
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Suppressing dimer formation by increasing conformational freedom in multi-carbazole thermally activated delayed fluorescence emitters†
Lubna
Salah
 a,
Marc K.
Etherington
*bc,
Ali
Shuaib
d,
Andrew
Danos
c,
Ahmed A.
Nazeer
a,
Basma
Ghazal
a,
Antonio
Prlj
e,
Andrew T.
Turley
e,
Abhijit
Mallick
e,
Paul R.
McGonigal
e,
Basile F. E.
Curchod
e,
Andrew P.
Monkman
c and
Saad
Makhseed
*a
a,
Marc K.
Etherington
*bc,
Ali
Shuaib
d,
Andrew
Danos
c,
Ahmed A.
Nazeer
a,
Basma
Ghazal
a,
Antonio
Prlj
e,
Andrew T.
Turley
e,
Abhijit
Mallick
e,
Paul R.
McGonigal
e,
Basile F. E.
Curchod
e,
Andrew P.
Monkman
c and
Saad
Makhseed
*a
aDepartment of Chemistry, Faculty of Science, Kuwait University, P.O. Box 5969, Safat-13060, Kuwait. E-mail: saad.makhseed@ku.edu.kw
bDepartment of Mathematics, Physics & Electrical Engineering, Northumbria University, Ellison Place, Newcastle upon Tyne, NE1 8ST, UK. E-mail: marc.k.etherington@northumbria.ac.uk
cDepartment of Physics, Durham University, South Road, Durham, DH1 3LE, UK
dBiomedical Engineering Unit, Department of Physiology, Faculty of Medicine, Kuwait University, P.O. Box 24923, Safat-13110, Kuwait
eDepartment of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK
First published on 18th November 2020
Abstract
Ideal emitters for organic light-emitting diodes (OLEDs) are capable of efficiently harvesting non-emissive triplet states, have high colour stabilities, and possess high photoluminescence quantum yields (PLQYs). Maintaining colour stability and PLQY is particularly challenging for multi-carbazole thermally activated delayed fluorescence (TADF) materials that form persistent dimers due to intermolecular interactions of their extended aromatic systems (with altered electronic states). Addressing this challenge, three new emitters are presented, which demonstrate that, somewhat counterintuitively, sterically uncrowded acceptor units can suppress these undesirable interactions. They do so by allowing the surrounding carbazole donors to be arranged with lower dihedral angles, which in turn limits their availability for dimerization. A new pyrazine-centered emitter 4CzPyz is contrasted directly with the cyanopyridine and terephthalonitrile analogues, 4CzCNPy and 4CzTPN respectively. The pyrazine derivative demonstrates enhanced colour stability in the solid-state compared to the cyanopyridine and terephthalonitrile acceptors, which we assign to its absence of intermolecular face-to-face aromatic interactions. This suppression of dimer formation is shared by two cyanopyrazine emitters 2Cz2CNPyz and 3CzCNPyz, each of which feature reduced steric pressure and flatter Cz-Pyz dihedral angles than non-heterocyclic analogues. Flatter dihedral angles consequently lead to C–H bonds of the Cz donors extending outwards at angles that prevent the stacking required for dimerization. This expanded understanding of dimer formation in TADF materials will guide future efforts to maintain colour stability in higher performance TADF materials by curbing the prevalence of face-to-face aromatic interactions.
Introduction
To meet the continuing technological demand for stable and efficient blue organic light-emitting diodes (OLEDs), synthetic chemists have been engaged in developing emissive organic compounds based on rapidly changing design principles. Towards this ultimate goal, in 2012, Uoyama et al.1 presented fluorescent OLEDs with superior external quantum efficiencies (EQEs) by virtue of triplet harvesting through a thermally activated delayed fluorescence (TADF) mechanism.2–5 The TADF mechanism allows an emissive material to achieve an internal quantum efficiency (IQE) of unity without the need to incorporate heavy elements in the structures. It provides a counterpoint to the phosphorescent mechanism that predominates in emitters based around iridium or other heavy metal atoms.6–9 The high IQEs of TADF emitters can translate to impressive EQEs. Consequently, the optimization of TADF-active structures has stimulated significant research interest in the past few years.4,10–14However, applying the TADF mechanism towards stable and efficient blue OLEDs has not been straightforward. Current state-of-the-art materials still struggle in terms of roll-off efficiency and device operational lifetime. As the experimental evidence and theoretical modeling surrounding TADF increases, a complex picture has emerged. The initial design principle of creating a small energy gap between the lowest excited singlet and triplet states is no longer accepted as being sufficient on its own.1,12,15,16 Charge transfer (CT) excitonic character, achieved by separation of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), is also required to reduce the exchange energy between the charge-transfer singlet (1CT) and triplet (3CT). There must also be a locally-excited triplet state (3LE) to mediate the reverse intersystem crossing (rISC) process between 3CT and 1CT via a spin-vibronic coupling mechanism.3,17–19 In the design of TADF systems utilizing CT states there must also be a compromise between complete spatial separation of the HOMO and LUMO (i.e. pure CT) and maintaining high oscillator strength, which vanishes in a pure CT system.20 The sensitivity of CT states to their surrounding medium enables host-mediated tuning of the CT states. As such, the CT states can be tuned to be isoenergetic with the 3LE state, which causes a significant enhancement in the TADF efficiency.21,22 This potential for external tuning makes the choice of host a crucial factor in designing efficient TADF OLEDs.
Equally crucial, though largely overlooked, is the physical behaviour of the emitter in the solid state. In recent work23 Etherington et al. have shown that the concentration of the emitter plays an important role in its TADF efficiency: supramolecular dimer formation brought about by intermolecular face-to-face aromatic interactions can fundamentally alter the TADF mechanism. This is surprisingly the case even at the low emitter concentrations used for device fabrication (including in samples prepared by thermal evaporation deposition methods). It was shown that two widely used multi-carbazole TADF systems, 4CzTPN and 4CzIPN (Fig. 1), are prone to dimerization, which may also be responsible for their mechanochromism24–28 and thermochromism.23 The investigation by Etherington et al. has been followed by further detailed investigation into the stimuli-responsive behaviour of TADF emitters13,29–34 and a recent mini-review.35
 | ||
| Fig. 1 The structures of 4CzTPN, 4CzIPN, 4CzCNPy, 2Cz2CNPyz, 3CzCNPyz and 4CzPyz. | ||
This previous work established that dimerization causes significant shifts in the photoluminescence and electroluminescence colour (1CT emission energy) of the TADF materials, beyond that expected by packing and polarizability effects alone. The presence of intermolecular aromatic interactions complicates attempts to (i) tune emission colour by changing the host and (ii) reliably reproduce colour and high efficiency in devices.23 The need to suppress dimerization and obtain more predictable emission tuning is, therefore, an additional emerging design criterion for future TADF emitters. Emitters must not only display efficient TADF, have a high PLQY, and perform well in a suitable host; they must also be designed to minimize intermolecular face-to-face aromatic interactions that can have unpredictable secondary impacts on their optical properties.
Here, we present three new compounds that show reduced susceptibility towards dimer formation. Firstly, 4CzPyz is a close structural analogue of both the initial phthalonitrile materials presented by Uoyama et al.,1 particularly 4CzTPN, as well as a previously published cyanopyridine 4CzCNPy.36–38 By comparing new measurements and reported data for this series, we analyze how the acceptor unit affects aromatic interactions in multi-carbazole TADF emitters. The study of acceptor effects on multi-carbazole functional properties such as TADF and room temperature phosphorescence is a topic of recent interest.23,39 We show that 4CzPyz does not exhibit the large emission shifts between high polarity solution and solid-state that 4CzCNPy and 4CzTPN are subject to, establishing 4CzPyz as the most colour stable of the three emitters.
Alongside 4CzPyz we also present 2Cz2CNPyz and 3CzCNPyz; related cyanopyrazine systems that also show resistance towards dimer formation. The additional strongly electron accepting nitrile groups impart higher ground state dipole moments and stronger CT character. Consequently, they are more susceptible to host tuning effects and show significant redshifts in dilute concentration films and low polarity solvents. Due to their better matched acceptor strength they also possess greater intersystem crossing (ISC) and rISC rates but lower PLQYs.
Rationalizing the full set of experimental observations, we put forward that careful design of steric space in the acceptors of multi-carbazole TADF materials can reduce their tendency to undergo dimer formation. The pyrazine motifs, in particular, appear promising for future design of more predictable TADF compounds.
Results and discussion
The structural formulas of the newly synthesized compounds 2Cz2CNPyz, 3CzCNPyz and 4CzPyz are shown in Fig. 1, alongside the previously published materials, 4CzTPN, 4CzIPN and 4CzCNPy. Synthetic procedures and characterization data for 2Cz2CNPyz, 3CzCNPyz, 4CzPyz and 4CzCNPy can be found in Scheme S1, Fig. S1–S12 and Table S1 in the ESI,† which have allowed comparison with procedures and data reported previously for 4CzTPN, 4CzIPN and 4CzCNPy.1,23,36Steady-state emission
Molecules exhibiting TADF often exhibit CT character and are sensitive to environmental factors, such as solution polarity or host polarizability. Some studies have attempted to rationalize emission shifts with increasing concentration by invoking solid-state solvation effects (SSSE), analogous to those experienced in the solution state, which are dependent on the high and low frequency polarizabilities of the medium.22,40–42However, Etherington et al.23 have recently shown that the large variation in emission colour of 4CzTPN (onset energies of 2.65 eV in methylcyclohexane (MCH) and 2.3 eV in neat film) cannot arise because of SSSE,43 but is instead caused by intermolecular face-to-face (π–π) aromatic interactions.44–47 The aromatic interactions account for both the low solubility and the drastic colour change 4CzTPN undergoes when transitioning from monomeric species (dilute in high polarity solutions such as acetonitrile, MeCN, 2.5 eV), to aggregated states such as thin films and crystals (2.3 eV).23 By comparing previously reported photophysical data we identify that the structurally similar cyanopyridine analogue 4CzCNPy also shows significant shifts in emission between dimethylformamide (DMF, 2.63 eV)38 and neat film (2.5 eV).36–38 These large spectral shifts indicate the presence of face-to-face aromatic interactions in this system too. In strong contrast, the emission of our newly synthesized compound, 4CzPyz (Table 1 and Fig. 2a), barely changes across a broad range of sample types and concentration. Revealingly, the emission onset in neat film (2.8 eV) is the same as that in highly polar MeCN solution (2.8 eV) precluding the presence of a dominant low-energy dimer species in film. The results are summarized in Table S8 (ESI†). As this colour stability of 4CzPyz is highly desirable in multi-carbazole TADF emitters, we have used a variety of techniques to explain its origin and probe additional effects of acceptor alteration.
| Compound | Zeonex | DPEPO | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PF | DF | PH | PF–PH | DF–PH | PF | DF | PH | PF–PH | DF–PH | |
| 2Cz2CNPyz | 2.79 | 2.65 | 2.53 | 0.26 | 0.12 | 2.66 | 2.61 | 2.50 | 0.15 | 0.11 |
| 3CzCNPyz | 2.77 | 2.69 | 2.45 | 0.32 | 0.24 | 2.72 | 2.60 | 2.41 | 0.29 | 0.19 |
| 4CzPyz | 2.92 | 2.81 | 2.52 | 0.40 | 0.29 | 2.84 | 2.73 | 2.50 | 0.34 | 0.23 |
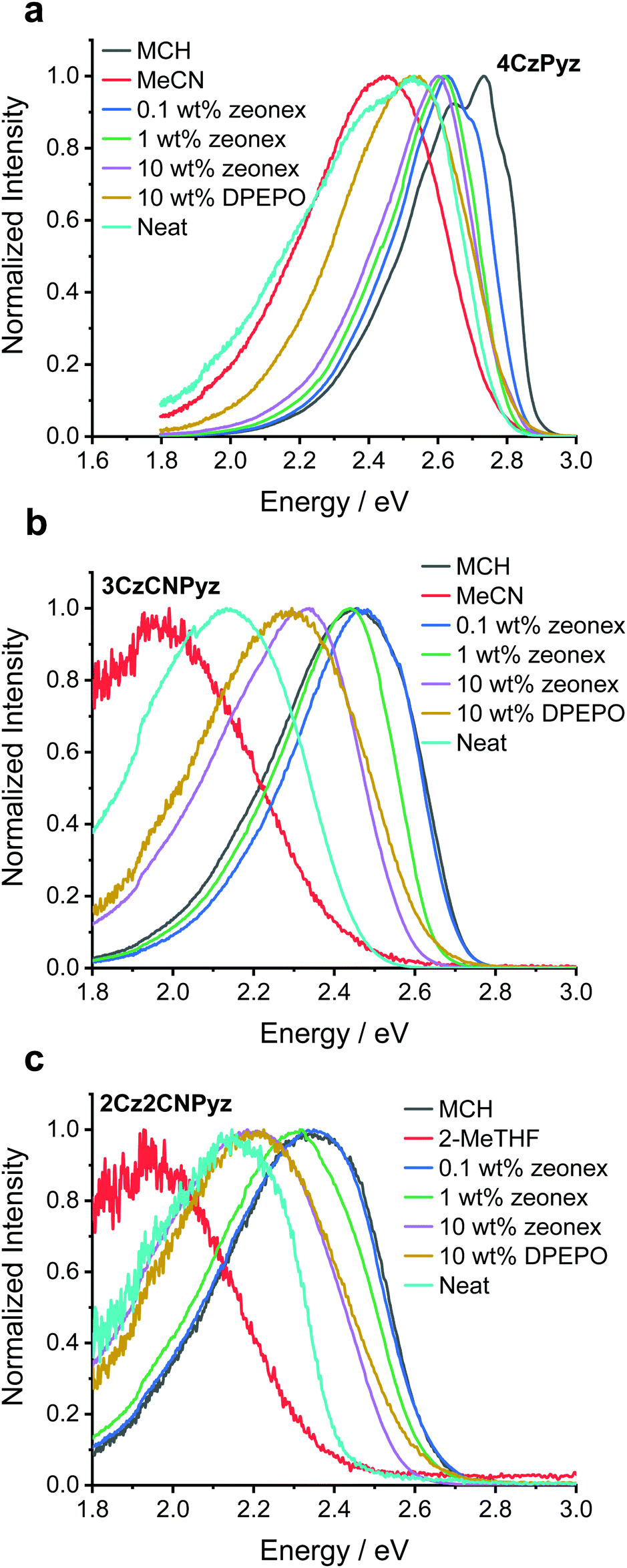 | ||
| Fig. 2 The photoluminescence of 10 μM solutions in methylcyclohexane (MCH) and acetonitrile (MeCN), doped zeonex and DPEPO films and neat films of (a) 4CzPyz (b) 3CzCNPyz and (c) 2Cz2CNPyz. [N.B. For 2Cz2CNPyz 2-MeTHF replaces MeCN as negligible signal-to-noise ratio was observed in the MeCN sample, most likely a result of severe relaxation of the CT state and associated increase in non-radiative decay]. | ||
Analysis of X-ray crystallographic data (Tables S2–S7 and Fig. S13–S27, ESI†) gives insights into the structural origins of the improved photophysical behaviour of 4CzPyz. For context the interlayer distance between sheets of graphite is 3.354 Å.48,49 The carbazole groups of 4CzTPN and 4CzCNPy take part (Fig. 3a–c and Fig. S27, ESI†) in offset, face-to-face (π–π) aromatic interactions in the solid state. For 4CzTPN the face-to-face (π–π) aromatic interactions occur intermolecularly and intramolecularly with average distances between centroids of the aromatic units in the range 3.63–3.80 Å and 4.01–4.21 Å respectively. These distances are consistent with close contacts between the aromatic groups similar to those seen in graphite, but with an offset between the centres of the rings. For 4CzCNPy only intramolecular face-to-face (π–π) aromatic interactions occur with an average distance in the range 3.97–4.15 Å. It is known that close π–π contacts between carbazoles are linked to emission redshift,47 and some of the interaction distances are short enough to allow intermolecular electronic communication, explaining the phase-dependent emission colours of 4CzTPN and 4CzCNPy and demonstrating the need to consider these interactions seriously when analysing photophysical properties. By contrast, the intermolecular contacts in the solid-state superstructure of 4CzPyz are edge-to-face (C–H⋯π) contacts (Fig. 3d–f), of which the closest distance is 3.99 Å. This packing by C–H⋯π interactions and the absence of any intermolecular π–π close contacts are supported by Hirshfeld analysis (Fig. S21, ESI†), which shows that the only contact closer than the sum of van der Waals radii (red regions) in the superstructure corresponds to the C–H⋯π interaction shown in Fig. 3e.50
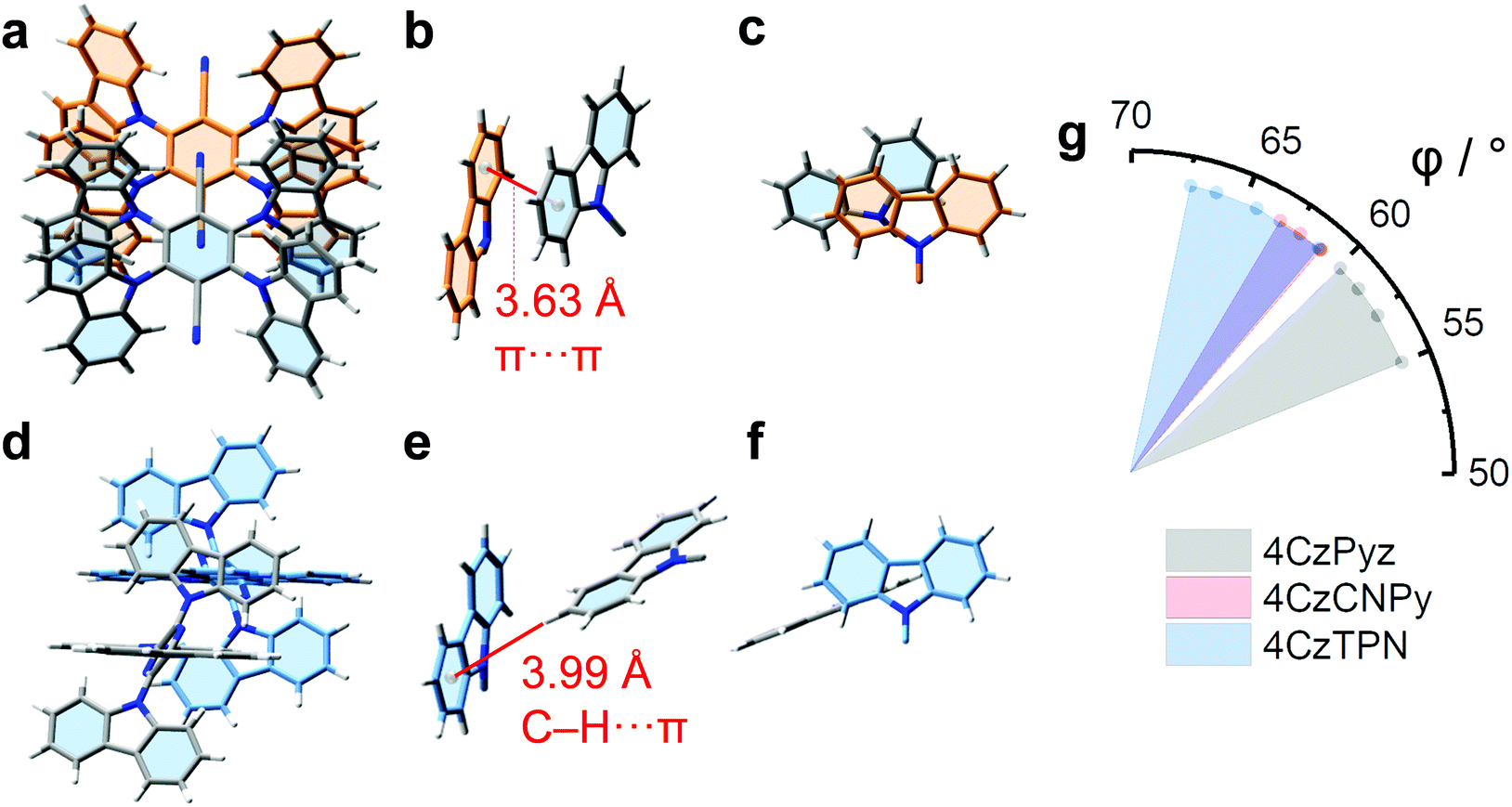 | ||
| Fig. 3 The crystal structures of 4CzTPN and 4CzPyz. (a) 4CzTPN packing behaviour, (b) two carbazoles of neighbouring molecules labelled to show the shortest distance between centroids of their aromatic rings and (c) a view showing their overlap. (d) 4CzPyz packing behaviour, (e) two carbazoles of neighbouring molecules labelled to show the C–H⋯π close contact and (f) their orthogonal orientations. (g) A plot of the angles (φ) between the acceptor unit plane (six-membered ring) and the carbazole unit planes (five-membered ring). The circular symbols represent the individual angles of the four carbazole units in each compound and the filled area highlights the range that they cover. | ||
Unlike the highly ordered environment of a single crystal, there is potential for molecules of 4CzPyz to pack with many different relative D–A orientations in amorphous neat films. Importantly for the maintaining of colour stability with increasing concentration we observe that the photophysical properties remain constant for 4CzPyz at a range of doping concentrations (from 0.1–10 wt%) in zeonex films. Colour stability is defined here as a molecule's ability to maintain spectral shape with respect to an external factor. In this particular work the external factor is concentration of the molecule in doped films. However, observation (Fig. 2a) of a low-energy shoulder (2.4 eV) in the emission spectrum of a neat film suggests amorphous phases of 4CzPyz may not be completely free from face-to-face aromatic interactions or conformational effects identified by density functional theory (DFT) calculations (Fig. S57, ESI†). This low-energy shoulder is also apparent by time-resolved emission spectroscopy (see Fig. S31, S34 and Table S11, ESI†) and has behaviour consistent with dimer (CTD) species observed elsewhere.23
The higher energy monomer CT emission (CTM) of 4CzPyz is confirmed through analysis of the spectral lineshape in toluene solution (Fig. S35, S38 and Table S14, ESI†). Although dimer formation has not been totally eliminated, this shoulder is still only a very small contribution to the emission, compared to the dominant and persistent dimers observed in 4CzTPN and 4CzIPN in earlier work.23 Thus, the pyrazine core of 4CzPyz effectively suppresses the undesirable aromatic interactions that afflict TADF emitters with isophthalonitrile (e.g., 4CzIPN), terephthalonitrile (e.g., 4CzTPN) and cyanopyridine (e.g., 4CzCNPy) acceptors. Crucially, the data also show that the presence of multiple carbazole units does not inevitably lead to deleterious intermolecular π–π interactions. Even at high concentrations, 4CzPyz maintains colour stability, making it an attractive design for new TADF emitters.
This immunity to dimer formation is shared across the series of 2Cz2CNPyz, 3CzCNPyz and 4CzPyz (Fig. 1) and is reflected in their 1931 CIE coordinates in comparison to 4CzIPN.23 For the three new compounds (Fig. 4) the shifts with increasing concentration in zeonex A–C are much more restrained compared to 4CzIPN A′–C (Fig. S30, ESI†). This again, affirms the lack of interactions in the new molecules and their improved colour stability. Similarly, to 4CzPyz, analyses of the crystal packing and Hirshfeld surfaces of the cyanopyrazines (see Fig. S15 and S18, ESI†) show an absence of significant π–π close contact points. The absence of interaction between the π–surfaces is demonstrated in the steady-state emission and time-resolved photoluminescence of the neat films (Fig. S31–S33 and Tables S9, S10, ESI†) i.e. no red-shifted species are observed, and the emission spectra are consistent with the time-resolved photoluminescence spectra of the compounds in toluene solution (Fig. S35–S37 and Tables S12, S13, ESI†). These observations imply that dimer formation does not occur to a significant extent in these two systems.
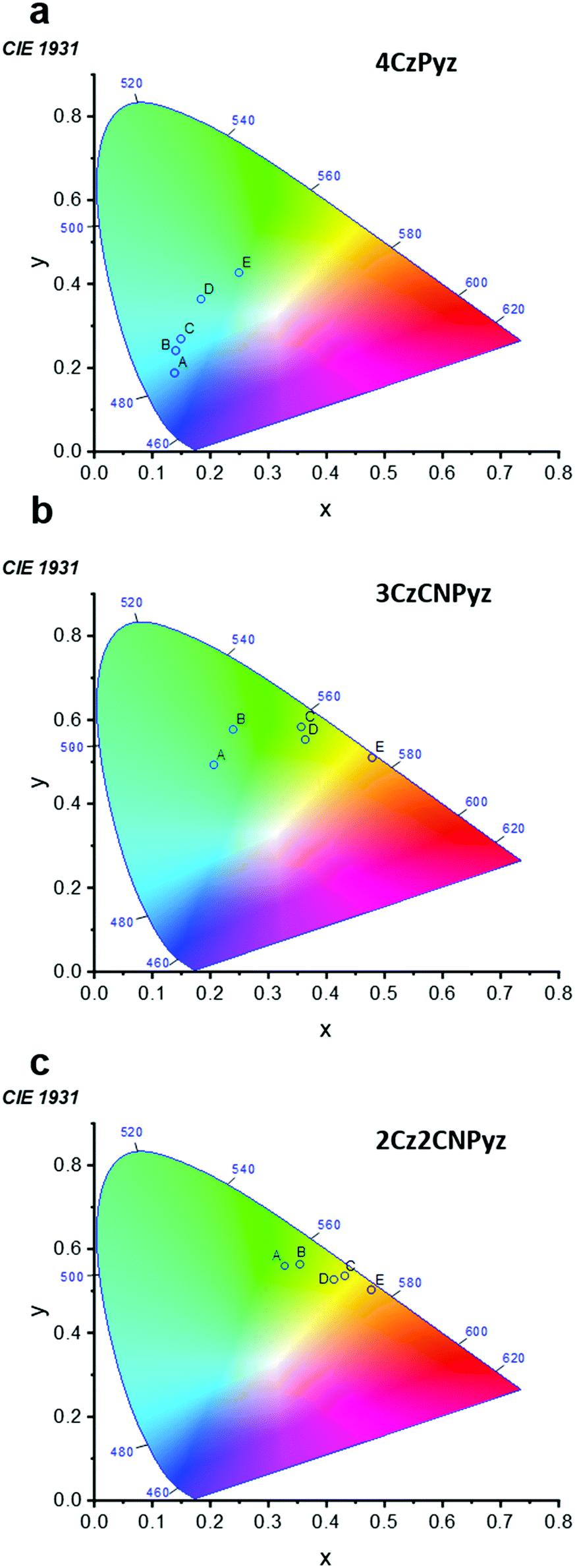 | ||
| Fig. 4 The CIE 1931 curves for the photoluminescence of (a) 4CzPyz, (b) 3CzCNPyz and (c) 2Cz2CNPyz in a series of films. A = 0.1 wt% zeonex, B = 1 wt% zeonex, C = 10 wt% zeonex, D = 10 wt% DPEPO and E = neat film. | ||
Inspection of the structural formulas shows the compounds with the greatest propensity to form dimer states share a common structural motif: carbazole groups that are flanked by two multiatom (nitrile or carbazole) substituents. We propose that this ortho relationship to two nitrile and/or carbazole groups (as opposed to neighbouring a less bulky ring nitrogen) limits the torsional freedom of the carbazole unit(s), which in turn influences the packing behaviour and intermolecular interactions. In the emitters with high colour stability with respect to concentration (2Cz2CNPyz, 3CzCNPyz, and 4CzPyz) each and every carbazole unit is positioned with an α relationship to a ring nitrogen. These carbazole units are therefore less hindered than the carbazole units in 4CzCNPy and 4CzTPN, so take up conformations with smaller dihedral angle. The torsion angles measured (Tables S3–S7, ESI†) between the carbazoles and central acceptor unit are consistent with this interpretation; the D–A dihedral angles of the carbazoles of 2Cz2CNPyz, 3CzCNPyz, and 4CzPyz are in the range 36–68°, whereas consistently higher torsion angles in the range 48–83° are found for 4CzCNPy and 4CzTPN. This interpretation is reinforced by DFT calculations of systems with unhindered carbazole units,51 including one next to a pyrazine spacer,52 which have estimated angles of <50° for the carbazole unit (similar to those measured for 2Cz2CNPyz, 3CzCNPyz, and 4CzPyz). In recent work by Woo et al.53 DFT calculations have shown that carbazole units around a highly congested central ring instead have dihedral angles of >60°, similar to those in 4CzCNPy and 4CzTPN. High torsion angles, >60°, of carbazole units have also been observed in the single crystal structure of a highly crowded TADF emitter in work by Yuan et al.34
The relaxed (flatter) dihedral angles of the carbazole units in 2Cz2CNPyz, 3CzCNPyz, and 4CzPyz mean that some C–H bonds will protrude at shallower angles from the compound preventing regular stacking of neighbouring molecules. For 4CzCNPy, 4CzTPN and 4CzIPN the carbazole units are more perpendicular meaning the C–H bonds protrude in such a way as to leave the π systems exposed to dimerization. Comparing the carbazole plane angles of 4CzPyz, 4CzCNPy and 4CzTPN (Fig. 3g) shows that 4CzPyz has lower angles between the central ring and its carbazoles than observed for the other two CN containing compounds. Engineering the steric environment of multi-carbazole emitters to enable these shallow angles and control the conformational freedom of carbazole units is therefore identified as crucial to suppressing face-to-face (π–π) aromatic interactions.
Having found that the pyrazine core alters the steric environment and reduces the π–π interactions in 2Cz2CNPyz, 3CzCNPyz and 4CzPyz, we now also consider the effect of additional nitrile groups on their emission properties and kinetics.
As demonstrated in the steady-state emission spectra (Fig. 2), 2Cz2CNPyz and 3CzCNPyz show significant shifts between dilute zeonex (a cyclo-olefin polymer, with large free space and dielectric constant similar to MCH) and neat films. However, these are much smaller than the solvatochromic shifts observed in solution (Fig. 2 and Fig. S29, ESI†) and we attribute these shifts in the solid-state to changes in packing and other host effects with increasing dopant concentrations.
In contrast, 4CzPyz shows limited shift in emission wavelength between zeonex and neat film, as well as a very small solvatochromic shift in solution. These results are consistent with its smaller dipole moment, low polarizability and weaker CT character (without any CN groups to augment its acceptor strength). The emission energies of 2Cz2CNPyz, 3CzCNPyz and 4CzPyz in MCH are supported by calculations (Tables S25–S29 and Fig. S57, S58, ESI†); the theoretically predicted emission energies for a single molecule in vacuum (Table S29, ESI†) are largely consistent with the emission data presented in Fig. 2. The calculations also identify two near degenerate conformers (Fig. S57, ESI†) which are likely the cause of the double peak that 4CzPyz displays in MCH solvent. The larger calculated ground state dipole moments (5.51 D and 4.50 D for 2Cz2CNPyz and 3CzCNPyz, respectively) and stronger excited state CT character mean that the cyanopyrazines emit much further in the red in the solid-state compared to 4CzPyz (0.01 D and 0.64 D for the two conformers).
The differences in dipole moments for these systems are related to the number of nitrile groups present in 2Cz2CNPyz and 3CzCNPyz. The nitrile groups induce stronger CT character. However, the trend is not straightforward, as an increasing number of carbazole donors also increases the CT character of the compound. Consequently, 3CzCNPyz has a higher calculated CT number54 than 2Cz2CNPyz (Table S27, ESI†). While the calculations do not take into account environmental effects that can influence the measured emission, they help explain the general trend observed. Similarly, this increase in the CT character of the system with increasing number of nitrile units is also demonstrated in the comparison between 4CzPyz, 4CzCNPy and 4CzTPN, where the increasing number of nitrile groups going from 4CzPyz to 4CzTPN cause redder emission, even in apolar solvents (see Fig. 2 and literature reports23,36,38). The differences in CT character due to different substituents has been widely discussed in the literature and we use this framework to rationalize how the additional nitrile groups going from 4CzPyz to 2Cz2CNPyz influence the delayed fluorescence, phosphorescence energy and photoluminescence quantum yield.
Delayed fluorescence
Considering the room temperature kinetics of the 1 wt% zeonex films, all three systems show delayed fluorescence (DF) (Fig. 5 and 6). The ratio of the DF component to the PF component increases across the pyrazine series from 4CzPyz to 3CzCNPyz to 2Cz2CNPyz (Fig. 6). The singlet–triplet gap was found to be smallest for 2Cz2CNPyz at 0.12 eV, whereas 3CzCNPyz and 4CzPyz have a singlet–triplet gap greater than 0.2 eV (see Fig. 5 and Table 1), which is consistent with the differences in kinetics and contribution of DF shown in Fig. 6.2Cz2CNPyz has a narrower singlet–triplet gap as a result of its higher CT character, due to the presence of the nitrile units. Accordingly, 2Cz2CNPyz exhibits the shortest DF lifetime of 10.9 μs compared to 1.62 ms and 1.47 ms for 3CzCNPyz and 4CzPyz, respectively. Additionally, 2Cz2CNPyz has a second prompt component which is attributed to the locally excited singlet state (1LE) of this compound (see Fig. 5 and Fig. S40, ESI†). Consulting the lineshapes of these three emitters and comparing them to the lineshapes observed by Etherington et al.,23 there is no significant shift in the emission energy with time. This observation is consistent with the suppressed dimer formation in 2Cz2CNPyz, 3CzCNPyz and 4CzPyz in the zeonex film at this concentration. The full time-resolved emission study in zeonex can be found in Fig. S39–S42 (ESI†) and the summary of the fits for both room temperature and 80 K measurements are shown in Tables S15–S17 (ESI†).
 | ||
| Fig. 5 The prompt and delayed emission of (a) 4CzPyz, (b) 3CzCNPyz and (c) 2Cz2CNPyz at 1 wt% (solid) and 0.1 wt% (dashed) in zeonex. There is also an additional 0.01 wt% concentration for 2Cz2CNPyz represented by dotted lines. | ||
Doping the compounds in a DPEPO host (bis[2-(diphenylphosphino)phenyl] ether oxide, a small molecule OLED host with a higher dielectric than zeonex) causes shifts in the singlet emission energies and changes in the singlet–triplet gaps (Table 1), as measured from Fig. S43 (ESI†). The full time-resolved spectra are shown in Fig. S44–S46 (ESI†). It is also immediately apparent that, compared to the zeonex decay lifetimes (Fig. 6), the rISC is more rapid for all systems in DPEPO with the DF components having lifetimes of 4.00 μs, 99.2 μs and 174 μs for 2C2CNPyz, 3CzCNPyz and 4CzPyz, respectively (Fig. 7).
 | ||
| Fig. 6 The time-resolved photoluminescence decays of (a) 4CzPyz, (b) 3CzCNPyz and (c) 2Cz2CNPyz in a 1 wt% zeonex film at room temperature (RT). | ||
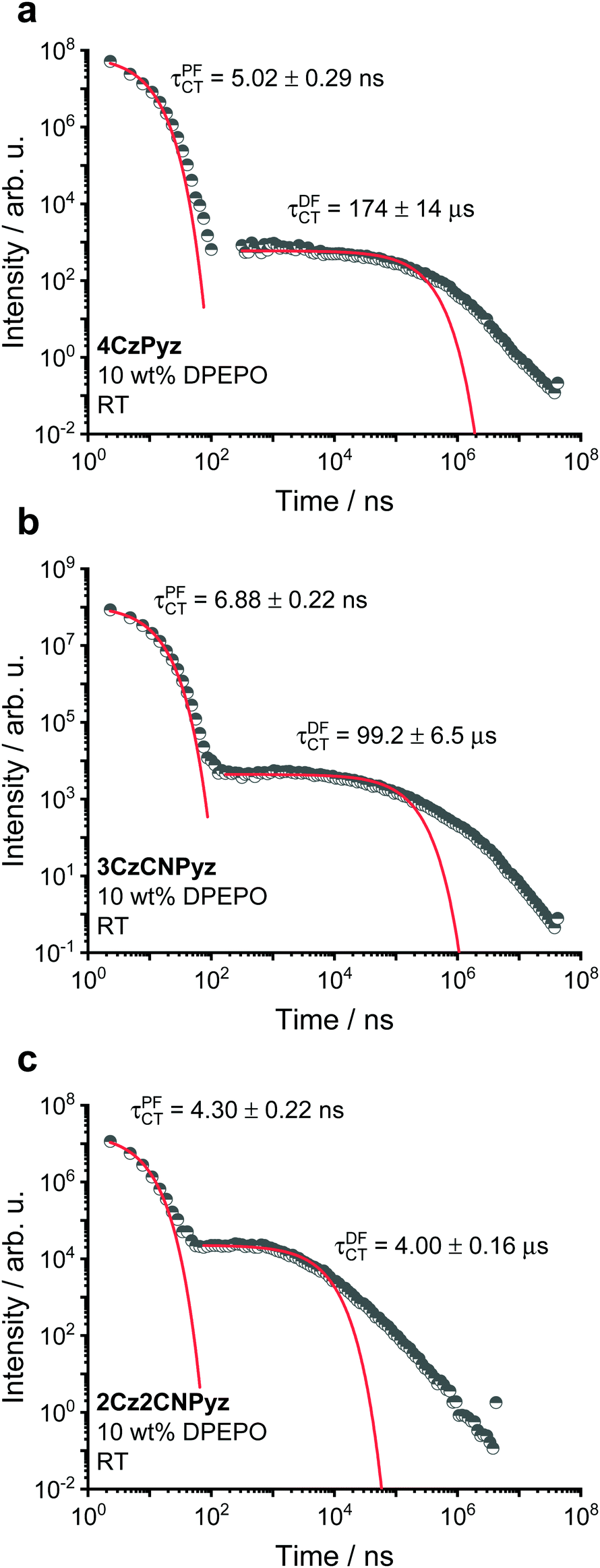 | ||
| Fig. 7 The time-resolved photoluminescence kinetics of 10 wt% DPEPO films of (a) 4CzPyz, (b) 3CzCNPyz and (c) 2Cz2CNPyz measured at room temperature. | ||
This improvement in TADF behaviour is consistent with a reduction in the singlet–triplet gap due to host tuning.55 The size of the emission shift from zeonex to DPEPO follows the trend of acceptor strength increasing from 4CzPyz to 2Cz2CNPyz. The emission kinetics for the three systems at 80 K for 10 wt% in DPEPO host are shown in Fig. S47 and a summary of the kinetic fittings for both room temperature and 80 K measurements are shown in Tables S18–S20 (ESI†).
To confirm that the DF in the films originates from TADF, the intensity of the emission as a function of laser energy was measured for the three pyrazine compounds in the various hosts (see Fig. S48 and S49, ESI†). The slope in each plot is close to unity, demonstrating that the delayed emission is a monomolecular process and, thus, the most likely origin is TADF.
Overall, the varying numbers of nitrile and carbazole groups around the pyrazine cores of 2Cz2CNPyz, 3CzCNPyz and 4CzPyz do not change the desirable packing properties as the carbazoles always have at least one α positioned ring nitrogen resulting in a relatively uncrowded steric environment. These changes do cause a change in CT character though, with the net result of smaller singlet–triplet gaps and the highest TADF activity in the most redshifted compound, 2Cz2CNPyz.
| Compound | 1 wt% zeonex | 10 wt% DPEPO |
|---|---|---|
| 2Cz2CNPyz | 34 | 11 |
| 3CzCNPyz | 68 | 24 |
| 4CzPyz | 75 | 36 |
Due to its weak accepting strength, negligible dipole moment and high LE character, 4CzPyz has a high emission oscillator strength. With a large singlet–triplet energy gap it is also not particularly ISC active (leading to its small DF emission component in time resolved decays). Absence of a rapid ISC channel means that excitons avoid cycling through the dark triplet state, allowing this material to retain a high PLQY (Table 2). In contrast, 3CzCNPyz and 2Cz2CNPyz have increased CT character (lower oscillator strength) and a smaller singlet–triplet gap (increased ISC rates) which give a greater proportion of DF. While rISC and TADF will recycle some of the resulting triplet states for emission, this process is unlikely to be efficient in these materials (evidenced by their long DF lifetimes). As a result, the overall PLQY is lowest for 2Cz2CNPyz, with its strong CT character also bestowing it with high TADF activity. In this way, TADF reduces and PLQY increases going from 2Cz2CNPyz to 3CzCNPyz to 4CzPyz. The differences in PLQYs between samples doped into zeonex or DPEPO are likely a result of the smaller singlet–triplet gaps and generally increased ISC rates in the DPEPO films. The DPEPO also stabilizes the CT state of the compounds, with the associated reduction in LE character leading to lower oscillator strength and, again, a lower PLQY. The experimental PLQYs are consistent with the trend in the calculated oscillator strengths shown in Table S25 (ESI†).
Conclusions
We report a series of pyrazine-centered TADF emitters that are resistant to intermolecular dimerization—a pernicious phenomenon that has previously limited the applicability of some otherwise promising TADF materials. The three new TADF materials were synthesized and investigated by X-ray diffraction and time-resolved luminescence spectroscopy. Our results reveal that managing steric space and D–A angles contributes to suppressing carbazole dimer interactions, leading to enhanced colour stability. 4CzPyz, in particular, exhibits greatly enhanced colour stability. The “uncrowded” pyrazine core, also present in 2Cz2CNPyz and 3CzCNPyz, is identified as an important motif for manipulating the steric space and dihedral angles in a way that prevents dimer formation. This investigation extends our understanding and control of molecular interactions in multi-carbazole TADF emitters and provides design rules for ‘turning off’ dimer formation, which will be invaluable in the future design of colour-stable TADF materials.Author contributions
S. M., M. K. E. and A. P. M. conceived the original idea for investigation. L. S. synthesized the pyrazine compounds under the direction of S. M. Further synthesis and characterization was performed by L. S., B. G., A. A. N., A. T. T., A. M. and P. R. M. L. S., M. K. E., A. S. and A. D. performed the photophysical characterization measurements and analyzed the data. A. T. T. performed the Hirshfeld analysis of the crystal structures. A. P. and B. F. E. C. performed the theoretical calculations. The manuscript was written through contributions of all authors and all authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr Dmitry Yufit for solving the crystal structure of 4CzCNPy. L. S. and S. M. would like to thank Kuwait University Research administration (grant no. SC10/18) for funding this work, and Kuwait Foundation for the Advancement of Science-KFAS; RSP unit general facilities of the Faculty of Science GFS (GS 01/01, GS 02/01, GS 03/01, GS 01/03, GS 01/05, GS 02/13). M. K. E., A. D. and A. P. M. would like to thank the EU's Horizon 2020 research and innovation programme for funding the HyperOLED project under grant agreement No. 732013. A. T. T. acknowledges the EPSRC for a Doctoral Training Grant. A. M. and P. R. M. thank UUKi and BEIS for funding through a Rutherford Strategic Partner Grant.Notes and references
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS.
- N. A. Kukhta, H. F. Higginbotham, T. Matulaitis, A. Danos, A. N. Bismillah, N. Haase, M. K. Etherington, D. S. Yufit, P. R. McGonigal, J. V. Gražulevičius and A. P. Monkman, J. Mater. Chem. C, 2019, 7, 9184–9194 RSC.
- P. Stachelek, J. S. Ward, P. L. dos Santos, A. Danos, M. Colella, N. Haase, S. J. Raynes, A. S. Batsanov, M. R. Bryce and A. P. Monkman, ACS Appl. Mater. Interfaces, 2019, 11, 27125–27133 CrossRef CAS.
- F. Kessler, Y. Watanabe, H. Sasabe, H. Katagiri, M. K. Nazeeruddin, M. Grätzel and J. Kido, J. Mater. Chem. C, 2013, 1, 1070–1075 RSC.
- C. Fan and C. Yang, Chem. Soc. Rev., 2014, 43, 6439–6469 RSC.
- A. K. Pal, S. Krotkus, M. Fontani, C. F. R. Mackenzie, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuel and E. Zysman-Colman, Adv. Mater., 2018, 30, 1804231 CrossRef.
- S. C. F. Kui, P. Keong Chow, G. Cheng, C. C. Kwok, C. Lam Kwong, K. H. Low and C. M. Che, Chem. Commun., 2013, 49, 1497–1499 RSC.
- S. W. Li, C. H. Yu, C. L. Ko, T. Chatterjee, W. Y. Hung and K. T. Wong, ACS Appl. Mater. Interfaces, 2018, 10, 12930–12936 CrossRef CAS.
- P. L. dos Santos, J. S. Ward, D. G. Congrave, A. S. Batsanov, J. Eng, J. E. Stacey, T. J. Penfold, A. P. Monkman and M. R. Bryce, Adv. Sci., 2018, 5, 1700989 CrossRef.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- M. Yang, I. S. Park, Y. Miyashita, K. Tanaka and T. Yasuda, Angew. Chem., Int. Ed., 2020, 59, 13955–13961 CrossRef CAS.
- E. Spuling, N. Sharma, I. D. W. Samuel, E. Zysman-Colman and S. Bräse, Chem. Commun., 2018, 54, 9278–9281 RSC.
- T. Ogiwara, Y. Wakikawa and T. Ikoma, J. Phys. Chem. A, 2015, 119, 3415–3418 CrossRef CAS.
- J. Eng and T. J. Penfold, Chem. Rec., 2020, 20, 831–856 CrossRef CAS.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS.
- C. M. Marian, J. Phys. Chem. C, 2016, 120, 3715–3721 CrossRef CAS.
- X. Chen, S. Zhang, J. Fan and A. Ren, J. Phys. Chem. C, 2015, 119, 9728–9733 CrossRef CAS.
- X. Chen, Y. Tsuchiya, Y. Ishikawa, C. Zhong, C. Adachi and J. Brédas, Adv. Mater., 2017, 29, 1702767 CrossRef.
- P. L. Santos, J. S. Ward, P. Data, A. S. Batsanov, M. R. Bryce, F. B. Dias and A. P. Monkman, J. Mater. Chem. C, 2016, 4, 3815–3824 RSC.
- B. L. Cotts, D. G. McCarthy, R. Noriega, S. B. Penwell, M. Delor, D. D. Devore, S. Mukhopadhyay, T. S. De Vries and N. S. Ginsberg, ACS Energy Lett., 2017, 2, 1526–1533 CrossRef CAS.
- M. K. Etherington, N. A. Kukhta, H. F. Higginbotham, A. Danos, A. N. Bismillah, D. R. Graves, P. R. McGonigal, N. Haase, A. Morherr, A. S. Batsanov, C. Pflumm, V. Bhalla, M. R. Bryce and A. P. Monkman, J. Phys. Chem. C, 2019, 123, 11109–11117 CrossRef CAS.
- R. Pashazadeh, P. Pander, A. Lazauskas, F. B. Dias and J. V. Grazulevicius, J. Phys. Chem. Lett., 2018, 9, 1172–1177 CrossRef CAS.
- E. Skuodis, O. Bezvikonnyi, A. Tomkeviciene, D. Volyniuk, V. Mimaite, A. Lazauskas, A. Bucinskas, R. Keruckiene, G. Sini and J. V. Grazulevicius, Org. Electron., 2018, 63, 29–40 CrossRef CAS.
- B. Huang, W.-C. Chen, Z. Li, J. Zhang, W. Zhao, Y. Feng, B. Z. Tang and C.-S. Lee, Angew. Chem., Int. Ed., 2018, 57, 12473–12477 CrossRef CAS.
- I. Hladka, D. Y. Volyniuk, O. Bezvikonnyi, V. Kinzhybalo, T. J. Bednarchuk, Y. Danyliv, R. Lytvyn, A. Lazauskas and J. V. Grazulevicius, J. Mater. Chem. C, 2018, 6, 13179–13189 RSC.
- K. Zheng, F. Ni, Z. Chen, C. Zhong and C. Yang, Angew. Chem., Int. Ed., 2020, 59, 9972–9976 CrossRef CAS.
- H. Bhatia and D. Ray, J. Phys. Chem. C, 2019, 123, 22104–22113 CrossRef CAS.
- J. Sun, J. Zhang, Q. Liang, Y. Wei, C. Duan, C. Han and H. Xu, Adv. Funct. Mater., 2019, 30, 1908568 CrossRef.
- Y. Zhan, Z. Yang, J. Tan, Z. Qiu, Y. Mao, J. He, Q. Yang, S. Ji, N. Cai and Y. Huo, Dyes Pigm., 2020, 173, 107898 CrossRef.
- R. Skaisgiris, T. Serevičius, K. Kazlauskas, Y. Geng, C. Adachi and S. Juršėnas, J. Mater. Chem. C, 2019, 7, 12601–12609 RSC.
- M. Kumar and L. Pereira, Nanomaterials, 2020, 10, 101 CrossRef CAS.
- W. Yuan, H. Yang, C. Duan, X. Cao, J. Zhang, H. Xu, N. Sun, Y. Tao and W. Huang, Chem, 2020, 6, 1998–2008 CAS.
- M. K. Etherington, Front. Chem., 2020, 8, 716 CrossRef.
- C. Tang, T. Yang, X. Cao, Y. Tao, F. Wang, C. Zhong, Y. Qian, X. Zhang and W. Huang, Adv. Opt. Mater., 2015, 3, 786–790 CrossRef CAS.
- D. Zhang, X. Cao, Q. Wu, M. Zhang, N. Sun, X. Zhang and Y. Tao, J. Mater. Chem. C, 2018, 6, 3675–3682 RSC.
- X. Cao, X. Zhang, C. Duan, H. Xu, W. Yuan, Y. Tao and W. Huang, Org. Electron., 2018, 57, 247–254 CrossRef CAS.
- H. T. Feng, J. Zeng, P. A. Yin, X. D. Wang, Q. Peng, Z. Zhao, J. W. Y. Lam and B. Z. Tang, Nat. Commun., 2020, 11, 2617 CrossRef CAS.
- V. Bulović, R. Deshpande, M. Thompson and S. Forrest, Chem. Phys. Lett., 1999, 308, 317–322 CrossRef.
- C. F. Madigan and V. Bulović, Phys. Rev. Lett., 2003, 91, 247403 CrossRef.
- H. S. Kim, S.-R. R. Park and M. C. Suh, J. Phys. Chem. C, 2017, 121, 13986–13997 CrossRef CAS.
- T. Northey, J. E. Stacey and T. J. Penfold, J. Mater. Chem. C, 2017, 5, 11001–11009 RSC.
- V. Jankus and A. P. Monkman, Adv. Funct. Mater., 2011, 21, 3350–3356 CrossRef CAS.
- S. A. Majeed, B. Ghazal, D. E. Nevonen, P. C. Goff, D. A. Blank, V. N. Nemykin and S. Makhseed, Inorg. Chem., 2017, 56, 11640–11653 CrossRef CAS.
- S. A. Majeed, B. Ghazal, D. E. Nevonen, V. N. Nemykin and S. Makhseed, Dyes Pigm., 2019, 170, 107593 CrossRef CAS.
- K. Tani, Y. Tohda, H. Takemura, H. Ohkita, S. Ito and M. Yamamoto, Chem. Commun., 2001, 1914–1915 RSC.
- O. E. Andersson, B. L. V. Prasad, H. Sato, T. Enoki, Y. Hishiyama, Y. Kaburagi, M. Yoshikawa and S. Bandow, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 16387–16395 CrossRef CAS.
- N. Iwashita, H. Imagawa and W. Nishiumi, Carbon N. Y., 2013, 61, 602–608 CrossRef CAS.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS.
- P. L. dos Santos, D. Chen, P. Rajamalli, T. Matulaitis, D. B. Cordes, A. M. Z. Slawin, D. Jacquemin, E. Zysman-Colman and I. D. W. Samuel, ACS Appl. Mater. Interfaces, 2019, 11, 45171–45179 CrossRef CAS.
- S.-J. Woo, Y.-H. Ha, Y.-H. Kim and J.-J. Kim, J. Mater. Chem. C, 2020, 8, 12075–12084 RSC.
- F. Plasser, J. Chem. Phys., 2020, 152, 084108 CrossRef CAS.
- P. L. dos Santos, J. S. Ward, M. R. Bryce and A. P. Monkman, J. Phys. Chem. Lett., 2016, 7, 3341–3346 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2007864–2007866 and 2022458. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0tc04222f |
| This journal is © The Royal Society of Chemistry 2021 |