
Solar-assisted co-electrolysis of glycerol and water for concurrent production of formic acid and hydrogen†
Zunjian
Ke
ab,
Nicholas
Williams
b,
Xingxu
Yan
c,
Sabrina
Younan
b,
Dong
He
a,
Xianyin
Song
e,
Xiaoqing
Pan
cd,
Xiangheng
Xiao
 *a and
Jing
Gu
*b
*a and
Jing
Gu
*b
aDepartment of Physics, Key Laboratory of Artificial Micro- and Nano-structures of Ministry of Education, Wuhan University, Wuhan 430072, Hubei, China. E-mail: xxh@whu.edu.cn
bDepartment of Chemistry and Biochemistry, San Diego State University, San Diego, California 92182-1030, USA. E-mail: jgu@sdsu.edu
cDepartment of Materials Science and Engineering, University of California, Irvine, Irvine, California 92697, USA
dDepartment of Physics and Astronomy, University of California, Irvine, Irvine, California 92697, USA
eCollege of Materials Science and Engineering, Hunan University, Changsha 410082, Hunan, China
First published on 30th June 2021
Abstract
Renewable electricity-driven water splitting provides a pathway to manufacturing hydrogen as a promising alternative to fossil fuels. A typical water electrolysis device is comprised of a cathodic hydrogen evolution reaction (HER) and an anodic oxygen evolution reaction (OER). Unfortunately, the OER consumes most of the overall electricity supply while generating negligible economic value, which inhibits the large-scale deployment of the water electrolysis technology. Here, we explored alternatives to the OER and demonstrated that electrooxidation of glycerol (a cheap byproduct of biodiesel and soap production) could lower anodic electricity consumption by up to 0.27 V while producing high-value formic acid with 96.2% faradaic efficiency (FE). Further, glycerol electrooxidation was combined with the photoelectrochemical HER to diminish the electricity requirement to 1.15 V, reducing the electricity consumption by ∼30% relative to typical water electrolysis. This study suggests that solar-assisted co-electrolysis of high-volume block chemicals and water may be an energy efficient and economically viable strategy to realize the sustainable production of value-added chemicals and hydrogen energy.
Introduction
The detrimental impact of global fossil fuel consumption on the climate and environment has instilled a tremendous interest in developing renewables-powered solutions to manufacturing clean energy.1 As a crucial carbon-neutral energy carrier and chemical reaction intermediate, hydrogen has been recognized as a renewable alternative to fossil fuels.2 At present, more than 100 Mt of hydrogen is manufactured per year. However, 95% is still produced by energy-intensive steam methane reformation, and only 4% through electricity-driven water splitting.3,4 Producing hydrogen by steam methane reformation renders the cost of hydrogen to be ∼$1.2–1.5/kg−1, whereas the cost of hydrogen produced via water electrolysis is more than $4/kg−1. This high cost of hydrogen production substantially diminishes the market share for water electrolysis. Consequently, the cost of hydrogen production via water electrolysis must reduce to ∼$2 per kg to achieve a large-scale application.5,6 Generally, costs associated with water electrolysis are dictated by operation expenses and capital input, with the former primarily dominated by electricity consumption.4 Unfortunately, a high overall electricity input (>1.5 V of the cell voltage) is required for typical water electrolysis devices (Scheme 1a), which limits the industrial deployment of these devices.7 The culprit behind this barrier is the thermodynamically sluggish OER, which consumes the majority of electricity input while producing negligible economic benefits (∼$0.1/kg−1 of oxygen price).8 As a consequence, exploring alternative strategies that circumvent sluggish OER thermodynamics while generating high-value products has the potential to lower energetic requirements and enable water electrolysis to replace traditional steam reforming processes. Recently, a few efforts have been reported that employ lower-energetic anodic reactions (such as electrooxidation reactions of ethanol,9 urea,10 hydrazine,11etc.) in water electrolysis. While these explorations are interesting, the oxidation of the reported chemicals may not be sustainable. For instance, the electrooxidation of urea will contribute to the emission of carbon dioxide.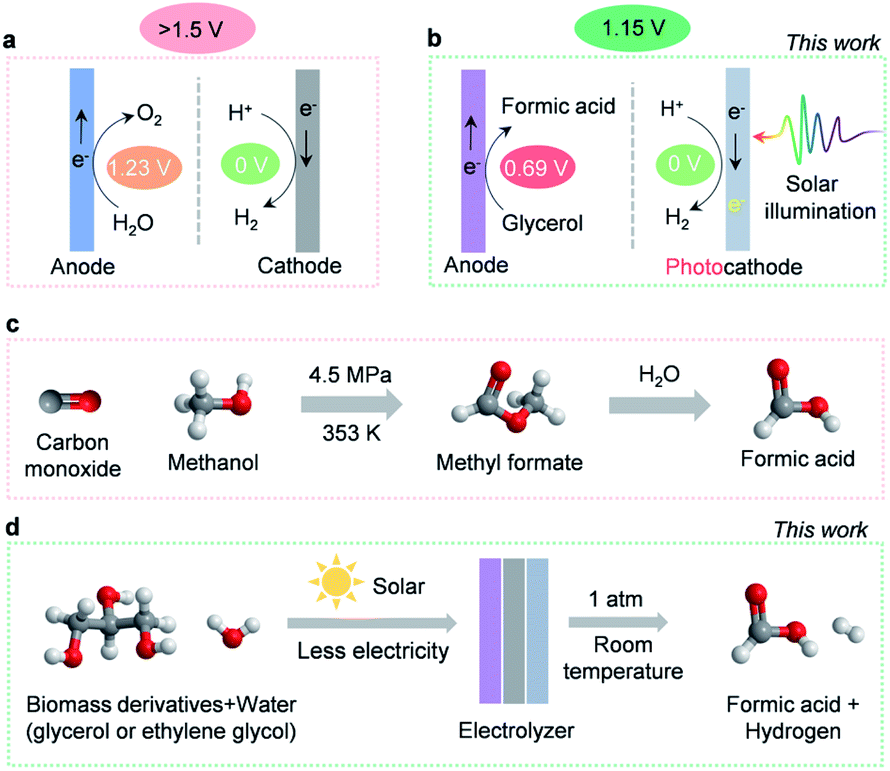 | ||
| Scheme 1 Overview of hydrogen production and formic acid manufacturing. (a) Typical water electrolysis device. (b) Energy-efficient and value-added hydrogen production system in this work. (c) Industrial formic acid production. (d) Solar-assisted formic acid electrosynthesis in this work. | ||
Herein, we investigate the electrooxidation of glycerol to formic acid as a promising substitute for the OER. Theoretically, the redox potential of glycerol to formic acid is 0.69 V, 44% lower than the 1.23 V required for the OER (Scheme 1a and b).12 This indicates that utilizing glycerol electrooxidation as an anodic reaction could significantly lower electricity requirements. More importantly, glycerol is a cheap by-product of biodiesel and soap production ($0.24/kg−1),13 whereas its major oxidation product, formic acid, is widely used in the fuel cell and hydrogen storage industries.14 As a high-value commodity chemical ($1.0 per kg),15,16 manufacturing formic acid by coupling glycerol electrooxidation with the HER retains high economic value ($17.25) upon producing 1 kg of hydrogen (Table S1, ESI†). In comparison, O2 produced by traditional water electrolysis (Scheme 1a) is only worth $0.80 to generate the same amount of hydrogen. In this regard, ambient glycerol electrooxidation will allow formic acid to be generated in a more sustainable manner than the industrial high temperature and pressure methyl formate hydrolysis (Scheme 1c).17 Therefore, highly efficient and selective electrocatalysts for glycerol oxidation are desired. However, most glycerol electro-oxidation catalysts so far are noble metals (like Pt,18 Au,19 Pd20) and their alloys (like PtRu,21 PtSb,22 PdxBi23). Moreover, the reaction pathways of glycerol oxidation involve various C1–C3 intermediates which can interconvert into many different oxidation products, thus suffering from low selectivity.24
In this work, we developed an efficient and cost-effective nitrogen-doped cobalt oxide (N-CoOx) electrocatalyst. The catalyst is derived from an organometallic cobalt complex and can achieve a highly selective formic acid synthesis by glycerol electrooxidation (96.2% FE). The anodic electricity consumption reduced up to 0.27 V relative to the OER, which is comparable with previous glycerol oxidation studies.32,33 In order to further diminish the electricity requirements, a cathodic photoelectrochemical HER was coupled with anodic glycerol oxidation (Scheme 1b). The hybrid system could be driven by solar energy with an additional electricity supply of 1.15 V to accomplish concurrent production of formic acid and hydrogen. This value, to our understanding, is the lowest electricity input reported for water electrolysis (Table S2, ESI†). In addition, the versatility of the N-CoOx catalyst was further confirmed by its ability to upgrade biomass-derived ethylene glycol into formic acid with a high FE of 62.3%. Overall, we have achieved highly selective, energy-efficient, and economically viable co-production of hydrogen fuel and high-value chemicals by solar-assisted co-electrolysis of water and cheap biomass waste (Scheme 1d).
Results and discussion
Material characterization
The nitrogen doped cobalt oxide electrocatalyst (N-CoOx) was directly deposited on carbon fibre paper by one-step facile atomic layer deposition (ALD). The precursor, a nitrogen-containing organometallic cobalt complex (bis(N,N′-di-i-propylacetamidinato) cobalt(II)) (Fig. 1a and S1, ESI†), was employed as the cobalt and nitrogen source for in situ nitrogen doping (see ESI†). The morphology of N-CoOx was characterized by scanning electron microscopy (SEM), in which the visibly smooth carbon surface confirms the uniform and ultrathin nature of the deposited catalyst film (Fig. S2a, ESI†). Atomic resolution high-angle annular dark-field (HAADF) scanning transmission electron microscopy (STEM) was employed to determine the structure and composition of N-CoOx (Fig. 1b). The resulting images clearly show lattice fringes and three characteristic crystal facets with interplanar spacings equal to 0.286, 0.463, and 0.241 nm. These crystal facets were indexed to the (220), (111), and (311) planes of Co3O4 with a standard spinel structure, respectively. The corresponding fast Fourier transform (FFT) patterns (Fig. 1b, inset) suggest the presence of multiple crystalline orientations of N-CoOx. Furthermore, the low magnification HAADF-STEM image and corresponding elemental mappings in Fig. 1c validate the uniform distribution of cobalt, oxygen, and nitrogen elements throughout the sample. These results are also confirmed by energy-dispersive X-ray spectroscopy of SEM (SEM-EDS) in Fig. S3, ESI.† The elemental compositions were further quantified by STEM-EDS (Fig. S4, ESI†), in which the presence of the non-stoichiometric ratio of cobalt and oxygen suggests the existence of multiple types of cobalt oxides in N-CoOx, corresponding effectively to the FFT patterns in Fig. 1b. X-ray diffraction (XRD) patterns were collected to identify the crystal structure of the catalysts (Fig. S2b, ESI†).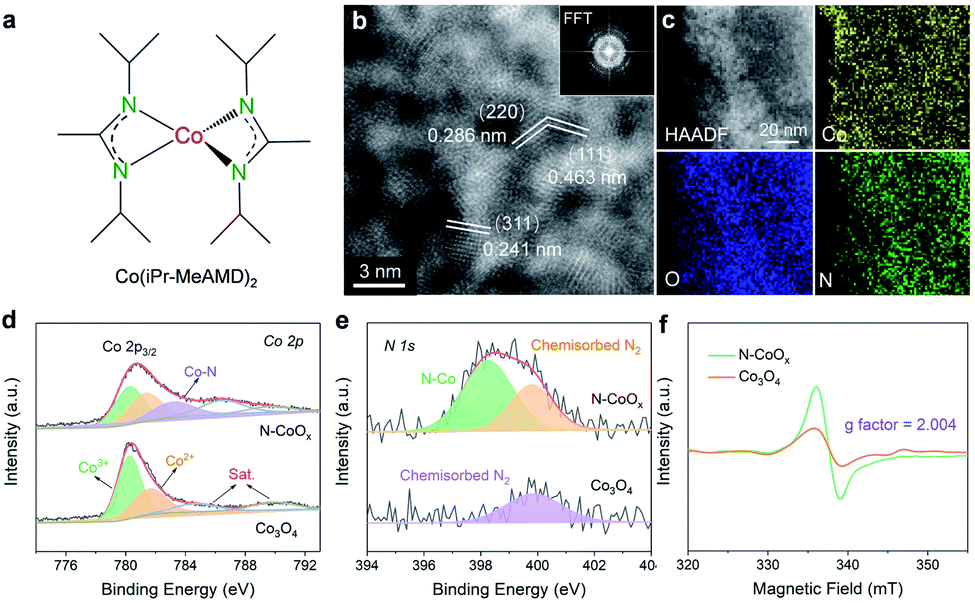 | ||
| Fig. 1 Structure and chemical states of N-CoOx. (a) Structure of the bis(N,N′-di-i-propylacetamidinato) cobalt(II) (Co(iPr-MeAMD)2) complex for N-CoOx synthesis. (b) and (c) HRTEM image and STEM-EDX elemental map of N-CoOx (inset: fast Fourier transform pattern). (d) XPS Co 2p, (e) XPS N 1s, and (f) EPR spectra of N-CoOx and Co3O4. | ||
In these results, only one broad peak, belonging to the amorphous substrate, was identified. The lack of distinct characteristic peaks in the XRD pattern may be attributed to the catalyst's ultrathin nature.25 On the other hand, the sample contains some amorphous regions in the HAADF-STEM image (Fig. 1b), which is evidenced by the amorphous ring in FFT patterns (Fig. 1b), correlating well with the XRD results. The surface chemical states of N-CoOx were elucidated by X-ray photoelectron spectroscopy (XPS) (Fig. 1d, e and S5, ESI†). The XPS survey spectrum in Fig. S5, ESI† revealed the presence of Co and O elements in N-CoOx and Co3O4 samples. Besides, an obvious N 1s signal at 399.1 eV in N-CoOx inevitably confirmed the existence of N in N-CoOx. In contrast, the N 1s signal in the Co3O4 control sample is negligible.26 The peak at 398.2 eV in the fitted high-resolution N 1s spectra (Fig. 1e) for N-CoOx references the existence of N–Co species, while the 399.9 eV peak identifies adsorbed N2 on both N-CoOx and Co3O4 samples.26,27 To analyse the chemical states of cobalt, the Co 2p XPS spectra are provided in Fig. 1d. The Co 2p3/2 peaks for Co3O4 may be fitted to the peaks at 780.4 eV and 781.8 eV and are assigned to tetrahedral and octahedral Co–O coordination geometries, respectively.28 Notably, the additional peak at 782.9 eV in Co 2p for N-CoOx further confirmed the presence of Co–N bonds in the catalyst.26,29 Here, it was worth noting that Co–Co interactions (around 778.8 eV) were not observed, precluding the presence of cobalt nitride species in N-CoOx.30 To further reveal the effects of the N dopant, the defects of catalysts were characterized by electron paramagnetic resonance (EPR) spectroscopy (Fig. 1f). The strong EPR signal located at g = 2.004 corresponds to the existence of oxygen vacancies. In addition, N dopant's potential to induce additional oxygen vacancies is indicated by the N-CoOx peak intensity being twice that of Co3O4.26,27,31
Glycerol electrooxidation
Electrochemical investigations were conducted to compare the performance of N-CoOx in glycerol oxidation to its performance in water oxidation. Compared to Co3O4, N-CoOx required a lower potential (1.58 V) to reach the current density of 10 mA cm−2 (Fig. S6a, ESI†). Likewise, the higher activity of N-CoOx was confirmed by its lower Tafel slope (179 mV dec−1) with respect to Co3O4 (187 mV dec−1) (Fig. S6b, ESI†). On average, the OER activities of both catalysts were very close and moderate, indicating that the N dopant did not contribute significantly to the OER. Next, systematic elucidation of the electrocatalytic activity of N-CoOx towards glycerol oxidation is provided in Fig. 2. Without glycerol, N-CoOx showed moderate OER activity by delivering an onset potential of 1.50 V and a potential of 1.58 V at 10 mA cm−2 (Fig. 2a). Addition of 1.0 M glycerol caused a negative shift in the onset potential and the potential at 10 mA cm−2 to 1.2 V and 1.31 V, respectively. Here, a visibly significant shift of 0.3 V and 0.27 V occurred for the onset potential and the potential at 10 mA cm−2, respectively. Upon evaluating the difference in potentials and current densities for N-CoOx before and after adding glycerol, lower energetic requirements are witnessed for each current density when glycerol is present (Fig. 2b). This result verifies the aptitude of N-CoOx for glycerol oxidation. It should be noted that the optimal glycerol concentration was determined to be 1.0 M by linear sweep voltammetry (LSV), where the lowest onset potential was determined by varying glycerol concentrations from 0 to 2.0 M. Notably, an extra peak at 1.45 V (pointed out by an arrow in Fig. S7, ESI†) appeared with the addition of 0.1 M glycerol, suggesting the ability of the OER to compete with glycerol oxidation at a higher potential (>1.45 V) while using a lower concentration of glycerol.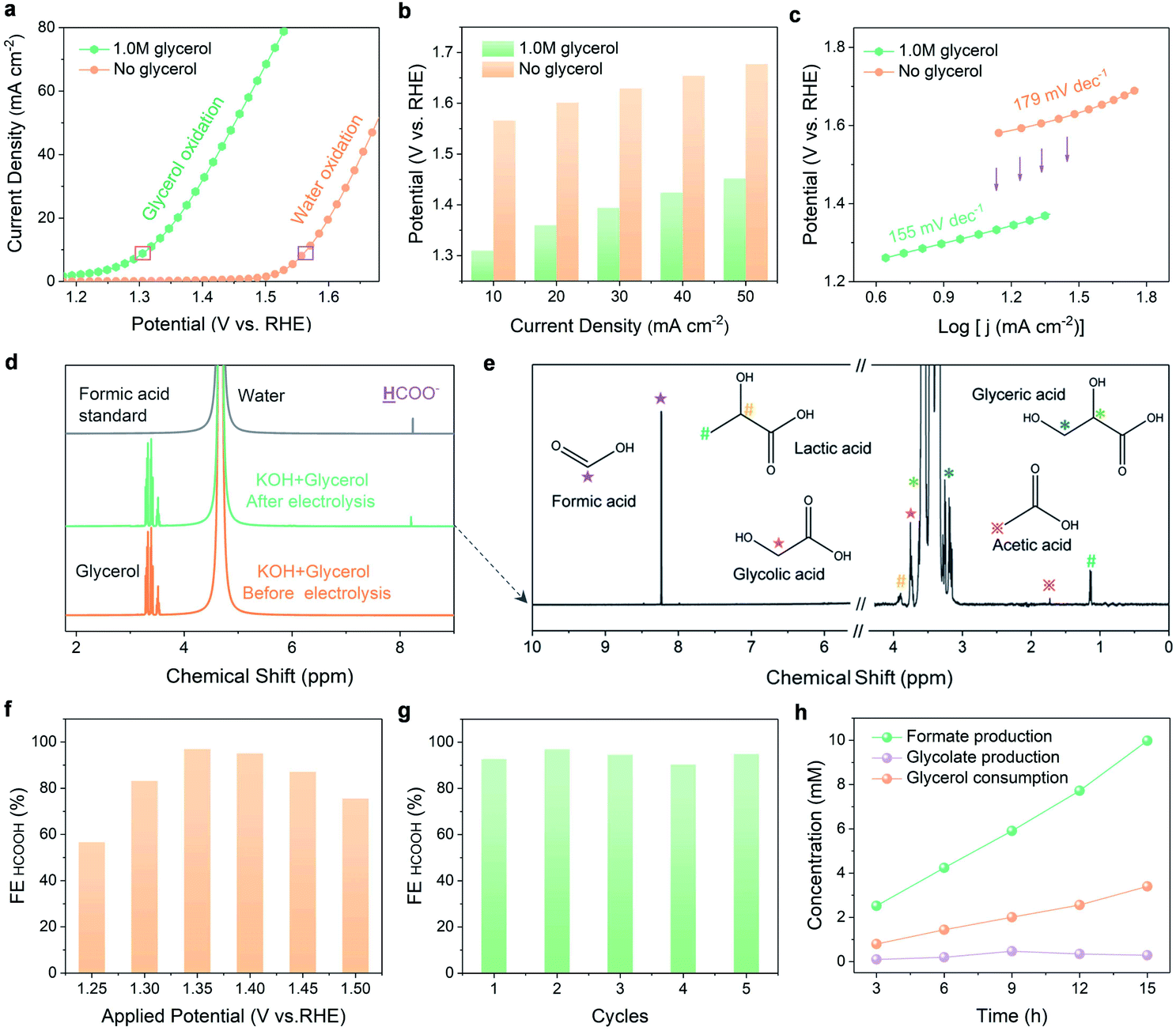 | ||
| Fig. 2 Glycerol electrooxidation performance. (a) Linear sweep voltammetry (LSV) plots (all LSV plots were collected with a scan rate of 5 mV s−1 in N2-saturated electrolyte with no I–R compensation). (b) Input potentials at various current densities. (c) Tafel slopes for the OER and glycerol oxidation in 1.0 M KOH solution both with and without 1.0 M glycerol. Purple arrows inset indicate the significant potential decrease at various current densities when 1.0 M glycerol is added. (d) 1H NMR spectra of the formic acid standard and electrolytes (1.0 M KOH + 1.0 M glycerol) before and after 12 h of electrolysis at 1.35 V vs. RHE. Due to the higher initial glycerol concentration, the intensity of 1H NMR peaks for glycerol did not decrease significantly after electrolysis. (e) High-resolution 1H NMR spectra (water peak was removed from 4 to 6 ppm) of the 1.0 M KOH + 1.0 M glycerol solution after 12 h of electrolysis (the symbols on the top of the peaks represent each type of proton present). (f) Faradaic efficiencies of formic acid at various potentials (each potential underwent 12 h of electrolysis). (g) Faradaic efficiencies of formic acid for five successive electrolysis cycles at 1.35 V vs. RHE. (h) Concentration changes of the reactant (glycerol), main product (formic acid), and representative intermediate species (glycolic acid) with electrolysis time. | ||
The role of the N dopant in enhancing kinetics for glycerol oxidation is demonstrated by the Tafel and impendence measurements. The Tafel slope (Fig. 2c) for glycerol oxidation was determined to be 155 mV dec−1, which is lower than that of the OER (179 mV dec−1). This indicates that N-CoOx achieves faster kinetics for glycerol oxidation relative to that of the OER. To clarify the influence of N dopants on N-CoOx catalytic activity, the oxidation performances of N-CoOx and Co3O4 were compared (Fig. S8, ESI†). Results show that Co3O4 requires a higher potential (1.43 V) than N-CoOx (1.31 V) to drive the current density of 10 mA cm−2 for the reaction forward. Co3O4 also exhibited a higher Tafel slope (173 mV dec−1) than N-CoOx (155 mV dec−1) (Fig. S8a and b, ESI†). Since the Tafel slopes indicate the catalytic reaction rate's dependence on applied potentials, these results show that the N-CoOx anode possesses much faster glycerol oxidation kinetics than that of Co3O4. This conclusion is further supported by the electrochemical impendence spectra (EIS), where N-CoOx showed much lower electric impedance than Co3O4 (Fig. S8c and Table S3, ESI†). Altogether, these results suggest that the incorporation of N leads to better electrical contact, lower electric impedance, and faster charge transfer kinetics for glycerol oxidation.
Product analysis
Glycerol oxidation products were collected and analyzed by 1H and 13C nuclear magnetic resonance (NMR) spectroscopy (Fig. 2d and e). A relatively low potential of 1.35 V was chosen here to limit the competitive OER. The 1H NMR spectra before and after 12 hours of electrolysis identify formic acid as the main product (Fig. 2d). Negligible changes in current density after 12 hours of electrolysis demonstrate the catalyst's high stability during extended operation (Fig. S9a, ESI†). LSV curves before and after 12 hours of electrolysis exhibit a slight deterioration in catalytic activity (Fig. S9b, ESI†). This may be attributed to a decrease in glycerol concentration and the possible change in the electrolyte pH.32 To further understand the stability of N-CoOx, post 12 h the LSV in a fresh electrolyte was compared to the LSV prior to electrolysis, where negligible changes in onset potential and overpotential are noticed (Fig. S10a, ESI†). Further, the intact Co 2p XPS spectrum after electrolysis confirms the superior stability of N-CoOx toward oxidizing glycerol (Fig. S10b, ESI†). The products of glycerol electrolysis were further confirmed by the 13C NMR. As shown in Fig. S11, ESI,† formic acid remained as the main product identified after 60 hours of electrolysis, which correlates well with 1H NMR findings. The small amount of carbonate detected in the 13C NMR spectra may be attributed to the high solubility of atmospheric CO2 in strongly alkaline media. A decrease in glycerol concentration was also detected by 13C NMR, suggesting a successful electrically-driven glycerol conversion. In addition to formic acid, glyceric acid, glycolic acid, lactic acid, and acetic acid were detected at concentrations much lower than what was seen for formic acid (Fig. 2e). These intermediate species will be used to unravel the glycerol oxidation mechanism, which is discussed in the mechanism investigation section.To determine the FE of formic acid, 1H NMR peaks of formic acid in a range of standard concentrations were collected with respect to the formic acid calibration curve (Fig. S12a and b, ESI†). From this analysis, the FE for formic acid production at 1.35 V was calculated to be 96.2%, which exceeds most glycerol upgrading studies reported (Table S2, ESI†).32,33 FEs at varied potentials were investigated in Fig. 2f, where the FE reaches a maximum (96.2%) at 1.35 V and gradually decreases to 58% and 75% at 1.25 V and 1.5 V, respectively. The FE and potential dependence may be explained by the strength of C–C bonds, which are harder to break at lower potentials. Unfortunately, at higher potentials, the OER competes with glycerol oxidation and diminishes glycerol conversion selectivity. N-CoOx also demonstrated great durability and recyclability by maintaining an average FE of 93.8% after five cycles of electrolysis (Fig. 2g). Lastly, concentration changes of the reactant (glycerol), intermediate (glycolic acid), and product (formic acid) were monitored during electrolysis (Fig. 2h). A linear relationship between the reaction time and the concentration of the product generated (or the reactant consumed) was observed, while the intermediate species demonstrated a nearly constant concentration throughout the reaction. Moreover, the FE of H2 at the cathode is found to be close to 100% in five successive electrolysis cycles (Fig. S13, ESI†). The result indicated that the HER is the only cathodic reaction, which also further indicates that replacing the anodic OER with glycerol oxidation will not affect the cathodic half-reaction.
To determine the potential of N-CoOx to oxidize other biomass-derived compounds, its catalytic activity towards ethylene glycol (EG) oxidation was investigated (Fig. S14a–c, ESI†). An onset potential of 1.25 V and a potential of 1.35 V at 10 mA cm−2 were observed (1.0 M EG), which saved 0.23 V of the potential (10 mA cm−2) compared to the OER (Fig. S12a, ESI†), thus proving that EG oxidation with N-CoOx reduces energy consumption at the anode. This potential shift was noticed at other current densities as well (10–40 mA cm−2) (Fig. S14b, ESI†). Regarding selectivity, an optimal FE of 62.3% at 1.40 V was obtained for formic acid production from EG (Fig. S14c, ESI†). From these results, we conclude that N-CoOx exhibits promising results towards converting glycerol and ethylene glycol into value-added formic acid. Currently, commercial production of formic acid involves reacting methanol with toxic carbon monoxide at an elevated pressure. This reaction produces the intermediate species methyl formate, which is then transformed into formic acid by a two-step hydrolysis reaction (Fig. 1c). This approach involves intensive energy and cost inputs.17 Instead, our work provides a facile route to synthesize formic acid by biomass electrolysis at room temperature and atmospheric pressure.
Mechanism investigation
The proposed reaction mechanism is provided in Fig. 3. In the first step, a primary hydroxyl group (–OH) on glycerol will be oxidized to an aldehyde via two-electron transfer.34 Further, the aldehyde will be oxidized to a carboxyl group, forming glyceric acid, which is supported by the 1H NMR (Fig. 2e). Then, we propose that the formic acid was produced with an oxidative cleavage of C–C bonds in glyceric and glycolic acid, where both intermediates are well identified by the NMR (Fig. 2e). Based on this route, one glycerol molecule may be completely converted into three formic acid molecules through an eight-electron transfer process. However, very small amounts of lactic acid and acetic acid are observed in the NMR results (Fig. 2e). Thus, we hypothesize that there is a minor reaction pathway (Fig. 3). In the minor reaction pathway, glyceraldehyde will reversibly react with OH– to form dihydroxy acetone. Dihydroxy acetone is then converted into 2-hydroxypropenal/pyruvaldehyde after a dehydration process. Further, 2-hydroxypropenal and pyruvaldehyde will convert to lactic acid after a Cannizzaro rearrangement. In the last step, the lactic acid will be oxidized to acetic acid and formic acid via C–C bond breakage.35,36 Similarly, the electrochemical oxidation mechanism for ethylene glycol conversion into formic acid is proposed in Fig. S15.† In this mechanism, a primary –OH group from ethylene glycol is oxidized to an aldehyde via a two-electron process, followed by oxidation into carboxyl. Finally, the glycolic acid intermediate will completely convert into formic acid via an oxidative cleavage of C–C bonds.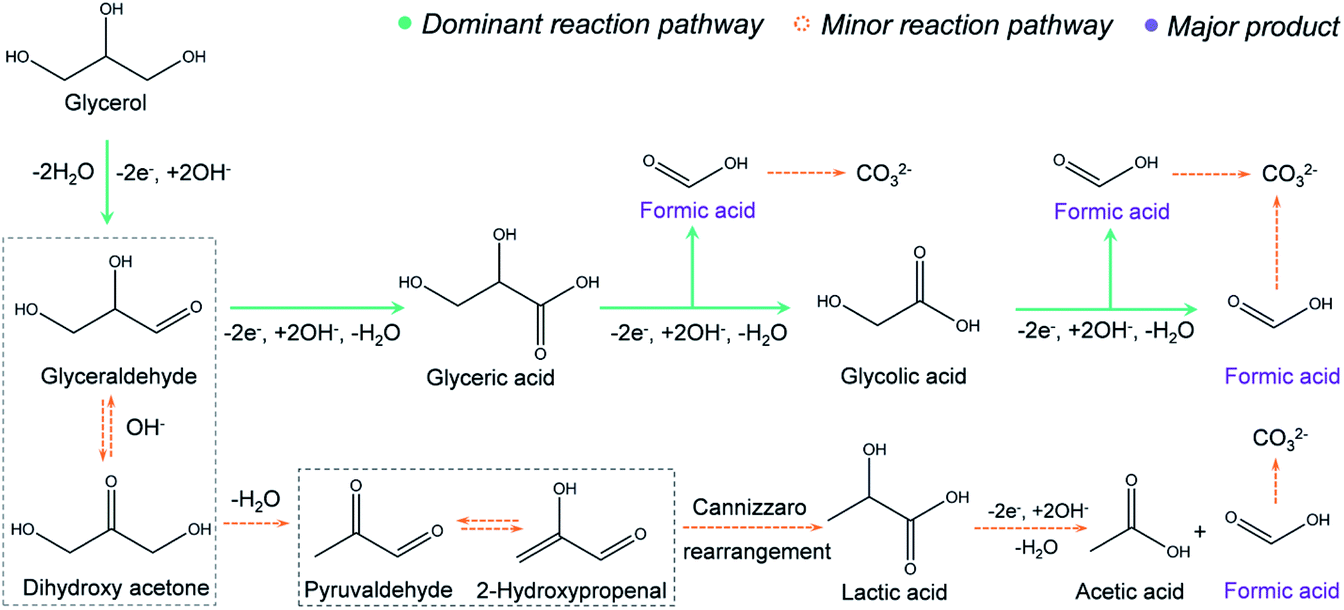 | ||
| Fig. 3 Proposed reaction pathway of glycerol electro-oxidation on N-CoOx. The solid green and dashed yellow arrows represent the major and minor reaction routes, respectively. Two-way dashed yellow arrows represent reversible processes. Formic acid was the major product (purple), while acetic acid and carbonate were minor products. Except for the unstable species that were difficult to detect under basic conditions (dashed rectangular boxes), NMR was employed to identify all intermediates and products and prove the feasibility of the proposed reaction routes. The route from glyceraldehyde to lactic acid is believed to undergo intermolecular dehydration and rearrangement under basic conditions, which does not involve electron transfer. All other routes involve electrochemical oxidation processes with related electron transfer pathways. | ||
Integrating glycerol electrooxidation with hydrogen generation
The results reported thus far are based on half-cell systems and demonstrate how well N-CoOx facilitates the conversion of biomass-derived alcohols. Aside from its critical role in oxidizing biomass-derived alcohols, N-CoOx also performs effectively as an HER catalyst under basic conditions. As displayed in Fig. S15a, ESI,† N-CoOx achieves an overpotential of 0.265 V at 10 mA cm−2, whereas Co3O4 requires 0.423 V of overpotential to reach 10 mA cm−2. Likewise, N-CoOx's 147 mV dec−1 Tafel slope implies N-CoOx's fast kinetics towards the HER (Fig. S16b, ESI†). To understand the catalyst's overall water splitting performance, N-CoOx was simultaneously used as an anode and cathode for water electrolysis in a two-electrode system (Fig. S16a and 18a, ESI†). A high cell voltage close to 1.9 V was required to reach 10 mA cm−2, which significantly contributes to OER's sluggish kinetics. Replacing the OER with glycerol oxidation dramatically reduces the cell voltage to 1.59 V (10 mA cm−2) (Fig. S17 and 18b, ESI†). O2 gas evolution was not detected at the anode upon adding 1.0 M glycerol, indicating the catalyst's preference for glycerol oxidation over the OER.To further reduce the dependence on electricity and promote renewable energy utilization, a PEC cell equipped with a Pt-coated silicon nanowire (Si NW-Pt) photocathode was coupled with the N-CoOx anode. The schematic diagram in Fig. 4a and digital photos in Fig. 4b and S22, ESI† for the hybrid system display how the Si NW-Pt photocathode may drive glycerol oxidation and the HER simultaneously under 1 sun illumination with a limited external bias. In this work, we utilized our previously reported method to fabricate Si NW-Pt photocathodes.37 SEM images and corresponding EDX elemental mapping of the Si NW-Pt photocathode are demonstrated in Fig. S18 and S20, ESI,† where the silicon nanowires are ∼2 µm long and Pt nanoparticles are uniformly presented across the surface of the silicon nanowires. Upon evaluating the HER performance of the Si NW-Pt photocathode under both basic (1.0 M KOH) and acidic (0.5 M H2SO4) conditions, the photocathode showed an onset potential of 0.25 V vs. RHE in both electrolytes (Fig. S21, ESI†). Si NW-Pt was then coupled with the N-CoOx anode (N-CoOx‖Si NW-Pt) into a two-electrode system for simultaneous glycerol oxidation and PEC hydrogen evolution. As shown in Fig. 4c, before adding glycerol, the N-CoOx‖Si NW-Pt hybrid system required a cell voltage of 1.34 V to reach 10 mA cm−2, which is much lower than the energy required in the N-CoOx‖N-CoOx system (1.81 V, no glycerol). This voltage was further reduced to 1.15 V after adding 1.0 M glycerol, much lower than that of the N-CoOx‖N-CoOx system (1.59 V, with glycerol). To the best of our knowledge, it is the first time that PEC hydrogen evolution has been coupled with glycerol oxidation, which demonstrates great success in saving the overall cell voltage (Fig. 4d and Table S2, ESI†). The cell voltage reported here is at least ∼0.2 V less compared with previous systems (Fig. 4d and Table S2, ESI†). Overall, our strategy has the potential to save at least 0.4 V in cell voltage compared to the conventional water splitting configurations, reducing ∼30% electricity consumption.
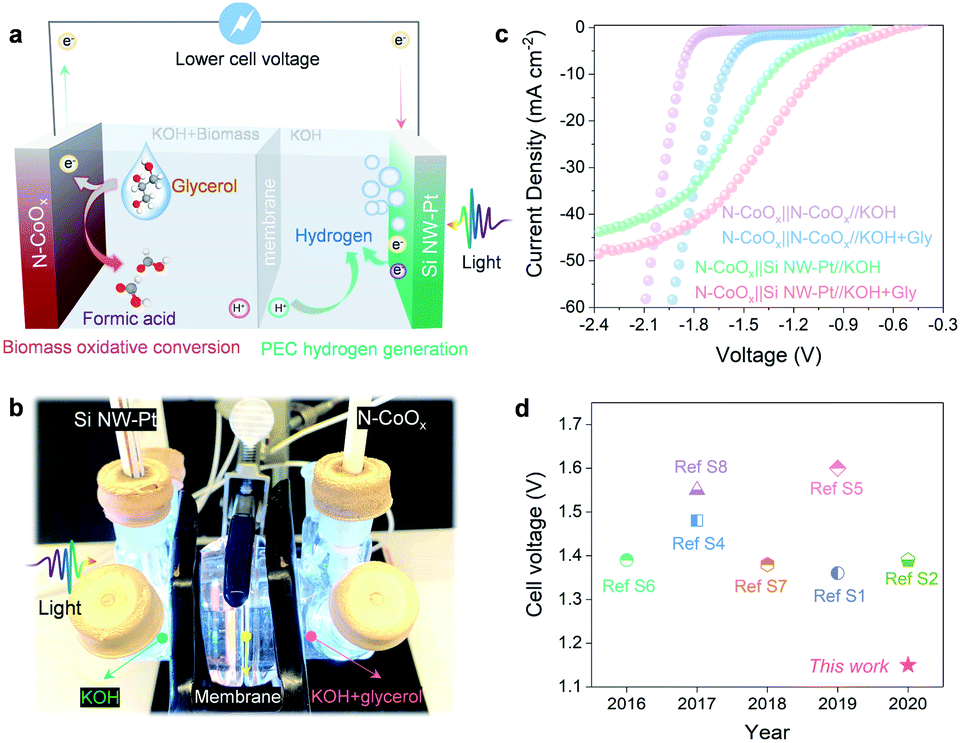 | ||
| Fig. 4 The N-CoOx‖Si NW-Pt hybrid system. (a) Schematic diagram and (b) digital photo of the N-CoOx‖Si NW-Pt hybrid system. (c) Cathodic current–voltage curves of the hybrid photoelectrochemical system. (d) A comparison of cell voltages in recently reported hybrid systems. | ||
Conclusion
In summary, we demonstrated an inexpensive N-CoOx electrocatalyst that could efficiently oxidize glycerol (a cheap byproduct of biodiesel production) and biomass-derived ethylene glycol. N-CoOx successfully converts glycerol into high-value formic acid at a low potential of 1.31 V (10 mA cm−2), which is 0.27 V less than the potential required to drive the OER. Additionally, selective conversion of glycerol to formic acid achieved FEs up to 96.2%. The catalyst also exhibited high activity for oxidizing ethylene glycol (1.35 V at 10 mA cm−2) to formic acid with 62.3% FE. Coupling the N-CoOx anode with a Si NW-Pt photocathode in a two-electrode PEC cell further reduced the cell voltage (only need 1.15 V to produce 10 mA cm−2) required to concurrently upgrade glycerol to formic acid and generate hydrogen fuel. These remarkable results revealed that glycerol electrooxidation would be a promising alternative to the traditional energy intensive OER process due to low electricity consumption. Besides, harvesting solar energy further reduced the system's dependence on electricity. More importantly, utilizing glycerol electrooxidation to replace the OER will generate significant economic profits relative to typical water electrolysis. As a consequence, solar-assisted co-electrolysis of water and high-volume chemicals may be an attractive pathway to achieve sustainable co-production of hydrogen energy and high-value chemicals due to improved techno-economics. This work provides a solid foundation to design devices for concurrent fuel generation and biomass upgradation in the future.Experimental
Materials
Bis(N,N′-di-i-propylacetamidinato) cobalt(II) (C16H34CoN4, 99.99%) was purchased from STREM. Cobalt(II) nitrate hexahydrate (Co (NO3)2·6H2O, 98%) and gallium indium alloy were purchased from Alfa Aesar. Ammonium chloride (NH4Cl, 99%), potassium hexachloroplatinate(IV) (K2PtCl6, 98%), glycerol (C3H8O3, 99%), formic acid (CH2O2, 98%), ethylene glycol (C2H6O2, 99%), and deuterium oxide (D2O, 99.9%) were purchased from Sigma-Aldrich. Glycolic acid (C2H4O3, 98%, TCI) and glyceric acid (C3H6O4, 98%) were purchased from TCI. The silicon wafer (p-type, boron doping) was obtained from WAFERPRO. Silver paste was from Ted Pella Inc. Copper wire was purchased from Fisher Scientific. Toray carbon fiber paper (TGP-H-60) was obtained from Advance Instruments. Deionized water (DIW) was used in all experiments. All chemicals and materials were directly used without further purification and treatment.Synthetic procedures
The N-CoOx was deposited using a GEM Star XT Atomic Layer Deposition System. In a typical process, a nitrogen-containing cobalt-based molecular compound (bis(N,N′-di-i-propylacetamidinato) cobalt(II)) was used as the cobalt and nitrogen precursor. Ozone and helium were employed as the oxidizer and carrier gas, respectively. The cobalt precursor was heated to 85 °C and the manifold was maintained at 115 °C to avoid condensation of the precursor in the gas lines. The temperature in the deposition chamber was 150 °C. Nitrogen doped CoOx (N-CoOx) was uniformly deposited on carbon fiber paper after 300 ALD cycles.Co3O4 nanosheets on carbon fiber paper were fabricated by the electrodeposition method. Typically, α-Co(OH)2 was first electrodeposited on carbon fiber paper in 100 ml of 0.02 M Co(NO3)2·6H2O solution with 0.1 M NH4Cl at −2.0 mA for 10 min. Then, Co3O4 was obtained by annealing α-Co (OH)2 nanosheets in air under 250 °C for 2 h. All electrodeposition experiments were conducted via a two-electrode system where the carbon fiber paper and graphite rod were used as the working and counter electrodes, respectively.
Si NW-Pt photoelectrode fabrication: a metal-assisted chemical etching strategy was employed to synthesize silicon nanowires (Si NW). Typically, the back side of a silicon wafer was firstly covered by polyimide to avoid being etched, and was then sonicated in acetone and DI water for 15 min to remove impurities. Further, the wafer was soaked in 5% HF for 90 s to etch the silicon oxide layer. Then, the washed clean wafer was soaked in 10% HF solution with 0.02 M AgNO3 for 60 s to deposit silver particles on the surface. Subsequently, the wafer was soaked into a mixture of 10% HF and 30% H2O2 (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, vol%) for 10 min and then into 35% HNO3 for 15 min to remove residual Ag. Finally, the wafer was used to fabricate the photoelectrode according to the following steps: (1) the silicon backside was cleaned with 5 wt% HF for 30 s; (2) ohmic contact was formed by coating the Ga–In eutectic on the backside; (3) the backside was fixed on a conductive copper coil with conductive silver paint; (4) the copper coil tail was passed through a glass tube for electric contact. (5) The photoelectrode assembly was encapsulated by coating Loctite 9462 epoxy and dried at room temperature overnight. For platinum deposition, the obtained photoelectrode was firstly cleaned in 5 wt% HF for 30 s and then soaked into 1 wt% HF solution with 5 mM K2PtCl6 for 60 s.
:1, vol%) for 10 min and then into 35% HNO3 for 15 min to remove residual Ag. Finally, the wafer was used to fabricate the photoelectrode according to the following steps: (1) the silicon backside was cleaned with 5 wt% HF for 30 s; (2) ohmic contact was formed by coating the Ga–In eutectic on the backside; (3) the backside was fixed on a conductive copper coil with conductive silver paint; (4) the copper coil tail was passed through a glass tube for electric contact. (5) The photoelectrode assembly was encapsulated by coating Loctite 9462 epoxy and dried at room temperature overnight. For platinum deposition, the obtained photoelectrode was firstly cleaned in 5 wt% HF for 30 s and then soaked into 1 wt% HF solution with 5 mM K2PtCl6 for 60 s.
Material characterization
The X-ray diffraction (XRD) pattern was collected on a PANalytical X' Pert with Cu-Kα radiation (λ = 1.5418 Å). X-ray photoelectron spectroscopy (XPS) measurements were performed using the Kratos AXIS Supra equipped with a monochromatic Al Kα X-ray source, running at a power of 300 W, and operating at 15 kV. All samples were measured with a 300 × 700 µm2 spot size and operating chamber pressure < 10−8 torr. XPS spectra were fitted with XPS peak software to analyse the atomic compositions and the possible chemical species. The binding energy scale was calibrated using the C 1s peak at 284.8 eV. Electron paramagnetic resonance (EPR) measurements were performed on a Magnettech MS 5000. Scanning transmission electron microscopy (STEM) was performed using the JEOL Grand and high-angle angular dark-filled (HAADF)-STEM images were acquired at a convergence semiangle of 22 mrad and inner and outer collection angles of 83 and 165 mrad, respectively. Energy dispersive X-ray spectroscopy (EDS) was conducted using JEOL dual EDS detectors and a specific high count analytical TEM holder. For TEM sample preparation, the copper TEM grid was directly put into an ALD chamber for N-CoOx deposition.Photo-/electro-chemical measurements
All electrochemical measurements were carried on CHI 600E and 660E workstations. We initially evaluated the performance of our samples for the HER, OER, glycerol oxidation, and ethylene glycol oxidation in a typical three-electrode system where the graphite rod and Ag/AgCl were used as the counter and reference electrodes, respectively. Then the N-CoOx anode and Si NW-Pt photocathode were coupled in a two-electrode system to investigate hybrid glycerol electrolysis and hydrogen evolution. A Nafion film was used to separate the oxidation and reduction reactions. A steady DC-powered 150 W Xe-arc lamp (Newport) was used as a simulated sunlight source. A water filter was used to block the infrared irradiation. The incident light intensity was calibrated to 100 mW cm−2 (one sun) using a Si photodiode. All LSV curves were collected at a scan rate of 5 mV s−1 and without IR compensation. All the electrochemical measurements were performed in N2-saturated electrolyte. The potentials were calibrated to the reversible hydrogen electrode (RHE) scale according to the equation: ERHE = EAg/AgCl + 0.059 pH + 0.197 V.Product quantification
A long-term electrolysis experiment was employed to determine products of glycerol oxidation and ethylene glycol oxidation. Electrolytes after electrolysis were collected and analysed using a nuclear magnetic resonance (NMR) spectrometer. All liquids to be tested contained 540 µl electrolyte and 60 µl D2O and were measured by 1H and 13C NMR spectra. 1H NMR and 13C NMR spectra were recorded on Varian spectrometers at 400 MHz. Formic acid standard was prepared by dissolving formic acid (15 mM for 1H NMR and 60 mM for 13C NMR) in 1.0 M KOH aqueous solution, and 1H and 13C NMR spectra of the standard were collected under the same conditions as the solution after biomass-polyol electrolysis. The gaseous products produced from the cathode after electrolysis were collected and analysed by gas chromatography (GC-2014C, SHIMADZU). According to the proposed reaction pathway, the conversion from glycerol to formic acid could be described by the following equation (eqn (1)).| C3H8O3 (glycerol) − 8e− + 8OH− − 5H2O = 3CH2O2 (formic acid) | (1) |
Hence, the faradaic efficiency of formic acid for glycerol oxidation can be determined by eqn (2),
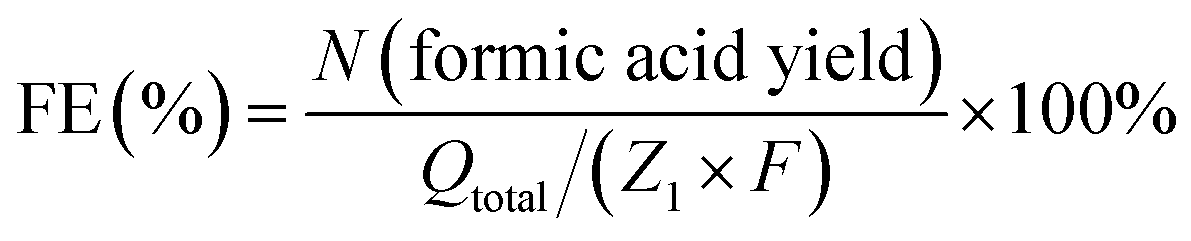 | (2) |
| C2H6O2 (ethylene glycol) − 6e− + 6OH− − 4H2O = 2CH2O2 | (3) |
Similarly, the faradaic efficiency of formic acid for ethylene glycol oxidation was described by eqn (4),
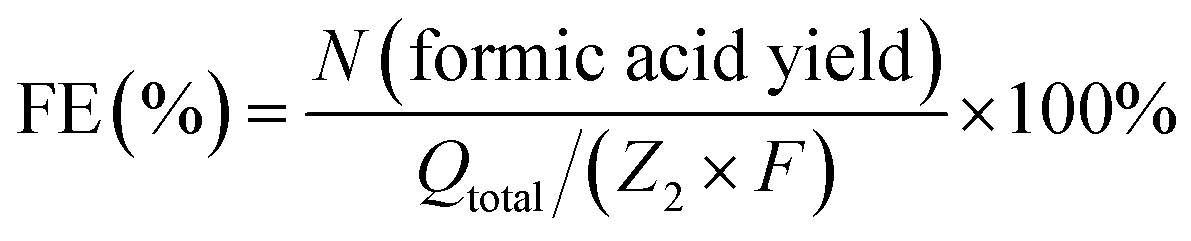 | (4) |
Author contributions
J. G. and X. X. lead the project. Z. K. designed and performed experiments. W. N. assisted with product analysis by NMR. X. Y. and X. P. conducted the STEM characterization. D. H. and X. S. helped with mechanism studies for this research. S.Y. helped with some electrochemical measurements and involved into writing this paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Jing Gu acknowledges San Diego State University (SDSU) start-up funds and NSF award (CEBT-1704992) to support this research. We gratefully acknowledge the UC Irvine Materials Research Institute (IMRI) for helping with STEM and XPS characterization. IMRI was funded in part by the National Science Foundation Major Research Instrumentation Program under grant no. CHE-1338173.References
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- A. Landman, H. Dotan, G. E. Shter, M. Wullenkord, A. Houaijia, A. Maljusch, G. S. Grader and A. Rothschild, Nat. Mater., 2017, 16, 646–651 CrossRef CAS PubMed.
- M. F. Lagadec and A. Grimaud, Nat. Mater., 2020, 19, 1140–1150 CrossRef CAS PubMed.
- Hydrogen Production Cost, https://www.sciencedirect.com/topics/engineering/hydrogen-production-cost, accessed Search PubMed.
- N. v. Hulst, The Clean Hydrogen Future has Already Begun, https://www.iea.org/commentaries/the-clean-hydrogen-future-has-already-begun, accessed Search PubMed.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 1–13 CrossRef.
- J. Wang, W. Cui, Q. Liu, Z. Xing, A. M. Asiri and X. Sun, Adv. Mater., 2016, 28, 215–230 CrossRef CAS PubMed.
- W. Wang, Y. B. Zhu, Q. Wen, Y. Wang, J. Xia, C. Li, M. W. Chen, Y. Liu, H. Li, H. A. Wu and T. Zhai, Adv. Mater., 2019, 31, e1900528 CrossRef PubMed.
- B. Zhu, Z. Liang and R. Zou, Small, 2020, 16, e1906133 CrossRef PubMed.
- Y. Liu, J. Zhang, Y. Li, Q. Qian, Z. Li, Y. Zhu and G. Zhang, Nat. Commun., 2020, 11, 1853 CrossRef CAS PubMed.
- S. Verma, S. Lu and P. J. A. Kenis, Nat. Energy, 2019, 4, 466–474 CrossRef CAS.
- R. Ciriminna, C. D. Pina, M. Rossi and M. Pagliaro, Eur. J. Lipid Sci. Technol., 2014, 116, 1432–1439 CrossRef CAS.
- N. Han, Y. Wang, H. Yang, J. Deng, J. Wu, Y. Li and Y. Li, Nat. Commun., 2018, 9, 1320 CrossRef PubMed.
- A. A. N. Afshar, Chemical Profile: Formic Acid, http://chemplan.biz/chemplan_demo/sample_reports/Formic_Acid_Profile.pdf, accessed Search PubMed.
- Formic Acid Prices, https://www.intratec.us/chemical-markets/formic-acid-price, accessed Search PubMed.
- D. A. Bulushev and J. R. H. Ross, ChemSusChem, 2018, 11, 821–836 CrossRef CAS PubMed.
- Z. Zhang, L. Xin and W. Li, Appl. Catal., B, 2012, 119–120, 40–48 CrossRef CAS.
- J. Qi, L. Xin, D. J. Chadderdon, Y. Qiu, Y. Jiang, N. Benipal, C. Liang and W. Li, Appl. Catal., B, 2014, 154–155, 360–368 CrossRef CAS.
- M. Simões, S. Baranton and C. Coutanceau, Appl. Catal., B, 2010, 93, 354–362 CrossRef.
- H. J. Kim, S. M. Choi, M. H. Seo, S. Green, G. W. Huber and W. B. Kim, Electrochem. Commun., 2011, 13, 890–893 CrossRef CAS.
- S. Lee, H. J. Kim, E. J. Lim, Y. Kim, Y. Noh, G. W. Huber and W. B. Kim, Green Chem., 2016, 18, 2877–2887 RSC.
- A. Zalineeva, A. Serov, M. Padilla, U. Martinez, K. Artyushkova, S. Baranton, C. Coutanceau and P. B. Atanassov, J. Am. Chem. Soc., 2014, 136, 3937–3945 CrossRef CAS PubMed.
- M. Simoes, S. Baranton and C. Coutanceau, ChemSusChem, 2012, 5, 2106–2124 CrossRef CAS PubMed.
- S. Wang, Y. Wang, S. L. Zhang, S. Q. Zang and X. W. D. Lou, Adv. Mater., 2019, 31, e1903404 CrossRef PubMed.
- Q. Yu, C. Liu, X. Li, C. Wang, X. Wang, H. Cao, M. Zhao, G. Wu, W. Su, T. Ma, J. Zhang, H. Bao, J. Wang, B. Ding, M. He, Y. Yamauchi and X. S. Zhao, Appl. Catal., B, 2020, 269, 118757 CrossRef CAS.
- Z. Wang, W. Xu, X. Chen, Y. Peng, Y. Song, C. Lv, H. Liu, J. Sun, D. Yuan, X. Li, X. Guo, D. Yang and L. Zhang, Adv. Funct. Mater., 2019, 29, 1902875 CrossRef.
- D. He, X. Song, W. Li, C. Tang, J. Liu, Z. Ke, C. Jiang and X. Xiao, Angew. Chem., Int. Ed., 2020, 59, 6929–6935 CrossRef CAS PubMed.
- M. Yu, Z. Wang, C. Hou, Z. Wang, C. Liang, C. Zhao, Y. Tong, X. Lu and S. Yang, Adv. Mater., 2017, 29, 1602868 CrossRef PubMed.
- Z. Chen, Y. Song, J. Cai, X. Zheng, D. Han, Y. Wu, Y. Zang, S. Niu, Y. Liu, J. Zhu, X. Liu and G. Wang, Angew. Chem., Int. Ed., 2018, 57, 5076–5080 CrossRef CAS PubMed.
- C. Yin, Y. Liu, Q. Xia, S. Kang, X. Li, Y. Wang and L. Cui, J. Colloid Interface Sci., 2019, 553, 427–435 CrossRef CAS PubMed.
- Y. Li, X. Wei, L. Chen, J. Shi and M. He, Nat. Commun., 2019, 10, 5335 CrossRef PubMed.
- X. Han, H. Sheng, C. Yu, T. W. Walker, G. W. Huber, J. Qiu and S. Jin, ACS Catal., 2020, 10, 6741–6752 CrossRef CAS.
- M. H. Haider, N. F. Dummer, D. W. Knight, R. L. Jenkins, M. Howard, J. Moulijn, S. H. Taylor and G. J. Hutchings, Nat. Chem., 2015, 7, 1028–1032 CrossRef CAS PubMed.
- C. Dai, L. Sun, H. Liao, B. Khezri, R. D. Webster, A. C. Fisher and Z. J. Xu, J. Catal., 2017, 356, 14–21 CrossRef CAS.
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. Della Pina, Angew. Chem., Int. Ed., 2007, 46, 4434–4440 CrossRef CAS PubMed.
- F. Yang, J. A. Aguiar, M. Fairchild, W. Vakki, S. Younan, Y. Zhou, L. Zhuo and J. Gu, Adv. Mater. Interfaces, 2019, 6, 1802085 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary SEM, XRD, TEM, XPS characterizations, and related electrochemical data. See DOI: 10.1039/d1ta02654b |
| This journal is © The Royal Society of Chemistry 2021 |