, N. Zarrabi, N. Cheng, M. Ghasemi, P. Chen, M. Lyu, D. He, J.-H. Yun, Y. Du, Y. Wang, S. Ding, A. Armin, P. Meredith, G. Liu, H.-M. Cheng and L. Wang, Nat. Energy, 2020, 5, 79–88 CrossRef CAS.
- W. van der Stam, J. J. Geuchies, T. Altantzis, K. H. W. van den Bos, J. D. Meeldijk, S. Van Aert, S. Bals, D. Vanmaekelbergh and C. de Mello Donega, J. Am. Chem. Soc., 2017, 139, 4087–4097 CrossRef CAS PubMed.
- B. Luo, F. Li, K. Xu, Y. Guo, Y. Liu, Z. Xia and J. Z. Zhang, J. Mater. Chem. C, 2019, 7, 2781–2808 RSC.
- S. Kumar, J. Jagielski, S. Yakunin, P. Rice, Y.-C. Chiu, M. Wang, G. Nedelcu, Y. Kim, S. Lin, E. J. G. Santos, M. V. Kovalenko and C.-J. Shih, ACS Nano, 2016, 10, 9720–9729 CrossRef CAS PubMed.
- H. J. An, Y. C. Kim, D. H. Kim and J.-M. Myoung, ACS Appl. Mater. Interfaces, 2020, 12, 16726–16735 CrossRef CAS PubMed.
- S. Kumar, J. Jagielski, T. Marcato, S. F. Solari and C.-J. Shih, J. Phys. Chem. Lett., 2019, 10, 7560–7567 CrossRef CAS PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635–5640 CrossRef CAS PubMed.
- D. Zhang, Y. Yang, Y. Bekenstein, Y. Yu, N. A. Gibson, A. B. Wong, S. W. Eaton, N. Kornienko, Q. Kong, M. Lai, A. P. Alivisatos, S. R. Leone and P. Yang, J. Am. Chem. Soc., 2016, 138, 7236–7239 CrossRef CAS PubMed.
- N. Pellet, J. Teuscher, J. Maier and M. Grätzel, Chem. Mater., 2015, 27, 2181–2188 CrossRef CAS.
- T. Chiba, Y. Hayashi, H. Ebe, K. Hoshi, J. Sato, S. Sato, Y.-J. Pu, S. Ohisa and J. Kido, Nat. Photonics, 2018, 12, 681–687 CrossRef CAS.
- Q. A. Akkerman, V. D’Innocenzo, S. Accornero, A. Scarpellini, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2015, 137, 10276–10281 CrossRef CAS PubMed.
- Y.-H. Suh, T. Kim, J. W. Choi, C.-L. Lee and J. Park, ACS Appl. Nano Mater., 2018, 1, 488–496 CrossRef CAS.
- B. A. Koscher, J. K. Swabeck, N. D. Bronstein and A. P. Alivisatos, J. Am. Chem. Soc., 2017, 139, 6566–6569 CrossRef CAS PubMed.
- N. Yarita, H. Tahara, M. Saruyama, T. Kawawaki, R. Sato, T. Teranishi and Y. Kanemitsu, J. Phys. Chem. Lett., 2017, 8, 6041–6047 CrossRef CAS PubMed.
- J. Pan, L. N. Quan, Y. Zhao, W. Peng, B. Murali, S. P. Sarmah, M. Yuan, L. Sinatra, N. M. Alyami, J. Liu, E. Yassitepe, Z. Yang, O. Voznyy, R. Comin, M. N. Hedhili, O. F. Mohammed, Z. H. Lu, D. H. Kim, E. H. Sargent and O. M. Bakr, Adv. Mater., 2016, 28, 8718–8725 CrossRef CAS PubMed.
- Y.-C. Chen, H.-L. Chou, J.-C. Lin, Y.-C. Lee, C.-W. Pao, J.-L. Chen, C.-C. Chang, R.-Y. Chi, T.-R. Kuo, C.-W. Lu and D.-Y. Wang, J. Phys. Chem. C, 2019, 123, 2353–2360 CrossRef CAS.
- X. Bao, M. Li, J. Zhao and Z. Xia, J. Mater. Chem. C, 2020, 8, 12302–12307 RSC.
- M. V. Kovalenko, L. Protesescu and M. I. Bodnarchuk, Science, 2017, 358, 745 CrossRef CAS PubMed.
- D. Parobek, Y. Dong, T. Qiao, D. Rossi and D. H. Son, J. Am. Chem. Soc., 2017, 139, 4358–4361 CrossRef CAS PubMed.
- Y. J. Yoon, K. T. Lee, T. K. Lee, S. H. Kim, Y. S. Shin, B. Walker, S. Y. Park, J. Heo, J. Lee, S. K. Kwak, G.-H. Kim and J. Y. Kim, Joule, 2018, 2, 2105–2116 CrossRef CAS.
- C. Guhrenz, A. Benad, C. Ziegler, D. Haubold, N. Gaponik and A. Eychmüller, Chem. Mater., 2016, 28, 9033–9040 CrossRef CAS.
- Z. Yuan, Y. Shu, Y. Tian, Y. Xin and B. Ma, Chem. Commun., 2015, 51, 16385–16388 RSC.
- J. De Roo, M. Ibáñez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS PubMed.
- Y. Ida, Phys. Earth Planet. Inter., 1976, 13, 97–104 CrossRef CAS.
- Y. Hassan, O. J. Ashton, J. H. Park, G. Li, N. Sakai, B. Wenger, A.-A. Haghighirad, N. K. Noel, M. H. Song, B. R. Lee, R. H. Friend and H. J. Snaith, J. Am. Chem. Soc., 2019, 141, 1269–1279 CrossRef CAS PubMed.
- J. Shamsi, A. L. Abdelhady, S. Accornero, M. Arciniegas, L. Goldoni, A. R. S. Kandada, A. Petrozza and L. Manna, ACS Energy Lett., 2016, 1, 1042–1048 CrossRef CAS PubMed.
- T. Baikie, Y. Fang, J. M. Kadro, M. Schreyer, F. Wei, S. G. Mhaisalkar, M. Graetzel and T. J. White, J. Mater. Chem. A, 2013, 1, 5628–5641 RSC.
- K.-H. Wang, L.-C. Li, M. Shellaiah and K. Wen Sun, Sci. Rep., 2017, 7, 13643 CrossRef PubMed.
- Y. Liu, Z. Yang, D. Cui, X. Ren, J. Sun, X. Liu, J. Zhang, Q. Wei, H. Fan, F. Yu, X. Zhang, C. Zhao and S. Liu, Adv. Mater., 2015, 27, 5176–5183 CrossRef CAS PubMed.
- C. Quarti, E. Mosconi, J. M. Ball, V. D'Innocenzo, C. Tao, S. Pathak, H. J. Snaith, A. Petrozza and F. De Angelis, Energy Environ. Sci., 2016, 9, 155–163 RSC.
- P. P. Boix, S. Agarwala, T. M. Koh, N. Mathews and S. G. Mhaisalkar, J. Phys. Chem. Lett., 2015, 6, 898–907 CrossRef CAS PubMed.
- E. Edri, S. Kirmayer, M. Kulbak, G. Hodes and D. Cahen, J. Phys. Chem. Lett., 2014, 5, 429–433 CrossRef CAS PubMed.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS PubMed.
- X. Liang, R. W. Baker, K. Wu, W. Deng, D. Ferdani, P. S. Kubiak, F. Marken, L. Torrente-Murciano and P. J. Cameron, React. Chem. Eng., 2018, 3, 640–644 RSC.
- F. Krieg, S. T. Ochsenbein, S. Yakunin, S. ten Brinck, P. Aellen, A. Süess, B. Clerc, D. Guggisberg, O. Nazarenko, Y. Shynkarenko, S. Kumar, C.-J. Shih, I. Infante and M. V. Kovalenko, ACS Energy Lett., 2018, 3, 641–646 CrossRef CAS PubMed.
- A. J. Knight and L. M. Herz, Energy Environ. Sci., 2020, 13, 2024–2046 RSC.
- T. Zhang, C. Hu and S. Yang, Small Methods, 2020, 4, 1900552 CrossRef CAS.
- N. Rybin, D. Ghosh, J. Tisdale, S. Shrestha, M. Yoho, D. Vo, J. Even, C. Katan, W. Nie, A. J. Neukirch and S. Tretiak, Chem. Mater., 2020, 32, 1854–1863 CrossRef CAS.
- D. Meggiolaro, E. Mosconi and F. De Angelis, ACS Energy Lett., 2019, 4, 779–785 CrossRef CAS.
- L. Yuan, R. Patterson, X. Wen, Z. Zhang, G. Conibeer and S. Huang, J. Colloid Interface Sci., 2017, 504, 586–592 CrossRef CAS PubMed.
- A. J. Knight, A. D. Wright, J. B. Patel, D. P. McMeekin, H. J. Snaith, M. B. Johnston and L. M. Herz, ACS Energy Lett., 2019, 4, 75–84 CrossRef CAS.
- Z. Xiao, L. Zhao, N. L. Tran, Y. L. Lin, S. H. Silver, R. A. Kerner, N. Yao, A. Kahn, G. D. Scholes and B. P. Rand, Nano Lett., 2017, 17, 6863–6869 CrossRef CAS PubMed.
- M. Abdi-Jalebi, Z. Andaji-Garmaroudi, S. Cacovich, C. Stavrakas, B. Philippe, J. M. Richter, M. Alsari, E. P. Booker, E. M. Hutter, A. J. Pearson, S. Lilliu, T. J. Savenije, H. Rensmo, G. Divitini, C. Ducati, R. H. Friend and S. D. Stranks, Nature, 2018, 555, 497 CrossRef CAS PubMed.
- J.-N. Yang, Y. Song, J.-S. Yao, K.-H. Wang, J.-J. Wang, B.-S. Zhu, M.-M. Yao, S. U. Rahman, Y.-F. Lan, F.-J. Fan and H.-B. Yao, J. Am. Chem. Soc., 2020, 142, 2956–2967 CrossRef CAS PubMed.
- R. G. Balakrishna, S. M. Kobosko and P. V. Kamat, ACS Energy Lett., 2018, 3, 2267–2272 CrossRef CAS.
- F. Zhang, H. Zhong, C. Chen, X.-G. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS PubMed.
- H. Huang, A. S. Susha, S. V. Kershaw, T. F. Hung and A. L. Rogach, Adv. Sci., 2015, 2, 1500194 CrossRef PubMed.
- Y. Tong, F. Ehrat, W. Vanderlinden, C. Cardenas-Daw, J. K. Stolarczyk, L. Polavarapu and A. S. Urban, ACS Nano, 2016, 10, 10936–10944 CrossRef CAS PubMed.
- T. Chiba, Y. Takahashi, J. Sato, S. Ishikawa, H. Ebe, K. Tamura, S. Ohisa and J. Kido, ACS Appl. Mater. Interfaces, 2020, 12, 45574–45581 CrossRef CAS PubMed.
- Y. Liu, F. Li, Q. Liu and Z. Xia, Chem. Mater., 2018, 30, 6922–6929 CrossRef CAS.
- H.-W. Chen, R.-D. Zhu, J. He, W. Duan, W. Hu, Y.-Q. Lu, M.-C. Li, S.-L. Lee, Y.-J. Dong and S.-T. Wu, Light: Sci. Appl., 2017, 6, e17043 CrossRef CAS PubMed.
- J.-H. Jou, S. Kumar, A. Agrawal, T.-H. Li and S. Sahoo, J. Mater. Chem. C, 2015, 3, 2974–3002 RSC.
- H. F. Haneef, A. M. Zeidell and O. D. Jurchescu, J. Mater. Chem. C, 2020, 8, 759–787 RSC.
- H. Liang, F. Yuan, A. Johnston, C. Gao, H. Choubisa, Y. Gao, Y.-K. Wang, L. K. Sagar, B. Sun, P. Li, G. Bappi, B. Chen, J. Li, Y. Wang, Y. Dong, D. Ma, Y. Gao, Y. Liu, M. Yuan, M. I. Saidaminov, S. Hoogland, Z.-H. Lu and E. H. Sargent, Adv. Sci., 2020, 7, 1903213 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tc04667a |
| This journal is © The Royal Society of Chemistry 2021 |