
Electrocatalytic hydrogen evolution using hybrid electrodes based on single-walled carbon nanohorns and cobalt(II) polypyridine complexes†
Elisabetta
Benazzi‡§
a,
Federico
Begato‡
 b,
Alessandro
Niorettini
a,
Lorenza
Destro
b,
Klaus
Wurst
c,
Giulia
Licini
b,
Stefano
Agnoli
b,
Cristiano
Zonta
*b and
Mirco
Natali
*ad
b,
Alessandro
Niorettini
a,
Lorenza
Destro
b,
Klaus
Wurst
c,
Giulia
Licini
b,
Stefano
Agnoli
b,
Cristiano
Zonta
*b and
Mirco
Natali
*ad
aDepartment of Chemical, Pharmaceutical, and Agricultural Sciences (DOCPAS), University of Ferrara, Via L. Borsari 46, Ferrara, 44121, Italy. E-mail: mirco.natali@unife.it
bDepartment of Chemical Sciences, University of Padova, Via F. Marzolo 1, 35131 Padova, Italy. E-mail: cristiano.zonta@unipd.it
cInstitute of General, Inorganic and Theoretical Chemistry, University of Innsbruck, Innrain 80-82, A-6020 Innsbruck, Austria
dCentro Interuniversitario per la Conversione Chimica dell'Energia Solare (SolarChem), Sez. Di. Ferrara, Via L. Borsari, 46, 44121 Ferrara, Italy
First published on 16th August 2021
Abstract
The generation of hydrogen from water represents an important task towards a carbon neutral economy. Within this context, the preparation of hybrid electrodes merging the versatility of solid-state porous substrates and the catalytic ability and tunability of molecular complexes represents a great challenge. In the present work, we report on the preparation of hybrid cathodes for the hydrogen evolution reaction (HER) through an unprecedented combination of single-walled carbon nanohorns (SWCNHs) and two novel cobalt(II) polypyridine complexes based on the tris(2-pyridylmethyl)amine (TPMA) ligand scaffold. Suitable pyrene groups are introduced in the ligand framework in different positions to provide a way for direct anchoring onto the carbonaceous substrate by exploiting non-covalent π–π interactions. The present systems behave as competent cathodes for the HER in neutral aqueous solution with overpotentials of η ∼ 0.5 V and stable current densities (within 1 h electrolysis) up to −0.50 mA cm−2, whose exact values depend on the catalyst used and are mainly related to the respective loading on the electrode surface. In both cases, hydrogen evolution is detected under continuous electrolysis for up to ca. 12 h leading to maximum turnover numbers (TONs) of 4700 and 9180 molH2 molCo−1 for the two different complexes. The progressive deactivation under electrolytic conditions is mainly ascribed to leaching of the metal centre from the polydentate ligand, likely occurring from the competent catalytic intermediates involved in the HER.
Introduction
The production of hydrogen from water splitting represents one of the most relevant, although challenging processes for the generation of clean fuels.1–5 During the last years, several efforts have been made towards the development of efficient catalysts with the potential ability of competing with and possibly replacing the expensive and rare platinum metal.6,7 In this respect, particular attention has been paid to the development of earth abundant molecular catalysts, whose structures and functions take inspiration from those of the active sites of natural hydrogenases.8–10 The main advantages in the use of molecular species arise from the fact that, although they are usually less stable than solid-state inorganic materials, molecular catalysts are prone to synthetic functionalization with the aim of tuning the catalytic activity towards the optimization of the performances.11,12 Hence, in this regard, many molecular catalysts based on iron,13–15 nickel,16–19 and cobalt20,21 metal centres have been indeed reported in the last years. Of peculiar relevance in this field is the use of polypyridine cobalt complexes featuring single chelating ligands11,22–26 which are able to promote hydrogen evolution at relatively low overpotentials, in fully aqueous solutions, and with good stability under operative conditions.The heterogenization of discrete molecular catalysts onto solid-state electrodes has received great attention in the last years as a means to obtain heterogeneous electrocatalysts with discrete active sites, combining the advantages of both material-based and molecular catalysis. However, the suitable coupling the two different catalytic realms remains a generally difficult task in the context of electrocatalysis. Several attempts have been made mainly involving covalent functionalization of mesoporous n-type semiconductors19a,27 and carbonaceous materials such as graphene and carbon nanotubes (CNTs).28,29 Furthermore, supramolecular interactions based on π–π stacking between pyrene units of molecular catalysts and CNTs have been also employed to prepare freestanding electrodes for electrocatalytic hydrogen evolution.30–33 However, in spite of their interesting catalytic properties, polypyridine cobalt complexes have been only rarely used for the preparation of hybrid solid-state electrodes.34,35
Single-walled carbon nanohorns (SWCNHs) have emerged in the last years as a novel carbon-based material with unique physical and chemical properties as well as potential applications in diverse fields such as energy conversion, energy storage, gas storage, and drug delivery.36,37 SWCNHs display conical-shaped structures with diameters between 2–5 nm and lengths in the order of 80–100 nm. These cones are organized in dahlia-type spherical structures with an average diameter of 100 nm.36 SWCNHs have several properties and features that make them highly attractive for catalytic applications:37,38 (i) they are produced with a metal-free synthesis thus avoiding interference on catalysis from exogenous species, (ii) they have a highly porous structure with large surface area (∼400 m2 g−1) while maintaining high electrical conductivity, (iii) they are easier to disperse in organic media than other carbon-based materials thus allowing for straightforward preparation protocols. Quite unexpectedly, while SWCNHs have been used for catalytic applications such as oxygen reduction and alcohol oxidation, their application in hydrogen evolving cathodes is limited to a very few cases.37
In the present work, we report for the first time the coupling of SWCNHs with molecular cobalt complexes 1–3 (Chart 1) based on the tris(2-pyridylmethyl)amine (TPMA) ligand scaffold39 for the preparation of hybrid cathodes for the hydrogen evolving reaction (HER). The ability of cobalt complexes of this kind to act as hydrogen evolving catalysts (HECs) in aqueous solution was already demonstrated under both electrochemical and photochemical homogeneous conditions.25 Interestingly, the introduction of pyrene moieties into the ligand framework provides a suitable way to efficiently functionalize the carbonaceous material through π–π interactions40,41 favouring pronounced electrocatalytic activity with respect to the unfunctionalized complex. The hybrid electrodes behave as competent cathodes for the HER in neutral aqueous solution with activity that depends on the type of metal complex used. Hydrogen is produced continuously for up to ca. 12 h upon electrolysis at a constant potential. The reasons for the progressive deactivation of these electrodes under turnover conditions has been examined.
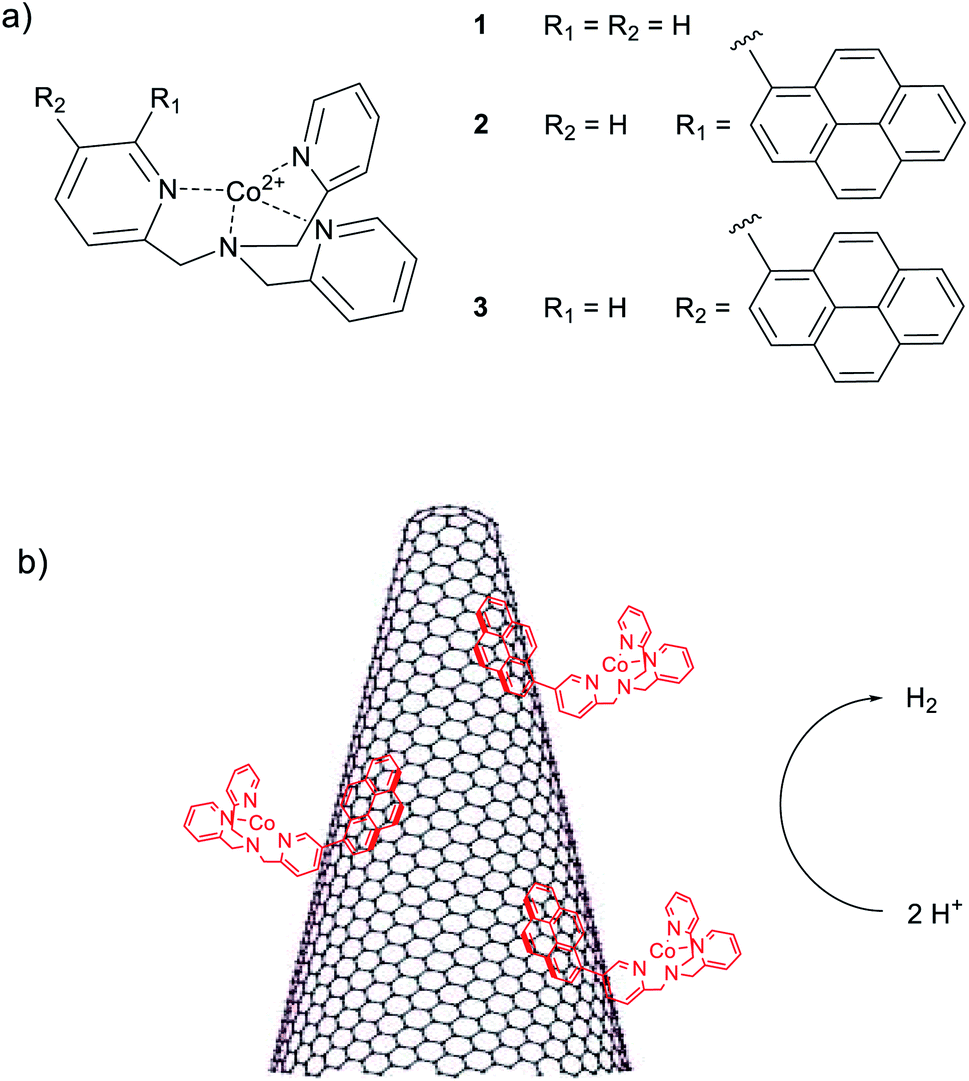 | ||
| Chart 1 (a) Molecular structures of the cobalt(II) complexes 1–3. Chloride counterions have been removed for clarity, (b) schematic representation of the electrocatalytic hydrogen evolution process using the hybrid cathode based on SWCNHs and the cobalt complexes. | ||
Experimental section
Materials and methods
Chemicals were purchased from Sigma Aldrich, TCI, or Apollo Scientific and used without further purification. Acetonitrile was of electrochemical grade. Milli-Q ultrapure water and related buffers were used. TEC 8 Fluorine-doped Tin Oxide (FTO) conductive glass slides were obtained from Pilkington. Single walled carbon nanohorns (SWCNHS) were produced by Carbonium s.r.l., Padova (Italy), by direct graphite evaporation in Ar flow, according to a patented method and used without purification.42NMR spectra were recorded at 301 K on a Bruker 400 Avance III BBi-z grad 5 mm or a Bruker Avance 300. All the 1H NMR spectra were referenced to residual isotopic impurity of CDCl3 (7.26 ppm). 13C-NMR spectra were referenced to the CDCl3 peak (77.0 ppm). The following abbreviations are used in reporting the multiplicity for NMR resonances: s = single, d = doublet, t = triplet, q = quartet, and m = multiplet. The NMR data were processed using MestReNova 12.0.2. High-resolution electrospray ionization mass spectrometry HRMS (ESI-TOF) analyses were performed in positive mode with Waters Xevo G2-S QTof. The analysis was performed with fast flow injection: 5 μL of the sample previously diluted in acetonitrile injected using methanol as mobile phase at 0.05 mL min−1: capillary: 2.0 kV, sample cone: 40 V, source temperature: 100 °C, desolvation temperature: 350 °C. The ESI crystallographic data were deposited as CCDC 2080990.†
SEM micrographs and EDS analysis were obtained using a scanning electron microscope (FE-SEM Jeol JSM 7600f) with an accelerating voltage of 15 kV.
Photoemission spectra were acquired with an Al Kα source (1486.7 eV) at room temperature after degassing the samples overnight. The electrodes were measured immediately after the preparation. The C 1s peak maximum was observed at 284.6 eV for all samples. The sample composition was determined by normalizing the photoemission intensity to the photoemission cross section43 and inelastic mean free path of electrons44 and corrected for the analyser transmission function.
The electrochemical studies were carried out with a PC-interfaced Eco Chemie Autolab/Pgstat 30 potentiostat. A conventional three-electrode cell assembly was adopted. A saturated calomel electrode (SCE Amel) and a platinum electrode were used as reference (RE) and counter (CE) electrodes, respectively. Conversion to NHE was made by the following relationship: E(NHE) = E(SCE) + 0.24 V. All experiments were performed in nitrogen-purged solutions. In cyclic voltammetry (CV) experiments in acetonitrile a glassy carbon electrode (7 mm2 surface area) was used as the working electrode (WE). Rapid assessment of the electrochemical properties (CV, short chronoamperometry) was made using functionalized SWCNHs electrodes of 1.5 cm2 surface area in a standard glass cell combining all electrodes (WE, CE, and RE) in a single compartment. Faradaic yield determination was carried out in a modular, custom-made polymethylmethacrylate (PMMA) cell. An ion exchange membrane (Nafion 117) divided the cell into two separated compartments. The WE (a round shaped SWCNHs-based cathode of 2 cm diameter, 3.1 cm2 surface area) and reference electrode (SCE) were in one side, while the Pt counter electrode in the other compartment. Both the anolyte and catholyte consisted in a phosphate aqueous solution (resulting pH = 7.4). The cathodic compartment of the cell was connected to a headspace, from which the GC pump automatically collected samples for gas detection and quantification. An Agilent Technologies 490 microGC equipped with a 5 Å molecular sieve column (10 m) and thermal conductivity detector was used. Ar was the carrier gas. 15 mL from the headspace was sampled by the internal GC pump and 9 μL was injected in the column, maintained at 60 °C. The unused gas was then reintroduced in the cell to minimize its consumption along the whole experiment. Hydrogen was quantified using a response factor obtained through galvanostatic electrolysis (1 mA, 1 h) of a 0.1 M H2SO4 solution in the same electrochemical cell, using a Pt working electrode and assuming 100% faradaic efficiency.
Preparation of functionalized SWCNHs electrodes
FTO cleaning was performed by sonicating the slides in Alconox® solution for 10 min and in 2-propanol for further 10 min and then placed in an oven for 30 min at 400 °C. A suspension of the pristine SWCNHs (1 mg mL−1) in ethanol was sonicated for 30 min at room temperature. This suspension was sprayed over masked FTO electrodes placed on a hot plate at 120 °C with an airbrush (FENGDA 0.33 mm) at distance of 10 cm from the plate, using N2 as carrier (1.5 Pa).38 Electrodes were subsequently annealed at 400 °C for 15 minutes. Subsequently, electrodes were soaked overnight in a 1 mM solution of the cobalt complex in acetonitrile.Results and discussion
Synthesis and coordination chemistry of Co(II) complexes
In recent years we have been working on the synthesis of functionalised tris(2-pyridylmethyl)amine complexes for applications in catalysis and molecular recognition.25,39,47 Interest in the literature on their catalytic activity is due to the ability to form tetradentate metal complexes that are stable under turnover conditions. In the context of this study, we recently reported that Co(II) complexes of these ligands are competent hydrogen evolving catalysts (HECs) also in light-assisted conditions.25 These findings have driven our attention to the possibility of developing a heterogeneous version of these systems. In particular, the substitution in the ligand backbone with a pyrene unit has been planned to furnish non-covalent grafting of our catalytic system to a hybrid cathode based on SWCNHs.For this reason, cobalt(II) chloride complexes 2 and 3 (Chart 1), have been prepared using a methodology developed in our group (see ESI, Section 1†).25,48 The obtained complexes have been characterized by ESI-HRMS and elemental analysis, confirming the purity of the desired compounds (see ESI, Section 3†). For comparative experiments complex 1 was also prepared according to literature protocols49 from commercially available TPMA ligand. Among the different structures, it was possible to obtain crystals of complex 3 suitable for X-ray diffraction (Fig. 1). The complex displays trigonal bipyramidal geometry with a chloride ion coordinated in the apical position. Relevant data obtained from structure refinement can be found in the ESI (Section 2 and Tables S1–S3†).
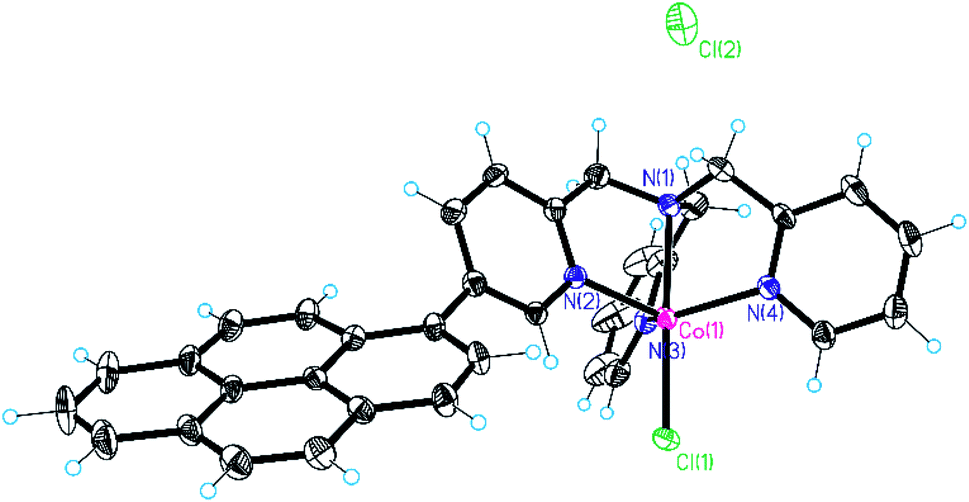 | ||
| Fig. 1 X-ray crystal structure of complex 3. | ||
Electrochemical experiments were then performed in nitrogen-purged acetonitrile solutions (0.1 M LiClO4) in order to assess the relevant redox processes associated with the metal complexes (Fig. S18†). Upon cathodic scan, an irreversible process with a peak potential of −1.06 V vs. NHE can be detected for 1, while two quasi-reversible reduction processes can be observed for 2 and 3 with half-wave potentials of −1.12 and −1.16 vs. NHE, respectively. These processes can be attributed to Co(II)/Co(I) reductions and fall at comparable values of those observed in related molecular analogues.25a Upon anodic scan, irreversible redox waves are observed for all cobalt complexes with peak potentials of +0.82, +0.92, and +0.90 V vs. NHE for 1, 2, and 3, respectively, attributable to Co(II)/Co(III) oxidation.
Cathode assembly and characterization
Deposition of SWCNHs on FTO glass slides was performed by spray coating at 120 °C of an ethanolic suspension followed by annealing at 400 °C, according to a previously optimized procedure.38 The use of a transparent FTO support was planned in view of future photoelectrochemical applications. SEM analysis (Fig. S19†) shows that the electrode displays a homogeneous and porous surface, which is suitable to promote efficient attachment of catalytic moieties such as compounds 2 and 3 through π–π stacking interactions.The hybrid electrodes based on SWCNHs and the cobalt complexes 1–3 were prepared by overnight soaking the functionalized glass slides into a 1 mM acetonitrile solution of the cobalt complex leading to SWCNHs-1, SWCNHs-2, and SWCNHs-3 electrodes. The effective functionalization with the metal complexes was subsequently checked by combined SEM-EDS analysis (Fig. S20–S22†). As expected, the adsorption process does not alter to an appreciable extent the morphology of the electrode surface (Fig. 2a for SWCNHs-2 and Fig. S20 and S22,† for SWCNHs-1 and SWCNHs-3, respectively). Furthermore, in the SWCNH-2 and SWCNH-3 samples detectable signals from the Co atoms were observed together with those arising from the FTO and SWCNHs substrate (Fig. S21 and S22†). On the other hand, with the SWCNHs-1 sample the signals associated to the metal centre were below the sensitivity of the EDS technique. These results, albeit qualitative, already suggest that the amount of cobalt complex on the SWCNHs surface is larger in the case of SWCNH-2 and SWCNH-3 than SWCNHs-1, as expected due to the presence of the pyrene groups in the former.
 | ||
| Fig. 2 SEM micrograph of (a) SWCNHs-2 electrode; (b) Co 2p and (c) N 1s signals obtained by XPS of SWCNHs-1, SWCNHs-2, and SWCNHs-3 electrodes. | ||
The three SWCNHs-based electrodes were then analysed by X-ray photoelectron spectroscopy (XPS). The survey spectrum is reported in Fig. S23 of the ESI.† In all samples, the Co 2p photoemission line (Fig. 2b) exhibits the maximum of the 2p3/2 peak at 781.7 eV, associated with an intense satellite shifted by ∼5 eV towards higher binding energies. This feature is typical of Co(II) paramagnetic compounds.45,46 No significant differences are observed among the various samples. Similar conclusions can be drawn inspecting the N 1s spectrum (Fig. 2c), which remains centred at a binding energy of 399.8 eV, compatible with the value expected for tertiary amines and pyridines coordinating metal cations.50,51 Importantly, for all the SWCNHs electrodes examined the Co/N atomic ratio is closed to the theoretical value of 0.25 (Table S4†) confirming the integrity of the cobalt complexes onto the carbonaceous substrate. Quantitative analysis on the different samples still highlights the larger loading onto the SWCNHs electrode for the pyrene-substituted samples 2 and 3, as previously inferred.
CVs of the hybrid electrodes were recorded under anodic scan (Fig. S24†). For all complexes, redox waves at ca. 0.7–0.8 V vs. NHE were observed, ascribable to the Co(III)/Co(II) process, as seen in acetonitrile solution (Fig. S18b†). Integration of the associated oxidation peaks provides a suitable way to estimate the amount of cobalt complex attached on the SWCNH electrodes.33 From this data, an average surface coverage of 0.6(±0.1), 4.2(±1.0), and 2.0(±0.4) nmol cm−2 can be calculated for 1, 2, and 3, respectively. These data clearly confirm that the presence of the pyrene substituents boosts the capability of the cobalt complex to be immobilized onto SWCNHs, as expected thanks to favourable π–π stacking interactions. Interestingly, a larger (by a factor of ca. 2) surface coverage is observed in the case of 2 onto SWCNHs than 3. As a possible explanation for such a difference, we can hypothesize that the molecular structures of the two complexes and the resulting molecular shapes may play a role, with the more spherical complex 2 being able to provide improved loading onto the carbonaceous surface than the more elongated analogue 3. In both cases, however, the amount of cobalt complex loaded onto the carbonaceous material is comparable to that found in similar hybrid electrodes, e.g., both covalently and non-covalently functionalized CNTs by either a cobaloxime HEC28 or a nickel DuBois catalyst.32
The hybrid electrodes were then tested as cathodes for the HER in a three-electrode cell, using 1 M phosphate buffer (pH = 7.4) as the electrolyte. Fig. 3a shows the resulting CV traces recorded at a scan rate of 10 mV s−1, while compensating for the ohmic drop. The performances of the three electrodes functionalized with the cobalt complexes were also compared to those of a bare SWCNHs electrode as a blank sample. The analysis of the CV curves revealed the onset of intense cathodic currents at potentials of −0.80 V vs. NHE for both the SWCNHs-2 and SWCNHs-3 electrodes, assignable to catalytic hydrogen evolution. This attribution is further confirmed in both cases by the pH dependence of the current density measured in a 1 M Britton–Robinson buffer solution (Fig. S25 and S26†). As a matter of fact, the potential measured at 1 mA cm−2 shifts towards negative values when the pH is increased with a slope of ca. −110 mV per pH unit, close to the theoretical value of −118 mV pH−1 characteristic of kinetically limiting hydrogen evolving reaction (HER) taking place at the electrode surface.52 Overpotentials (η) of 0.52 V and 0.54 V were determined in 1 M phosphate buffer (pH = 7.4) for the SWCNHs-2 and SWCNHs-3 electrodes, respectively, defined as the difference between the thermodynamic potential for the HER at this pH and the experimental value (Fig. 3a) required to reach a current density of 1 mA cm−2.28,53 Interestingly, a similar but less intense current rise was observed even in the case of the SWCNHs-1 electrode, even if at more negative potentials than both SWCNHs-2 and SWCNHs-3 (onset potential of −0.90 V vs. NHE). On the other hand, negligible current was released by the bare SWCNHs electrode at such potential values, confirming the relevant role of the cobalt catalyst in enhancing catalytic proton reduction to dihydrogen.
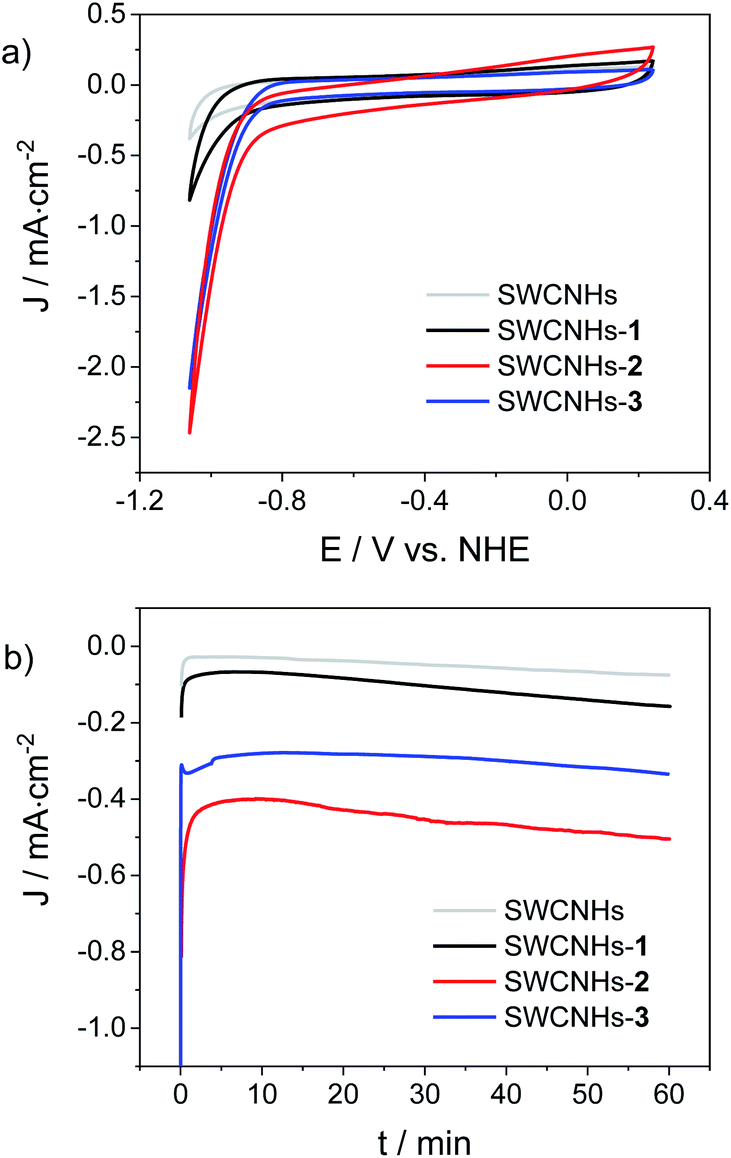 | ||
| Fig. 3 (a) CV (scan rate of 10 mV s−1) and (b) chronoamperometry (−0.96 V vs. NHE) of SWCNHs electrodes (1.5 cm2 surface area) functionalized with cobalt complexes 1–3 measured in 1 M phosphate buffer pH 7.4 (Pt as counter electrode, SCE as reference). | ||
Chronoamperometric measurements at a constant potential of −0.96 V vs. NHE were then performed under the same experimental conditions (Fig. 3b). Stable current densities in the order of −0.12, −0.50, and −0.33 mA cm−2 were observed over a timescale of 1 h for the SWCNHs-1, SWCNHs-2, and SWCNHs-3 electrodes, respectively. These data, once subtracted by the background current of the bare SWCNHs electrode (−0.07 mA cm−2) can be used to extract the average turnover frequencies (TOFs) per grafted cobalt catalyst, considering the surface concentration of the cobalt complex in each sample (see above) and the two-electron nature of the HER.28,53 These estimates yield average TOFs of 0.43, 0.56, and 0.66 s−1 for SWCNHs-1, SWCNHs-2, and SWCNHs-3 electrodes, respectively. These values are ca. ¼ of that reported for a cobaloxime HEC covalently attached onto CNTs.28
The observation of comparable TOF values is consistent with the catalytic activity being promoted by the attached cobalt complex onto the SWCNHs surface and thus supports the molecular nature of the HER catalyst. Hence, the improvements in terms of current densities in the case of the SWCNHs-2 and SWCNHs-3 electrodes mainly stems from the presence of a higher number of catalytic sites, still highlighting the pivotal role of the pyrene group in providing a stable and effective anchoring onto SWCNHs.
Tafel analysis was then applied on the electrocatalytic response of SWCNHs-2 and SWCNHs-3 at pH 7.4 to attain further insights into the HER catalysed by the hybrid cathodes (Fig. S27†). Tafel slopes of 190 mV dec−1 and 160 mV dec−1 and exchange current densities of 10−5.9 and 10−6.3 A cm−2 were calculated for SWCNHs-2 and SWCNHs-3, respectively. These values are comparable to those measured for a hybrid electrode based on CNTs covalently functionalized with a cobaloxime HEC.28 Interestingly, the exchange current densities are in the order of those reported for MoS2 catalysts,53 which are known as potential candidates for replacing Pt.
Chronoamperometric measurements over a longer time window coupled to hydrogen gas detection were then performed to test the full catalytic ability of the SWCNHs-2 and SWCNHs-3 electrodes (Fig. 4). Hydrogen is produced immediately upon application of a constant potential of −0.96 V vs. NHE, as confirmed by visual observation of gas bubbles at the surface of the electrode. A faradaic efficiency (FE) of ca. 60% can be estimated after one hour electrolysis for both samples. Interestingly, this value progressively decreases upon continuous application of the cathodic potential (e.g., a FE of ca. 30% is measured after 5 h electrolysis) and hydrogen production levels off after ca. 8–12 h (Fig. 4a). The final loss in catalytic activity is associated to the detachment of the SWCNHs film from the FTO substrate, as confirmed by visual inspection, which is accompanied by a sharp rise in current (Fig. S28†). This possibly arises from mechanical stress related to extensive catalytic activity as well as to the not perfect adhesion of the SWCNHs onto the FTO substrate. Maximum quantities of 61 and 57 μmol hydrogen gas were measured for the SWCNHs-2 and SWCNHs-3 electrodes, respectively. Importantly, from the amount of cobalt complex attached on the electrode surface (see above), maximum turnover numbers (TONs) of 4700 and 9180 molH2 molCo−1 can be estimated for the SWCNHs-2 and SWCNHs-3 electrodes, respectively (Fig. 4b). Interestingly, these values are higher than those observed for a poly (cobaloxime) CNT electrode, although the durability of the system is lower.33 On the other hand, both activity and durability of the present system are apparently lower than those reported for CNTs decorated with a pyrene-based nickel DuBois catalyst,32 although the latter study was conducted at acidic pH where the HER catalysis usually displays both thermodynamic and kinetic advantages.
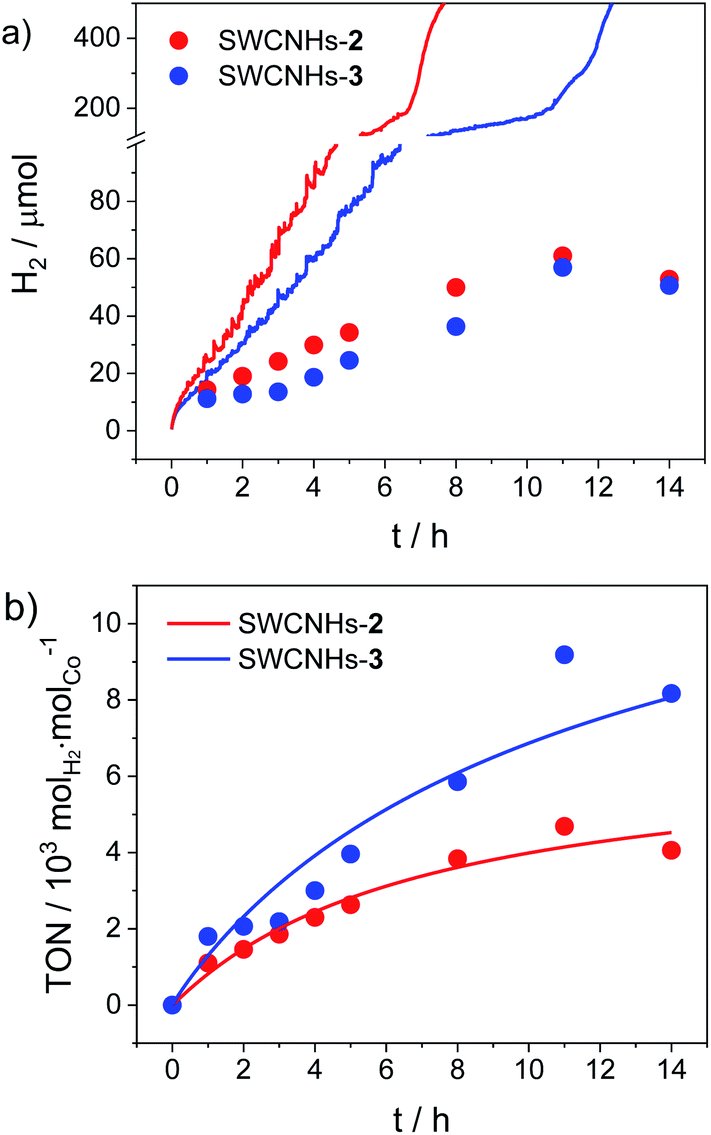 | ||
| Fig. 4 (a) Theoretical (lines, from chronoamperometry, Fig. S27†) vs. experimental (dots, from gas chromatography) amount of hydrogen produced upon electrolysis at −0.96 V vs. NHE of SWCNHs-2 and SWCNHs-3 electrodes (3.1 cm2 surface area) in 1 M phosphate buffer pH 7.4 (Pt as counter electrode, SCE as reference); (b) TONs of hydrogen production for SWCNHs-2 and SWCNHs-3 electrodes. | ||
XPS analysis was then performed on SWCNHs-2 and SWCNH-3 electrodes after 1 h and 6 h continuous electrolysis at −0.96 V vs. NHE to understand possible reasons for the progressive inactivation of the hybrid catalytic material. After 1 h electrolysis, no relevant changes are observed in terms of binding energies for both the Co 2p and N 1s signals (Fig. S29†) suggesting that the electrochemical work does not alter to an appreciable extent the chemical composition of the catalytic film. It should be noticed, however, that the Co/N ratio significantly decreases (Table S5†) supporting a modest leaching of the metal centre from the tetradentate chelating TPMA ligand during continuous electrocatalysis. This hypothesis is further confirmed by the subsequent XPS analysis performed after 6 h electrolysis (Fig. S30†). Negligible signals associated with the Co 2p core levels are indeed detectable, suggesting that the amount of cobalt left onto the electrode is lower than the XPS detection limit. Importantly, this evidence indicates that the attached cobalt complexes in both SWCNHs-2 and SWCNH-3 electrodes are not precursors for heterogeneous catalytic species, such as metallic cobalt or related oxides,54,55 and still confirms the molecular nature of the cobalt catalyst within the hybrid assemblies. These observations are also corroborated by CV analysis on the same electrodes (Fig. S31†) which reveals the net decrease of the anodic wave associated to the Co(III)/Co(II) process and the absence of additional oxidation processes potentially ascribable to electrogenerated heterogeneous species. Interestingly, a sharp abatement of the Co(III)/Co(II) wave is observed in the case of SWCNHs-2, whereas a residual signal (ca. 5% with respect to the sample before electrolysis) is still detectable in the case of SWCNHs-3 (Fig. S31†). This agrees with the greater stability of the latter as confirmed by chronoamperometry (Fig. S28†) and hydrogen gas detection (Fig. 4b). On the other hand, after 6 h electrolysis the N 1s photoemission signals (Fig. S30†) are still detectable and centred at a slightly lower binding energy (399.6 eV) compared to the pristine, pre-electrolysis electrodes. This confirms that nitrogen is no longer bonded to the metal centre and demonstrates that the ligand framework remains attached onto the carbonaceous surface thanks to favourable π–π stacking interactions.41
Overall, these results clearly point out that the progressive abatement of the catalytic properties in both SWCNHs-2 and SWCNH-3 arises from demetallation of the complexes on the electrode surface. This decomposition route markedly differs from that of other cobalt catalysts of molecular nature (for which inactivation usually occurs via competitive hydrogenation of the ligand pool)20 and seems thus characteristic of this kind of molecular systems. These observations are indeed fully consistent with those made by Zhao and co-workers for light-driven hydrogen evolution using cobalt complexes featuring pentadentate polypyridine ligands.56 In this respect, demetallation likely originate under turnover conditions from the relevant cobalt(I) catalytic intermediate which is known to display longer Co–N bonds as well as lower coordination numbers than cobalt(II) species.23,57 The progressive loss of the metal ions from the TPMA scaffolds can be thus considered as the main reason for the gradual decrease in faradaic yield for electrochemical hydrogen evolution by our hybrid systems. Hence, stabilization of the metal centre via the polydentate ligand turns out to be a critical task to obtain a hybrid cathode based on polypyridine cobalt complexes as well as for potential applications of this class of molecular catalysts for (photo)electrochemical HER.
Conclusions
Hybrid cathodes for the HER have been realized on FTO by combining a porous material such as SWCNHs and molecular polypyridine cobalt complexes as catalysts. Two different complexes 2 and 3 have been synthesized featuring pyrene groups in the ligand framework in two different positions. This group has been introduced to promote catalyst attachment onto the carbonaceous substrate. The hybrid electrodes have been prepared by soaking of the SWCNHs into acetonitrile solutions of the cobalt complex. The position of the pyrene moiety is observed to affect the amount of cobalt catalyst on the electrode surface. The as-prepared systems behave as competent hydrogen evolving cathodes in aqueous solution at neutral pH with an overpotential of η ∼ 0.5 V and stable current densities (within 1 h electrolysis) of −0.50 and −0.33 mA cm−2 for SWCNHs-2 and SWCNHs-3, respectively. Hydrogen gas is produced continuously for up to ca. 12 h leading to maximum TONs of 4700 and 9180 molH2 molCo−1 for SWCNHs-2 and SWCNHs-3 electrodes, respectively. The progressive abatement in the hydrogen evolving activity has been attributed to catalyst deactivation occurring via metal detachment from the tetradentate ligand. Strategies to better stabilize the cobalt complexes onto the carbonaceous electrode as well as the possible extension of this hybrid approach to other challenging redox reactions58,59 are currently underway in our labs.Author contributions
M. N. and C. Z. designed the research, analysed the data, and wrote the manuscript. G. L. co-supervised the research. E. B., F. B., A. N., and L. D. performed the experiments and analysed the data. K. W. collected the crystallographic data and refined the structure. S. A. performed the XPS characterization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Prof. Maurizio Prato (University of Trieste) and Dr Zois Syrgiannis (Northwestern University) are gratefully acknowledged for the generous gift of SWCNHs. M. N. acknowledges financial support from the University of Ferrara (FAR 2020). The Department of Chemical Sciences, University of Padova, is acknowledged for funding (P-DiSC#10BIRD2020-UNIPD).Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474 CrossRef CAS PubMed.
- J. A. Turner, Science, 1999, 285, 687 CrossRef CAS PubMed.
- V. Balzani, A. Credi and M. Venturi, ChemSusChem, 2008, 1, 26 CrossRef CAS PubMed.
- A. Agosti, M. Natali, L. Amirav and G. Bergamini, ChemSusChem, 2020, 13, 4894 CrossRef CAS PubMed.
- J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Chem. Sci., 2014, 5, 865 RSC.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752 CrossRef CAS PubMed.
- V. Artero, G. Berggren, M. Atta, G. Caserta, S. Roy, L. Pecqueur and M. Fontecave, Acc. Chem. Res., 2015, 48, 2380 CrossRef CAS PubMed.
- (a) W. T. Eckenhoff, W. R. McNamara, P. Du and R. Eisenberg, Biochim. Biophys. Acta, 2013, 1827, 958 CrossRef CAS PubMed; (b) W. T. Eckenhoff, Coord. Chem. Rev., 2018, 373, 295 CrossRef CAS.
- L. Tong, L. Duan, A. Zhou and R. P. Thummel, Coord. Chem. Rev., 2020, 402, 213079 CrossRef CAS.
- R. S. Khnayzer, V. S. Thoi, M. Nippe, A. E. King, J. W. Jurss, K. A. El Roz, J. R. Long, C. J. Chang and F. N. Castellano, Energy Environ. Sci., 2014, 7, 1477 RSC.
- R. W. Hogue, O. Schott, G. S. Hanan and S. Brooker, Chem.–Eur. J., 2018, 24, 9820 CrossRef CAS PubMed.
- C. Tard and C. J. Pickett, Chem. Rev., 2009, 109, 2245 CrossRef CAS PubMed.
- (a) A. C. Cavell, C. L. Hartley, D. Liu, C. S. Tribble and W. R. McNamara, Inorg. Chem., 2015, 54, 3325 CrossRef CAS PubMed; (b) Z. R. Schiffman, C. M. Margonis, A. Moyer, M. Ott and W. R. McNamara, Inorg. Chim. Acta, 2020, 503, 119394 CrossRef CAS.
- M. Tagliapietra, A. Squarcina, N. Hickey, R. De Zorzi, S. Geremia, A. Sartorel and M. Bonchio, ChemSusChem, 2017, 10, 4430 CrossRef CAS PubMed.
- (a) Z. Han, W. R. McNamara, M. S. Eum, P. L. Holland and R. Eisenberg, Angew. Chem., Int. Ed., 2012, 51, 1667 CrossRef CAS PubMed; (b) Z. Han, L. Shen, W. W. Brennessel, P. L. Holland and R. Eisenberg, J. Am. Chem. Soc., 2013, 135, 14659 CrossRef CAS PubMed.
- (a) M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863 CrossRef CAS PubMed; (b) U. J. Kilgore, J. A. S. Roberts, D. H. Pool, A. M. Appel, M. P. Stewart, M. R. DuBois, W. G. Dougherty, W. S. Kassel, R. M. Bullock and D. L. DuBois, J. Am. Chem. Soc., 2011, 133, 5861 CrossRef CAS PubMed.
- (a) M. A. Gross, A. Reynal, J. R. Durrant and E. Reisner, J. Am. Chem. Soc., 2014, 136, 356 CrossRef CAS PubMed; (b) B. C. M. Martindale, G. A. M. Hutton, C. Caputo and E. Reisner, J. Am. Chem. Soc., 2015, 137, 6018 CrossRef CAS PubMed.
- (a) G. Bergamini and M. Natali, Dalton Trans., 2019, 48, 14653 RSC; (b) E. Benazzi, V. C. Coni, M. Boni, R. Mazzaro, V. Morandi and M. Natali, Dalton Trans., 2019, 49, 10212 RSC; (c) E. Benazzi, V. Cristino, R. Boaretto, S. Caramori and M. Natali, Dalton Trans., 2021, 50, 696 RSC.
- (a) V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238 CrossRef CAS PubMed; (b) T. M. McCormick, B. D. Calitree, A. Orchard, N. D. Kraut, F. V. Bright, M. R. Detty and R. Eisenberg, J. Am. Chem. Soc., 2010, 132, 15480 CrossRef CAS PubMed; (c) M. Natali, R. Argazzi, C. Chiorboli, E. Iengo and F. Scandola, Chem.–Eur. J., 2013, 19, 9261 CrossRef CAS PubMed; (d) M. Natali, ACS Catal., 2017, 7, 1330 CrossRef CAS.
- (a) S. Varma, C. E. Castillo, T. Stoll, J. Fortage, A. G. Blackman, F. Molton, A. Deronzier and M.-N. Collomb, Phys. Chem. Chem. Phys., 2013, 15, 17544 RSC; (b) S. Grau, M. Schilling, D. Moonshiram, J. Benet-Buchholz, S. Luber, A. Llobet and C. Gimbert-Suriñach, ChemSusChem, 2020, 13, 2745 CrossRef CAS PubMed.
- N. Queyriaux, R. T. Jane, J. Massin, V. Artero and M. Chavarot-Kerlidou, Coord. Chem. Rev., 2015, 304, 3 CrossRef PubMed.
- (a) F. Lucarini, J. Fize, A. Morozan, M. Marazzi, M. Natali, M. Pastore, V. Artero and A. Ruggi, Sustainable Energy Fuels, 2020, 4, 589 RSC; (b) F. Lucarini, D. Bongni, P. Schiel, G. Bevini, E. Benazzi, E. Solari, F. Fadaei-Tirani, R. Scopelliti, M. Marazzi, M. Natali, M. Pastore and A. Ruggi, ChemSusChem, 2021, 14, 1874 CrossRef CAS PubMed.
- (a) E. Deponti, A. Luisa, M. Natali, E. Iengo and F. Scandola, Dalton Trans., 2014, 43, 16345 RSC; (b) W. M. Singh, M. Mirmohades, R. T. Jane, T. A. White, L. Hammarström, A. Thapper, R. Lomoth and S. Ott, Chem. Commun., 2013, 49, 8638 RSC.
- (a) M. Natali, E. Badetti, E. Deponti, M. Gamberoni, F. A. Scaramuzzo, A. Sartorel and C. Zonta, Dalton Trans., 2016, 45, 14764 RSC; (b) N. A. Carmo dos Santos, M. Natali, E. Badetti, K. Wurst, G. Licini and C. Zonta, Dalton Trans., 2017, 46, 16455 RSC.
- (a) S. Schnidrig, C. Bachmann, P. Müller, N. Weder, B. Spingler, E. Joliat-Wick, M. Mosberger, J. Windisch, R. Alberto and B. Probst, ChemSusChem, 2017, 10, 4570 CrossRef CAS PubMed; (b) E. Joliat-Wick, N. Weder, D. Klose, C. Bachmann, B. Spingler, B. Probst and R. Alberto, Inorg. Chem., 2018, 57, 1651 CrossRef CAS PubMed.
- (a) N. M. Muresan, J. Willkomm, D. Mersch, Y. Vaynzof and E. Reisner, Angew. Chem., Int. Ed., 2012, 124, 12921 CrossRef; (b) T. E. Rosser, M. A. Gross, Y. H. Lai and E. Reisner, Chem. Sci., 2016, 7, 4024 RSC.
- E. S. Andreiadis, P. A. Jacques, P. D. Tran, A. Leyris, M. Chavarot-Kerlidou, B. Jousselme, M. Matheron, J. Pécaut, S. Palacin, M. Fontecave and V. Artero, Nat. Chem., 2013, 5, 48 CrossRef CAS PubMed.
- A. Le Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Métaye, A. Fihri, S. Palacin and M. Fontecave, Science, 2009, 326, 1384 CrossRef CAS PubMed.
- (a) H. Lei, C. Liu, Z. Wang, Z. Zhang, M. Zhang, X. Chang, W. Zhang and R. Cao, ACS Catal., 2016, 6, 6429 CrossRef CAS; (b) X. Li, H. Lei, X. Guo, X. Zhao, S. Ding, X. Gao, W. Zhang and R. Cao, ChemSusChem, 2017, 10, 4632 CrossRef CAS PubMed.
- T. T. Li, J. Qian, Q. Zhou, J. L. Lin and Y. Q. Zheng, Dalton Trans., 2017, 46, 13020 RSC.
- P. D. Tran, A. Le Goff, J. Heidkamp, B. Jousselme, N. Guillet, S. Palacin, H. Dau, M. Fontecave and V. Artero, Angew. Chem., Int. Ed., 2011, 50, 1371 CrossRef CAS PubMed.
- B. Reuillard, J. Warnan, J. J. Leung, D. W. Wakerley and E. Reisner, Angew. Chem., Int. Ed., 2016, 55, 3952 CrossRef CAS PubMed.
- X. Chen, H. Ren, W. Peng, H. Zhang, J. Lu and L. Zhuang, J. Phys. Chem. C, 2014, 118, 20791 CrossRef CAS.
- Z. Kap, E. Ulker, S. V. K. Nune and F. Karadas, J. Appl. Electrochem., 2018, 48, 201 CrossRef CAS.
- S. Zhu and G. Xu, Nanoscale, 2010, 2, 2538 RSC.
- Z. Zhang, S. Han, C. Wang, J. Li and G. Xu, Nanomaterials, 2015, 5, 1732 CrossRef CAS PubMed.
- S. Carli, L. Casarin, Z. Syrgiannis, R. Boaretto, E. Benazzi, S. Caramori, M. Prato and C. A. Bignozzi, ACS Appl. Mater. Interfaces, 2016, 8, 14604 CrossRef CAS PubMed.
- C. Bravin, E. Badetti, G. Licini and C. Zonta, Coord. Chem. Rev., 2021, 427, 213558 CrossRef CAS.
- C. Ehli, G. M. A. Rahman, N. Jux, D. Balbinot, D. M. Guldi, F. Paolucci, M. Marcaccio, D. Paolucci, M. Melle-Franco, F. Zerbetto, S. Campidelli and M. Prato, J. Am. Chem. Soc., 2006, 128, 11222 CrossRef CAS PubMed.
- (a) G. Pagona, A. S. D. Sandanayaka, A. Maigné, J. Fan, G. C. Papavassiliou, I. D. Petsalakis, B. R. Steele, M. Yudasaka, S. Iijima, N. Tagmatarchis and O. Ito, Chem.–Eur. J., 2007, 13, 7600 CrossRef CAS PubMed; (b) G. Pagona, J. Fan, A. Maigné, M. Yudasaka, S. Iijima and N. Tagmatarchis, Diamond Relat. Mater., 2007, 16, 1150 CrossRef CAS.
- M. Schiavon, Device and Method for Production of Carbon Nanotubes, Fullerene and Their Derivatives, Patent EP1428794 (A2), 2006.
- J. J. Yeh and I. Lindau, At. Data Nucl. Data Tables, 1985, 32, 1 CrossRef CAS.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 1991, 17, 927 CrossRef CAS.
- Y. G. Borod'ko, S. I. Vetchinkin, S. L. Zimont, I. N. Ivleva and Y. M. Shul'ga, Chem. Phys. Lett., 1976, 42, 264 CrossRef.
- C. A. Strydom and H. J. Strydom, Inorg. Chim. Acta, 1989, 159, 191 CrossRef CAS.
- (a) C. Bravin, E. Badetti, F. A. Scaramuzzo, G. Licini and C. Zonta, J. Am. Chem. Soc., 2017, 139, 6456 CrossRef CAS PubMed; (b) C. Bravin, A. Guidetti, G. Licini and C. Zonta, Chem. Sci., 2019, 10, 3532 Search PubMed; (c) C. Bravin, G. Mason, G. Licini and C. Zonta, J. Am. Chem. Soc., 2019, 141, 11963 CrossRef CAS PubMed.
- F. A. Scaramuzzo, G. Licini and C. Zonta, Chem.–Eur. J., 2013, 19, 16809 CrossRef CAS PubMed.
- Z. Li, J. D. Xiao and H. L. Jiang, ACS Catal., 2016, 6, 5359 CrossRef CAS.
- J. J. Leung, J. Warnan, K. H. Ly, N. Heidary, D. H. Nam, M. F. Kuehnel and E. Reisner, Nat. Catal., 2019, 2, 354 CrossRef CAS.
- K. Artyushkova, J. Vac. Sci. Technol., A, 2020, 38, 031002 CrossRef CAS.
- D. S. Miller, A. J. Bard, G. McLandon and J. Ferguson, J. Am. Chem. Soc., 1981, 103, 5336 CrossRef CAS.
- J. D. Benck, T. R. Hellstern, J. Kibsgaard, P. Chakthranont and T. F. Jaramillo, ACS Catal., 2014, 4, 3957 CrossRef CAS.
- N. Kaeffer, A. Morozan, J. Fize, E. Martinez, L. Guetaz and V. Artero, ACS Catal., 2016, 6, 3727 CrossRef CAS.
- E. Anxolabéhère-Mallart, C. Costentin, M. Fournier, S. Nowak, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2012, 134, 6104 CrossRef PubMed.
- (a) M. Vennampalli, G. Liang, L. Katta, C. E. Webster and X. Zhao, Inorg. Chem., 2014, 53, 10094 CrossRef CAS PubMed; (b) P. Wang, G. Liang, N. Smith, K. Hill, B. Donnadieu, C. E. Webster and X. Zhao, Angew. Chem., Int. Ed., 2020, 59, 12694 CrossRef CAS PubMed.
- J. Xie, Q. Zhou, C. Li, W. Wang, Y. Hou, B. Zhang and X. Wang, Chem. Commun., 2014, 50, 6520 RSC.
- (a) S. L. Chan, T. L. Lam, C. Yang, S.-C. Yan and N. M. Cheng, Chem. Commun., 2015, 51, 7799 RSC; (b) C. Yang, F. Mehmood, T. L. Lam, S. L. Chan, Y. Wu, C.-S. Yeung, X. Guan, K. Li, C. Y. Chung, C.-Y. Zhou, T. Zou and C.-M. Che, Chem. Sci., 2016, 7, 3123 RSC.
- (a) H. Wang, Y. Lu, E. Mijangos and A. Thapper, Chin. J. Chem., 2014, 32, 467 CrossRef CAS; (b) S. Liu, Y. Lei, Z. Xin, R. Xiang, S. Styring, A. Thapper and H. Wang, Int. J. Hydrogen Energy, 2017, 42, 29716 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2080990. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ta03645a |
| ‡ These authors contributed equally. |
| § Current address: Department of Chemical Sciences, University of Padova, Via F. Marzolo 1, 35131 Padova, Italy. |
| This journal is © The Royal Society of Chemistry 2021 |