
A PEGylated alternating copolymeric prodrug of sulfur dioxide with glutathione responsiveness for Irinotecan delivery†
Yu
Zhang
a,
Hongyu
Zhang
a,
Pan
He
*b,
Xuan
Yi
ac,
Xinming
Liu
a and
Chunsheng
Xiao
 *ac
*ac
aKey Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China. E-mail: xiaocs@ciac.ac.cn
bSchool of Materials Science and Engineering, Changchun University of Science and Technology, Changchun 130022, P. R. China. E-mail: hepan@cust.edu.cn
cJilin Biomedical Polymers Engineering Laboratory, Changchun 130022, P. R. China
First published on 16th November 2020
Abstract
In this study, an enhanced anticancer strategy combining the chemotherapy from antineoplastics with the oxidative damage from a sulfur dioxide (SO2) prodrug is presented. Based on the characteristics of a high glutathione (GSH) level in the tumor microenvironment, a novel GSH-responsive SO2 polymeric prodrug mPEG-b-P(PA-alt-GDNs) was designed and synthesized via a ring-opening alternating copolymerization and “click” reaction. The GSH-sensitive mechanism of the polymer was investigated in detail. Furthermore, Irinotecan was loaded into the polymeric prodrug nanoparticles by a self-assembly method with a drug loading content of 12.3 wt% and a loading efficiency of 42.2%. The drug-loaded nanoparticles showed a sensitive response to high concentrations of GSH in the tumor cells and rapidly released both Irinotecan and SO2. The depletion of GSH and the release of SO2 were supposed to increase the level of reactive oxygen species in the tumor cell, which, in combination with the released Irinotecan, exerted an enhanced anti-proliferative effect against HepG2 cells. Finally, Irinotecan-loaded nanoparticles exhibited a stronger antitumor effect than free antineoplastics in HepG2 cells. Thus, these results indicated that our polymeric prodrug SO2 is a promising candidate for chemotherapeutic drug delivery and would be a new weapon in anticancer treatment.
Introduction
Malignant tumors, commonly known as cancers, are the second leading cause of death worldwide.1 Emerging cancer therapy techniques include immunotherapy, monoclonal antibody therapy, and stem cell transplantation. Chemotherapy is still the prevalent treatment option for cancer therapy.2–4 Most chemotherapeutics used in the clinic are hydrophobic, including doxorubicin, docetaxel, and camptothecin.5 Irinotecan, a hydrophilic derivative of camptothecin, is a US Food and Drug Administration (FDA)-approved anticancer drug.6,7 Its metabolite, 7-ethyl-10-hydroxycamptothecin (SN-38), interacts with DNA Topoisomerase I and thereby inhibits DNA replication to rapidly induce cancer cell death.8 Irinotecan has been regarded as the second line drug for the treatment of late-stage colorectal cancer.9 However, similar to other chemotherapeutics, free Irinotecan by intravenous injection exhibits severe side effects (toxic to bone marrow and the gastrointestinal tract).10 Therefore, it is of utmost importance to develop novel drug delivery systems (DDS) to selectively release chemotherapeutics to the tumor site and to treat cancer more effectively using a lower drug dose.11,12Inspired by the distinct physiological microenvironment in tumor cells compared to that in normal tissue, such as an acidic pH, hypoxia, and a higher intracellular concentration of glutathione (GSH), various stimuli-responsive DDS have been developed.13–15 Specifically, taking advantage of the higher intracellular concentration of GSH in cancer cells, various GSH-responsive polymeric DDS have been developed for effective anticancer treatment.16,17 For example, Shen et al. designed a series of reduction sensitive platinum(IV)-coordinate polymeric micelles with dicarboxylate backbone groups, which could be reduced after endocytosis to release cisplatin.18 Interestingly, these polymer–drug conjugates showed a stronger growth inhibitory effect against SKOV-3, MDA-MB-468 and MCF-7 cancer cells and better tumor accumulation than free platinum(IV). Furthermore, the combination of two or more chemotherapeutic drugs with a different mechanism of action is an emerging approach to overcome drug resistance and achieves better anti-tumor efficacy.19 Kim et al. developed pH-responsive poly(ethylene oxide)-b-poly(acrylic acid) micelles that were coated with a lecithin lipid layer to improve their in vivo stability. DOX and Irinotecan co-encapsulated nanoparticles exhibited synergistic anti-proliferative effects against several drug-sensitive and drug-resistant cancer cell lines and showed superior antitumor efficacy in an SCC-7 xenograft tumor-bearing nude mouse model.20 In another study, thermal and pH dual responsive nanoparticles consisting of four FDA-approved polymers were produced for the delivery of DOX and Irinotecan. Due to the synergistic anticancer activities, the two-drug-loaded nanoparticles eliminated cancer stem-like cells with high efficacy both in vitro and in vivo.21 Fabricating multi-component nanoparticles is complicated. Therefore, developing DDS with inherent cancer therapy effects is a promising alternative method.
Sulfur dioxide (SO2), which is known as an air pollutant, has been shown to be a vasorelaxant substance and an antimycobacterial agent in recent studies.22–24 Our group pioneered the application of SO2 polymeric prodrug micelles for DOX delivery. The intracellular GSH triggered simultaneous release of SO2 and DOX exhibited an enhanced anticancer effect against drug-resistant MCF-7 ADR cell lines and a better chemotherapeutic effect in a nude mice model.25 Furthermore, SO2 polymeric prodrug nanoparticles were used for improving photodynamic therapy (PDT) through the regulation of redox balance in tumor cells.26 Song et al. also developed rattle-structured upconversion@silica systems (RUCSNs) loaded with a a photolabile SO2 prodrug for cancer therapy.27 Upon near-infrared (NIR) light irradiation, the RUCSNs could convert NIR photons to high-energy UV photons, which efficiently stimulate SO2 release through the photolysis of the loaded prodrug. The released SO2 was found to elevate the intracellular oxidation level and thereby resulted in cancer cell death through apoptosis.
Our group focused on the development of various polyester and polypeptide-based copolymers such as DDS in the past two decades.28–30 We aimed to enhance the therapeutic effect of chemotherapeutics in cancer therapy; therefore, we designed an alternating polyester-based sulfur dioxide prodrug, mPEG-b-P(PA-alt-GDNs), for the delivery of Irinotecan (Scheme 1). The GSH-responsive polymer was synthesized via a ring-opening alternating copolymerization (ROAP) and click reaction. Next, Irinotecan was encapsulated into the polymeric nanoparticles via self-assembly. The obtained Irinotecan-loaded nanoparticles exhibited the GSH-responsive release of SO2 and Irinotecan, thereby upregulating synergistic anticancer effects in HepG2 human hepatoma cells.
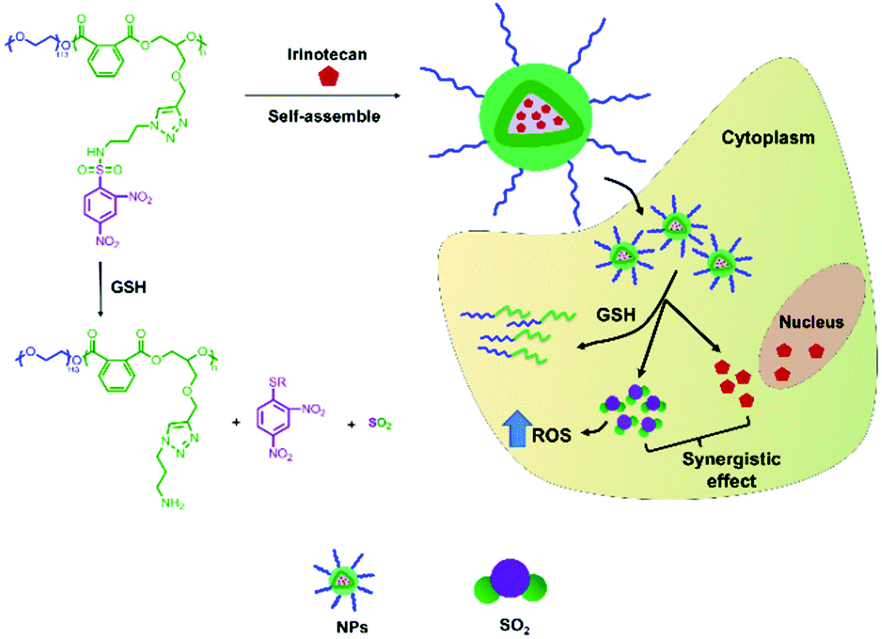 | ||
| Scheme 1 Illustration of the thiol-induced production of SO2 from mPEG-b-P(PA-alt-GDNs) and the GSH-responsive intracellular release of Irinotecan and SO2. | ||
Experimental procedures
Materials and characterization
Irinotecan was purchased from Dalian Meilun Biotechnology Co., Ltd (Dalian, China). Epichlorohydrin, carbonyldiimidazole (CDI), phthalic anhydride (PA), 1-amino-3-chloropropane hydrochloride, sodium ascorbate, copper sulfate pentahydrate, and sodium azide were obtained from Aladdin Bio-Chem Technology Co., Ltd (Shanghai, China). Methoxy poly(ethylene glycol) (mPEG-OH, Mn = 5000 g mol−1), 2,4-dinitrobenzenesulfonyl chloride (DNs-Cl), benzoylacetone, 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA), 4,6-diamidino-2-phenylindole dihydrochloride (DAPI), GSH, and IR-783 were obtained from Sigma-Aldrich LLC (Shanghai, China). Anhydrous tetrahydrofuran (THF) was purified using a solvent purification system (MB SPS-800, MBRAUN, Germany). Dialysis membranes were purchased from Greenbird Technology Co., Ltd (Shanghai, China). Sodium azide and propargyl alcohol were from Duodian Chemical Co., Ltd (Nanjing, China). L-Cysteine (Cys) was from Shanghai Macklin Biochemical Co., Ltd (Shanghai, China). Fluorescamine (FA) was from Alfa-Aesar Chemistry Co., Ltd (Shanghai, China). 7-Diethylaminocoumarin-3-aldehyde (DEACA) was synthesized according to the literature.31 Deuterated dimethyl sulfoxide (DMSO-d6), deuterated trifluoroacetic acid (TFA-d), deuterated chloroform (CDCl3), deuterated water (D2O) with TMS as a reference, and all other chemicals were used as received.Nuclear magnetic resonance (NMR) spectra were performed on a Bruker AV-300 NMR spectrometer. Gel permeation chromatography (GPC) was conducted in a Waters instrument (515 HPLC pump) equipped with a Wyatt interferometric refractometer. A series of polystyrene standards were used as standards to calculate the molecular weights. Dynamic light scattering (DLS) measurement was conducted on a Zetasizer (Nano-ZS, Malvern Instruments, UK). The morphology of nanoparticles was observed using a field emission scanning electron microscope (Zeiss Merlin, Germany).
Synthesis of mPEG-b-P(PA-alt-GPE)
Glycidyl propargyl ether (GPE) and poly(ethylene glycol)-block-poly(phthalic anhydride-alter-glycidyl propargyl ether) (mPEG-b-P(PA-alt-GPE)) block copolymers were synthesized according to our previously reported method.32Synthesis of N-(3-azidopropyl)-2,4-dinitrobenzenesulfonamide (AP-DNs)
Sodium hydrogen carbonate (2.52 g, 0.029 mol) and 3-azido-propylamine (APA) (1.36 g, 0.013 mol) were dispersed in 20 mL of anhydrous THF at 0 °C. Then, DNs-Cl (4.00 g, 0.015 mol) solution in THF was added dropwise. After stirring for 16 h at room temperature, the pH of the mixture was adjusted to about 1.0 using hydrochloric acid (1 M). Subsequently, THF was removed and the residual aqueous mixture was extracted with 100 mL of dichloromethane. The organic phase was washed with 50 mL brine. After drying, 3.1 g of AP-DNs was obtained (yield: 70.2%).Synthesis of mPEG-b-P(PA-alt-GDNs)
The target copolymer mPEG-b-P(PA-alt-GDNs) was synthesized via a “click” reaction. In brief, 2.0 g of mPEG-b-P(PA-alt-GPE) (3.5 mmol alkyne) and 2.5 g of AP-DNs (7.6 mmol) were dissolved in 40 mL of anhydrous DMSO. After the removal of oxygen by bubbling with nitrogen for 0.5 h, sodium ascorbate (0.35 g, 1.8 mmol) and copper sulfate pentahydrate (0.2 g, 0.8 mmol) were added. The mixture was stirred at 40 °C for 72 h and purified by dialysis. A total of 1.93 g of pure mPEG-b-P(PA-alt-GDNs) polymer was obtained after freeze-drying (yield: 61.4%).Preparation of mPEG-b-P(PA-alt-GDNs) micelles
To prepare blank micelles, 200 mg of mPEG-b-P(PA-alt-GDNs) was dissolved in 20 mL of DMF. The polymer solution was dropped into 150 mL of phosphate buffered saline (PBS, pH = 7.4) under vigorous stirring for 2 h. Then, the mixture was transferred into a dialysis bag (MWCO 3500) and dialyzed against water for 24 h. The fresh micelle solution was characterized via DLS and scanning electron microscopy (SEM). Blank micelles were obtained after lyophilization. The critical micelle concentration (CMC) of polymeric micelles was determined via the UV absorption of the benzoylacetone probe.30 The UV-vis spectrum was recorded using a Lambda 365 spectrophotometer (PerkinElmer, USA).Thiol-responsiveness of mPEG-b-P(PA-alt-GDNs)
The thiol-responsiveness of mPEG-b-P(PA-alt-GDNs) micelles was investigated via1H NMR spectroscopy, primary amine production, and thiol-triggered SO2 release. Firstly, 4.0 mg of lyophilized micelles were suspended in 0.5 mL of D2O, to which 9.7 mg of cysteine was added. Then, 1H NMR spectra were recorded at 15, 30, 60, 90, 120, and 180 min. The structure of polymers after complete conversion from pendant DNs to amino groups was also investigated via1H NMR spectroscopy. The primary amine production after the thiol treatment was determined by using fluorescamine as a fluorescent probe.33The thiol-triggered release of SO2 was quantified by measuring bisulfite (HSO3−) in PBS using DEACA as a probe.22,28 In brief, blank polymeric micelles were dissolved in PBS with a pH of 5.8. After cysteine (20 molar equivalents of disulfide group) was added, DEACA solution in ethanol was dropped into the above mixtures. The concentration of DEACA was fixed at 5 mmol in each solution. Finally, the fluorescence intensity at 483.5 nm was measured on a Fluorescence Master System (Photon Technology International, USA) at different time points.
Irinotecan encapsulation and in vitro release
Briefly, 200 mg of mPEG-b-P(PA-alt-GDNs) and 50 mg of Irinotecan were dissolved in 20 mL of DMF. The mixed solution was dropped into 150 mL of PBS under stirring. The following protocols were the same as those for blank micelles. Finally, the drug-loaded nanoparticles, denoted as NPs-Irinotecan, were obtained through lyophilization. The drug loading content (DLC) and drug loading efficiency (DLE) were calculated according to the following formulas:| DLC (wt%) = (weight of loaded Irinotecan/weight of NPs-Irinotecan) × 100% |
| DLE (%) = (weight of loaded Irinotecan/weight of feeding Irinotecan) × 100% |
For the drug release study, dialysis bags containing 4 mL of the drug-loaded nanoparticles were immersed in 50 mL PBS with different concentrations of GSH (0, 1, 5, and 10 mmol). At predetermined times, 2 mL of the release solution was withdrawn and 2 mL of the fresh PBS solution was added. The concentration of Irinotecan was determined via HPLC at a wavelength of 254 nm using a mixture of methanol, acetonitrile, and PBS (pH 4) (35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:55, v/v/v) as the eluting solution.
:10:55, v/v/v) as the eluting solution.
Cell culture, MTT assay, and cellular uptake
The human hepatocellular carcinoma (HepG2) cell line was obtained from American Type Culture Collection (Rockville, MD, USA). The cytotoxicity of mPEG-b-P(PA-alt-GDNs) micelles (denoted as NPs), NPs-Irinotecan, and free Irinotecan toward HepG2 cells was measured to evaluate the in vitro anticancer effects. Firstly, HepG2 cells were cultured under normal conditions (5% CO2 at 37 °C) in DMEM (GIBCO) buffer which contained 10% inactivated fetal bovine serum, penicillin (50 U mL−1) and streptomycin (50 U mL−1). For cytotoxicity evaluation, HepG2 cells at a density of 1 × 104 per well were seeded into 96 well plates in 180.0 μL of DMEM. After overnight incubation, 20.0 μL of blank NPs, NPs-Irinotecan, or free Irinotecan was added. The maximum concentrations of blank NPs, Irinotecan in NPs-Irinotecan and free Irinotecan were 2.0 mg mL−1, 20.0 μg mL−1 and 20.0 μg mL−1, respectively. Three parallel tests were performed for each concentration. After 24 h or 48 h, 20.0 μL of MTT stock solution (5 mg mL−1) was added into each well and cultured for another 4 h. The supernatant was discarded and replaced with 150.0 μL of DMSO. After shaking for 5 minutes, the absorbance of the above-mentioned solutions was measured at 490 nm using a Bio-Rad 680 microplate reader. Cell viability (%) was calculated using the following equation: Cell viability (%) = (Asample/Acontrol) × 100%, where Asample and Acontrol are the absorbance of the sample and control wells, respectively.The cellular uptake of free Irinotecan and NPs-Irinotecan by HepG2 cells was investigated via HPLC. Briefly, HepG2 cells were seeded in a 6-well plate at a density of 1 × 104 cells mL−1 After 24 h, predetermined amounts of NPs-Irinotecan or free Irinotecan were added and incubated for 3 and 6 h. Next, the cells were collected and homogenized. The uptake drug content was determined via HPLC.
Cellular ROS level, GSH concentration, and SO2 release study
HepG2 cells were seeded in glass plates and incubated for 24 h. Fresh DMEM (control group) or DMEM containing NPs were added and incubated for 10 h. Then 10 μM of DEACA (for the determination of SO2, λex = 390 nm) or DCFH-DA (for the determination of ROS, λex = 488 nm) was added and incubated for 30 min. Subsequently, HepG2 cells were washed with PBS four times, then fixed with 4% formaldehyde, and observed via confocal laser scanning microscopy (CLSM, Carl Zeiss, LSM 780). The variation of cellular GSH concentration was measured using the same protocol as in our previous report.25Data analysis
All experiments were conducted independently at least three times, and the results were expressed as mean value ± standard deviation (SD). SPSS (version 18.0, Chicago, IL, USA) was used for statistical analysis. p < 0.05 was considered statistically significant. p < 0.01 and p < 0.001 were considered highly statistically significant.Results and discussion
Synthesis of mPEG-b-P(PA-alt-GDNs)
The synthetic route is depicted in Scheme 2. Firstly, PA and GPE were prepared according to our previous protocols.32 The block copolymer, mPEG-b-P(PA-alt-GPE), with pendant alkyne groups was synthesized by the ROAP method. Using this strategy, mPEG-OH was used as the macroinitiator and tert-butylimino-tris(dimethylamino)phosphorane (t-BuP1) was employed as the phosphazene base catalyst for the alternating copolymerization of PA and GPE, yielding mPEG-b-P(PA-alt-GPE) with a molecular weight of 20.9 kg mol−1 and a polydispersity of 1.29. Subsequently, a standard copper(I)-catalyzed click chemistry reaction was conducted to conjugate side alkynes with the azide compound AP-DNs (Fig. S1, ESI†). After dialysis, the target polymeric prodrug of SO2, mPEG-b-P(PA-alt-GDNs), was obtained via lyophilization at a yield of 61.4%. The chemical structure of this polymer prodrug of SO2 was characterized via1H NMR and 13C NMR spectroscopy (Fig. S2, ESI†), which showed all the characteristic proton and carbon signals of mPEG-b-P(PA-alt-GDNs). The new proton resonance peaks (a, b, d, e, and f) showed the successful conjugation of AP-DNs onto the side chains of mPEG-b-P(PA-alt-GPE). Moreover, the GPC profiles (Fig. S3, ESI†) showed a number-average molar mass of 27.0 kg mol−1 (Đ = 1.33) for mPEG-b-P(PA-alt-GDNs), which was higher compared to that of the precursor polymer mPEG-b-P(PA-alt-GPE) (Mn = 20.9 kg mol−1, Đ = 1.27) (Table S1, ESI†) and further confirmed the successful preparation of the polymeric prodrug of SO2, mPEG-b-P(PA-alt-GDNs).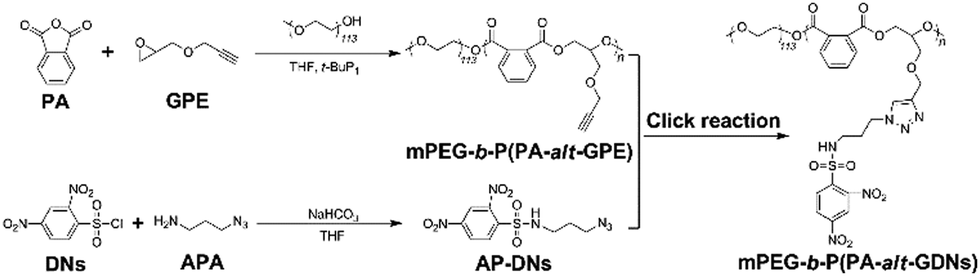 | ||
| Scheme 2 Synthetic route to mPEG-b-P(PA-alt-GDNs). | ||
Self-assembly and the thiol-responsive property of mPEG-b-P(PA-alt-GDNs)
By using benzoylacetone as a probe, the critical micelle concentration (CMC) of the self-assembled mPEG-b-P(PA-alt-GDNs) micellar nanoparticles (denoted as NPs) was determined to be 0.841 μg mL−1 (Fig. 1A). The average hydrodynamic diameter of blank NPs was 85.4 nm as determined by DLS, while the corresponding Irinotecan-loaded nanoparticles (denoted as NPs-Irinotecan) exhibited a larger hydrodynamic size of 106.6 nm (Fig. 1B). SEM images showed spherical morphologies for NPs-Irinotecan (Fig. 1C) with narrow size distribution. To investigate the stability after in vivo injection, the diameter of drug-loaded NPs incubated with bovine serum albumin (BSA, 0.1 mg mL−1) was monitored. No significant changes in the average size of NPs-Irinotecan were observed during the 48 h of incubation, thereby indicating the good stability of NPs-Irinotecan (Fig. 1D).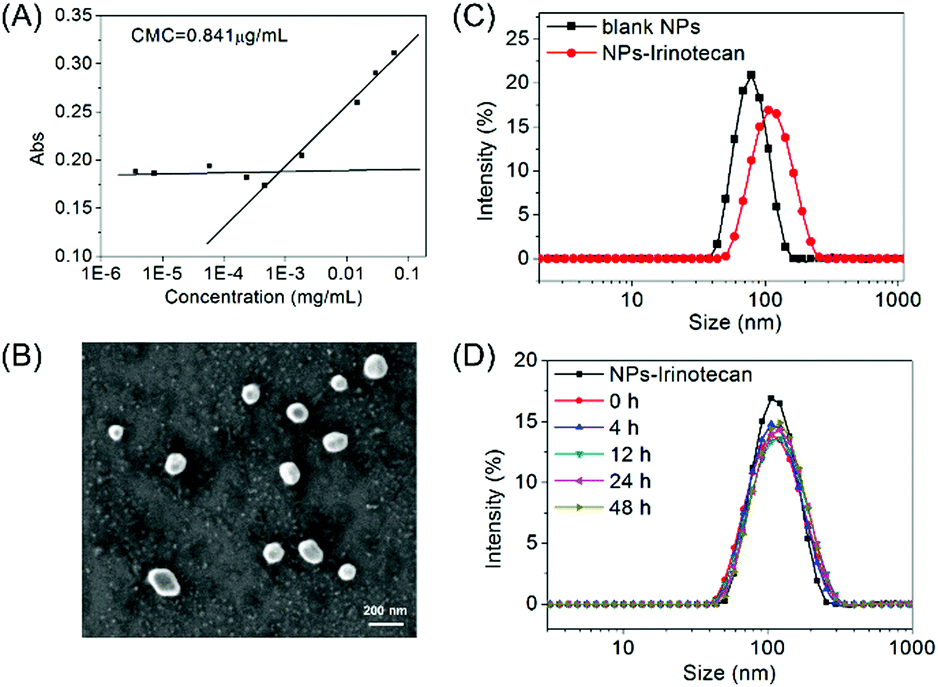 | ||
| Fig. 1 Characterization of mPEG-b-P(PA-alt-GDNs) micelles (blank NPs) and Irinotecan-loaded micelles (NPs-Irinotecan). (A) Determination of the CMC of mPEG-b-P(PA-alt-GDNs) micelles in aqueous media; (B) DLS profiles of blank NPs and NPs-Irinotecan; (C) SEM image of NPs-Irinotecan; (D) DLS profiles of NPs-Irinotecan treated with bovine serum albumin (BSA, 0.1 mg mL−1) for 48 h. | ||
The thiol-responsive mechanism of 2,4-dinitrobenzenesulfonamide (DNs) compounds was verified in our previous report.25 In this study, we investigated the thiol-responsive process by monitoring the in situ1H NMR spectra of NPs after adding cysteine (Cys). As shown in Fig. S4 (ESI†), no obvious signal was observed, except those of PEG protons in the 1H NMR spectrum of NPs suspended in D2O (0 min), due to the self-assembly of mPEG-b-P(PA-alt-GDNs) into core–shell structured micelles. At 30 min after the addition of Cys, three proton resonance peaks of the degradation small molecules (a, b and c) and the signal of the proton (e) neighbored to the newly generated amino group appeared, which gradually increased in the following 3 h, thereby indicating the formation of a free amino group in the side chain of the polymer and the release of the Cys-2,4-dinitrophenyl conjugate into D2O. The 1H NMR spectrum of the resultant polymer treated using an excess amount of Cys for 24 h and purified by dialysis is shown in Fig. 2A, which demonstrated the detachment of the DNs group to liberate the primary amino group in the side chain of mPEG-b-P(PA-alt-GDNs).
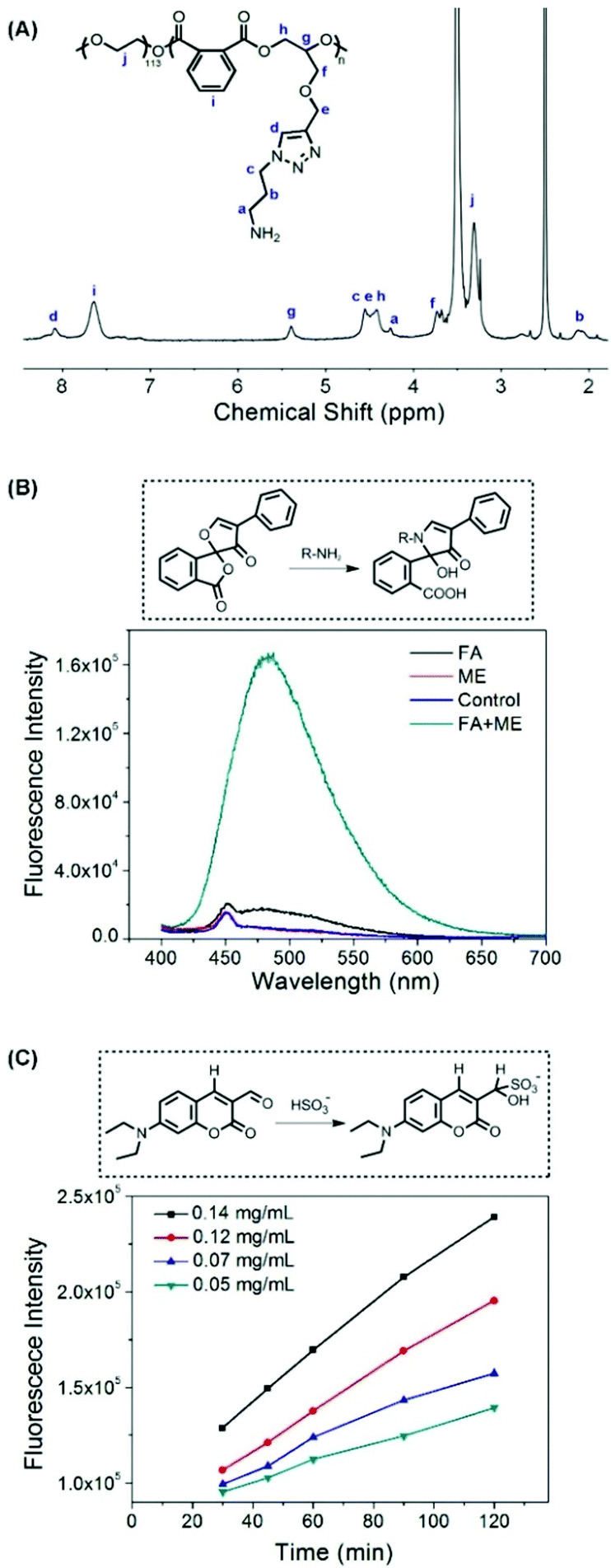 | ||
| Fig. 2 (A) 1H NMR spectrum of NPs treated using an excess amount of cysteine in DMSO-d6. (B) Detection of the free amino group in the NP solution after treatment with 2-mercapto-ethanol (ME) by using fluorescamine (FA) as a fluorescent probe. (C) Release of SO2 from Cys-trigged NPs of different concentrations by using DEACA as a fluorescent probe. | ||
To further verify the thiol-induced detachment of the DNs group in the polymer side chain, the newly generated amino group was checked using fluorescamine as a probe. The dramatic increase of the fluorescence intensity in Fig. 2B confirmed the formation of an amino group after treatment with 2-mercapto-ethanol (ME). The production and release of SO2 was quantitatively characterized using the DEACA probe. After the addition of Cys, the fluorescence intensity from HSO3− conjugating DEACA was almost proportional to time and increased with the increasing concentration of cysteine (Fig. 2C). In fact, more than 90% of SO2 was released within 2 h for the 0.14 mg mL−1 cysteine treated NP solution. The thiol-responsive property of blank NPs was further investigated by monitoring the size changes of NPs in the presence of 5 mM GSH. As displayed in Fig. S5 (ESI†), the diameter of NPs increased significantly and the size distribution became much broader after incubation for 8 h, indicating the swelling/dissociation of NPs after treatment with GSH.
In summary, mPEG-b-P(PA-alt-GDNs) formed NPs in aqueous solution, which produced a free amino group in the side chain and released SO2 by the trigger of thiol compounds. Thus, the generated primary amino groups in the side chain could make the resultant polymer more hydrophilic, which further triggered the swelling/dissociation of NPs.
In vitro release of Irinotecan and cellular uptake of NPs-Irinotecan
Irinotecan was encapsulated into the polymeric prodrug micellar nanoparticles with a DLC of 12.3 wt% and a DLE of 42.2%. The relatively low DLE may be attributed to the slight hydrophilicity of Irinotecan compared to other extremely hydrophobic anti-cancer drugs. In this study, the in vitro release of Irinotecan was investigated in PBS with different concentrations of GSH. As illustrated in Fig. 3A, less than 10% of the encapsulated Irinotecan was released for 3 days in PBS (pH 7.4) in the absence of GSH, which could be attributed to the rapid diffusion of Irinotecan located at the outside layer of the micelle core. When 1 mM GSH was added to this neutral buffer, a more rapid Irinotecan release was observed and a cumulative drug release of 25.2% was achieved at 24 h. Moreover, 5 mM and 10 mM GSH triggered the burst release of Irinotecan in the first 10 h, reaching 67.3% and 75.3% drug release at 72 h in the presence of 5 mM and 10 mM GSH, respectively. This should be ascribed to the thiol-triggered detachment of DNs groups and the simultaneous generation of amino groups in the side chains, which resulted in the conversion of the hydrophobic P(PA-alt-GDNs) segment into a relatively high hydrophilic segment and subsequently triggered the swelling/dissociation of NPs and a rapid drug release from the drug-loaded nanoparticles.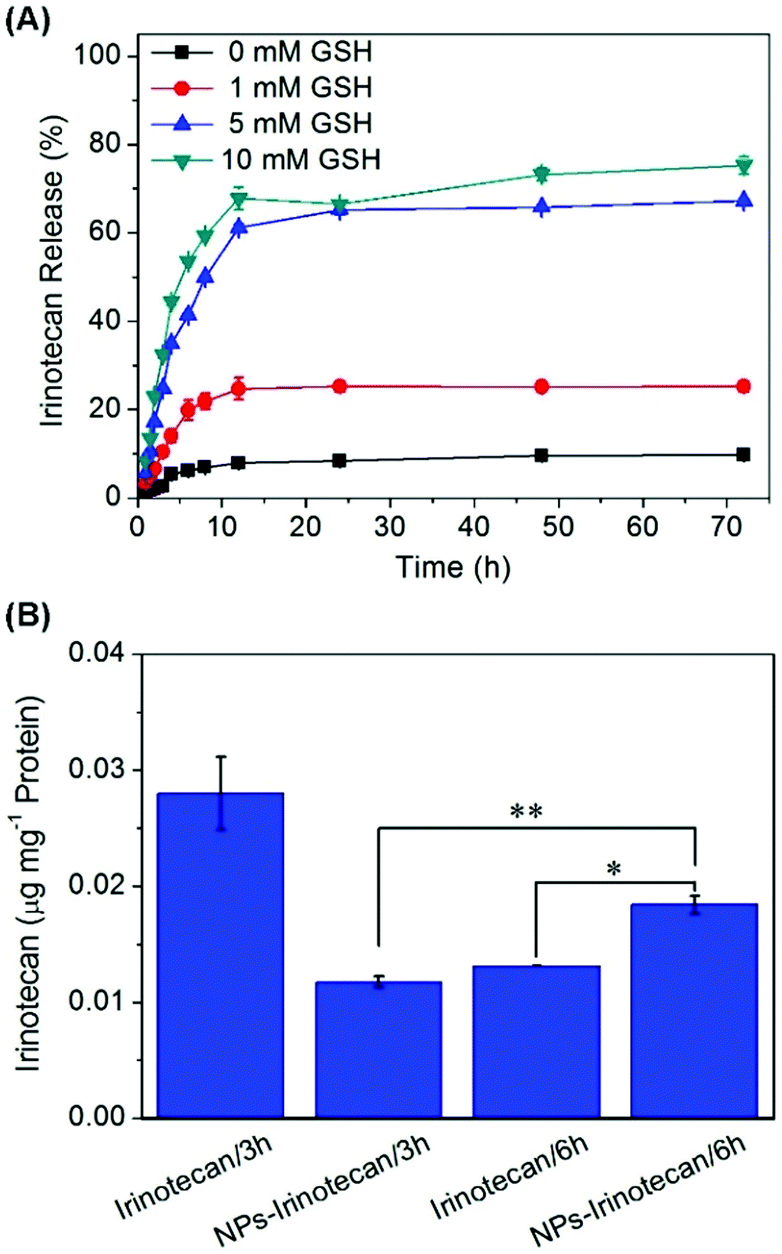 | ||
| Fig. 3 (A) Drug release behavior of NPs-Irinotecan tested in PBS (pH 7.4) containing 0, 1, 5, or 10 mM GSH. (B) Cellular uptake of Irinotecan after incubation with free Irinotecan and NPs-Irinotecan for 3 or 6 h in HepG2 cells. **p < 0.01, *p < 0.05. | ||
Next, the cellular uptake of NPs-Irinotecan was evaluated by measuring the concentration of Irinotecan in HepG2 cells. When NPs-Irinotecan and free drug were incubated with HepG2 cells, more Irinotecan was detected in the free Irinotecan-treated group in the first 3 h due to the fast diffusion of the free drug into cancer cells (Fig. 3B). However, after incubation for 6 h, a higher cellular uptake (0.018 μg Irinotecan per mg protein) was observed in the NPs-Irinotecan group due to the time-dependent endocytosis of NPs. Taken together, these results indicated that NPs-Irinotecan could be effectively internalized into cancer cells and release the loaded drugs into cells in an intracellular reductive environment.
Anticancer effect in vitro
To evaluate the anticancer effect, the in vitro cytotoxicity of NPs, NPs-Irinotecan, and free Irinotecan toward HepG2 cells was investigated. As shown in Fig. 4, the cell viability of the NPs-Irinotecan group was lower compared to that of the other two groups, when the equivalent concentrations of Irinotecan were higher than 10 μg mL−1. The IC50 values of NPs-Irinotecan and free Irinotecan groups were 11.4 and 39.1 μg mL−1, respectively, thereby indicating an enhanced anticancer effect by using SO2 prodrug NPs as the delivery vehicle.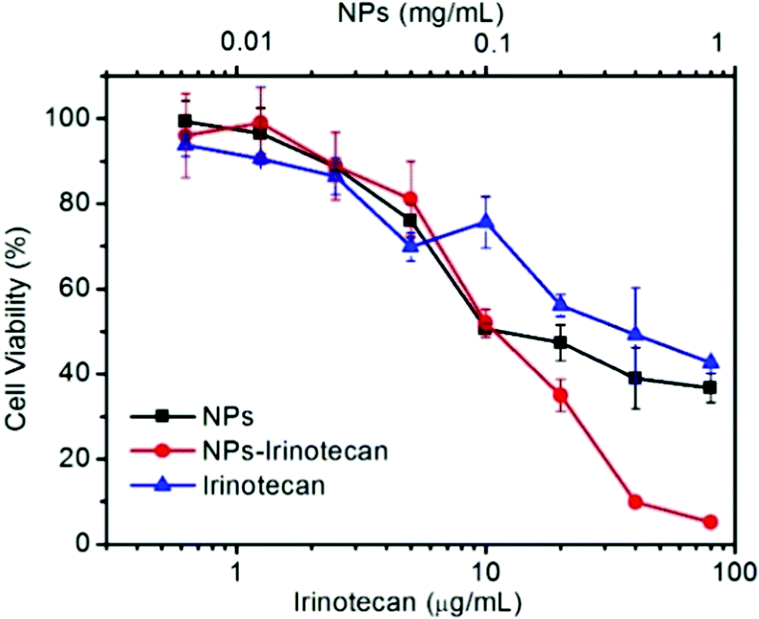 | ||
| Fig. 4 Cell viabilities of HepG2 cells co-cultured with different concentrations of NPs, Irinotecan, or NPs-Irinotecan for 48 h. | ||
Reduction of GSH, intracellular release of SO2 and up-regulation of ROS in HepG2 cells
To further understand the anticancer mechanism of SO2 prodrug NPs, the changes of GSH and ROS levels in cancer cells and the intracellular release of SO2 were investigated. As presented in Fig. 5, the GSH level in HepG2 cells was significantly decreased after treatment with NPs for 12 h, which should be ascribed to the reaction of GSH with the DNs group in NPs. The GSH-induced detachment of the DNs group in NPs would result in the release of SO2. As shown in Fig. 6A, HepG2 cells incubated with NPs exhibited homogeneous blue fluorescence throughout the cells, which originated from the SO2-conjugated fluorescent probe DEACA, indicating the successful internalization of NPs into tumor cells and the intracellular GSH-triggered release of SO2. The fluorescence density in control cells was only one percent of that of NP-treated cells (Fig. S6A, ESI†). Due to the oxidative nature of the released SO2 and the consumption of GSH during the release of SO2, it was anticipated that the ROS level would increase in NP-treated cancer cells. To verify this theory, the ROS level in HepG2 cells with or without treatment using NPs was determined via CLSM using DCFH-DA as the fluorescent probe. As shown in Fig. 6B, green fluorescence was clearly observed in the NP-treated cells, which was about fivefold higher when compared to that in the control group (Fig. S6B, ESI†), and indicated a significant up-regulation of the ROS level in HepG2 cells after incubation with NPs. The released SO2 and increased ROS levels would ultimately induce high oxidative stress and cause damages to nuclear DNA in tumor cells, leading to the apoptosis of cancer cells.34,35 Meanwhile, the synchronously released Irinotecan could also induce cell death by blocking topoisomerase I. As a result, NPs-Irinotecan showed an enhanced anticancer effect (Fig. 4) through the combined effect of the released SO2 and Irinotecan. Therefore, our P(PA-alt-GDNs) NPs should be a promising nanocarrier for anticancer drug delivery, which can boost the anticancer effect by elevating the ROS level in cancer cells by SO2 and thereby exerting the combined therapeutic effect with Irinotecan.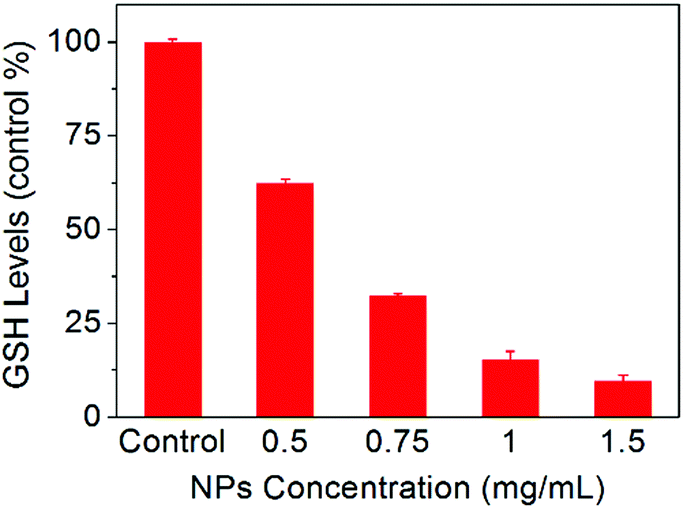 | ||
| Fig. 5 Changes of GSH levels in HepG2 cells after incubation with different concentrations of NPs for 12 h. | ||
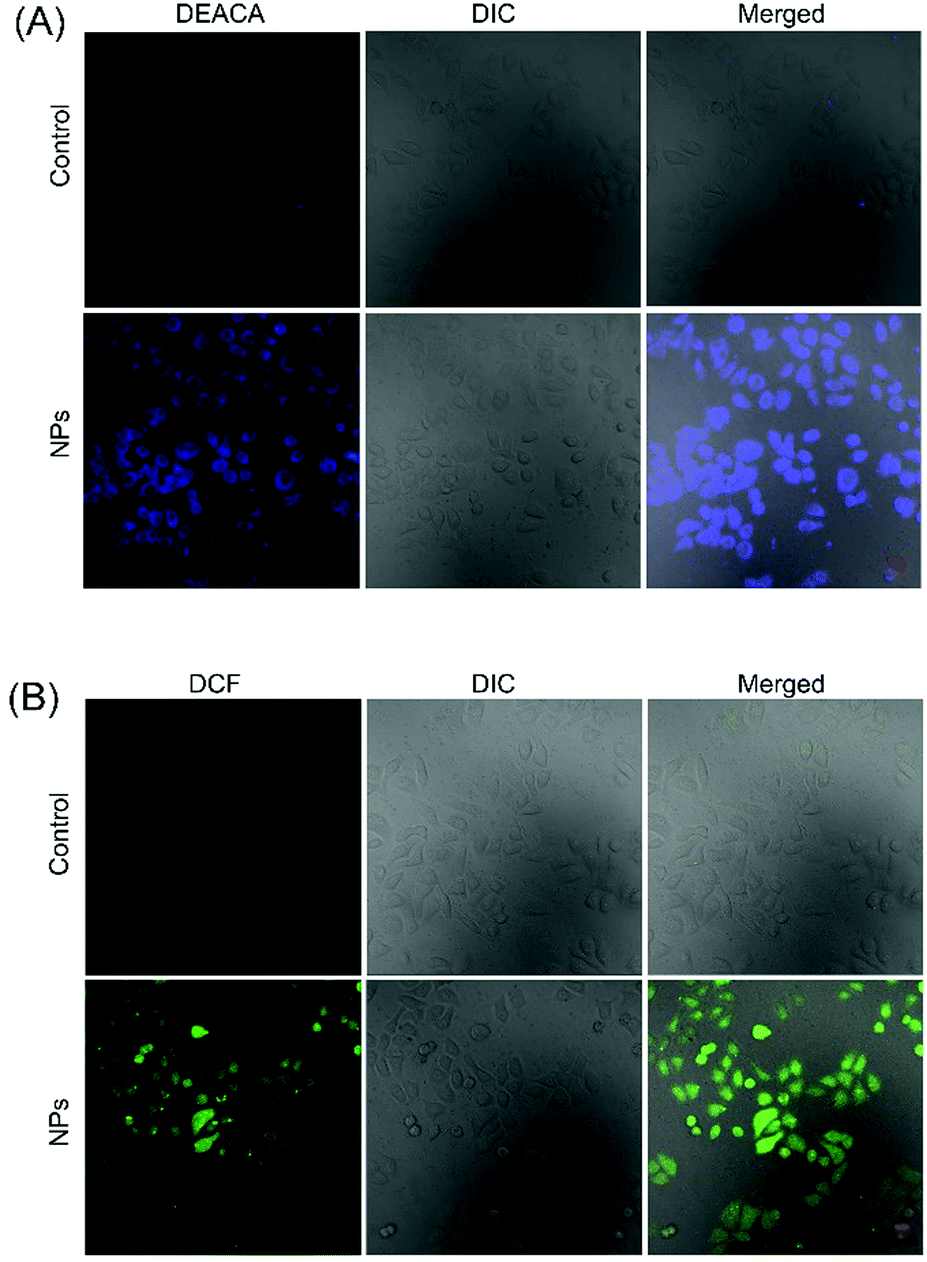 | ||
| Fig. 6 CLSM observation of SO2 release (A) and the increase of ROS (B) in HepG2 cells. The scale bar is 20 μm. | ||
Conclusions
In summary, a novel GSH-responsive polymeric prodrug of SO2, mPEG-b-P(PA-alt-GDNs), was successfully synthesized through the combination of the ROAP and click chemistry reaction. The obtained PEGylated alternating copolymer could self-assemble into micellar nanoparticles with narrow size distribution in an aqueous solution. The resultant prodrug NPs exhibited the thiol-triggered release of SO2 and the simultaneous generation of primary amine groups in the side chains. As a result, Irinotecan-loaded NPs displayed a GSH-responsive release of both SO2 and Irinotecan. The released SO2 increased the ROS level in cancer cells and caused cell death through oxidation damage, thereby boosting the therapeutic effect of Irinotecan. The GSH sensitive Irinotecan-loaded mPEG-b-P(PA-alt-GDNs) nanoparticles were expected to be a promising strategy for clinical anti-tumor therapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (51803014, 51573184, 52003268, 51520105004 and 51833010), the Jilin Provincial Science and Technology Development Program (20190103022JH, 20200801006GH), and the Youth Innovation Promotion Association of Chinese Academy of Sciences (2017266).References
- S. Hossen, M. K. Hossain, M. K. Basher, M. N. H. Mia, M. T. Rahman and M. J. Uddin, J. Adv. Res., 2019, 15, 1–18 CrossRef CAS.
- B. De Angelis, N. Depalo, F. Petronella, C. Quintarelli, M. L. Curri, R. Pani, A. Calogero, F. Locatelli and L. De Sio, J. Mater. Chem. B, 2020, 8, 1823–1840 RSC.
- S. Ezrahi, A. Aserin and N. Garti, Adv. Colloid Interface Sci., 2019, 263, 95–130 CrossRef CAS.
- W. Song, L. Shen, Y. Wang, Q. Liu, T. J. Goodwin, J. Li, O. Dorosheva, T. Liu, R. Liu and L. Huang, Nat. Commun., 2018, 9, 2237 CrossRef.
- T. M. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. X. Yang and Y. N. Xia, Angew. Chem., Int. Ed., 2014, 53, 12320–12364 CAS.
- X. S. Liu, A. Situ, Y. A. Kang, K. R. Villabroza, Y. P. Liao, C. H. Chang, T. Donahue, A. E. Nel and H. Meng, ACS Nano, 2016, 10, 2702–2715 CrossRef CAS.
- T. Matsuzaki, T. Yokokura, M. Mutai and T. Tsuruo, Cancer Chemother. Pharmacol., 1988, 21, 308–312 CrossRef CAS.
- Y. Pommier, Nat. Rev. Cancer, 2006, 6, 789–802 CrossRef CAS.
- J. Weekes, A. K. Y. Lam, S. Sebesan and Y. H. Ho, World J. Gastroenterol., 2009, 15, 3597–3602 CrossRef CAS.
- Y.-E. Gao, S. Bai, X. Shi, M. Hou, X. Ma, T. Zhang, B. Xiao, P. Xue, Y. Kang and Z. Xu, Colloids Surf., B, 2018, 170, 488–496 CrossRef CAS.
- Y. Wang, A. Santos, A. Evdokiou and D. Losic, J. Mater. Chem. B, 2015, 3, 7153–7172 RSC.
- J. Ding, X. Feng, Z. Jiang, W. Xu, H. Guo, X. Zhuang and X. Chen, Biomacromolecules, 2019, 20(12), 4258–4271 CrossRef CAS.
- H. S. El-Sawy, A. M. Al-Abd, T. A. Ahmed, K. M. El-Say and V. P. Torchilin, ACS Nano, 2018, 12, 10636–10664 CrossRef.
- Y. Zhang, P. He, X. Liu, H. Yang, H. Zhang, C. Xiao and X. Chen, Eur. Polym. J., 2018, 107, 308–314 CrossRef CAS.
- R. M. Uda, N. Yoshida, T. Iwasaki and K. Hayashi, J. Mater. Chem. B, 2020, 8(36), 8242–8248 RSC.
- M. Huo, J. Yuan, L. Tao and Y. Wei, Polym. Chem., 2014, 5, 1519–1528 RSC.
- J. Yao, P. He, Y. Zhang, H. Zhang, P. Zhang, M. Deng and C. Xiao, Polym. Int., 2019, 68, 1817–1825 CrossRef CAS.
- J. Yang, W. Liu, M. Sui, J. Tang and Y. Shen, Biomaterials, 2011, 32, 9136–9143 CrossRef CAS.
- K. Chen, X. Xin, L. Qiu, W. Li, G. Guan, G. Li, M. Qiao, X. Zhao, H. Hu and D. Chen, Acta Biomater., 2019, 100, 118–131 CrossRef CAS.
- T. Ramasamy, H. B. Ruttala, J. Y. Choi, T. H. Tran, J. H. Kim, S. K. Ku, H. G. Choi, C. S. Yong and J. O. Kim, Chem. Commun., 2015, 51, 5758–5761 RSC.
- H. Wang, P. Agarvval, S. T. Zhao, R. X. Xu, J. H. Yu, X. B. Lu and X. M. He, Biomaterials, 2015, 72, 74–89 CrossRef CAS.
- Z. Meng, J. Li, Q. Zhang, W. Bai, Z. Yang, Y. Zhao and F. Wang, Inhalation Toxicol., 2009, 21, 1223–1228 CrossRef.
- S. R. Malwal, D. Sriram, P. Yogeeswari, V. B. Konkimalla and H. Chakrapani, J. Med. Chem., 2012, 55, 553–557 CrossRef CAS.
- K. A. Pardeshi, S. R. Malwal, A. Banerjee, S. Lahiri, R. Rangarajan and H. Chakrapani, Bioorg. Med. Chem. Lett., 2015, 25, 2694–2697 CrossRef CAS.
- W. Shen, W. Liu, H. Yang, P. Zhang, C. Xiao and X. Chen, Biomaterials, 2018, 178, 706–719 CrossRef CAS.
- Y. Zhang, W. Shen, P. Zhang, L. Chen and C. Xiao, Chem. Commun., 2020, 56(42), 5645–5648 RSC.
- S. Li, R. Liu, X. Jiang, Y. Qiu, X. Song, G. Huang, N. Fu, L. Lin, J. Song, X. Chen and H. Yang, ACS Nano, 2019, 13, 2103–2113 CAS.
- J. Jiang, N. Shen, T. Ci, Z. Tang, Z. Gu, G. Li and X. Chen, Adv. Mater., 2019, 31, 1904278 CrossRef CAS.
- G.-q. C. Xue-si Chen, Y.-h. Tao, Y.-z. Wang, X.-b. Lv, L.-q. Zhang, J. Zhu, J. Zhang and X.-h. Wang, Acta Polym. Sin., 2019, 50, 1068–1082 Search PubMed.
- C. Xiao, J. Ding, C. He and X. Chen, Acta Polym. Sin., 2018, 1, 45–55 Search PubMed.
- Y. Yang, F. Huo, J. Zhang, Z. Xie, J. Chao, C. Yin, H. Tong, D. Liu, S. Jin, F. Cheng and X. Yan, Sens. Actuators, B, 2012, 166-167, 665–670 CrossRef CAS.
- Y. Zhang, P. He, X. Liu, H. Yang, H. Zhang, C. Xiao and X. Chen, Biomater. Sci., 2019, 7, 3898–3905 RSC.
- S. Udenfriend, S. Stein, P. Böhlen, W. Dairman, W. Leimgruber and M. Weigele, Science, 1972, 178, 871 CrossRef CAS.
- S. C. Langley-Evans, G. J. Phillips and A. A. Jackson, Comp. Biochem. Physiol., Part C: Pharmacol., Toxicol. Endocrinol., 1996, 114, 89–98 CAS.
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary Fig. S1–S6. See DOI: 10.1039/d0tb02097d |
| This journal is © The Royal Society of Chemistry 2021 |