
The metal halide structure and the extent of distortion control the photo-physical properties of luminescent zero dimensional organic-antimony(III) halide hybrids†‡
Anupam
Biswas
ab,
Rangarajan
Bakthavatsalam
 ab,
Bhupendra P.
Mali
ab,
Vir
Bahadur
ab,
Chinmoy
Biswas
c,
Sai Santosh Kumar
Raavi
c,
Rajesh G.
Gonnade
ab and
Janardan
Kundu
*d
ab,
Bhupendra P.
Mali
ab,
Vir
Bahadur
ab,
Chinmoy
Biswas
c,
Sai Santosh Kumar
Raavi
c,
Rajesh G.
Gonnade
ab and
Janardan
Kundu
*d
aCSIR-National Chemical Laboratory, Pune, India
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad, India
cIndian Institute of Technology Hyderabad, Kandi, India
dIndian Institute of Science Education and Research (IISER) Tirupati, Tirupati, India. E-mail: janardan@iisertirupati.ac.in
First published on 17th November 2020
Abstract
Antimony(III) halide based zero dimensional hybrids have gained attention as broadband emitters. Until now, quadrangular pyramidal SbX5 based and octahedral SbX6 based 0D hybrids have been reported utilizing different organic ligands demonstrating some structural tunability affecting their emissive properties. Utilizing a common organic ligand, here we demonstrate the structural tunability (quadrangular pyramidal, octahedral, or a combination thereof) of the metal halide unit in Sb(III)Cl 0D hybrids with contrasting photo-physical properties (broadband, Stokes shift, strong/weak colored emission). The structure–property–mechanism correlation of the synthesized compounds [1 (C12H52Cl18N8O4Sb3; tris Sb green); 2 (C12H50Cl14N8O3Sb2; tris Sb red); 3 (C24H88Cl25N16O4Sb3; tris Sb yellow)] identifies crucial factors that control their emissive properties. The X-ray analysis reveals the structure (1-octahedral; 2-quadrangular pyramidal; 3-combination thereof) and the order of the extent of structural distortion as 1–3 ≪ 2. The metal halide coordination environment asymmetry and its structure are observed to dictate PL emission energy (1-green; 2-red; 3-yellow) as supported by a qualitative Molecular Orbital scheme. The extent of structural distortion guides the observed Stokes shifts (1–165 nm; 2–290 nm; 3–200 nm; 1–3 < 2). Interestingly, the extent of distortion is found to be well correlated with the observed PLQY (1–45%; 2–6%; 3–43%; 1–3 ≫ 2). This report clearly demonstrates the structural tunability and the effect of the metal halide unit structure/distortion in shaping the emissive properties of 0D organic Sb(III) halide hybrids.
Introduction
Recently, low dimensional organic-metal halide hybrids (OMHH) have emerged as a new class of materials with exquisite properties enabling optoelectronic applications in photovoltaics and solid state lighting.1–6 Dimensionality in such organic–metal halide hybrid materials refers to the electronic dimensionality/networked structure of the constituent metal halide inorganic unit (i.e. 2D, 1D, 0D). For zero dimensional (0D) variants of such materials, the semiconducting metal halide unit is isolated and surrounded by organic ligands. Photo-excitation of such low dimensional hybrids leads to the generation of strongly bound excitons confined within the metal halide unit.7 Strong electron–phonon coupling in such materials allows transient localization of the charge carriers (electrons, holes) in the metal halide unit by introducing local distortions of the lattice. The self-trapped excitons (STEs), thus produced, lead to phonon emission that alters the energy of the photoluminescence (PL) emission. This primarily leads to Stokes shifted broadband visible emission.8–12 Various factors that govern the PL emission energy and photoluminescence quantum yield (PLQY) of such broadband emission are not clearly understood.1,13–15 However, it is generally observed that the PLQY tends to be enhanced as the dimensionality is lowered.2,14,16 Typically, lead halide based low dimensional (2D, 1D, 0D) hybrids have been reported to manifest emissive properties corroborating to the STE based broadband emission with a modest/high PLQY.17 Noteworthily, for a given metal ion, the accessibility of a desired dimensionality in such systems is largely dictated by the choice of halides, organic ligands, and experimental reaction conditions.15,18,19The toxicity20 of Pb(II) has instigated many research initiatives for the development of the lead free variant of low dimensional OMHH materials.21–23 Suitable replacements for Pb2+ ions in such hybrids must retain the ns2 electronic configuration of the valence shell that has been touted as the key role player in conferring enabling properties to the lead-based low dimensional hybrids.24 Recently, Sb3+ and Bi3+ ions that are less susceptible to oxidation while retaining the ns2 valence electronic configuration have been introduced as replacements for Pb2+ ions.7,25–27 There have been few reports on low dimensional Sb(III) chloride based organic hybrids that have strong, long lived, Stokes shifted, and broadband STE based ambient emission.7,26–32 Interestingly, all of these 0D antimony chloride hybrids show triplet STE based broadband emission with a high/modest PLQY. They all demonstrate the structural commonality of having individual metal-halide units ([SbCl5]2−, [SbCl6]3−) that are completely isolated from each other and surrounded by the respective organic ligands. The quadrangular pyramidal metal halide unit in these reports has Sb–Cl equatorial bonds with very similar bond lengths and a shorter Sb–Cl axial/apical bond with a low/modest variation of bond angles from their ideal values. Similar bond length and bond angle distortions are observed for the octahedral metal-halide units. Such distortion of the metal halide unit likely arises due to the presence of an Sb centred stereochemically active lone pair.33,34 This might imply that the strong PL emission properties may be correlated to the ground state structure/distortion of the metal-halide unit. Antimony(III) halides are known to exist in different stoichiometric polyhedral units ([SbX4]−, [SbX5]2−, [SbX6]3−, [Sb2X9]3−) in the solid state.35 Given the various possible polyhedral unit types in these hybrids, is there any correlation that exists between the luminescence properties and the geometric structure/distortion of the metal-halide unit? How does the presence/absence of distortion in the metal-halide unit affect the PLQY and Stokes shift of the emission band? Research efforts aimed at answering these questions (tunability of the metal halide unit structure: octahedral or quadrangular pyramidal, and factors that affect emissive properties) are of current importance.29,36–38 In an effort to demonstrate structural tunability (octahedral, quadrangular pyramidal) and to rationalize the photo-physical origins of the observed emissive properties, we have synthesized various Sb(III) chloride 0D hybrids utilizing a common organic ligand and have analyzed the correlation between the specific structural features of the metal-halide unit and their luminescence properties. Here, we have utilized Tris(2-aminoethyl)amine as the common ligand to synthesize different 0D hybrids:
1 [(C6H22N4)2 (Sb2Cl10) (SbCl6) (Cl)2 (H3O)]·(3H2O); tris Sb green); 2 [(C6H22N4)2 (SbCl6)2 (Cl)2]·3(H2O); tris Sb red; 3 [(C6H22N4)4 (SbCl6)3 (Cl)7]·4(H2O); tris Sb yellow; with different structural and emissive properties. These compounds show long lived broadband emission, likely due to the self-trapping of excitons (STEs). However, the peak of the broadband emission and their PLQYs are markedly different (1: green emission with PLQY∼ 45%; 2: red emission with PLQY ∼ 6%; 3: yellow emission with PLQY∼ 43%). The single crystal structure analysis of the synthesized compounds reveals that the hybrids have different metal-halide unit structures with different extents of distortion: 1 has a combination of isolated, undistorted octahedron and distorted edge shared dimer octahedra; 2 has a combination of isolated heavily distorted octahedron and isolated heavily distorted pyramid; 3 has a combination of isolated undistorted octahedron, slightly distorted octahedron and slightly distorted quadrangular pyramid. The estimated extent of metal halide unit distortions (bond lengths, bond angles) follows: 1–3 ≪ 2 clearly highlighting the role of the metal halide unit structural symmetry of the coordination environment (octahedron vs. quadrangular pyramid) and the absence of distortion of the metal halide unit in affecting the PL emission energy, and the PLQY of their emission, respectively. The utilized reaction chemistry here allows us to crystallize different 0D hybrids wherein any crucial electronic contribution from the organic ligand towards photoluminescence remains constant. Noteworthily, demonstrated here is structural tunability (SbX5, SbX6 unit) with its concomitant effect on photo-physical properties that allow us to identify the crucial role played by ground state structural symmetry and the extent of distortion of the metal halide unit in dictating their emissive properties.
Results and discussion
Antimony(III)chloride 0D hybrids were synthesized utilizing Sb2O3 and Tris(2-aminoethyl)amine as the ligand in concentrated HCl acid. Single crystals of 1 tris Sb green can be obtained when the reaction temperature is set at 60 °C while clear single crystals of 2 tris Sb red result when the reaction temperature is set at 120 °C (see Scheme 1). Extensive details of the utilized synthetic conditions are provided in the Experimental section.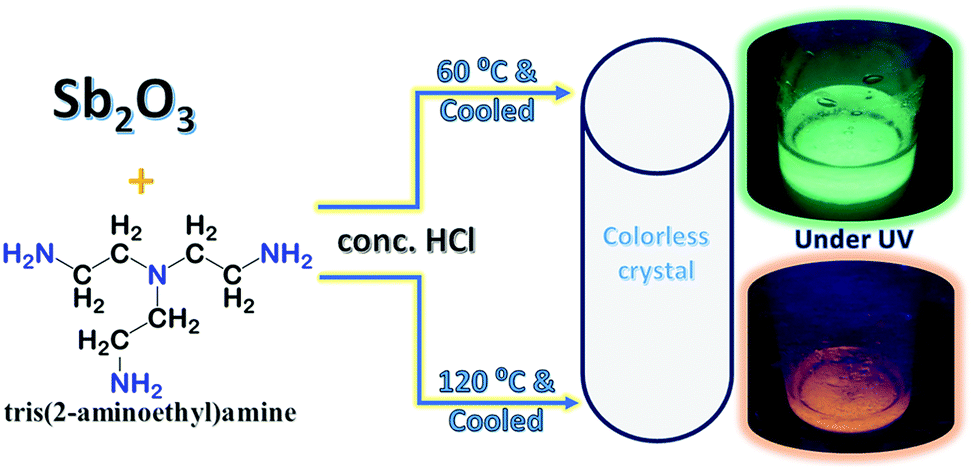 | ||
Scheme 1 Reaction scheme utilized for the synthesis of single crystals of 1 tris Sb green and 2 tris Sb red samples wherein metal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ligand ratio = 1:1. The structure of the ligand is shown. :ligand ratio = 1:1. The structure of the ligand is shown. | ||
Product 1 tris Sb green shows strong, green emission while the 2 tris Sb red sample shows weak, red emission when viewed inside the UV chamber. The presence of Sb ions in products 1 and 2 was verified using SEM/EDS analysis as shown in Fig. S1, ESI.‡ Furthermore, XPS analysis confirms the presence of Sb(III) metal ions in 1, 2 as shown in Fig. S2, ESI.‡ The 1H NMR spectra of 1, 2 reveal the presence of phase pure products with peaks attributed to the cationic ligand moiety (Fig. S3(a–d), ESI‡). The thermogravimetric analysis (TGA) of 1 and 2, as shown in Fig. S4, ESI,‡ shows a multistep weight loss profile. Here, the onset of weight loss observed at ∼100 °C for both 1, 2 is attributed to the loss of water molecules, followed by ligand loss at ∼200 °C and plausibly halogen loss at temperatures above 300 °C (see below for a discussion on the structure of products). The optical properties of 1 tris Sb green, presented in Fig. 1a, show a strong and broad photoluminescence (PL) emission band centered at 517 nm with a full width at half maximum (FWHM) of ∼110 nm. The photoluminescence excitation (PLE) spectra, collected at 530 nm, match well with the absorption spectra which show multiple features in the 300–400 nm range. The estimated “Stokes shift” (difference between the peak positions of PL and PLE) is ∼165 nm. The photoluminescence quantum yield (PLQY) for 1 tris Sb green is estimated to be ∼45%. On the other hand, the optical properties of 2 tris Sb red, presented in Fig. 1b, show a weak and broad emission band centered at 638 nm with a FWHM of ∼160 nm. The PLE spectra, collected at 650 nm, match well with the absorption spectra which show multiple features in the 300–400 nm range. The estimated “Stokes shift” is ∼290 nm and the PLQY for 2 tris Sb red is estimated to be ∼6%.
 | ||
| Fig. 1 Absorbance (Abs), photoluminescence (PL) and photoluminescence excitation (PLE) spectra of (a) 1 tris Sb green and (b) 2 tris Sb red single crystal samples. | ||
The absorption features in halogen antimonite(III) systems can be attributed to the electronic transitions between the sp excited states and the s2 ground state. The observed absorbance features for products 1 and 2 can be tentatively attributed to the A band (low energy, spin-forbidden transition 1S0 → 3P1), B band (intermediate energy, spin-forbidden transition 1S0 → 3P2), and C band (high energy, spin-allowed transition 1S0 → 1P1).39 Rationalization of the differences in the observed PL emission peak position, Stokes shift, and PLQYs for products 1 and 2 is discussed below.
The lifetime decay profiles (collected using the TCSPC set-up) and the extracted lifetimes across the broadband emission for 1 and 2, as presented in Fig. S5(a–d) (ESI‡), show a dominant lifetime component of ∼1 microsecond that remains unchanged across the emission band for 1 and 2. In order to better estimate the longer lifetimes, PL decay profiles were collected across the broadband utilizing a microsecond (μs) flash lamp source and are presented in Fig. S6 and Table S1; ESI.‡ Clearly, lifetime components (and relative %) of 4.8 μs (73%) and 71.9 μs (27%) were observed for 1 tris Sb green while for 2 tris Sb red the lifetime components (and relative %) were 5.6 μs (90%) and 94.4 μs (10%). The observed long emission lifetime components in products 1 and 2 highlight the role played by the triplet excited state (3P1) from which the radiative emission (phosphorescence) originates (Fig. S5, S6 and Table S1; ESI‡). Given the broad nature of the PL emission peak for both 1 and 2, it is important to decipher if defect emission leads to the observed broad emission bandwidth. The dependence of the emission band shape/profile on excitation wavelength was studied for products 1 and 2 as presented in Fig. S7a and b, ESI.‡ The PL emission band shape/profile remains unchanged for products 1 and 2 (albeit with a concomitant change in PL intensity) as across the excitation wavelength range of 320–380 nm. The PLE spectra collected across the broad emission band of products 1 and 2 (Fig. S7a and b, ESI‡) also remain unchanged. Furthermore, the estimated lifetime components for products 1 and 2 across the broad emission band were found to remain unchanged (Fig. S5, S6 and Table S1; ESI‡). These observations suggest that unique emissive species are responsible for the broad emission in products 1 and 2 and very likely do not involve extrinsic defects. Furthermore, PL/PLE studies were performed for products 1 and 2 wherein the single crystal samples were ground. If defects are the cause for the broadband emission, sample grinding would cause a substantial change of the PL/PLE profile.10 However, no changes in the PL/PLE band profile for both the products were observed when the samples were ground thoroughly as shown in Fig. S8, ESI.‡ Also, the ground samples of both the products were annealed at different temperatures, followed by their PL/PLE characterizations, as shown in Fig. S8, ESI.‡ Again here, no changes in the PL/PLE band profile are observed (albeit with a concomitant change in PL intensity@100–120 °C attributed to solvent loss as discussed later). These observations clearly suggest that the broad emission is not due to the presence of the defects in products 1 and 2 but is due to the presence of unique emissive species that lead to intrinsic broadband emission.
The broadband emissions in low dimensional metal halide hybrid materials have been attributed to the self-trapping of excitons (STE) due to strong electron–phonon coupling that produces transiently localized charges (holes/electrons) that distort the metal halide unit. The PL emission of these self-trapped excitons is phonon assisted that broadens the radiative bandwidth.9 Upon photoexcitation, the low lying transient STE states can accept carriers from the excited 3P1 state and allow slow and phonon assisted radiative decay to the 1S0 ground state, thereby broadening the emission bandwidth. Necessarily, this relay of the radiative decay channel leads to finite energy losses through non-radiative state hopping and accounts for a finite Stokes shift in broad emitting low dimensional materials. Importantly, this is in addition to any Stokes shift that might arise due to the changes in the excited state geometry/structure relative to the ground state structure (as discussed later).
Low temperature PL measurements were carried out to gain further insights into the phonon assisted radiative recombination of STEs leading to the broadband emission in 1 and 2. Steady state photoluminescence spectra were collected over the temperature range of 300–77 K as shown in Fig. S9, ESI.‡ Integrated PL peak area and FWHM were calculated for both products (1, 2) and are presented in Fig. 2(a and b). For both products, the PL intensity is initially observed to increase reaching the maxima (∼230 K), followed by a decrease, as the temperature is lowered (to 80 K). Such dependence of PL intensity can be ascribed to the thermally activated trapping–detrapping of excitons from STE states.10 However, the dependence in the low temperature regime can get complicated due to tunneling and defect bound excitons.10 Moreover, the phonon modes that couple to electronic excitation to generate STE have their own temperature dependence.40 Bandwidths (FWHMs) of the broad emission of 1 and 2 are observed to decrease monotonically as the temperature is lowered. As the temperature is lowered fewer phonon modes are thermally accessible to couple to the STEs assisting radiative recombination, thereby reducing the bandwidth of the PL emission. Furthermore, the lifetime of the emission for both products 1 and 2 are observed to lengthen at a lower temperature (Fig. S10; ESI‡). This could suggest thermal equilibrium between the triplet excitons and self-trapped excitons that governs the thermally activated trapping of triplet excitons in the STE states. These results cumulatively indicate intrinsic broadband STE based emission in accord with many other reports on low dimensional organic–inorganic (Sb, Pb, Sn) hybrids showing broadband emission.7,10,14,27,28,36,41,42 Further detailed experiments and analyses are needed to characterize43 the nature of the STE in these systems.
 | ||
| Fig. 2 Integrated PL peak area and FWHM of the broad PL peak for (a) 1 tris Sb green and (b) 2 tris Sb red powder samples as a function of temperature (300–80 K). | ||
A clear understanding of the structure of the obtained products can potentially provide an insight into the PL peak positions (green vs. red), large Stokes shift, and markedly different PLQYs of 1 and 2. Fig. 3 shows an overview of the single-crystal structure of the products 1 (CCDC 2017736), and 2 (CCDC 2017737)† with the details of the bond angles, bond lengths, etc. provided in Tables S3–S8; ESI.‡ Both the products are zero-dimensional in nature, wherein the organic ligands surround the metal halide unit. 1 tris Sb green crystallized in a triclinic centrosymmetric space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) containing one ligand (C6H22N4 with +4 charge), one unit of SbCl5 (with −2 charge), a half unit of SbCl6 (−1.5 charge), one Cl anion (−1 charge), one water molecule, and one H2.5O molecule (+1/2 charge) in the asymmetric unit. The H atom of the charged water molecule (H2.5O) is located at the inversion center (0.5, 0.5, 0.5) and shared between the two water molecules, which are related by inversion symmetry across the inversion center. The moiety formula of 1 tris Sb green is represented as [(C6H22N4)2(Sb2Cl10)(SbCl6)(Cl)2(H3O)]·(3H2O). The structure of 1 tris Sb green is composed of two basic building units of the antimony chloride framework: isolated octahedron and isolated edge shared octahedra (Fig. 3b and c). In the isolated octahedron, the Sb1 atom occupies a special position (0.5, 1.0, 1.0) across the inversion center and hence only half of the SbCl6 moiety is present in the asymmetric unit and the other half unit is generated by inversion operation, whereas the other SbCl5 moiety generates an edge-shared dimeric octahedral unit by inversion operation. These metal halide units, periodically embedded in the organic ligand matrix, also incorporate solvent water molecules. The crystal structure is governed by strong N–H⋯Cl hydrogen bonding interactions engaging the ligand N–H moieties and Cl atoms (Table S9, ESI‡). The view of the molecular packing down the b-axis shows the layered assembly of the ligand molecules along the a-axis, which accommodate the octahedra and the edge-shared dimeric octahedral units between its layers alternately parallel to the c-axis. The arrangement of the neighboring edge-shared dimeric octahedral units along the channel encapsulates the water molecules between them which interact with these metal halide units through O–H⋯Cl hydrogen bonding interactions (Table S9, ESI‡). Conversely, the non-bonded Cl ions are located in the space between the two octahedral units along the channel parallel to the a-axis and forms a N–H⋯Cl hydrogen bond with the ammonium protons. These strong hydrogen bonding interactions govern the crystal packing (Fig. S11a, ESI‡) and play a crucial role in optoelectronic responses. The loss of these water molecules at ∼100–120 °C, as evidenced in the TGA profile (Fig. S4, ESI‡), could be due to their encapsulation in the open channel leading to the drastic loss of the PL intensity of the annealed sample 1 (Fig. S8, ESI‡). The extent of distortion (bond lengths, bond angles) in the monomeric and dimeric octahedra was estimated for 1 tris Sb green. The monomeric octahedron is almost undistorted with very similar Sb–Cl bond lengths (largest difference in Sb–Cl bond length = 0.01 Å, largest deviation of the Cl–Sb–Cl bond angle from the ideal value = 3°). The edge shared dimeric octahedron with six different Sb–Cl bond lengths shows slight distortions (largest difference in Sb–Cl bond length = 0.6 Å, largest deviation of the Cl–Sb–Cl bond angle from the ideal value = 6°). Such distortions are not unexpected in the edge shared (μ-Cl) dimeric octahedral metal halide unit. The phase purity of 1 was verified using the powder X-ray diffraction (PXRD) patterns, which matched well with the simulated ones in terms of the single-crystal X-ray data as shown in Fig. S12; ESI.‡
containing one ligand (C6H22N4 with +4 charge), one unit of SbCl5 (with −2 charge), a half unit of SbCl6 (−1.5 charge), one Cl anion (−1 charge), one water molecule, and one H2.5O molecule (+1/2 charge) in the asymmetric unit. The H atom of the charged water molecule (H2.5O) is located at the inversion center (0.5, 0.5, 0.5) and shared between the two water molecules, which are related by inversion symmetry across the inversion center. The moiety formula of 1 tris Sb green is represented as [(C6H22N4)2(Sb2Cl10)(SbCl6)(Cl)2(H3O)]·(3H2O). The structure of 1 tris Sb green is composed of two basic building units of the antimony chloride framework: isolated octahedron and isolated edge shared octahedra (Fig. 3b and c). In the isolated octahedron, the Sb1 atom occupies a special position (0.5, 1.0, 1.0) across the inversion center and hence only half of the SbCl6 moiety is present in the asymmetric unit and the other half unit is generated by inversion operation, whereas the other SbCl5 moiety generates an edge-shared dimeric octahedral unit by inversion operation. These metal halide units, periodically embedded in the organic ligand matrix, also incorporate solvent water molecules. The crystal structure is governed by strong N–H⋯Cl hydrogen bonding interactions engaging the ligand N–H moieties and Cl atoms (Table S9, ESI‡). The view of the molecular packing down the b-axis shows the layered assembly of the ligand molecules along the a-axis, which accommodate the octahedra and the edge-shared dimeric octahedral units between its layers alternately parallel to the c-axis. The arrangement of the neighboring edge-shared dimeric octahedral units along the channel encapsulates the water molecules between them which interact with these metal halide units through O–H⋯Cl hydrogen bonding interactions (Table S9, ESI‡). Conversely, the non-bonded Cl ions are located in the space between the two octahedral units along the channel parallel to the a-axis and forms a N–H⋯Cl hydrogen bond with the ammonium protons. These strong hydrogen bonding interactions govern the crystal packing (Fig. S11a, ESI‡) and play a crucial role in optoelectronic responses. The loss of these water molecules at ∼100–120 °C, as evidenced in the TGA profile (Fig. S4, ESI‡), could be due to their encapsulation in the open channel leading to the drastic loss of the PL intensity of the annealed sample 1 (Fig. S8, ESI‡). The extent of distortion (bond lengths, bond angles) in the monomeric and dimeric octahedra was estimated for 1 tris Sb green. The monomeric octahedron is almost undistorted with very similar Sb–Cl bond lengths (largest difference in Sb–Cl bond length = 0.01 Å, largest deviation of the Cl–Sb–Cl bond angle from the ideal value = 3°). The edge shared dimeric octahedron with six different Sb–Cl bond lengths shows slight distortions (largest difference in Sb–Cl bond length = 0.6 Å, largest deviation of the Cl–Sb–Cl bond angle from the ideal value = 6°). Such distortions are not unexpected in the edge shared (μ-Cl) dimeric octahedral metal halide unit. The phase purity of 1 was verified using the powder X-ray diffraction (PXRD) patterns, which matched well with the simulated ones in terms of the single-crystal X-ray data as shown in Fig. S12; ESI.‡
 | ||
| Fig. 3 Single crystal structures of zero dimensional chloro antimonite(III)–organic hybrids for (a–c) 1 tris Sb green and (d–f) 2 tris Sb red. Shown also are the metal halide units (octahedra and dimer octahedra for 1 tris Sb green; distorted octahedra and distorted pyramid for 2 tris Sb red) for both the products. | ||
Product 2 tris Sb red crystallizes in a monoclinic chiral space group P21 containing two ligands (tetra positive), two [SbCl6]3− anions, two [Cl]− anions, and three H2O molecules in the asymmetric unit having the formula [(C6H22N4)2(SbCl6)2(Cl)2]·3(H2O). The structure of 2 is composed of two basic building units of antimony chloride with a hexa-coordinated motif: isolated very distorted octahedron and isolated extremely distorted octahedron (Fig. 3e and f). For the later motif, the sixth coordination site is occupied by a distant Cl atom (3.156 Å) as highlighted in Fig. 3f and may be thought of as an extremely distorted ‘quadrangular pyramidal’ unit. The isolated very distorted octahedron with a lengthened apical Sb–Cl bond length (3.042 Å) may also be thought of as a distorted ‘quadrangular pyramidal’ structure. The presence of such long Sb–Cl bond distances is not uncommon and has been attributed to the secondary bonding interaction.44 These distortions highlight the site asymmetry in the coordination environment of the hexacoordinated Sb motifs. These metal halide units are again periodically embedded in the inert matrix of the organic ligand incorporating water molecules forming the 0D structure. The view of the molecular packing down the a-axis revealed the layered arrangement of both metal halides parallel to the b-axis which are embedded between the channels formed by symmetry independent organic ligands. The respective layers of metal halides are arranged alternately along the c-axis. The crystal structure is stabilized by strong intermolecular N–H⋯Cl hydrogen bonding interactions engaging ammonium H-atoms of the organic ligand and Cl atoms (of metal halides) or Cl ions (Fig. S11b, ESI‡). The three water molecules also form strong H-bonding interactions (O–H⋯Cl and N–H⋯O) involving Cl atoms and ammonium protons of metal halides and organic ligands, respectively. The water molecules also interact with each other through O–H⋯O hydrogen bonds (Table S9, ESI‡). The loss of the water molecules at ∼100–120 °C, as evidenced in the TGA profile (Fig. S4, ESI‡), leads to a drastic loss of the PL intensity of the annealed sample 2 (Fig. S8, ESI‡). The extent of distortion in the heavily distorted octahedron and the extremely distorted octahedron was estimated for 2 tris Sb red. The heavily distorted octahedron shows high site asymmetry with six different Sb–Cl bond lengths (largest difference in Sb–Cl bond length = 0.6 Å, largest deviation of the Cl–Sb–Cl bond angle from ideal value = 23°), thereby approaching a ‘quadrangular pyramidal’ structure. The extremely distorted octahedron shows extreme site asymmetry with very different Sb–Cl bond lengths (largest difference in Sb–Cl bond length = 0.45 Å, largest deviation of Cl–Sb–Cl bond angle from the ideal value = 10°). The ‘apical’ Sb–Cl bond length in this unit is 2.448 Å. The phase purity of 2 was confirmed using the PXRD patterns which matched well with the simulated ones in terms of the single-crystal X-ray data as shown in Fig. S12; ESI.‡
Comparison of the above estimated ground state structural distortions of the photoactive metal halide units reveals that the average distortion is appreciably higher in 2 tris Sb red and is relatively lower in 1 tris Sb green product (Fig. S13, ESI‡). In essence, 1 tris Sb green has undistorted octahedral metal halide units, while 2 tris Sb red has distorted quadrangular pyramid units. The presence of the common organic ligand in 1 and 2 allows us to perform a structure–property–mechanism correlation that can provide us with a deeper understanding of the crucial factors that control their photo-physical properties. Understandably, the extent of distortion and the ground state structure affect Stokes shifts and PL emission energies. It is clear from Fig. 1 that both products (1, 2) show a large Stokes shift implying strong structural re-organization in the excited state (3P1). The observed Stokes shift is much higher for 2 tris Sb red (∼290 nm) than that of 1 tris Sb green (∼165 nm) suggesting stronger excited state reorganization for 2 than that for 1. Clearly, Stokes shift depends not only on the ground state structure (and its distortion) but also on the structure in the excited state of the metal halide unit. Within the theoretical framework of ns2 metal ions, as proposed by Blasse et al., the luminescence properties are directly related to the structure and the extent of distortion of the ns2 metal-halide unit.45–47 Upon photo-excitation of these ns2 metal-halide units, the ground state s2 configuration is transformed into sp excited state with higher symmetry.39,45 Gaining this structural symmetry due to photo-excitation from the distorted ground state structure results in the observed Stokes shift. Clearly, the ground state structural distortion in 2 tris Sb red is relatively higher than that in 1 tris Sb green and is in accord with the observed larger Stokes shift in the former. A similar approach has been successfully utilized by Blasse et al. for ns2 metal ions doped in host lattices that show large Stokes shifts if the metal ions occupy off-center positions in large interstices as these ions can move towards the center in the excited state, thereby gaining symmetry.45 The difference in the PL emission energies (peak positions) for the two products (1, 2) can again be rationalized in terms of the ground state structure. Typically, the structures of these s2 metal halide units are affected by the presence of the metal centered, stereochemically active lone pair. However, s2 complexes with a coordination number of six have octahedral geometry and appear as an exception from the VSEPR model.39,48 Complexes with coordination numbers lower than six are in general agreement with the VSEPR model and show distortions/asymmetry in their structures due to the stereochemical activity of the lone pair.39,49,50 These considerations are also applicable to halogen antimonite(III) polyhedral units here that constitute the photoactive building units for products 1, 2. For 1 tris Sb green, the metal halide coordination sphere has octahedral geometry with slight differences in the Sb–Cl bond lengths (undistorted octahedron and distorted dimeric octahedra). However, for 2 tris Sb red, the metal halide coordination sphere shows extreme site asymmetry (‘quadrangular pyramidal’ structure) with modest differences in the Sb–Cl bond lengths (both octahedral units approaching the quadrangular pyramidal structure). Clearly, the ‘effective’ interaction strength of Cl− anions (3s, 3p orbitals) with the Sb3+ (5s, 5p orbitals) metal ion will be stronger for 1 tris Sb green (near ideal octahedral units with less bond length distortions) compared to 2 tris Sb red (heavy/extreme distortions in octahedral units that approach the ‘quadrangular pyramidal’ structure) due to the degree of asymmetry in the metal-halide coordination environment. A symmetrically stronger ‘effective’ interaction between the metal and the halide in 1 will lead to a higher energy gap between the HOMO and LUMO. An asymmetric and weaker metal halide interaction in 2 will result in a smaller energy difference between the HOMO and LUMO (the qualitative molecular orbital diagrams for octahedral and square pyramidal structural cases are shown in Fig. S14, ESI‡). Hence, it is anticipated that 1 tris Sb green, with a symmetrical octahedral structure, would display a high energy PL emission (LUMO → HOMO) peak while 2 tris Sb red, with an asymmetric quadrangular pyramidal structure, will emit at lower energies. This is in qualitative agreement with the observed high energy PL emission peak of 1 tris Sb green (λem = 517 nm) and low energy PL emission peak of 2 tris Sb red (λem = 638 nm). Such a ground state structure dependent molecular orbital scheme has been successfully utilized in attributing differences in the PL emission energies of [SbCl6]3− and [SbCl4]− ions in solution.39 It is worth noting here that a symmetrical (undistorted) octahedral halide ligand field will lead to a higher HOMO–LUMO energy gap than an undistorted square pyramidal ligand field. This again is in qualitative agreement with the reported low PL emission energies for many [SbCl5]2− based metal halide organic 0D hybrids (with emission energies in the orange region of 600–650 nm) in solid state and higher PL emission energies in the green region for [SbCl6]3− based ions in solution.39
The photoluminescence quantum yield (PLQY) of 1 tris Sb green is (∼45%) distinctly higher than that of 2 tris Sb red (∼6%). Note that the observed PLQY for 1 tris Sb green is lower than those reported in the literature7,26–28 on 0D antimony chloride hybrids that have a near unity PLQY. However, a direct comparison of the PLQY of 1 tris Sb green with these other reports including different organic ligands might not lead to the required understanding of the rationale behind the observed differences in their PLQYs. This is due to the surrounding environment, provided by the organic ligand to the photoactive metal halide unit, that can appreciably affect the PL emission intensity. There can be effective energy transfer of electron excitation from the molecular levels of the surrounding organic moiety to the 3P1 luminescence level of the s2 metal ion, thereby enhancing the PLQY.51,52 ‘Loan’ of luminescence intensity from the ligands to the metal has also been reported wherein ligand to metal charge transfer interactions lead to additional thermal population of the luminescent excited state of metal ions leading to an enhanced PLQY.53,54 This clearly suggests the electronic contribution of the surrounding organic ligand in affecting the photoluminescence intensity in the low dimensional OMHH.54 Interestingly, the synthetic strategy utilized here allows the crystallization of both 1 and 2 incorporating the same organic ligand. This, to a large extent, helps maintain the same electronic contribution (if any) to the metal ion, thereby similarly affecting the photoluminescence intensity from 1 and 2. Hence, the PLQYs of 1 and 2 can be compared and possibly correlated to their structure/distortion to gain some insights into the observed differences in their PLQYs. This effort of finding any existent correlation between structure/distortion and PLQY is extremely important for further design and synthesis of broadband, lead-free emitters and is reported for the first time here for Sb halide based 0D OMHH. Although there have been few reports on antimony chloride based 0D hybrids that show near unity PLQYs, none of the reports provide any insight into the observed PL peak position and the observed high PLQY. The commonality in all of these high PLQY low dimensional antimony chloride based hybrids is the presence of isolated, almost undistorted metal halide unit (particularly the SbX5 unit).7,26–28
Moreover, antimony chloride based low dimensional hybrids that have polymeric metal halide units (showing distortions) have a low/modest emission strength.51,52,55 In the present comparison of PLQYs of 1 and 2, metal halide polyhedral units for 1 (isolated octahedra, dimer octahedra) show small distortions while metal halide polyhedral units for 2 (octahedra and quadrangular pyramid) show relatively higher distortions. The survey of the existing literature on antimony chloride based low dimensional materials, revealing that the isolated undistorted monomeric units demonstrate high emission intensities, suggests the importance of distortions in affecting the PLQY.51–54,56 Clearly, the extent of distortion in 1 is relatively lower than that in 2. This implies a correlation of the structure/distortion with the PLQY for 0D Sb halide-based hybrids. Noteworthily, the presence of distorted dimer octahedra (plausibly suppressing the PLQY) along with almost undistorted octahedra (plausibly enhancing the PLQY) in 1 shows a higher PLQY compared to 2 having all isolated monomeric units (plausibly enhancing the PLQY) with heavy distortions (plausibly suppressing the PLQY). This again indicates the important role played by structural distortions in the ground state. The presence of the dimer octahedra with some distortions in 1 can then explain the observed lower than unity PLQY which is in accord with earlier reports52 on low dimensional antimony chloride hybrids with dimeric/polymeric metal halide units. The observed correlation of the structure/distortion with the PLQY could be rationalized in terms of photoexcitation of ns2 metal ions transiting from the ground state to the excited state having higher symmetry. Hence, the smaller the distortions in the ground state structure of the metal halide polyhedra, the lower the amount of electronic excitation energy that will be utilized in the process of excited state structural reorganization. This will minimize non-radiative losses of the electronic excitation energy thereby enhancing the possibility of stronger luminescence. The above proposed rationalization providing insight to the experimentally observed correlation between structure/distortion and PLQY for 0D antimony chloride-based hybrids is by no means universal and needs to be further tested/verified. In order to clearly understand the role of distortion in affecting the PLQY, efforts are now being devoted to the design and synthesis of antimony chloride based 0D hybrids (utilizing a common organic ligand) that have only one type of isolated metal halide polyhedral unit but with different extents of distortion. This needs further exploratory synthetic efforts that are currently underway.
However, we have been successful in modifying our synthetic strategy that allows us to crystallize 0D hybrids with an isolated monomeric metal-halide unit (albeit with a mixture of octahedral and pyramidal units). This is achieved by increasing the metal to ligand ratio to 1:10 as detailed in the Experimental section. The obtained product, 3 tris Sb yellow, appears to emit bright yellow light when viewed inside the UV chamber. The 1H NMR spectra of 3 reveal the presence of phase pure products with peaks attributed to the cationic ligand moiety (Fig. S3(a–d), ESI‡). The TGA data of 3, as shown in Fig. S4, ESI,‡ show a multistep weight loss profile. Here, the onset of weight loss observed at ∼100 °C for 3 is attributed to the loss of water molecules, followed by ligand loss at ∼200 °C and plausibly halogen loss at temperatures above 300 °C. Notably, the continuous nature of the weight loss curve without clearly visible step-like features in the TGA plot for 3 and 2 might indicate the similarity of the binding interaction of the ligands and the halogens in the structures. The presence of water of crystallization in (1, 2, 3) allows us to assign the weight loss peaks at ∼100 °C to the loss of water molecules from the structures. Furthermore, free halogens in the structures (1, 2, 3) allow us to tentatively attribute the weight loss at ∼300 °C to be due to halogen loss as has been suggested in a recent report.30 The steady state optical characterization of 3 tris Sb yellow, as shown in Fig. 4a, reveals a strong, broad yellow emission band centered at 580 nm with a full width at half maximum (FWHM) of ∼140 nm. The PLE spectra, collected at 580 nm, match well with the absorption spectra which show multiple features in the 300–400 nm range. The estimated “Stokes shift” is ∼200 nm and the PLQY for 3 tris Sb yellow is estimated to be ∼43%. The broad yellow emission profile remains unchanged as the excitation wavelength is changed. Furthermore, the PLE profile remains unchanged across the broadband emission (Fig. S15; ESI‡). The PL and PLE profile remain unchanged upon solid state grinding and annealing (Fig. S16; ESI‡). Moreover, the analysis of the collected decay profiles across the broad emission band of 3 tris Sb yellow, as presented in Fig. 4b, provides lifetime components (and relative%) of 5.2 μs (85%) and 84.6 μs (15%) that largely remain unchanged across the emission band (Table S10, ESI‡). This indicates the presence of unique emissive species responsible for the observed phosphorescence in 3 tris Sb yellow. Notably, the PL emission energy of 3 tris Sb yellow sample represents an intermediate value when compared to those of 1 tris Sb green and 2 tris Sb red while the PLQY value is close to that of 1 tris Sb green.
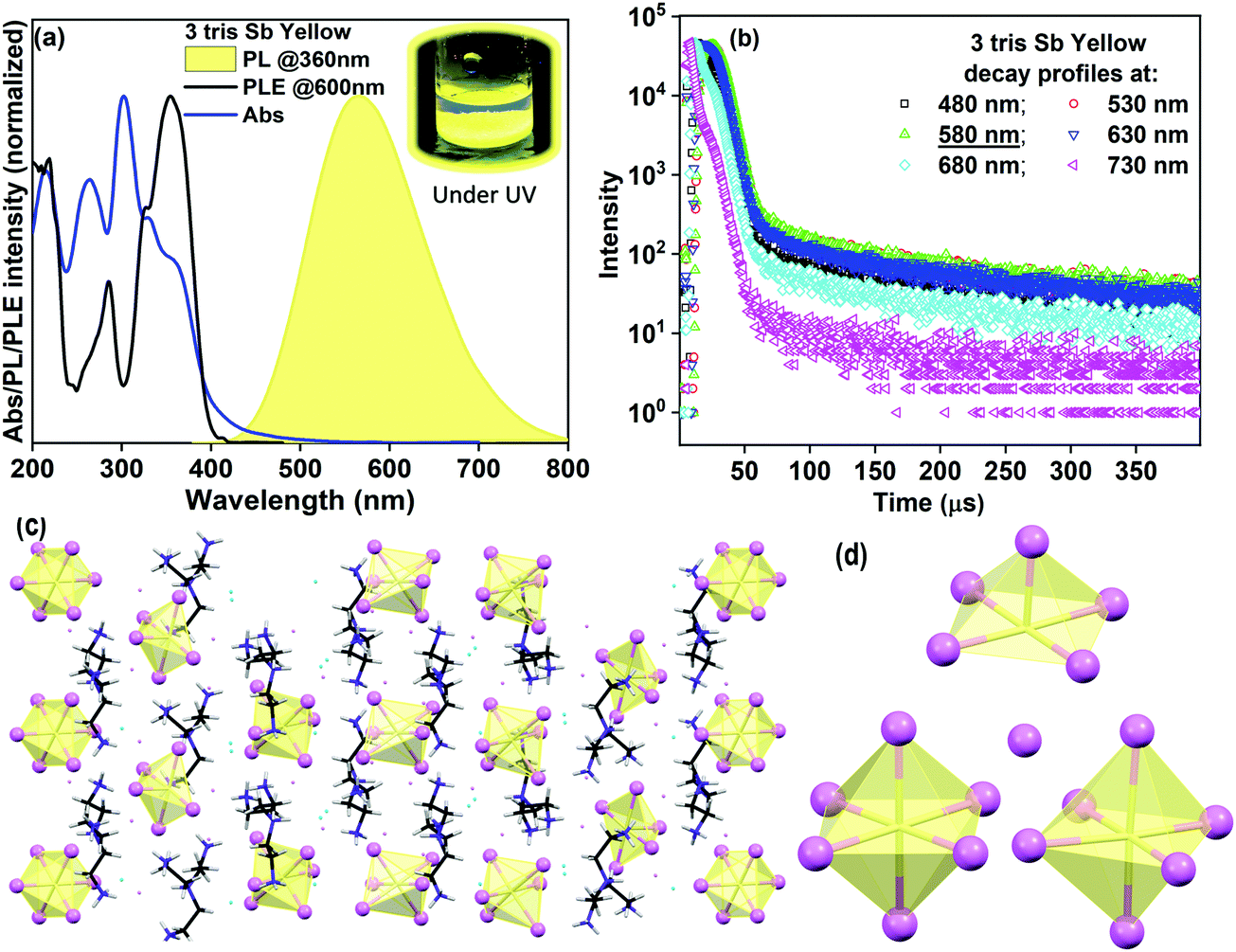 | ||
| Fig. 4 Optical and structural characterization of 3 tris Sb yellow: (a) absorbance (Abs), photoluminescence (PL) and photoluminescence excitation (PLE) spectra; (b) lifetime decay profiles collected across the yellow emission band using microsecond flash lamp excitation (360 nm); (c) overview of the single crystal structure of 0D 3 tris Sb yellow product; and (d) metal halide units (distorted quadrangular pyramid, octahedra, and distorted octahedra) comprising the 0D structure. | ||
Now, the pertinent question here is can we utilize the same structure/distortion correlation to understand the observed photo-physical properties for 3 tris Sb yellow. Such rationalization would then fall in line with the proposition made for products 1 and 2 and would support generality. The single crystal structure of 3 tris Sb yellow (CCDC 2017738) belongs to a triclinic space group P containing four ligands (tetra positive), two units of half of the [SbCl6]3− anion, two units of the [SbCl6]3− anion, seven [Cl]− anions, and four [H2O] molecules in the asymmetric unit leading to [(C6H22N4)4 (SbCl6)3 (Cl)7]·4(H2O) as the formula moiety (Fig. 4(c and d)). Two of the Sb atoms of the half units of the [SbCl6]3− anion occupy a special position (inversion center), Sb1 (0.0, 0.5, 0.0) and Sb2 (0.0, 0.5, 0.5), and hence only a half unit is present in the asymmetric unit and the other half is generated by inversion operation. The structure of 3 is composed of isolated octahedra and an isolated heavily distorted octahedron (approaching the quadrangular pyramidal structure) as the basic building units (Fig. 4c, d and Fig. S14; ESI‡). These metal halide units are periodically embedded in the channels of the organic ligand matrix and interact mostly with its ammonium H-atoms through N–H⋯Cl hydrogen bonds. The four water molecules are also encapsulated in the crystal lattice which interact with the ammonium H-atoms of the organic ligand through N–H⋯O hydrogen bonds (Table S9 and Fig. S11c, ESI‡). In addition to the presence of the distorted ‘quadrangular pyramidal’ unit, the crystal structure shows two types of isolated octahedron – one that is almost undistorted and the other with slight distortions. The undistorted octahedron has very similar Sb–Cl bond lengths (largest difference in Sb–Cl bond length = 0.028 Å, largest deviation of the Cl–Sb–Cl bond angle from the ideal value = 3°). The distorted octahedron has six different Sb–Cl bond lengths (largest difference in Sb–Cl bond length = 0.46 Å, largest deviation of the Cl–Sb–Cl bond angle from the ideal value = 4.6°). The distorted ‘quadrangular pyramid’ has the sixth coordination by a distant Cl atom (3.06 Å) as highlighted in Fig. 4c and d. This ‘quadrangular pyramidal’ unit has five different Sb–Cl bond lengths (largest difference in Sb–Cl bond length = 0.21 Å, largest deviation of Cl–Sb–Cl bond angle from the ideal value = 3.2°). The ‘apical’ Sb–Cl bond length here is 2.452 Å. A detailed list of bond angles and bond lengths describing the crystal structure is provided in Tables S11–S13, and Fig. S17, ESI.‡ Such distortions within the hexa-coordinated metal halide environment depict the extent of site asymmetry. The phase purity of 3 was verified using the PXRD patterns which matched well with the simulated ones from the single-crystal X-ray data as shown in Fig. S12; ESI.‡
A comparison of the structure/extent of distortions in the comprising metal halide units of 1 tris Sb green, 2 tris Sb red, and 3 tris Sb yellow in the light of their observed photo-physical properties is crucial here. The bond length variances (λoct) of Sb–Cl and bond angle variances (σ2) of Cl–Sb–Cl in the various structural units for products 1, 2, and 3 were calculated (as presented in Table S2, ESI‡), which exhibit the following order of the extent of distortions: 1–3 ≪ 2. Qualitatively, the building unit type of isolated metal halide polyhedra in 1 is octahedra, in 2 is ‘quadrangular pyramid’, and in 3 is a mixture of octahedron and ‘quadrangular pyramid’. The degree of asymmetry of the coordination environment in the metal halide structural unit, as discussed earlier (MO diagram, Fig. S12, ESI‡), largely dictates the PL emission energy with a high energy emission band for the symmetric octahedral case (1 tris Sb green) and a low energy emission band for the asymmetric ‘pyramidal structure’ (2 tris Sb red). Now, for 3 tris Sb yellow, the presence of a mixture of octahedral and ‘quadrangular pyramidal’ units would cause the PL emission band to appear in the intermediate energy range. Indeed, for 3 tris Sb yellow, the PL band appears at an energy in between the emission bands of 1 tris Sb green and 2 tris Sb red. This strongly suggests the active role of the metal-halide polyhedral unit structure and the degree of symmetry in the metal-halide coordination environment in dictating the PL emission band position. As discussed earlier, the extent of distortion present in 1 tris Sb green is modest and is appreciably lower than that in 2 tris Sb red. This difference in the extent of distortion reflected in the drastic difference of the observed PLQY. Interestingly, the extent of distortion present in 3 tris Sb yellow is comparable to that in 1 tris Sb green (Table S2; ESI‡) and follows 1–3 ≪ 2. The similarity of the extent of distortion in 1 tris Sb green and 3 tris Sb yellow would indicate similar PLQY values which are much higher than that of 2 tris Sb red. This is in accord with the measured PLQY of 3 tris Sb yellow which is close to that of 1 tris Sb green. Such observed correlation between the extent of distortion and the PLQY clearly highlights the role of the extent of distortion in affecting the photo-physical properties. The observed Stokes shifts of three products can also be then positively correlated to the extent of distortion (Stokes shifts: 1–3 ≪ 2). Thus, the above proposed factors (metal-halide unit structure; extent of distortion) are observed to directly impact the photo-physical properties (PL emission energy; PLQY and Stokes shift, respectively) for the different 0D Sb(III) chloride hybrids (1, 2, 3) synthesized here utilizing a common organic ligand. Further efforts are underway to ascertain the effect of the metal-halide polyhedral type and different extents of distortion in a given type of polyhedral unit on the emissive properties of 0D organic-antimony chloride hybrids.
Conclusion
We demonstrate metal halide structural tunability (SbX6 octahedron, SbX5 quadrangular pyramidal, or a combination thereof) through the synthesis of different Sb(III) chloride 0D hybrids (1, 2, 3) utilizing a common organic ligand [tris(2-aminoethyl)amine]. The crystal structure analysis reveals that the site asymmetry leads to different metal halide unit structures that may be regarded as (i) octahedral in product 1, (ii) quadrangular pyramidal in product 2, and (iii) a combination of octahedra and quadrangular pyramidal in product 3. Furthermore, the structure analysis reveals that the extent of distortion of the metal halide units in these products follows the order: 1–3 ≪ 2. The emissive properties of these products that need a rational understanding include Stokes shifted broadband visible emission with distinctly different PL emission energies (λem) and markedly different PLQYs: 1: λem = 517 nm, Stokes shift = 165 nm, PLQY = 45%; 2: λem = 638 nm, Stokes shift = 290 nm, PLQY = 6%; and 3: λem = 590 nm, Stokes shift = 200 nm, PLQY = 43%. A structure–property correlation analysis performed here provides us with the following deeper understanding of the origins of the above emissive properties: (i) qualitative molecular orbital scheme on metal halide bonding (with an asymmetric metal halide coordination environment) allows the observed PL emission energies to be rationalized based on the ground state structure (octahedral vs. pyramidal) of 1, 2, 3; (ii) structural reorganization that accompanies while transiting from the distorted ground state to the symmetric excited 3P1 state of Sb3+ ions accounts for the large Stokes shifts in the order of 1–3 < 2; (iii) the extent of ground state structural distortion (1–3 ≪ 2) is well correlated to the PLQY (1–3 ≫ 2). A lower (higher) extent of ground state structural distortion would minimize (maximize) the non-radiative loss of the excitation energy in the process of excited state reorganization into the symmetric structure, thereby enhancing (suppressing) the possibility of stronger luminescence. This report demonstrates the structural tunability of the metal halide units (octahedral, pyramidal) and highlights the importance of the ground state structure/extent of distortions in affecting PL emission energies, Stokes shifts, and PLQY for 0D antimony chloride hybrids incorporating common organic ligands.Experimental
Materials
Antimony(III) oxide (99%), hydrochloric acid (37%) and acetone were purchased from Sigma-Aldrich. Tris(2-aminoethyl)amine was purchased from TCI Chemicals. Diethyl ether was purchased from HiMedia. All chemicals were used as purchased without further purification.Synthesis of powdered 1 tris Sb green:
For the preparation of 1 tris Sb green powder sample, 0.1 mmol (29.1 mg) of antimony(III) oxide was dissolved in 5 mL of hydrochloric acid. To this, 0.1 mmol (14.6 mg) tris(2-aminoethyl)amine was added. The solution turned turbid white after some time. The resulting precipitate was filtered and washed with diethyl ether repeatedly and dried in vacuum for further characterization.Synthesis of single crystals of 1 tris Sb green:
To obtain single crystals of 1 tris Sb green, the same procedure was followed with the following details: 0.1 mmol (29.1 mg) of antimony(III) oxide was dissolved in 5 mL of hydrochloric acid. To this, 0.1 mmol (14.6 mg) tris(2-aminoethyl)amine was added. The resultant mixture was heated in a preheated oil bath at 60 °C until dissolution. The solution is slowly cooled to obtain white crystals. Crystallization led to the formation of plate shaped pale yellow crystals which appear bright green under UV (365 nm) light. The crystals were filtered using a vacuum pump and washed repeatedly with acetone and diethyl ether for further characterization.Synthesis of single crystals of 2 tris Sb red:
In order to synthesize single crystals of 2 tris Sb red, the same procedure was followed as that for 1 described above. However, the oil bath temperature was set at 120 °C for dissolving the crystal and cooled naturally to room temperature. After cooling the needle shaped colorless crystals formed which emit red color under UV (365 nm) light. Here we have used the same filtration and washing procedure as mentioned above.Synthesis of powdered 3 tris Sb yellow
For the preparation of the 3 tris Sb yellow powder sample, 0.1 mmol (29.1 mg) of antimony(III) oxide was dissolved in 5 mL of hydrochloric acid. To this, 1 mmol (146 mg) tris(2-aminoethyl)amine was added. The solution turned turbid white after some time. The precipitate appeared bright yellow under UV (365 nm) light.Synthesis of single crystals of 3 tris Sb yellow
To obtain single crystals of 3 tris Sb yellow, a 1:10 equivalent metal vs. ligand ratio was used. Typically for the reaction 0.1 mmol (29.1 mg) of antimony(III) oxide was dissolved in 5 mL of hydrochloric acid. To this, 1 mmol (146 mg) tris(2-aminoethyl)amine was added. The resultant mixture was kept in a preheated oil bath at 120 °C until dissolution. The solution is slowly cooled to room temperature to obtain crystals. Crystallization led to the formation of cube shaped pale yellow crystals which appear bright yellow under UV (365 nm) light. The crystals were filtered using a vacuum pump and washed repeatedly with acetone and diethyl ether for further characterization.
Methods
UV-Vis absorbance was performed in a Shimadzu UV-3600 Plus UV-Vis-NIR spectrometer. Steady state PL and lifetime were measured using an Edinburgh FS5 spectrophotometer. TGA measurements were performed using a TAG system (Mettler-Toledo, Model TGA/SDTA851e) and samples were heated in the range of 25–800 °C at a heating rate of 5 °C min−1 under a nitrogen atmosphere. 1H NMR spectra were recorded on a Bruker A-400 MHz system using DMSO-d6 as the solvent at room temperature. Powder X-ray diffraction (XRD) patterns were recorded using a PANalytical X’Pert Pro equipped with Cu Kα radiation (λ = 1.5406 Å). Absolute quantum yield measurements were carried out in a Horiba JOBIN YVON Fluoromax-4 spectrometer with a calibrated integrating sphere attachment. Scanning Electron Microscopy (SEM) imaging and mapping were performed using a Zeiss™ Ultra Plus field-emission scanning electron microscope. X-ray photoelectron spectroscopy (XPS) characterization was performed with an ESCALab spectrometer having an Al Kα X-ray source (hν = 1486.6 eV) operating at 150 W using a Physical Electronics 04–548 dual Mg/Al anode and in a UHV system with a base pressure of ≤5 × 10−9 torr. Low temperature PL of the crystals was performed using an Edinburgh FLS 1000 photoluminescence spectrometer, attached with an OptistatDN cryostat and the temperature was controlled using a Mercury iTC temperature controller (Oxford Instruments). The sample was excited using a xenon lamp and emission was collected from 320 nm to 800 nm. Single crystal X-ray intensity measurements of compounds 1 and 2 were carried out on a Bruker D8 VENTURE Kappa Duo PHOTON II CPAD diffractometer.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank Milan K Bisai (NCL Pune), Dr J. Nithyanandhan (NCL Pune), Dr S. S. Sen (NCL Pune), and Dr R. Vaidhyanathan (IISER Pune) for insightful discussion. This work was financially supported through the DST Grant No. CRG/2019/000252. A. B. and R. B. thank the CSIR for Senior Research Fellowship.References
- B.-B. Cui, Y. Han, B. Huang, Y. Zhao, X. Wu, L. Liu, G. Cao, Q. Du, N. Liu, W. Zou, M. Sun, L. Wang, X. Liu, J. Wang, H. Zhou and Q. Chen, Nat. Commun., 2019, 10, 1–8 CrossRef CAS.
- R. Gautier, F. Massuyeau, G. Galnon and M. Paris, Adv. Mater., 2019, 31, 1807383 CrossRef.
- H. Lin, C. Zhou, Y. Tian, T. Siegrist and B. Ma, ACS Energy Lett., 2017, 3, 54–62 CrossRef.
- M. I. Saidaminov, O. F. Mohammed and O. M. Bakr, ACS Energy Lett., 2017, 2, 889–896 CrossRef CAS.
- H. Tsai, W. Nie, J.-C. Blancon, C. C. Stoumpos, R. Asadpour, B. Harutyunyan, A. J. Neukirch, R. Verduzco, J. J. Crochet, S. Tretiak, L. Pedesseau, J. Even, M. A. Alam, G. Gupta, J. Lou, P. M. Ajayan, M. J. Bedzyk, M. G. Kanatzidis and A. D. Mohite, Nature, 2016, 536, 312–316 CrossRef CAS.
- C. Zhou, H. Lin, S. Lee, M. Chaaban and B. Ma, Mater. Res. Lett., 2018, 6, 552–569 CrossRef CAS.
- Z. Li, Y. Li, P. Liang, T. Zhou, L. Wang and R.-J. Xie, Chem. Mater., 2019, 31, 9363–9371 CrossRef CAS.
- K. Huang and A. Rhys, Selected Papers Of Kun Huang: (With Commentary), World Scientific, 2000, pp. 74–92 Search PubMed.
- K. M. McCall, C. C. Stoumpos, S. S. Kostina, M. G. Kanatzidis and B. W. Wessels, Chem. Mater., 2017, 29, 4129–4145 CrossRef CAS.
- M. D. Smith and H. Karunadasa, Acc. Chem. Res., 2018, 51, 619–627 CrossRef CAS.
- X. Wang, W. Meng, W. Liao, J. Wang, R.-G. Xiong and Y. Yan, J. Phys. Chem. Lett., 2019, 10, 501–506 CrossRef.
- S. Li, J. Luo, J. Liu and J. Tang, J. Phys. Chem. Lett., 2019, 10, 1999–2007 CrossRef CAS.
- H. Shi, D. Han, S. Chen and M.-H. Du, Phys. Rev. Mater., 2019, 3, 034604 CrossRef.
- L. Mao, P. Guo, M. Kepenekian, I. Hadar, C. Katan, J. Even, R. D. Schaller, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2018, 140, 13078–13088 CrossRef CAS.
- A. Biswas, R. Bakthavatsalam, S. R. Shaikh, A. Shinde, A. Lohar, S. Jena, R. G. Gonnade and J. Kundu, Chem. Mater., 2019, 31, 2253–2257 CrossRef CAS.
- S. Brochard-Garnier, M. Paris, R. Génois, Q. Han, Y. Liu, F. Massuyeau and R. Gautier, Adv. Funct. Mater., 2019, 29, 1806728 CrossRef.
- D. Cortecchia, J. Yin, A. Petrozza and C. Soci, J. Mater. Chem. C, 2019, 7, 4956–4969 RSC.
- R. Bakthavatsalam, M. P. Haris, S. R. Shaikh, A. Lohar, A. Mohanty, D. Moghe, S. Sharma, C. Biswas, S. S. K. Raavi, R. G. Gonnade and J. Kundu, J. Phys. Chem. C, 2020, 124, 1888–1897 CrossRef CAS.
- M. P. Haris, R. Bakthavatsalam, S. Shaikh, B. P. Kore, D. Moghe, R. G. Gonnade, D. Sarma, D. Kabra and J. Kundu, Inorg. Chem., 2018, 57, 13443–13452 CrossRef CAS.
- D. Fabini, J. Phys. Chem. Lett., 2015, 6, 3546–3548 CrossRef CAS.
- S. Chakraborty, W. Xie, N. Mathews, M. Sherburne, R. Ahuja, M. Asta and S. G. Mhaisalkar, ACS Energy Lett., 2017, 2, 837–845 CrossRef CAS.
- A. H. Slavney, L. Leppert, A. Saldivar Valdes, D. Bartesaghi, T. J. Savenije, J. B. Neaton and H. I. Karunadasa, Angew. Chem., Int. Ed., 2018, 57, 12765–12770 CrossRef CAS.
- Q. Fan, G. V. Biesold-McGee, J. Ma, Q. Xu, S. Pan, J. Peng and Z. Lin, Angew. Chem., Int. Ed., 2020, 59, 1030–1046 CrossRef CAS.
- A. M. Ganose, C. N. Savory and D. O. Scanlon, Chem. Commun., 2017, 53, 20–44 RSC.
- M. Leng, Z. Chen, Y. Yang, Z. Li, K. Zeng, K. Li, G. Niu, Y. He, Q. Zhou and J. Tang, Angew. Chem., Int. Ed., 2016, 55, 15012–15016 CrossRef CAS.
- Z.-P. Wang, J.-Y. Wang, J.-R. Li, M.-L. Feng, G.-D. Zou and X.-Y. Huang, Chem. Commun., 2015, 51, 3094–3097 RSC.
- C. Zhou, H. Lin, Y. Tian, Z. Yuan, R. Clark, B. Chen, L. J. van de Burgt, J. C. Wang, Y. Zhou, K. Hanson, Q. J. Meisner, J. Neu, T. Besara, T. Siegrist, E. Lambers, P. Djurovich and B. Ma, Chem. Sci., 2018, 9, 586–593 RSC.
- C. Zhou, M. Worku, J. Neu, H. Lin, Y. Tian, S. Lee, Y. Zhou, D. Han, S. Chen, A. Hao, P. I. Djurovich, T. Siegrist, M.-H. Du and B. Ma, Chem. Mater., 2018, 30, 2374–2378 CrossRef CAS.
- Z. Wang, Z. Zhang, L. Tao, N. Shen, B. Hu, L. Gong, J. Li, X. Chen and X. Huang, Angew. Chem., Int. Ed., 2019, 58, 9974–9978 CrossRef CAS.
- D. Chen, F. Dai, S. Hao, G. Zhou, Q. Liu, C. Wolverton, J. Zhao and Z. Xia, J. Mater. Chem. C, 2020, 8, 7322–7329 RSC.
- F. Lin, H. Wang, W. Liu and J. Li, J. Mater. Chem. C, 2020, 8, 7300–7303 RSC.
- G. Song, M. Li, S. Zhang, N. Wang, P. Gong, Z. Xia and Z. Lin, Adv. Funct. Mater., 2020, 30, 2002468 CrossRef CAS.
- G. Laurita, D. H. Fabini, C. C. Stoumpos, M. G. Kanatzidis and R. Seshadri, Chem. Sci., 2017, 8, 5628–5635 RSC.
- A. Walsh, D. J. Payne, R. G. Egdell and G. W. Watson, Chem. Soc. Rev., 2011, 40, 4455–4463 RSC.
- L. Sobczyk, R. Jakubas and J. Zaleski, Pol. J. Chem., 1997, 71, 265–300 CAS.
- X. Liu, W. Wu, Y. Zhang, Y. Li, H. Wu and J. Fan, J. Phys. Chem. Lett., 2019, 10, 7586–7593 CrossRef CAS.
- B. Febriansyah, T. Borzda, D. Cortecchia, S. Neutzner, G. Folpini, T. M. Koh, Y. Li, N. Mathews, A. Petrozza and J. England, Angew. Chem., Int. Ed., 2020, 132, 10883–10888 CrossRef.
- V. Morad, S. Yakunin and M. Kovalenko, ACS Mater. Lett., 2020, 2, 845–852 CrossRef CAS.
- H. Nikol and A. Vogler, J. Am. Chem. Soc., 1991, 113, 8988–8990 CrossRef CAS.
- K. Thirumal, W. K. Chong, W. Xie, R. Ganguly, S. K. Muduli, M. Sherburne, M. Asta, S. Mhaisalkar, T. C. Sum, H. S. Soo and N. Mathews, Chem. Mater., 2017, 29, 3947–3953 CrossRef CAS.
- Z. Yuan, C. Zhou, Y. Tian, Y. Shu, J. Messier, J. C. Wang, L. J. Van De Burgt, K. Kountouriotis, Y. Xin, E. Holt, K. Schanze, R. Clark, T. Siegrist and B. Ma, Nat. Commun., 2017, 8, 1–7 CrossRef.
- B.-B. Zhang, J.-K. Chen, J.-P. Ma, X.-F. Jia, Q. Zhao, S.-Q. Guo, Y.-M. Chen, Q. Liu, Y. Kuroiwa, C. Moriyoshi, J. Zhang and H.-T. Sun, J. Phys. Chem. Lett., 2020, 11, 2902–2909 CrossRef CAS.
- J. Yu, J. Kong, W. Hao, X. Guo, H. He, W. R. Leow, Z. Liu, P. Cai, G. Qian, S. Li, X. Chen and X. Chen, Adv. Mater., 2019, 31, 1806385 Search PubMed.
- J. F. Sawyer and R. J. Gillespie, Prog. Inorg. Chem., 1986, 34, 65–114 CAS.
- G. Blasse, Prog. Solid State Chem., 1988, 18, 79–171 CrossRef CAS.
- G. Blasse, Rev. Inorg. Chem., 1983, 5, 319–381 CAS.
- A. Ranfagni, D. Mugnai, M. Bacci, G. Viliani and M. Fontana, Adv. Phys., 1983, 32, 823–905 CrossRef CAS.
- A. d. Bois and W. Abriel, Z. Naturforsch., 1990, 45, 573–578 Search PubMed.
- U. Ensinger, W. Schwarz and A. Schmidt, Z. Naturforsch., B: Chem. Sci., 1983, 38, 149–154 Search PubMed.
- B. Blažič and F. Lazarini, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, 41, 1619–1621 CrossRef.
- A. Mirochnik, A. Udovenko, T. Storozhuk, V. Karasev and B. Bukvetskii, Russ. J. Coord. Chem., 2003, 48, 961–971 Search PubMed.
- T. Sedakova, A. Mirochnik and V. Karasev, Opt. Spectrosc., 2008, 105, 517–523 CrossRef CAS.
- M. Bely, I. Zakharchenko, B. Okhrimenko and V. Skryshevsky, Ukr. Fiz. Zh., 1980, 25, 1785–1788 Search PubMed.
- N. Petrochenkova, T. Storozhuk, A. Mirochnik and V. Karasev, Russ. J. Coord. Chem., 2002, 28, 468–472 Search PubMed.
- Z. Tan, M. Hu, G. Niu, Q. Hu, J. Li, M. Leng, L. Gao and J. Tang, Sci. Bull., 2019, 64, 904–909 CrossRef CAS.
- T. V. Storozhuk, A. G. Mirochnik, N. V. Petrochenkova and V. E. Karasev, Opt. Spectrosc., 2003, 94, 920–923 CrossRef CAS.
Footnotes |
| † This paper is dedicated to all the brave individuals working tirelessly to win the battle against COVID-19 Pandemic. |
| ‡ Electronic supplementary information (ESI) available: Experimental details and additional characterizations of compounds 1, 2, and 3. CCDC Single crystal structure file for 1 (CCDC 2017736), 2 (CCDC 2017737), 3 (CCDC 2017738). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0tc03440a |
| This journal is © The Royal Society of Chemistry 2021 |