
Phase modulation of 1T/2H MoSe2 nanoflowers for highly efficient bifunctional electrocatalysis in rechargeable Li–O2 batteries†
Qing
Xia‡
a,
Lanling
Zhao‡
 b,
Deyuan
Li
a,
Jun
Wang
*ac,
Lili
Liu
d,
Chuanxin
Hou
e,
Xiaomeng
Liu
a,
Haoran
Xu
a,
Feng
Dang
a and
Jintao
Zhang
c
b,
Deyuan
Li
a,
Jun
Wang
*ac,
Lili
Liu
d,
Chuanxin
Hou
e,
Xiaomeng
Liu
a,
Haoran
Xu
a,
Feng
Dang
a and
Jintao
Zhang
c
aKey Laboratory for Liquid-Solid Structural Evolution and Processing of Materials (Ministry of Education), Shandong University, Jinan 250061, China. E-mail: jw707@sdu.edu.cn
bSchool of Physics, Shandong University, Jinan, 250100, P. R. China
cKey Laboratory for Colloid and Interface Chemistry (Ministry of Education), School of Chemistry and Chemical Engineering, Shandong University, Jinan 250061, China
dSchool of Energy Science and Engineering, Institute for Advanced Materials, Nanjing Tech University, Nanjing 211816, Jiangsu Province, China
eSchool of Environmental and Material Engineering, Yantai University, No. 30 Qingquan Road, Yantai, Shandong 264005, China
First published on 15th June 2021
Abstract
Li–O2 batteries with outstanding energy density are considered to be promising next-generation power sources for various future electric devices. However, the battery performance is limited by the inferior electrocatalytic activity of catalysts in the cathode. In this work, MoSe2 catalysts with hybrid 1T and 2H phases were prepared through an efficient two-step hydrothermal strategy. The combination of the 1T phase with the 2H phase could synergically enhance the catalytic activities towards both oxygen reduction reaction and oxygen evolution reaction, and the built-in heterojunction between them also dramatically facilitated the interfacial charge transfer kinetics. Additionally, the hierarchical flower-like architecture with multidimensional channels would promote the rapid electron and mass diffusion, as well as provide enough space for storing discharge products. Thanks to the above merits, MoSe2 cathodes delivered improved electrocatalytic properties for Li–O2 batteries. This present work may pave a new avenue for highly efficient bifunctional electrocatalyst construction for Li–O2 batteries.
Introduction
Increasing global warming issues and reduction of fossil energy have promoted the development of energy storage and conversion technologies.1–3 Among them, non-aqueous Li–O2 batteries (LOBs) based on the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) have attracted extensive attention because of their ultrahigh energy density (3500 W h kg−1), which is 10 times that of Li-ion batteries.4–6 Their further development, however, is currently hindered by low electrocatalytic efficiency and poor cycling stability, which are associated with sluggish reaction kinetics of insulated and insoluble Li2O2 formation and decomposition.7–9 In response, development of efficient bifunctional cathode catalysts for promoting electrocatalytic reactions of LOBs is urgently needed.Noble metals have been proven to exhibit high-efficiency catalytic properties in LOBs.10–12 Unfortunately, because of the expensive and scarce precious metal resources, their large-scale application is restricted, and it is necessary to develop cost-effective efficient catalysts. So far, a variety of earth-rich metals and corresponding compounds have been intensively investigated as alternatives, including alloys,13,14 transition metal oxides,15–17 sulfides,18–20 carbides,21,22, carbon based materials,23–25etc. In recent years, it is evident that two-dimensional transition metal chalcogenides (2D-TMCs) could exhibit favorable electrocatalytic activities and hold great promise as cathode catalyst candidates for LOBs.26–28 Typically, they show a layered X–M–X sandwich structure constructed from the crystal units connected by weak van der Waals forces along the c-axis, in which M mainly represents Mo, W or Re, and X can be S, Se or Te. As a member of TMCs, MoSe2 displays tunable structures and low Gibbs free energy (ΔGH ∼ 0.05 eV),29 and their unique layered architecture could facilitate the insertion and extraction of ions,30,31 making it widely applied in the energy-related research fields, such as supercapacitors,32,33 batteries,34,35 water splitting,36,37 hydrogen catalysis38,39 and so on. In fact, MoSe2 appears in the form of 2H and 1T phases according to different arrangement of Se atoms. Notably, the 2H phase (triangular prism structure) shows semiconductor characteristics with a band gap between 1.3 and 1.9 eV, and the exposed edge sites were proved to endow it with remarkable eletrocatalytic ability.40,41 Zhang et al.42 successfully grew 2H MoSe2 nanosheets on the surfaces of hollow carbon nanofibers with rich edge sites exposed, delivering a charge voltage platform of 3.69 V at 0.1 mA cm−2 in LOBs. Long's group43 also synthesized 2H MoSe2 nanosheets as cathode catalysts for LOBs, showing good cycle performance of 86 cycles at 500 mA g−1 with a fixed specific capacity of 4000 mA h g−1. However the 1T phase (octahedral structure) could deliver metallic properties with a good electrical conductivity (107 times that of the 2H phase), which can accelerate charge injection/transfer and thus lead to advanced electrocatalytic activities.44,45 To the best of our knowledge, reports on 1T MoSe2 catalysts for LOBs are rare. This is because traditional preparation methods of 1T MoSe2 are complicated and usually accompanied by high risks of using metallic Li,29 and 1T MoSe2 is also thermodynamically metastable, which can be easily transformed into stable 2H MoSe2 by repacking.46 In this regard, it is a rewarding strategy to construct a heterostructured MoSe2 catalyst for LOBs, combining the conductive 1T phase with abundant active sites with the environmentally stable 2H phase. In recent years, it is found that 1T phase MoSe2 nanosheets could be prepared by hydrothermal synthesis routes, but excessive strong reducing agent should be involved, such as N2H4, NaBH4, making the fabrication process violent and poisonous.39,46–48 Jiang et al.49 also successfully prepared 1T-MoSe2 nanosheets by a hydrothermal method, but the octylamine solvent used is poisonous and could lead to the coverage of electrocatalytically inactive organic groups on the resulting products. Therefore, there is an urgent need for a green and controllable synthesis method to obtain MoSe2 composites with 1T and 2H phases.
Herein, nanoflower-like 2H MoSe2 nanosheets were prepared by a one-step hydrothermal method, and their 2H phase could be controllably and effectively transformed into 1T and 2H hybrid phases by changing the reaction time of hydrothermal treatment. Benefitting from unique architecture with large surface area and rich heterogeneous interfaces of the hybrid phases, 1T/2H MoSe2 nanoflowers with abundant active sites could promote fast transport of oxygen, ions and electrons to facilitate the ORR and OER during cycling and provide enough space for storing discharge products, and they thus show large specific capacities, low overpotentials, good rate performance and excellent cycle stability.
Experimental
Material synthesis processes
2H MoSe2 nanoflowers were synthesized by a one-step hydrothermal method. Specifically, 0.111 g of SeO2 and 0.106 g of Na2MoO4·2H2O were dissolved in 28 mL deionized water under vigorous stirring. Afterwards, 1 mL NaBH4 aqueous solution (0.1 M) was gradually added into the solution, which was then transferred into a 50 mL Teflon-lined autoclave and kept at 180 °C for 24 hours. A black precipitate was obtained and collected by centrifugation, which was washed four times with alcohol and deionized water. After drying at 60 °C for 12 hours, 2H MoSe2 nanoflowers (MS) were finally obtained. In order to prepare 1T/2H MoSe2 nanoflowers, 60 mg of MS were ultrasonically treated for 20 minutes in 50 mL alcohol and transferred into an 80 mL Teflon-lined autoclave. After keeping at 220 °C for 5, 10, 15 and 20 hours, 1T/2H MoSe2 nanoflowers were yielded, which are denoted as MS-5, MS-10, MS-15 and MS-20, respectively.Material characterization
The morphology of the samples was observed by using a thermal field emission scanning electron microscope (SEM, Hitachi, S-4800, Japan) and transmission electron microscope (TEM, JEOL, JEM-2100F, Japan) operating at an acceleration voltage of 200 kV. Energy dispersive spectroscopy (EDS) images were collected using an EDS system (Oxford Materials Analysis, UK) equipped on a SEM. The phase investigation was conducted by X-ray diffraction (XRD, Rigaku D/max 2500, Japan) with Cu Kα radiation (λ = 1.54056 Å) with a scan rate of 2° per minute in the 2θ degree range from 20–80°. X-ray photoelectron spectroscopy (XPS, ESCALAB 250, USA) measurements were carried out to study the surface nature of the elements on the samples. Raman spectroscopy (LabRam HR Evolution, France) was used to study the component of samples. The specific surface area and pore size distribution of samples were measured using a nitrogen adsorption surface area and porosity analyzer (Micromeritics ASAP2020, USA).Electrochemical measurements
To fabricate cathodes, the as-prepared materials, Ketjen black (KB) and polytetrafluoroethylene (PTFE) were ultrasonically dispersed in an isopropanol solution for 20 minutes according to a mass ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:2, which were subsequently dropped evenly onto carbon papers and dried in a vacuum oven at 120 °C for 12 hours. Li–O2 cells for electrochemical measurements were assembled using the prepared cathode, a glass fiber separator, lithium metal foil as the counter anode and an electrolyte of 1 M LiTFSI in TEGDME. They were galvanostatically discharged and charged in a hermetic container filled with high-purity oxygen using a Land battery tester. The specific capacities and currents were calculated based on the weight of the active catalysts supported on the cathodes. A CHI 660E electrochemical workstation was employed to record cyclic voltammograms (CV) at a scan rate of 0.15 mV s−1 in the voltage range of 2.35–4.50 V (vs. Li+/Li) and perform electrochemical impedance spectroscopy (EIS) with a sine wave of 10 mV and a frequency range of 100–0.01 Hz. The evolution rate profiles of different gas species were measured by in situ differential electrochemical mass spectrometry (DEMS).
:4:2, which were subsequently dropped evenly onto carbon papers and dried in a vacuum oven at 120 °C for 12 hours. Li–O2 cells for electrochemical measurements were assembled using the prepared cathode, a glass fiber separator, lithium metal foil as the counter anode and an electrolyte of 1 M LiTFSI in TEGDME. They were galvanostatically discharged and charged in a hermetic container filled with high-purity oxygen using a Land battery tester. The specific capacities and currents were calculated based on the weight of the active catalysts supported on the cathodes. A CHI 660E electrochemical workstation was employed to record cyclic voltammograms (CV) at a scan rate of 0.15 mV s−1 in the voltage range of 2.35–4.50 V (vs. Li+/Li) and perform electrochemical impedance spectroscopy (EIS) with a sine wave of 10 mV and a frequency range of 100–0.01 Hz. The evolution rate profiles of different gas species were measured by in situ differential electrochemical mass spectrometry (DEMS).
Results and discussion
The synthesis diagram of 1T/2H MoSe2 samples is shown in Fig. 1. In the first step, Na2MoO4 and SeO2 powders were ultrasonically dispersed and mixed in deionized water under magnetic stirring, into which a certain amount of NaBH4 aqueous solution was added, and the MS sample was achieved after a hydrothermal treatment. In the second step, the obtained product was subjected to another hydrothermal treatment for various reaction times, and MoSe2 samples with different compositions were obtained. 2H MoSe2 phase configuration tends to be energetically more stable than its 1T counterpart, and it is evident that sufficient thermal activation energy introduction could effectively result in phase transition from 2H to 1T.50,51 In this work, post hydrothermal treatment for the as-prepared 2H MoSe2 offered the driving force to promote this phase transition with atomic replacement, during which Mo atoms partially converted from the state of the prismatic coordination to the antiprismatic coordination,52,53 evidenced by the following TEM, Raman and XPS results.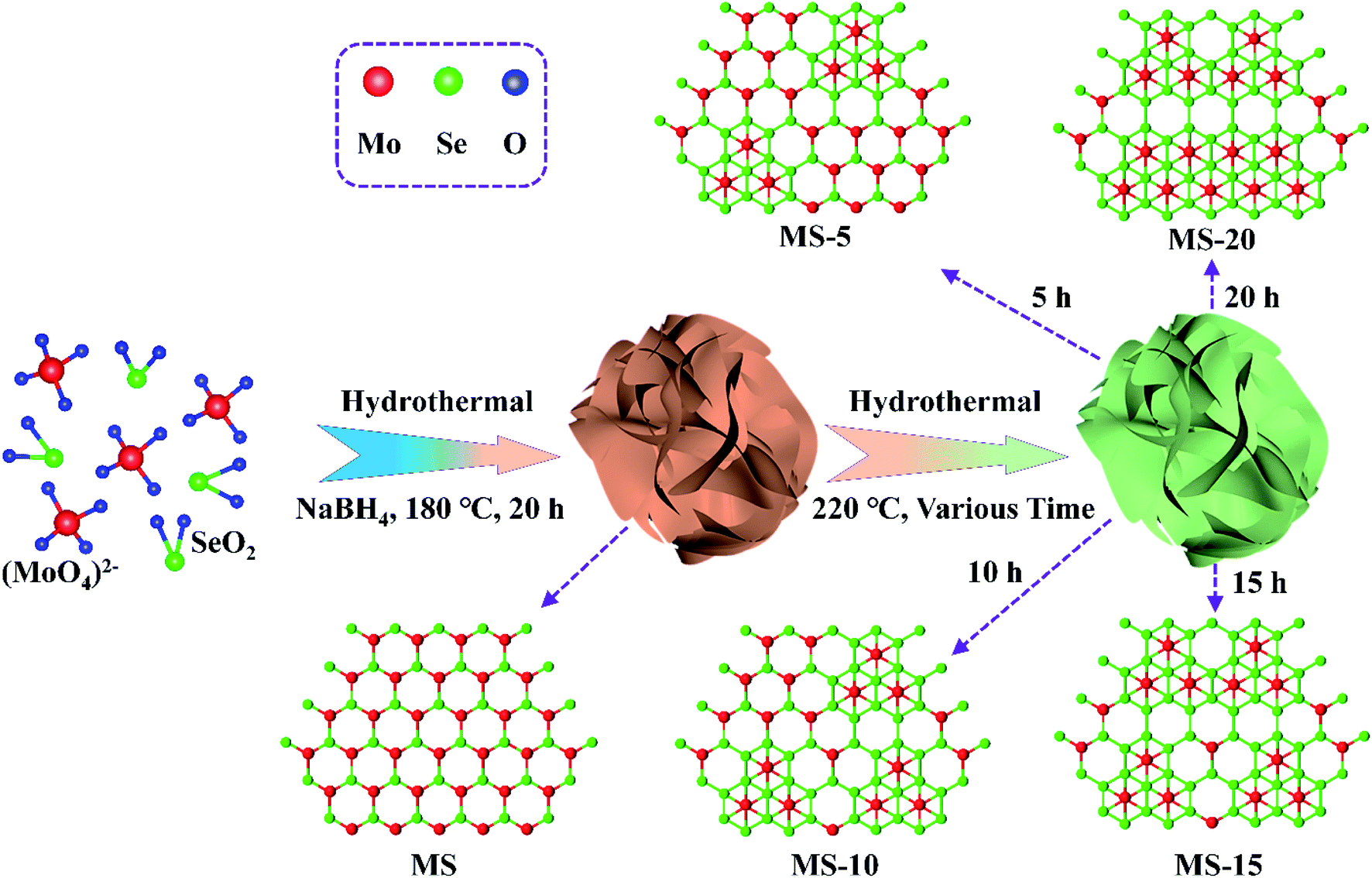 | ||
| Fig. 1 Schematic illustration of the preparation process of 1T/2H MoSe2 nanoflowers. | ||
SEM images of MS-10 (Fig. 2a) and MS, MS-5, MS-15 and MS-20 (Fig. S1†) show a flower-like structure with sizes of 1–2 μm. The TEM image in Fig. 2c indicates that this structure is actually composed of nanosheets with a thickness of about 50 nm (Fig. 2b). The elemental mapping image and EDS analysis show the uniform distribution of Mo and Se elements, as displayed in Fig. 2g and h. Its crystal structure was characterized by selective area electron diffraction (SAED). As shown in Fig. S2,† the diffraction rings with the radii of 0.283, 0.240, 0.192, 0.169, 0.144, 0.110 and 0.105 nm correspond to the (100), (103), (105), (110), (200), (1011) and (213) planes of the hexagonal MoSe2, respectively. Compared with the standard 2H MoSe2 crystal plane spacing, the slightly larger crystal plane spacings of (103), (105) and (110) for MS-10 indicate that MS-10 may be closely related to the introduction of 1T phase MoSe2. To confirm this conclusion, the high-resolution transmission electron microscope (HRTEM) image of MS-10 was recorded and shown in Fig. S3.† It can be clearly seen that the interplanar distance in the yellow rectangle is 0.316 nm, which is indexed to (100) planes of 2H MoSe2. The lattice spacing in the red rectangle is 0.739 nm, larger than the maximum interplanar spacing of 2H MoSe2 ((002), 0.646 nm), which is considered to be corresponding to the (002) crystal plane of 1T MoSe2 in previous reports.29,46 It is worth noting that there are two different atomic arrangements in Fig. 2d, which are associated with the 1T and the 2H phases, respectively, and Fig. 2e and f are enlarged HRTEM images of Fig. 2d. Obviously, the Mo atoms of the 2H phase display trigonal prismatic coordination in Fig. 2e, while the 1T phase shows a different arrangement with an octahedral or trigonal antiprismatic symmetry in Fig. 2f.53,54 All the above results proved that 2H and 1T phases coexist in MS-10.
 | ||
| Fig. 2 (a and b) SEM images of MS-10; (c) TEM image of MS-10; (d–f) HRTEM images showing the trigonal prismatic and octahedral unit cell structures, indicating the 2H and 1T phases, respectively; (g) elemental mapping images and (h) EDS analysis of MS-10. | ||
In order to study the compositions and phase states of different samples, XRD, Raman and XPS measurements were conducted. As shown in Fig. 3a, the sharp peaks at 31.7, 37.9, 47.5 and 56.1° can be assigned to (100), (103), (105) and (110) faces of MoSe2 (JCPDS no. 29-0914), respectively. This means that there are no other crystalline impurities in the as-prepared MoSe2 samples. Raman spectroscopy is a powerful tool for distinguishing 1T and 2H phases, and the corresponding results are listed in Fig. 3b. There are only two vibration modes at 240 and 288 cm−1 in the MS, corresponding to the A1g and E2g phonon modes,47 respectively. In other samples, there are other strong peaks found at 195 (J1) and 350 (J2) cm−1, indicating that the 1T and 2H phases coexist in these samples. This is well consistent with the SAED and HRTEM results. More importantly, it can be clearly seen that as the reaction time extended in the second hydrothermal treatment, the 1T phase ratios in the samples continuously increased, which demonstrates that longer reaction time can be helpful for transforming the 2H phase into the 1T phase. Fig. S4† provides the XPS survey spectra of these samples. It is evident that only Mo, Se and C elements were detected on the surfaces of the samples, and the appearance of the C element may be due to the adhesive on the conductive tape. What is more, the ratios of 1T and 2H phases can be further obtained by fitting and analyzing the high-resolution XPS spectra of Mo 3d in Fig. 3d–h. Clearly, there are two characteristic peaks in each spectrum, including the higher energy peak and the lower energy peak, corresponding to Mo 3d3/2 and Mo 3d5/2 orbitals, respectively. The higher strength peaks at 228.5 and 231.6 eV were attributed to Mo 3d5/2 and Mo 3d3/2 of 1T-MoSe2, and the weaker peaks at 228.9 and 232 eV were related to Mo 3d5/2 and Mo 3d3/2 of 2H-MoSe2, respectively. Based on these results, the corresponding 1T and 2H phase contents can be obtained by integrating the areas under the different peaks. As shown in Fig. 3i, it was calculated that the 1T phase ratios of the MS, MS-5, MS-10, MS-15 and MS-20 samples are respectively 1.2, 25.6, 42.3, 59.6 and 81.1%, which implies that with the increase of the reaction time, the proportion of the 1T phase gradually increases, which is in good agreement with the Raman results.
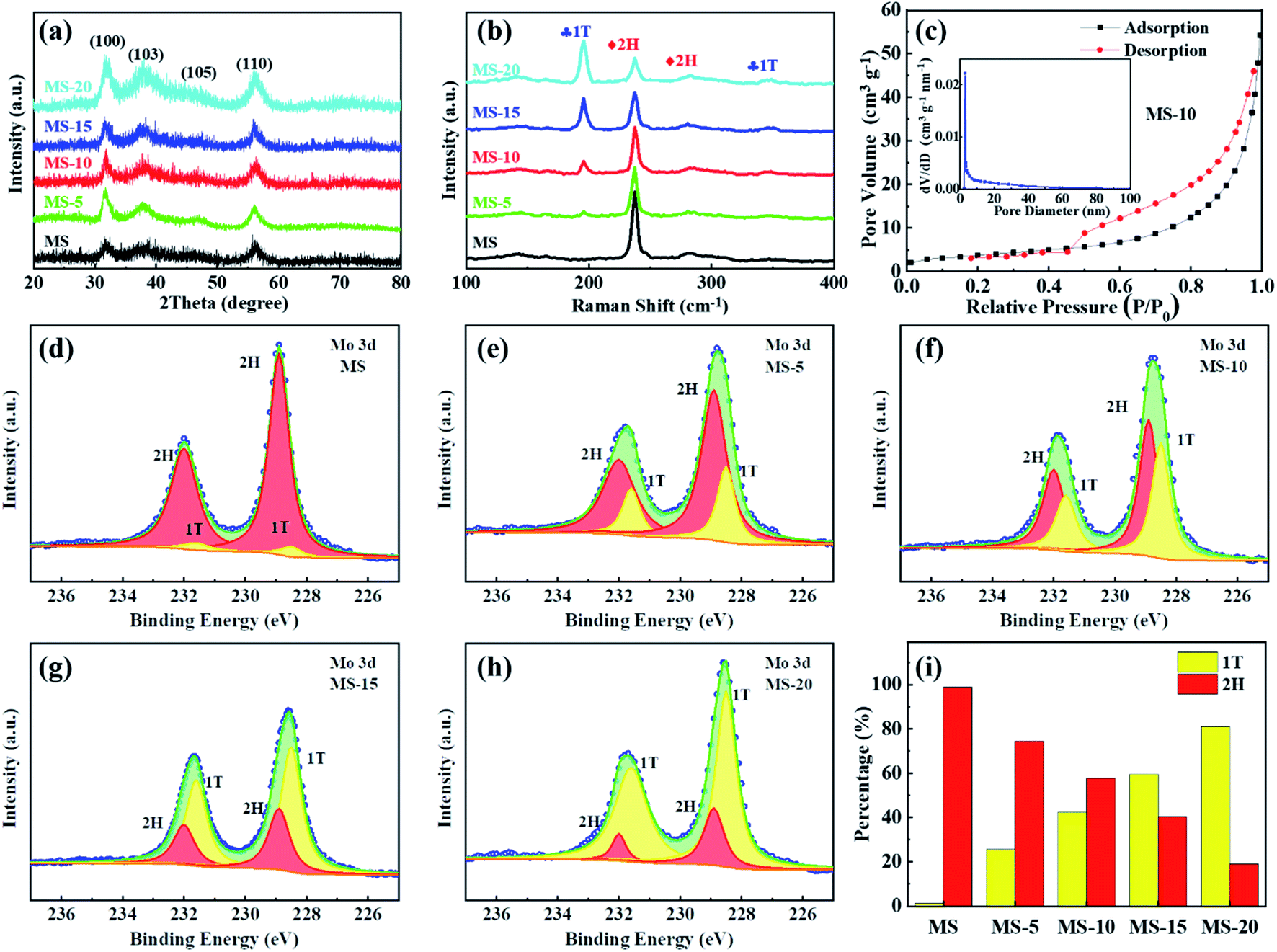 | ||
| Fig. 3 (a) XRD patterns, (b) Raman spectra, (d–h) high-resolution XPS spectra of Mo 3d and (i) ratios of 1T and 2H phases of different samples; (c) nitrogen adsorption–desorption isotherms with pore size distribution curve of MS-10. | ||
N2 adsorption–desorption isotherm and pore size distribution analyses were carried out on different MoSe2 samples, and the results are presented in Fig. 3c and S5.† All the N2 adsorption isotherms of these samples show that the IV type H3 hysteresis loop is in the P/P0 range of 0.4 to 1.0. Among the observed isotherms, the hysteresis loop exhibits a saturated adsorption platform, indicating the uniform pore formation. It is evident that the BET specific surface areas of different samples are 13.5, 16.4, 16.6, 17.1 and 18.3 m2 g−1, and the total pore volumes are 0.084, 0.079, 0.082, 0.910 and 0.881 cm3 g−1, respectively. By analyzing their pore size distributions, it is concluded that the pore sizes of the samples are mainly around 3–20 nm. This demonstrated that for different treatment durations, although the composition of the 1T and 2H phases has changed greatly, the microscopic morphology and pore size distributions are actually similar. It would also ensure that the difference in electrochemical performance of different samples is mainly caused by the difference in phase compositions, but not affected by their microstructures. Electrocatalytic activities of different samples as the cathode catalysts were evaluated in a hermetic container purchased from NJ Scientific Ltd in Fig. S6,† shown in Fig. 4. LOB cells with carbon paper (Fig. S7†) and KB cathodes were also assembled and included to eliminate the influence and confusion caused by their own electrocatalytic properties. Fig. 4a presents the initial discharge/charge curves of different cathodes at 100 mA g−1 with a voltage window of 2.35–4.4 V. Compared with the KB counterpart, MoSe2 cathodes show significantly improved coulombic efficiency and discharge/charge specific capacities. Specifically, discharge/charge specific capacities of KB, MS, MS-5, MS-15 and MS-20 cathodes are 3842.2/1301.3, 8088.5/6032.2, 18150.8/17805.6, 15056.2/14941.8 and 12385/10228 mA h g−1, and their coulombic efficiencies are 34%, 75%, 98%, 99% and 82% respectively. In contrast, the MS-10 cathode exhibits the highest discharge/charge specific capacities of 21112.4/20740.7 mA h g−1 with the coulombic efficiency of 98%. The reason for the better performance of the MoSe2 cathode was attributed to the unique nanoflower architecture composed of abundant nanosheets, and they expose a large number of active edge sites (Fig. 2a–c), favorable for delivering excellent electrocatalytic activities.42,54,55 Moreover, it is worth noting that the 1T/2H MoSe2 cathodes show lower charge and discharge overpotentials than those of the MS cathode. This is closely related to the high ORR/OER bifunctional electrocatalytic activity of the 1T phase, and the abundant heterojunctions formed by the two phases will also accelerate the electron transport and promote the reaction kinetics. In addition, MS-5 and MS-10 cathodes deliver obvious two voltage platforms at around 3.5 and 4.2 V during charging, which are considered to be related to the decomposition of the Li-deficient solid solution (Li2−xO2) phase and the oxidation of Li2O2, respectively.56 In order to further illustrate the ORR/OER electrocatalytic performance of different cathodes, CV tests were performed in the range of 2.35–4.5 V, and the results are given in Fig. 4b. It is clearly revealed that the MS-10 cathode displays lower OER potentials and higher ORR potentials, and it also delivers much higher peak current densities than those of other cathodes. More importantly, obvious OER peaks appeared at around 3.5 V for MS-5 and MS-10 cathodes, which have not been detected in the CV plots of other cathodes, and the reason can be clarified by XPS analysis in Fig. S8† for different cathodes charged to 3.5 V at 100 mA g−1. Based on high-resolution Li 1s XPS spectra of different cathodes, the discharge products of MS-5 and MS-10 cathodes contain 14 and 18% Li2−xO2, respectively, while those of MS, MS-15 and MS-20 cathodes contain Li2−xO2 less than 5%. The oxidation peaks at around 3.5 V are related to the reaction of Li2O2 → Li2−xO2 + xLi+ + xe−,18,57 and thus MS-5 and MS-10 cathodes show specific oxidation peaks, which were not observed for other samples. Fig. 4c and S9† show the rate performance of different cathodes at 100–800 mA g−1 for each 3 cycles under a specific capacity limit of 1000 mA h g−1. Fig. 4c gives the information on the discharge/charge terminal voltages with the variation of current densities. It was also traced that even when the current density increases from 100 to 800 mA g−1, the discharge and charge terminal voltages of the MS-10 cathode exhibit only a decrease and increase of 0.22 and 0.21 V, respectively, as shown in Fig. S9.† When the current density returned to 100 mA g−1, its discharge/charge terminal voltages are almost unchanged, compared with those at the initial stages with lower overpotentials in Fig. 4c, which is far more stable than other cathode counterparts.
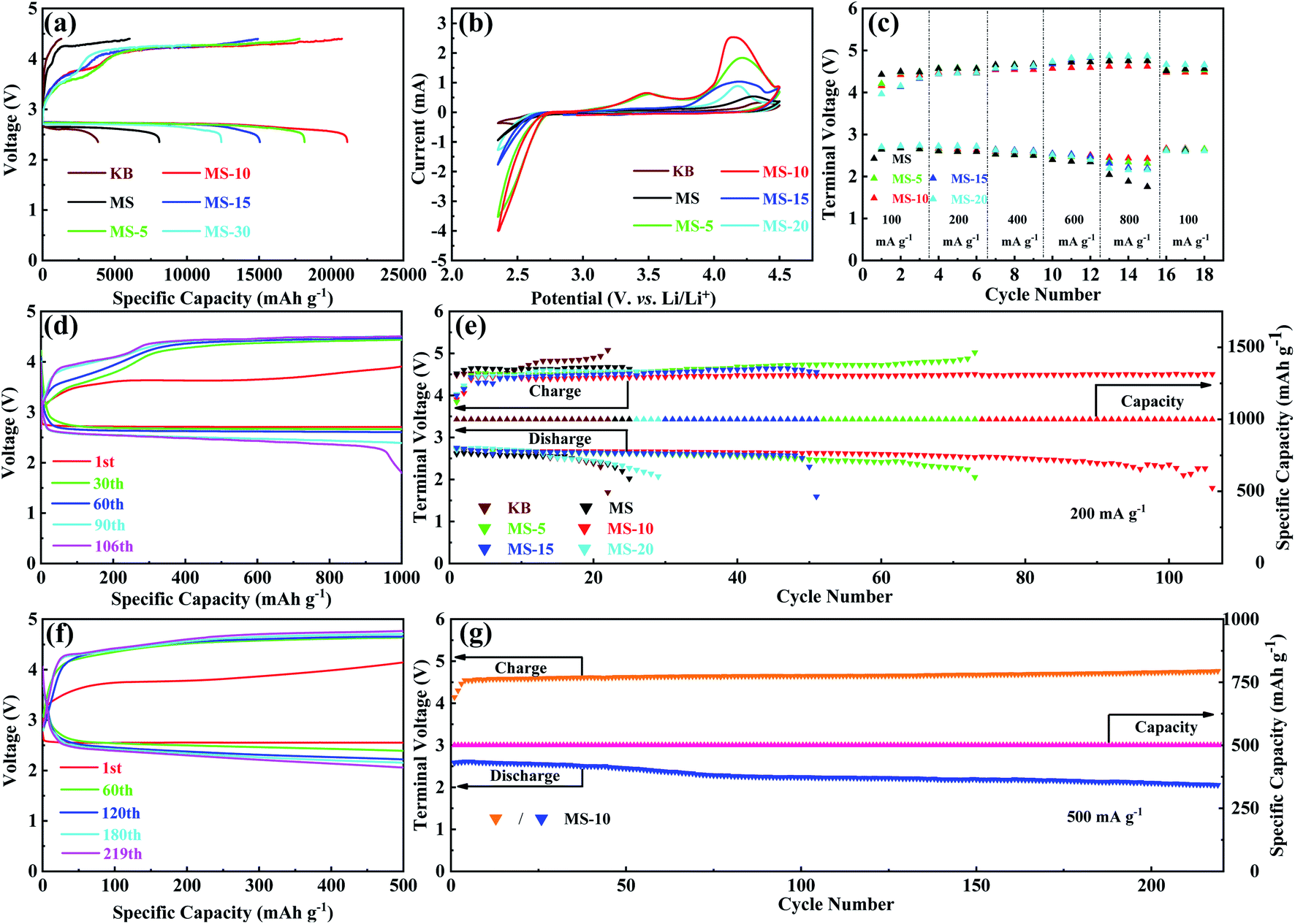 | ||
| Fig. 4 (a) Initial discharge/charge profiles of different cathodes at 100 mA g−1 from 2.35–4.4 V; (b) CV curves of different cathodes at 0.15 mV s−1; (c) comparison of rate performance of different cathodes; (d) typical discharge/charge profiles of the MS-10 cathode at 200 mA g−1 under a specific capacity limit of 1000 mA h g−1; (e) cycling performance of different cathodes at 200 mA g−1; (f) typical discharge/charge profiles of the MS-10 cathode at 500 mA g−1 under a specific capacity limit of 500 mA h g−1; (g) cycling performance of the MS-10 cathode at 500 mA g−1. | ||
The cycling performance of different cathodes was compared at 200 mA g−1 with a cut-off specific capacity of 1000 mA h g−1 in Fig. 4d, e and S10.† It is evident that KB, MS and MS-20 cathodes cannot remain stable within 30 cycles. However MS-5, MS-10, and MS-15 cathodes show extended cycling life, and the MS-10 cathode could be even discharged and charged for more than 100 cycles, demonstrating that introducing a certain amount of the 1T phase into the MoSe2 cathode could effectively activate its electrocatalytic activity and improve the cycling stability. What is more, MS-5 shows higher overpotentials than those of MS-15 and MS-20 during cycling, but it cycled more times, benefiting from the excellent stability of the 2H phase. In addition, as shown in Fig. 4f and g, the MS-10 cathode could be circulated stably for 219 cycles at 500 mA g−1 with a cut-off specific capacity of 500 mA h g−1, showing a long cycle life at the high current density. In addition, compared with the most of the reported TMC based cathode catalysts (Table S1†), it is worth mentioning that the 1T/2H MoSe2 cathodes deliver better performance than them under similar testing conditions, including specific capacities, coulombic efficiency and cycling stability.
In order to reveal the effect of the heterogeneous interface in the 1T/2H MoSe2 on the LOB performance, EIS measurements were conducted, and mass diffusion rates for different cathodes could be obtained from the Warburg elements (W) at open circuit potentials,58,59 which were fitted using an equivalent circuit in Fig. S11a.† Specifically, MS-5, MS-10, MS-15 and MS-20 cathodes respectively deliver mass diffusion rates of 2.084 × 10−2, 2.522 × 10−2, 2.488 × 10−2 and 2.485 × 10−2 Ω−1 cm−2 s0.5, much higher than that of the MS (1.466 × 10−2 Ω−1 cm−2 s0.5) cathode, as depicted in Fig. S11b.† This indicates that rich heterogeneous interfaces in the 1T/2H MoSe2 cathodes can greatly increase the mass diffusion rates, thus accelerating the ORR/OER kinetics. In addition, the charge transfer resistances are also summarized in Fig. S11c,† and it is found that the charge transfer resistances of MS-5, MS-10, MS-15 and MS-20 cathodes are 30.67, 20.38, 36.70 and 43.41 Ω, which are much smaller than that of the MS cathode (73.23 Ω). This is due to the introduction of the highly metallic 1T phase in the composites, and the rich heterogeneous interfaces between the two phases also contribute to the improvement of their electrical conductivity.
The synergetic effect from 1T and 2H phases played a critical role in increasing the electrocatalytic activities of MoSe2 cathodes. Notably, the 1T phase features high intrinsic catalytic activity, which could largely reduce the overpotentials of the ORR/OER and optimize battery efficiency, while the 2H phase is helpful for maintaining the structural stability of the cathodes during cycling. Besides, it is believed that the in situ incorporation of 1T and 2H phases would generate a heterostructure to enable intimate electrocontact between each other, aiding in increasing electrical conductivity and boosting Li+ diffusion kinetics for LOBs. Additionally, the nanoflower-like structure composed of porous nanosheets provides enough three-phase interfaces and free spaces to facilitate the deposition and storage of discharge products, optimizing the electrocatalytic performance of MoSe2 cathodes.
To further interpret the excellent eletrocatalytic activity and cycling stability of MS-10, the cathodes at various stages were characterized by XRD, SEM and EIS measurements. Compared with the fresh cathode in Fig. 5a, obvious film-like substances appeared on the surface of the MS-10 cathode after 1st discharging in Fig. 5b. At this stage, two peaks could be traced at 32.7 and 34.8° in Fig. 5e, corresponding to the (200) and (201) crystal planes of Li2O2 (JCPDS 73-1640), respectively. With discharge and charge processes being repeated for the 1st and 100th cycles, Li2O2 is not detected in the cathode (Fig. 5c–e), which implies that the MS-10 cathode can effectively catalyze the formation and decomposition of Li2O2 and almost no insoluble by-products were produced during cycling. The EIS plots of the MS-10 cathode at different stages were recorded and shown in Fig. 5f, and the values of each parameter were fitted in Table S2† using the equivalent circuit shown in the inset. Rohm represents ohmic resistance of the Li–O2 cell. Rint1 and Rint2 denote the charge transfer resistance between the cathode/electrolyte and Li anode/electrolyte, respectively. W denotes mass diffusion rate, which is related to the speed of transporting reaction species. Rint1 increased from 20.38 to 80.24 Ω after 1st discharging, which would be caused by the deposition of insulated Li2O2 on the surface of the cathodes. It then obviously decreased to 28.22 Ω after 1st charging, mainly due to the decomposition of Li2O2. Notably, the Rint1 increase of the MS-10 cathode after the 100th cycle is not obvious, indicating its good reversibility. In addition, XPS measurement for the MS-10 cathode after cycling test was conducted, and the result in Fig. S13† indicates that the ratio of 1T and 2H phases shows almost no changes, compared with that of the as-prepared material, which proves the good structural stability of the MS-10 cathode during cycling.
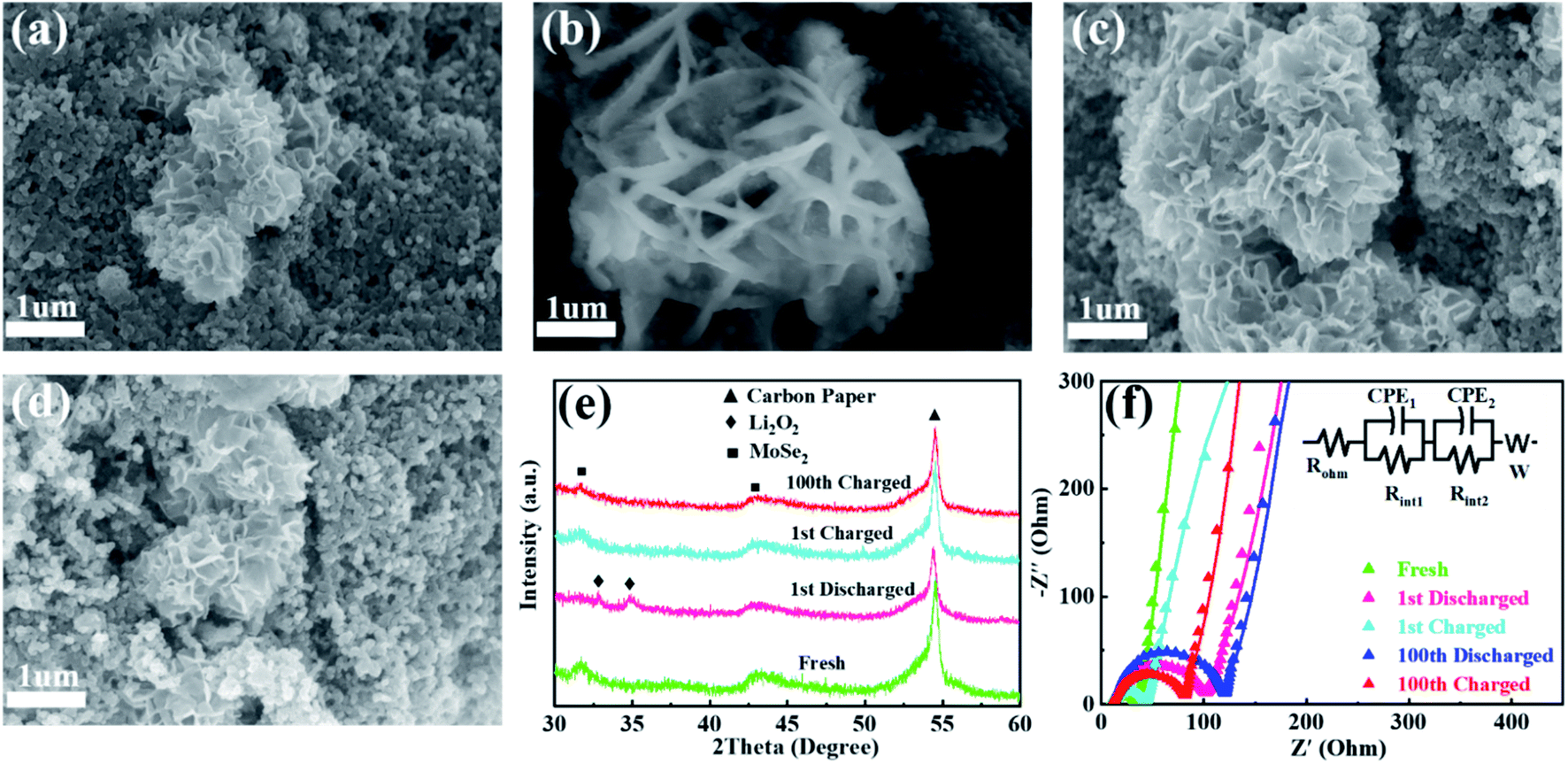 | ||
| Fig. 5 SEM images of the MS-10 cathodes at different stages: (a) fresh, (b) after 1st discharging, (c) after 1st recharging and (d) after 100th recharging under a limited specific capacity of 1000 mA h g−1; (e) XRD patterns and (f) EIS plots of the MS-10 cathodes at different stages. | ||
Ex situ XPS and SAED of MS-10 cathodes were performed to study the reaction mechanism of MS-10 samples during cycling. Fig. 6b–f show the high-resolution Li 1s XPS spectra at different stages in Fig. 6a. For the pristine cathode, there is no corresponding Li compound peak found in Fig. 6b. When the discharge specific capacity reaches 200 mA h g−1 (state I), two peaks can be obtained by fitting the high-resolution XPS spectrum of Li 1 s (Fig. 6c). The peak at 56.1 eV may be related to the Li2−xO2 discharge products, while the other peak at 54.8 eV is caused by the Li2O2 deposition on the cathode surfaces.60,61 When the discharge specific capacity reaches 800 mA h g−1 (state II), only one peak of Li2O2 can be traced in Fig. 6d, which indicates that Li2−xO2 at state II had been further oxidized to Li2O2 totally. This was verified by the corresponding SAED spectrum in Fig. S12† with only diffraction rings of Li2O2 and MoSe2, indicating that rare by-products formed at this stage. Based on the above discussion, it can be proposed that the growth mechanism of Li2O2 in discharging processes occurred through a typical surface-nucleation route, and the corresponding electrochemical equations are listed as follows:
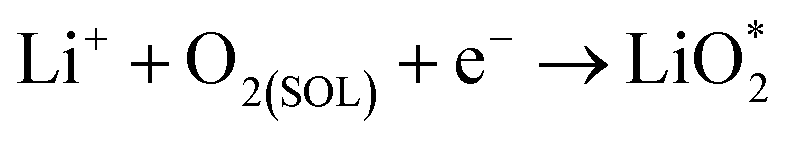 | (1) |
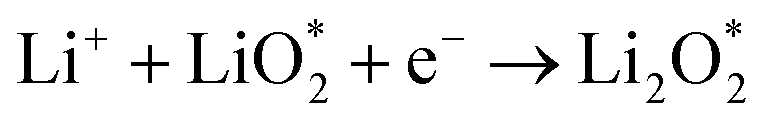 | (2) |
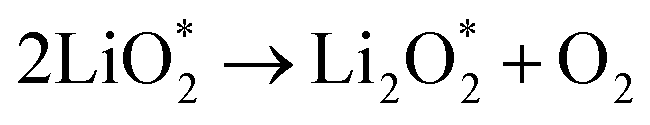 | (3) |
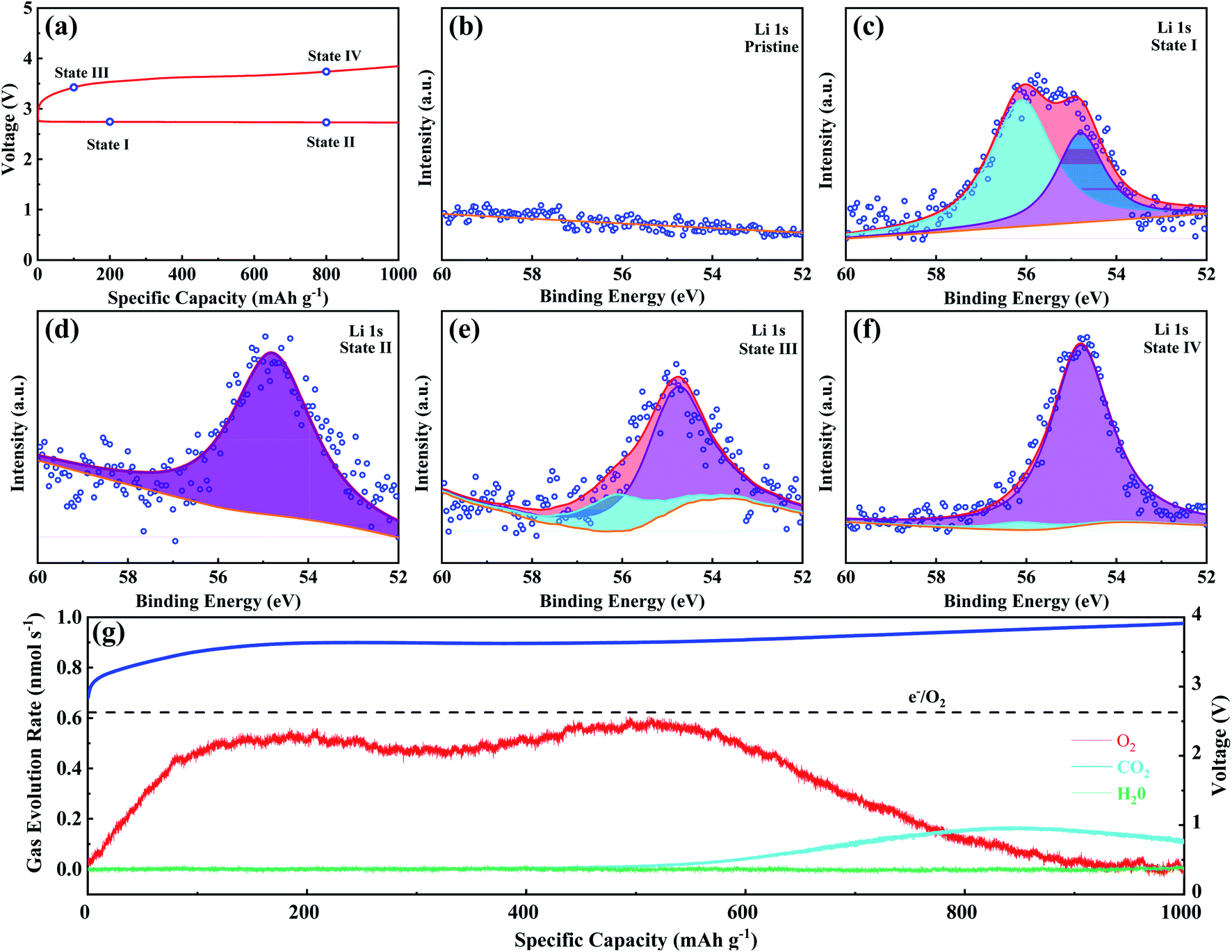 | ||
| Fig. 6 (a) Discharge and charge curves for the 1st cycle at 200 mA g−1 under a cutoff specific capacity of 1000 mA h g−1; (b–f) high-resolution Li 1s XPS spectra at different states of MS-10 cathodes; (g) in situ DEMS profiles for the MS-10 cathode with charge curves at 1000 mA g−1. | ||
In addition, when charging to a small amount of specific capacity (100 mA h g−1, state III), the corresponding Li 1s XPS signal can be fitted into two peaks associated with Li2O2 and Li2−xO2 (Fig. 6e). Fig. 6g shows the in situ DEMS spectrum during the corresponding charging process, which is a typical “M”-shaped curve. According to previous studies, the O2 release at the beginning of the curve is attributed to decomposition of the non-stoichiometric Li2−xO2 on the cathode with lower overpotentials.62–64 The corresponding electrochemical equations are given as follows:
| Li2O2 → LiO2 + Li+ + e− | (4) |
| LiO2 → Li+ + O2 + e− | (5) |
At state IV, it can be concluded from Fig. 6f that the product at this stage is almost Li2O2, corresponding to the second peak of the DEMS curve, and O2 in the second mountain escapes from the higher crystallinity Li2O2, which could result in a slightly higher overpotential during charging. This means that at a higher voltage of 4.35 V, Li2O2 directly turned into Li+ and O2 through a one-step two-electron transfer process, and the electrochemical equation is given as follows:
| Li2O2 → 2Li+ + O2 + 2e− | (6) |
Conclusions
In summary, MoSe2 catalysts with tunable 1T/2H phases were successfully constructed by simply varying the reaction time in the hydrothermal process. Their unique hierarchical framework with large surface area and modified intrinsic structures could not only expose abundant active sites to boost the electron and mass transport, but also provide sufficient storage for discharge products, thus exhibiting superior ORR/OER performance in LOBs. The MoSe2 cathode with 42.3% 1T phase could deliver discharge/charge specific capacities of 21210.4/20740.7 mA h g−1 at 100 mA g−1, and it also shows good high rate capability, even at 800 mA g−1. Excellent cyclabilities had been achieved with low polarization, of more than 100 cycles at 200 mA g−1 with a fixed capacity of 1000 mA h g−1 and 219 cycles at 500 mA g−1 with a fixed capacity of 500 mA h g−1. This rational synthetic strategy with effective modulation of hybrid phases and electrocatalytic capability could provide guidance for the application of other TMC materials in LOBs, which is also expected to be extended to various high specific energy storage and conversion systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding support from the China Postdoctoral Science Foundation (2020M672054), Natural Science Foundation of Shandong Province (ZR2020QB122), Young Scholars Program of Shandong University (2019WLJH21), Taishan Scholars Programme of Shandong Province (No. tsqn20161004) and Project for Scientific Research Innovation Team of Young Scholar in Colleges and Universities of Shandong Province (2019KJC025) is gratefully acknowledged.Notes and references
- N. Kittner, F. Lill and D. M. Kammen, Nat. Energy., 2017, 2, 17125 CrossRef.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef PubMed.
- Y. Hou, J. Wang, C. X. Hou, Y. Q. Fan, Y. J. Zhai, H. Y. Li, F. Dang and S. L. Chou, J. Mater. Chem. A, 2019, 7, 6552–6561 RSC.
- Y. Yu, G. Huang, J. Z. Wang, K. Li, J. L. Ma and X. B. Zhang, Adv. Mater., 2020, 32, 2004157 CrossRef CAS PubMed.
- C. T. Zhao, C. Yu, S. H. Liu, J. Yang, X. M. Fan, H. W. Huang and J. S. Qiu, Adv. Funct. Mater., 2015, 25, 6913–6920 CrossRef CAS.
- C. X. Hou, W. Yang, X. Xie, X. Sun, J. Wang, N. Naik, D. Pan, X. Mai, Z. Guo, F. Dang and W. Du, J. Colloid Interface Sci., 2021, 596, 396–407 CrossRef CAS.
- Z. W. Chang, J. J. Xu and X. B. Zhang, Adv. Energy Mater., 2017, 7, 1700875 CrossRef.
- Y. J. Kang, S. C. Jung, H. J. Kim, Y. K. Han and S. H. Oh, Nano Energy, 2016, 27, 1–7 CrossRef CAS.
- Y. Wang and Y. Xia, Nat. Chem., 2013, 5, 445–447 CrossRef.
- D. U. Lee, P. Xu, Z. P. Cano, A. G. Kashkooli, M. G. Park and Z. W. Chen, J. Mater. Chem. A, 2016, 4, 7107–7134 RSC.
- B. Liu, Y. L. Sun, L. Y. Liu, J. T. Chen, B. J. Yang, S. Xu and X. B. Yan, Energy Environ. Sci., 2019, 12, 887–922 RSC.
- A. J. Hu, C. Z. Shu, C. X. Xu, R. X. Liang, J. B. Li, R. X. Zheng, M. L. Li and J. P. Long, J. Mater. Chem. A, 2019, 7, 21605–21633 RSC.
- Y. C. Lu, Z. C. Xu, H. A. Gasteiger, S. Chen, K. Hamad-Schifferli and Y. Shao-Horn, J. Am. Chem. Soc., 2010, 132, 12170–12171 CrossRef PubMed.
- J. Suntivich, Z. Xu, C. E. Carlton, J. Kim, B. Han, S. W. Lee, N. Bonnet, N. Marzari, L. F. Allard, H. A. Gasteiger, K. Hamad-Schifferli and Y. Shao-Horn, J. Am. Chem. Soc., 2013, 135, 7985–7991 CrossRef.
- Q. C. Zhu, S. M. Xu, M. M. Harris, C. Ma, Y. S. Liu, X. Wei, H. S. Xu, Y. X. Zhou, Y. C. Cao, K. X. Wang and J. S. Chen, Adv. Funct. Mater., 2016, 26, 8514–8520 CrossRef.
- J. J. Xu, Z. L. Wang, D. Xu, F. Z. Meng and X. B. Zhang, Energy Environ. Sci., 2014, 7, 2045–2384 RSC.
- H. L. Wang, Y. Yang, Y. Y. Liang, G. Y. Zheng, Y. G. Li, Y. Cui and H. J. Dai, Energy Environ. Sci., 2012, 5, 7931–7935 RSC.
- Q. S. Huang, F. Dang, H. T. Zhu, L. L. Zhao, B. He, Y. Wang, J. Wang and X. M. Mai, J. Power Sources, 2020, 451, 227738 CrossRef CAS.
- J. P. Long, Z. Q. Hou, C. Z. Shu, C. Han, W. J. Li, R. Huang and J. Z. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 3834–3842 CrossRef CAS.
- X. F. Wang, Y. J. Li, X. X. Bi, L. Ma, T. P. Wu, M. Sina, S. Wang, M. H. Zhang, J. Alvarado, B. Y. Lu, A. Banerjee, K. Amine, J. Lu and Y. S. Meng, Joule, 2018, 2, 2381–2392 CrossRef CAS.
- M. M. Ottakam Thotiyl, S. A. Freunberger, Z. Q. Peng, Y. H. Chen, Z. Liu and P. G. Bruce, Nat. Mater., 2013, 12, 1050–1056 CrossRef CAS.
- H. J. Zhang, K. X. Wang, X. Y. Wu, Y. M. Jiang, Y. B. Zhai, C. Wang, X. Wei and J. S. Chen, Adv. Funct. Mater., 2014, 24, 3399–3404 CrossRef CAS.
- C. Z. Shu, Y. M. Lin and D. S. Su, J. Mater. Chem. A, 2016, 4, 2128–2136 RSC.
- Y. F. Li, Z. P. Huang, K. Huang, D. Carnahan and Y. C. Xing, Energy Environ. Sci., 2013, 6, 3339–3345 RSC.
- J. H. Han, X. W. Guo, Y. Ito, P. Liu, D. Hojo, T. Aida, A. Hirata, T. Fujita, T. Adschiri, H. S. Zhou and M. W. Chen, Adv. Energy Mater., 2016, 6, 1501870 CrossRef.
- M. Song, H. Tan, X. L. Li, A. L. Y. Tok, P. Liang, D. L. Chao and H. J. Fan, Small Methods, 2019, 4, 1900274 CrossRef.
- P. P. Zhang, X. Y. Lu, Y. Huang, J. W. Deng, L. Zhang, F. Ding, Z. Q. Su, G. Wei and O. G. Schmidt, J. Mater. Chem. A, 2015, 3, 14562–14566 RSC.
- Z. M. Sun, J. L. He, M. W. Yuan, L. Lin, Z. Zhang, Z. Kang, Q. L. Liao, H. F. Li, G. B. Sun, X. J. Yang, R. Long and Y. Zhang, Nano Energy, 2019, 65, 103996 CrossRef.
- N. Li, J. J. Wu, Y. T. Lu, Z. J. Zhao, H. C. Zhang, X. T. Li, Y. Z. Zheng and X. Tao, Appl. Catal., B, 2018, 238, 27–37 CrossRef.
- J. J. Zhang, W. P. Kang, M. Jiang, Y. You, Y. L. Cao, T. W. Ng, D. Y. W. Yu, C. S. Lee and J. Xu, Nanoscale, 2017, 9, 1484–1490 RSC.
- T. Xiang, S. Tao, W. Y. Xu, Q. Fang, C. Q. Wu, D. B. Liu, Y. Zhou, A. Khalil, Z. Muhammad, W. S. Chu, Z. H. Wang, H. F. Xiang, Q. Liu and L. Song, ACS Nano, 2017, 11, 6483–6491 CrossRef PubMed.
- Z. Jiang, Y. Wang, S. G. Yuan, L. Shi, N. Wang, J. Xiong, W. H. Lai, X. Y. Wang, F. Y. Kang, W. Lin, C. P. Wong and C. Yang, Adv. Funct. Mater., 2018, 29, 1807116 CrossRef.
- P. Pazhamalai, K. Krishnamoorthy, V. K. Mariappan, S. Sahoo, S. Manoharan and S. J. Kim, Adv. Mater. Interfaces, 2018, 5, 1800055 CrossRef.
- J. M. Ge, L. Fan, J. Wang, Q. F. Zhang, Z. M. Liu, E. J. Zhang, Q. Liu, X. Z. Yu and B. G. Lu, Adv. Energy Mater., 2018, 8, 1801477 CrossRef.
- P. Ge, L. M. Zhang, Y. Yang, W. Sun, Y. H. Hu and X. B. Ji, Adv. Mater. Interfaces, 2019, 7, 1901651 CrossRef.
- N. K. Oh, C. Kim, J. Lee, O. Kwon, Y. Choi, G. Y. Jung, H. Y. Lim, S. K. Kwak, G. Kim and H. Park, Nat. Commun., 2019, 10, 1723 CrossRef PubMed.
- N. Li, Y. F. Zhang, M. L. Jia, X. D. Lv, X. T. Li, R. Li, X. Q. Ding, Y. Z. Zheng and X. Tao, Electrochim. Acta, 2019, 326, 134976 CrossRef.
- L. Najafi, S. Bellani, R. Oropesa-Nuñez, A. Ansaldo, M. Prato, A. E. Del Rio Castillo and F. Bonaccorso, Adv. Energy Mater., 2018, 8, 1801764 CrossRef.
- Y. Yin, Y. M. Zhang, T. L. Gao, T. Yao, X. H. Zhang, J. C. Han, X. J. Wang, Z. H. Zhang, P. Xu, P. Zhang, X. Z. Cao, B. Song and S. Jin, Adv. Mater., 2017, 29, 1700311 CrossRef PubMed.
- C. Xu, S. J. Peng, C. L. Tan, H. X. Ang, H. T. Tan, H. Zhang and Q. Y. Yan, J. Mater. Chem. A, 2014, 2, 5597–5601 RSC.
- H. Tang, K. P. Dou, C. C. Kaun, Q. Kuang and S. H. Yang, J. Mater. Chem. A, 2014, 2, 360–364 RSC.
- Y. Q. Lai, W. Chen, Z. A. Zhang, Y. Q. Gan, X. Yang and J. Li, RSC Adv., 2016, 6, 19843–19847 RSC.
- M. L. Li, C. Z. Shu, A. J. Hu, J. B. Li, Y. Yan, M. He and J. P. Long, J. Alloys Compd., 2021, 855, 157484 CrossRef.
- Y. Y. Feng, T. Zhang, J. H. Zhang, H. Fan, C. He and J. X. Song, Small, 2020, 16, 2002850 CrossRef PubMed.
- Y. F. Yu, G. H. Nam, Q. Y. He, X. J. Wu, K. Zhang, Z. Z. Yang, J. Z. Chen, Q. L. Ma, M. T. Zhao, Z. Q. Liu, F. R. Ran, X. Z. Wang, H. Li, X. Huang, B. Li, Q. H. Xiong, Q. Zhang, Z. Liu, L. Gu, Y. H. Du, W. Huang and H. Zhang, Nat. Chem., 2018, 10, 638–643 CrossRef.
- W. P. Xiao, D. Bukhvalov, Z. Y. Zou, L. Zhang, Z. X. Lin and X. F. Yang, ChemSusChem, 2019, 12, 5015–5022 CrossRef PubMed.
- Y. H. Li, Y. Zhang, X. L. Tong, X. L. Wang, L. J. Zhang, X. H. Xia and J. P. Tu, J. Mater. Chem. A, 2021, 9, 1418–1428 RSC.
- J. T. Zhang, Y. L. Chen, M. Liu, K. Du, Y. Zhou, Y. P. Li, Z. J. Wang and J. Zhang, Nano Res., 2018, 11, 4587–4598 CrossRef.
- M. Jiang, J. J. Zhang, M. H. Wu, W. J. Jian, H. T. Xue, T. W. Ng, C. S. Lee and J. Xu, J. Mater. Chem. A, 2016, 4, 14949–14953 RSC.
- S. J. Deng, C. Z. Ai, M. Luo, B. Liu, Y. Zhang, Y. Y. Li, S. W. Lin, G. X. Pan, Q. Q. Xiong, Q. Liu, X. L. Wang, X. H. Xia and J. P. Tu, Small, 2019, 15, 1901796 CrossRef.
- H. Y. Shi, H. Zhang, M. J. Li, Y. Wang and D. Z. Wang, J. Alloys Compd., 2021, 878, 160381 CrossRef.
- Z. Z. Wu, R. B. Zhang, H. Fei, R. Q. Liu, D. Z. Wang and X. L. Liu, Appl. Surf. Sci., 2020, 532, 147372 CrossRef.
- J. Y. Zhang, T. T. Wang, P. T. Liu, Y. G. Liu, J. Ma and D. Q. Gao, Electrochim. Acta, 2016, 217, 181–186 CrossRef.
- Y. J. Chung, C. S. Yang, J. T. Lee, G. H. Wu and J. M. Wu, Adv. Energy Mater., 2020, 10, 2002082 CrossRef.
- S. J. Deng, C. Z. Ai, M. Luo, B. Liu, Y. Zhang, Y. Y. Li, S. W. Lin, G. X. Pan, Q. Q. Xiong, Q. Liu, X. L. Wang, X. H. Xia and J. P. Tu, Small, 2019, 15, 1901796 CrossRef PubMed.
- W. G. Fan, B. Z. Wang, X. X. Guo, X. Y. Kong and J. J. Liu, Nano Energy, 2016, 27, 577–586 CrossRef.
- S. Ganapathy, B. D. Adams, G. Stenou, M. S. Anastasaki, K. Goubitz, X. F. Miao, L. F. Nazar and M. Wagemaker, J. Am. Chem. Soc., 2014, 136, 16335–16344 CrossRef CAS.
- K. Cheng, D. P. He, T. Peng, H. F. Lv, M. Pan and S. C. Mu, Electrochim. Acta, 2014, 132, 356–363 CrossRef CAS.
- J. J. Xu, Z. L. Wang, D. Xu, L. L. Zhang and X. B. Zhang, Nat. Commun., 2013, 4, 2438 CrossRef.
- G. Y. Li, N. Li, S. T. Peng, B. He, J. Wang, Y. Du, W. B. Zhang, K. Han and F. Dang, Adv. Energy Mater., 2020, 11, 2002721 CrossRef.
- J. H. Lin, Y. H. Wang, X. H. Zheng, H. Y. Liang, H. N. Jia, J. L. Qi, J. Cao, J. C. Tu, W. D. Fei and J. C. Feng, Dalton Trans., 2018, 47, 8771–8778 RSC.
- D. W. Kim, S. M. Ahn, J. W. Kang, J. D. Suk, H. K. Kim and Y. K. Kang, J. Mater. Chem. A, 2016, 4, 6332–6341 RSC.
- D. P. Kundu, R. Black, E. J. Berg and L. F. Nazar, Energy Environ. Sci., 2015, 8, 1292–1298 RSC.
- Y. Bae, D. H. Ko, S. Lee, H. D. Lim, Y. J. Kim, H. S. Shim, H. Park, Y. Ko, S. K. Park, H. J. Kwon, H. Kim, H. T. Kim, Y. S. Min, D. M. Im and K. Kang, Adv. Energy Mater., 2018, 8, 1702661 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: SEM images and BET results of MS, MS-5, MS-15 and MS-20, SAED pattern, HRTEM image and the intensity profile of MS-10, XPS results and rate performance of MS, MS-5, MS-10, MS-15 and MS-20, image of the hermetic container for Li–O2 batteries, galvanostatic discharge–charge curve of the pure carbon cathode, SAED pattern of the discharged MS-10 cathode, and cycling performance of KB, MS, MS-5, MS-15 and MS-20 cathodes. See DOI: 10.1039/d1ta03584c |
| ‡ These authors contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2021 |