
Controlling the Co–S coordination environment in Co-doped WS2 nanosheets for electrochemical oxygen reduction†
Wei
Hong‡
 ,
Erika
Meza‡
and
Christina W.
Li
*
,
Erika
Meza‡
and
Christina W.
Li
*
Department of Chemistry, Purdue University, West Lafayette, IN 47907, USA. E-mail: christinawli@purdue.edu
First published on 28th May 2021
Abstract
Cobalt sulfide nanomaterials are among the most active and stable catalysts for the electrocatalytic oxygen reduction reaction in pH 7 electrolyte. However, due to the complexity and dynamism of the catalytic surfaces in cobalt sulfide bulk materials, it is challenging to identify and tune the active site structure in order to achieve low overpotential oxygen reduction reactivity. In this work, we synthesize isolated Co sites supported on colloidal WS2 nanosheets and develop a synthetic strategy to rationally control the first-shell coordination environment surrounding the adsorbed Co active sites. By studying Co–WS2 materials with a range of Co–S coordination numbers, we are able to identify the optimal active site for pH 7 oxygen reduction catalysis, which comprises cobalt atoms bound to the WS2 support with a Co–S coordination number of 3–4. The optimized Co–WS2 material exhibits an oxygen reduction onset potential of 0.798 V vs. RHE, which is comparable to the most active bulk phases of cobalt sulfide in neutral electrolyte conditions.
Introduction
Recent interest in coupling fuel cells and electrolyzers to biological systems has led to an effort to develop electrocatalysts that operate efficiently under biologically-compatible conditions: pH 7 aqueous buffer and ambient pressure and temperature.1–9 The oxygen reduction reaction (ORR) in neutral media presents catalytic challenges that are distinct from those present in acidic and alkaline electrolytes. The state-of-the-art catalysts in acidic and alkaline media, supported noble metal nanoparticles and metal oxide nanostructures, are susceptible to deactivation in neutral electrolyte due to surface poisoning and catalyst dissolution, respectively.10–12First-row transition metal sulfides have emerged as alternatives to Pt-based catalysts in ORR under a wide range of pH conditions due to their high activity, stability, and low cost.9,13–18 Literature studies have focused primarily on tuning the structure of nickel and cobalt sulfides to understand the role that metal sulfide phase and composition play in dictating ORR reactivity.19–25 The Co9S8 phase, in particular, has been identified as uniquely active amongst the first-row transition metal sulfides for alkaline and neutral ORR and has been studied in a wide range of composite nanostructures.26–33 Computational studies have postulated that the M–S coordination environment at the catalytic surface influences oxygen adsorbate binding energies and thus ORR catalytic turnover.34,35 However, when experimentally altering the crystal structure or composition of a bulk metal sulfide phase, it is challenging to isolate the role that coordination environment plays amidst the multiple geometries and oxidation states that exist within any given phase as well as the dynamic nature of the surface under electrocatalytic conditions.35,36
Supported single atom catalysts (SACs) are intriguing model systems for catalytic surface sites on bulk materials because their active site structure can be studied at the atomic level.37–39 Previous work on single metal atoms supported on metal oxide and heteroatom-doped carbon materials has shown the importance of local coordination environment and metal–support interaction in influencing catalyst selectivity and reactivity.40–44 Particular effort has been invested in active site characterization for metal single atoms supported on N-doped carbon.45–49 Recent work on Co SACs have postulated that the number and chemical nature of nitrogen atoms in the first coordination shell may have an impact on catalytic activity in the alkaline oxygen reduction reaction.50,51 While different single atom coordination environments have been observed in these examples, it remains challenging to synthetically control the coordination environment surrounding a single atom catalyst. Single atoms supported on MoS2 have also been studied extensively, primarily for use as electrocatalysts in the hydrogen evolution reaction (HER).52 However, doped first-row transition metal single atoms in MoS2 are not themselves catalytically active sites for HER but rather serve to tune the electronic and catalytic properties of the MoS2 surface.53–55 In this work, we put forward a synthetic strategy to generate cobalt single atoms supported on WS2 nanosheets, in which the Co–S coordination environment can be explicitly tuned through controlled introduction of excess sulfur. These Co–WS2 nanosheets provide the basis for a systematic study on how the local coordination environment of surface cobalt sulfide active sites influence oxygen reduction reactivity in neutral electrolyte.
Experimental methods
Synthesis of colloidal Co-doped WS2 nanosheets
Colloidal 1T-WS2 nanosheets capped with oleylamine were synthesized based on a previously reported method.56,57 To the dried WS2 nanosheets (26.2 mg, 0.007 mmol), a 0.1 M solution of n-BuLi in hexanes (10 mL) was added under inert atmosphere and stirred for 2 hours. The Li-treated nanosheets (Li–WS2) were then rinsed with excess hexanes and redispersed in NMF (1 mL). To a 1 mL solution of Li–WS2, a Co precursor solution (30 μL or 100 μL of 70 mM CoCl2 in NMF) was then added and stirred for 24 hours at room temperature under nitrogen. After the Co functionalization step, the nanosheets were precipitated with ethanol (10 mL) and hexanes (20 mL) followed by centrifugation at 8700 rpm for 10 minutes. The supernatant was decanted and the cleaning step was repeated one time.K2S impregnation and annealing treatment for Co–WS2
The colloidal Co–WS2 nanosheets were first supported on carbon black by mixing 1 mL of a 7 mM solution of Co–WS2 nanosheets with 11.6 mg carbon in 4 mL ethanol at room temperature. The supported Co–WS2 samples were centrifuged at 8700 rpm for 10 min and resuspended in 2 mL of ethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :IPA (v/v; 1:3) solution. Next, varying amounts of K2S solution (70 mM K2S in EtOH) were added, ranging from 0.5 eq. to 10 eq. with respect to Co. The mixture was left stirring in air at room temperature for 24–36 hours until the solvent fully evaporated. The dried powder was then annealed at 300 °C for 2 hours under N2. After annealing, the sample was successively rinsed with ethanol, water, and IPA to remove unincorporated K2S.
:IPA (v/v; 1:3) solution. Next, varying amounts of K2S solution (70 mM K2S in EtOH) were added, ranging from 0.5 eq. to 10 eq. with respect to Co. The mixture was left stirring in air at room temperature for 24–36 hours until the solvent fully evaporated. The dried powder was then annealed at 300 °C for 2 hours under N2. After annealing, the sample was successively rinsed with ethanol, water, and IPA to remove unincorporated K2S.
Scanning/transmission electron microscopy
Low-resolution TEM images were acquired using an FEI Tecnai T20 TEM equipped with a 200 kV LaB6 filament. Low-resolution HAADF-STEM imaging and EDS mapping were obtained on an FEI Talos F200X S/TEM with a 200 kV X-FEG field-emission source and a super X-EDS system. High-resolution HAADF-STEM images were collected using a Thermo Scientific Themis Z, a spherical aberration corrected S/TEM with a 300 kV X-FEG field-emission source. Associated EDS spectra were obtained with a quad-silicon FEI Super X drift detector.In order to obtain atomic-resolution HAADF-STEM images, it is critical to remove all organic ligand and solvent residue from the TEM sample. Colloidal Co–WS2 samples for HR-STEM were cleaned four additional times with ethanol and hexanes prior to drop casting onto a Au grid coated with an ultrathin carbon film. The Au grid was then submerged in a suspension of carbon black in hexanes, and N2 gas was bubbled into the solution for 1 min to agitate the suspension.58 The TEM grid was air-dried and stored under inert atmosphere prior to imaging.
X-ray absorption spectroscopy
X-ray absorption spectroscopy experiments were carried out at the 10-ID and 10-BM beamlines at the Advanced Photon Source at Argonne National Laboratory. Samples were pressed into a self-supporting pellet, and spectra were obtained in transmission mode at room temperature. The Demeter software package was used to analyze the collected data.59 Data was collected at the Co K-edge (7.7089 keV) using metallic Co foil for energy calibration. EXAFS coordination parameters were obtained by a least-squares fit in R-space of the k2-weighted Fourier transform data from 2.5 to 8.5 Å−1 for Co–WS2 samples and 2.5 to 9.0 Å−1 for control samples. A Co foil reference sample was first fit to its known crystallographic parameters to obtain an amplitude reduction factor (S02) for the Co K-edge. EXAFS fitting of the first coordination shell was carried out between 1.1 and 2.3 Å in R-space. Fittings were done by refining bond distances (R), coordination numbers (CN) and energy shift (E0). The Debye–Waller factor (σ2) was kept constant for each sample.Electrochemical measurements
Electrochemical experiments were conducted on a Pine WaveDriver 20 bipotentiostat. A catalyst ink was obtained by sonicating the catalyst powder in a solution containing 75.6% water, 24% IPA, and 0.4% Nafion (v/v) to obtain a nominal concentration of 7 mM based on Co–WS2. The working electrode was prepared by drop casting 10 μL of the catalyst ink onto a polished glassy carbon electrode with 5 mm diameter. The catalyst film was dried in air for 25 min at a rotation speed of 700 rpm. The counter electrode was a graphite rod. The electrolyte used for electrochemical experiments was 1.0 M sodium phosphate buffer solution (PBS, pH 7) or 0.1 M KOH (pH 13). Currents are reported with anodic current as positive and cathodic current as negative. Potentials were measured against a Ag/AgCl reference electrode (3 M NaCl) and converted to the RHE reference scale using:| E (vs. RHE) = E (vs. Ag/AgCl) + 0.210 V + 0.0591 V × pH |
Cyclic voltammetry (CV) scans for Co–WS2 samples were performed in N2-saturated 0.1 M KOH electrolyte with a scan rate of 50 mV s−1. Oxygen reduction reaction voltammetry was carried out using a rotating ring-disk electrode in a single-compartment glass cell containing 150 mL of 1.0 M PBS electrolyte. The solution was purged with O2 for at least 30 min prior to the start of the experiment. For all ORR experiments, a rotation rate of 1600 rpm was utilized. One cathodic linear sweep voltammetry scan was collected at 10 mV s−1 prior to the reported anodic LSV, obtained by scanning at 1 mV s−1 from 0.0 V to 0.9 V vs. RHE. The onset potential (Eonset) for ORR is defined to be the potential at which the ORR current density reaches −0.1 mA cm−2. All Eonset values are obtained from an average of three sample runs to account for variability in the electrode drop-drying process, and the error bar reflects the standard deviation (Fig. S11 and S12†). The Pt ring in the rotating-ring disk electrode was held at 1.26 V vs. RHE during ORR linear sweep voltammetry.
Uncompensated resistances were measured for a set of Co–WS2 samples, which ranged from 20–36 Ω in 1.0 M PBS electrolyte. Catalytic measurements are reported without iR compensation. To assess the stability of Co–WS2 catalysts in ORR, galvanostatic electrolysis was performed at −0.25 mA cm−2 in the standard electrolyte (1 M PBS) as well as with added methanol (1 M MeOH + 1 M PBS). Accelerated CV scanning was performed in a high-purity 1.0 M sodium phosphate electrolyte (pH 7, 99.9%). During the CV stability test, anodic LSV scans at 1 mV s−1 were obtained in the 1st, 5th, 20th, and 200th scan while all intervening CV scans were collected at 200 mV s−1 (Fig. S16†).
Results and discussion
Synthesis
The synthetic strategy to generate Co single atoms with tunable Co–S coordination environment, depicted in Scheme 1, is based on previous work from our group on the synthesis of single atom Ni-doped WS2 nanosheets.57 Colloidal WS2 nanosheets were synthesized via a literature method and activated using n-butyllithium (n-BuLi) to generate dangling sulfide defects on the basal planes of WS2 (Li–WS2).56 A dilute solution of CoCl2 in N-methylformamide (NMF) was then introduced into the colloidal Li–WS2 nanosheets under an inert atmosphere and permitted to passively adsorb over 24 hours. Any excess CoCl2 remaining in solution was readily removed through precipitation and centrifugation of the Co-doped WS2 nanosheets (colloidal Co–WS2). Two loadings of Co on WS2 were synthesized in order to vary the degree of dopant clustering and aggregation – a low loading (0.3 equiv.) to access more isolated Co atoms and a high loading (1.0 equiv.) to generate larger Co aggregates.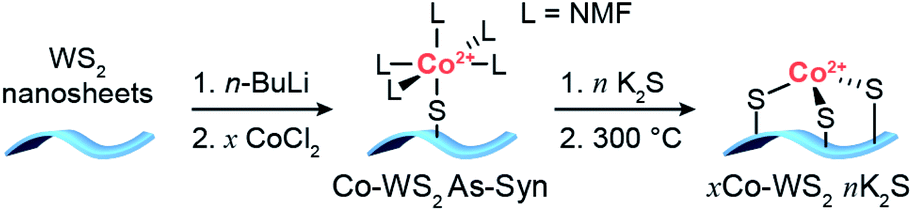 | ||
| Scheme 1 Synthetic strategy for the deposition of Co single atoms onto colloidal WS2 nanosheets followed by controlled sulfidation and annealing steps. | ||
In our previous work, we showed that Ni dopants adsorbed relatively weakly and in a monodentate fashion to the WS2 surface during the colloidal synthesis. In order to tune the Co–S coordination and more strongly anchor the Co dopants to the WS2 surface, we develop a method herein for controlled sulfidation of the adsorbed Co atoms. The colloidal Co–WS2 nanosheets are supported on carbon black (xCo–WS2 As-Syn, where x refers to the nominal Co loading) and impregnated with a variable amount of potassium sulfide, ranging from 0.5 to 10 equivalents with respect to the adsorbed Co atoms. The impregnated samples are then annealed at 300 °C under an inert atmosphere and rinsed repeatedly with solvent to remove unreacted sulfides (xCo–WS2nK2S).
Scanning transmission electron microscopy
We utilize high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) coupled to energy-dispersive X-ray spectroscopy (EDS) to characterize the morphology and elemental distribution of Co and W on the Co–WS2 nanosheets. Colloidal WS2 comprises circular nanosheets with ∼100 nm diameter and 10–14 layer thickness (Fig. S1a†).57 After n-BuLi activation and Co adsorption, the nanosheet morphology is retained, and Co atoms are evenly distributed across the WS2 based on the low-resolution HAADF-STEM image and EDS maps (Fig. S1b–f†). Based on EDS, Co loadings on the WS2 nanosheets follow closely with the amount introduced in solution, ranging from 0.2 up to 1.0 equivalents with respect to W (Table S1†). At high Co loading, we anticipate that a significant fraction of Co species may be loosely intercalated or physisorbed within the WS2 layers rather than coordinatively bound to the surface.High-resolution HAADF-STEM imaging allows us to confirm the presence of Co single atoms in the low loading 0.3Co–WS2 colloidal nanosheets in two surface adsorption sites (Fig. 1a). Due to the n-BuLi treatment, the underlying WS2 nanosheets exhibit variable thickness, ranging from monolayer to multilayer, and regions where W vacancies and structural disorder are present (Fig. 1b and S2a†). In the monolayer regions, the ordered hexagonal lattice of W atoms for the WS2 nanosheet is clearly visible, and deviations to the hexagonal array in terms of both atomic position and contrast provide evidence for the presence of Co single atoms (Fig. 1c and d). Atoms interstitial to the hexagonal W array, highlighted in Fig. 1c, S2b and c,† represent Co atoms bound on-top of a S atom or in a S vacancy (S-top). In addition, analysis of annular-dark field (ADF) Z-contrast shows that Co atoms are also present on-top of W atoms in the hexagonal lattice, highlighted in Fig. 1d, S2d and e (W-top). Interestingly, Co atoms bound in W-top sites are frequently found adjacent to W vacancies in the lattice, suggesting that dangling sulfide moieties may be responsible for Co coordination in these positions.
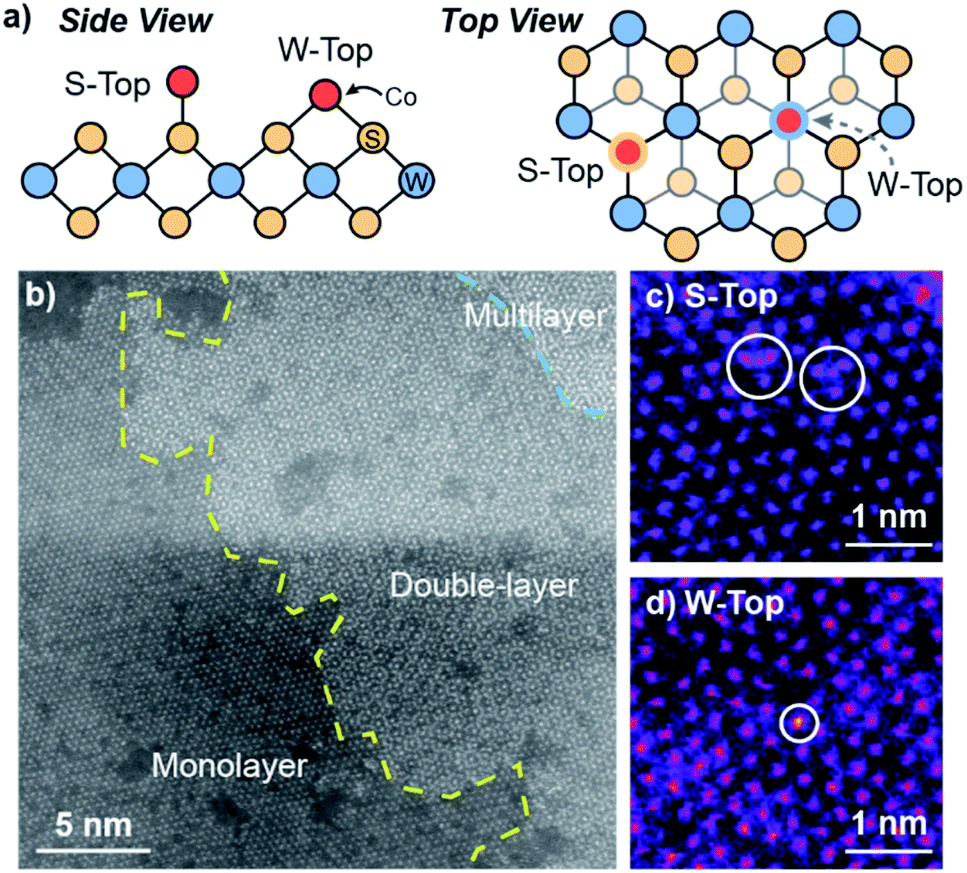 | ||
| Fig. 1 (a) Schematic depicting side and top views for S-top and W-top binding of Co atoms on WS2, (b) high-resolution STEM image of colloidal 0.3Co–WS2, (c) HR-STEM region containing S-top Co sites, and (d) HR-STEM region containing W-top sites. | ||
In addition to ADF Z-contrast analysis, we also obtained EDS spectra in small regions throughout the high-resolution STEM image to ascertain that Co atoms can be spectroscopically detected on colloidal 0.3Co–WS2. While the EDS signal is quite weak when focused on 3 × 3 nm square regions of the monolayer Co–WS2 nanosheets, we can clearly distinguish peaks associated with the W M, S Kα, and Co Kα transitions (Fig. S2f and g†). In addition, when the EDS spectra is obtained over a slightly larger region, the atomic ratio of Co:W is ∼0.3:1, similar to the elemental ratios obtained in the lower resolution EDS data.
Prior to sulfur addition, colloidal Co–WS2 nanosheets are supported on carbon black, which causes the circular nanosheets to fold onto themselves rather than lie flat on the hydrophobic carbon surface (Fig. 2a and b). Impregnation of 0.5–10 equiv. of K2S (with respect to Co) and annealing at 300 °C under N2 does not alter the overall nanosheet morphology or the average loading of Co relative to W (Fig. 2c, d, S3, S4 and Table S1†). However, we begin to see evidence of Co clustering in the STEM-EDS elemental maps after K2S annealing. In 0.3Co–WS2 samples treated with K2S, Co atoms remain colocalized with W and S and evenly distributed across the WS2 nanosheets in the lower-resolution STEM-EDS maps (Fig. 2c). Obtaining EDS spectra and quantifying the Co:W ratios at higher resolution reveals spatial variation in the Co distribution on WS2 (Fig. S5†). In 8 × 8 nm regions across the 0.3Co–WS2 3K2S sample, we observe Co:W (X:1) ratios ranging from 0.14–0.29, suggesting that Co atoms are beginning to cluster but larger cobalt sulfide nanoparticles have not yet formed on the nanosheets. At the higher loading of Co (1.0Co–WS2nK2S), cobalt sulfide nanoparticles are observed even in the low-resolution STEM-EDS maps (Fig. 2d). Aggregates of Co atoms (10–30 nm) that are not co-localized with the underlying W are clearly present.
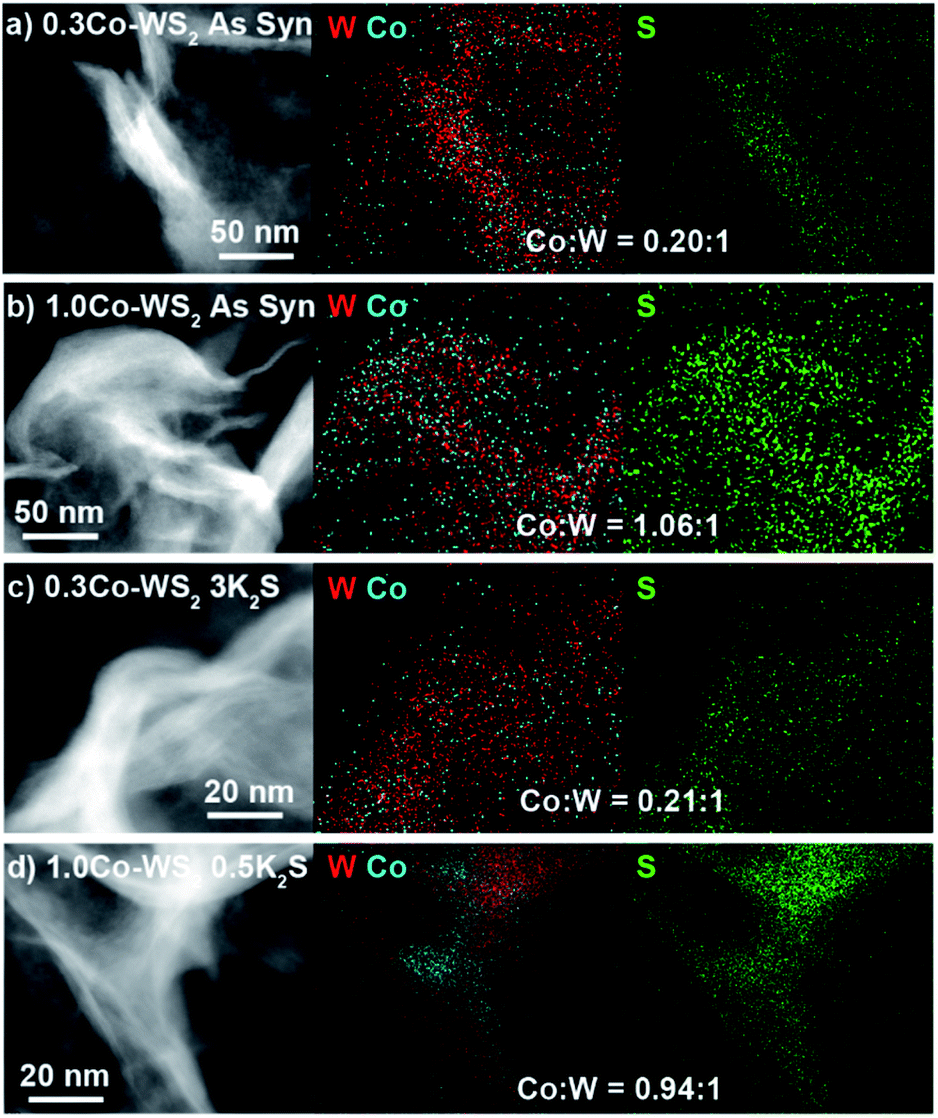 | ||
| Fig. 2 STEM-EDS image and elemental maps of W, Co, and S for (a and b) as-synthesized 0.3Co and 1.0Co–WS2 supported on C as well as (c and d) 0.3Co and 1.0Co–WS2 after annealing with K2S. | ||
Powder X-ray diffraction (XRD) patterns show the expected WS2 phase transition from 1T to 2H after thermal annealing and no Co-containing crystalline phases (Fig. S6 and S7†). In the 1.0Co–WS2nK2S samples, the CoOx or CoSx aggregates observed in the STEM-EDS maps must therefore be amorphous or comprise many small nanocrystalline domains.
Oxygen reduction reaction
We began by evaluating the oxygen reduction reactivity of the low and high loading Co–WS2 catalysts as a function of the amount of K2S incorporated during thermal treatment. The ORR catalytic activity of all samples was measured using linear scan voltammetry (LSV) on a rotating ring-disk electrode in O2-saturated 1 M phosphate buffer solution (PBS, pH = 7). Negligible ring current is detected using any of the Co–WS2 catalysts, which indicates quantitative selectivity toward the 4 e− reduction of O2 to H2O under these conditions (Fig. S13†). In order to compare the reactivity of various Co–WS2 and bulk control samples to one another, we utilize the ORR onset potential (Eonset), the potential at which the catalyst attains −0.1 mA cm−2 of ORR current density, as the primary catalytic metric due to differences in sample mass-transport properties at higher current densities (Fig. S8 and S9†).The annealed Li–WS2 nanosheets alone show poor ORR activity with Eonset of 0.575 V vs. RHE (Fig. S8†). The addition of Co to the WS2 sample at low loading (0.3Co–WS2) in the absence of K2S (0 K2S) induces a modest increase in ORR onset potential to 0.614 V vs. RHE (Fig. 3a and c). Introduction of varying equivalents of K2S to the 0.3Co–WS2 sample has a dramatic effect on ORR catalysis. Upon mild sulfidation at 0.5 equiv. K2S, the onset potential for ORR immediately increases by 54 mV to 0.668 V vs. RHE. From 0.5 to 3.0 equiv. of K2S relative to Co, a steady increase in ORR onset potential is observed with the highest Eonset of 0.798 V vs. RHE occurring at 3.0 equiv. K2S. Further increase in K2S loading causes a decay in the ORR activity, dropping back down to 0.731 V vs. RHE at 10 equiv. K2S. A modest improvement in catalytic activity is also observed when Li–WS2 alone is annealed in the presence of excess sulfur (Fig. S8 and Table S2†). However, the onset potential of Li–WS2 peaks at 0.639 V vs. RHE in the absence of doped Co atoms.
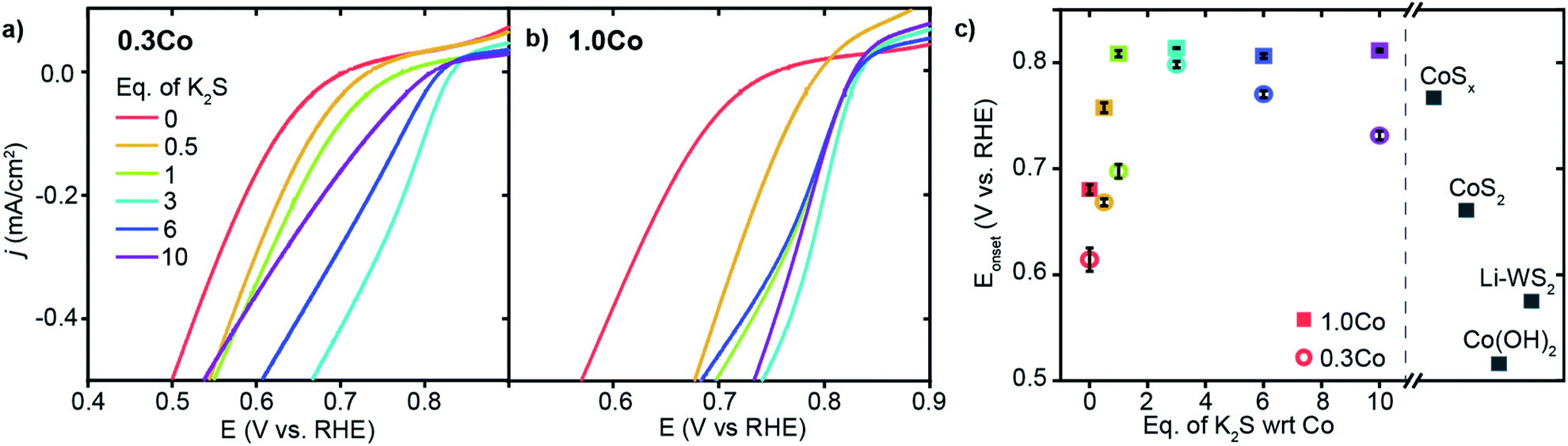 | ||
| Fig. 3 Oxygen reduction reaction activity in O2-saturated 1 M PBS (pH = 7) electrolyte. (a and b) ORR LSVs for the full series of 0.3Co and 1.0Co–WS2 samples treated with K2S, (c) Eonsetvs. equivalents of K2S for Co–WS2 catalysts compared to Co and WS2 control samples. | ||
At the higher Co loading (1.0Co–WS2), the ORR activity is also sensitive to K2S loading but with a somewhat different trend relative to the lower Co loading (Fig. 3b). In the absence of K2S, 1.0Co–WS2 exhibits an Eonset of 0.680 V vs. RHE. The Eonset rises rapidly with the addition of K2S and reaches a peak at 1 equiv. K2S, significantly earlier than on the 0.3Co–WS2 sample (Fig. 3c). Intriguingly, the Eonset plateaus at this point and stays relatively constant at ∼0.81 V vs. RHE even up to 10 equiv. K2S. The fast rise and lack of a peak in the onset potential trend is consistent with the presence of aggregated CoSx nanoparticles rather than isolated Co atoms or small Co clusters. The lower utilization of Co in the aggregated sample means a smaller amount of K2S is required to fully sulfidize the catalytic surface, and excess K2S serves only to convert more of the nanoparticle core into sulfided species without further influencing the surface.
Comparing the high and low loading Co–WS2 at their most active sulfidation point, we find that the ORR onset potentials are quite similar between the two samples despite the very different amounts of Co present (Fig. 3c). These data further support that the low loading 0.3Co–WS2 displays excellent catalytic utilization of Co atoms due to the relatively unaggregated structure of supported Co atoms on WS2. For this reason, we believe that the 0.3Co–WS2 treated with varying equivalents with K2S serves as a useful structural model to understand the role of Co–S coordination in dictating ORR reactivity.
To evaluate the catalytic stability of Co-doped WS2 catalysts, we undertook galvanostatic bulk electrolysis at −0.25 mA cm−2 on the 0.3Co–WS2 3K2S sample and a Pt/C control (Fig. S17†). While Pt/C is more active, Co–WS2 is somewhat more stable in 1 M PBS electrolyte. Over 60 min of electrolysis, the 0.3Co–WS2 3K2S catalyst decays by 47 mV while Pt/C decays by 90 mV. Notably, when 1 M MeOH is added to the electrolyte, the overpotential required to drive ORR increases by 180 mV on Pt/C due to carbon monoxide poisoning of the Pt surface while remaining essentially unchanged on 0.3Co–WS2.
X-ray absorption spectroscopy
To probe the local coordination environment and electronic state of adsorbed Co atoms in Co–WS2, we obtain Co K-edge X-ray absorption spectroscopy (XAS) for Co–WS2 samples in three states: as-synthesized (As-Syn), K2S-treated (K2S), and post-catalysis (PBS). These spectra are compared to the precursor molecular complex, Co(NMF)xCl2, as well as a control sample of amorphous cobalt sulfide (CoSx) with approximate composition of CoS1.2 (Fig. S14 and S15†).23,24 Fitting of the Fourier-transformed X-ray absorption fine structure (EXAFS) data was carried out on a subset of samples that showed sufficient Co K-edge absorption, and coordination numbers (CN) and bond distances (R) for both Co–O and Co–S scattering were obtained. Full fitting parameters and overlays of fitted and experimental data are provided in Table S4 and Fig. S20–S22.†The X-ray absorption near edge spectroscopy (XANES) data for the precursor Co(NMF)xCl2 complex exhibits Co K-edge energy of 7721 eV and high white line intensity, consistent with a Co2+ ion primarily coordinated to oxygen atoms, as anticipated for a CoCl2 precursor dissolved in the coordinating solvent NMF (Fig. 4a).60 Likewise, the EXAFS spectrum shows a scattering peak at a short radial distance due to pure Co–O coordination. Fitting of the EXAFS data shows a Co–O bond length of 2.08 Å with CN ∼6, consistent with an octahedral molecular complex (Table 1). On the opposite end of the spectrum, amorphous CoSx exhibits a completely suppressed XANES white line and a significantly lower Co K-edge energy of 7714 eV with a strong pre-edge feature at 7710 eV, indicative of mixed tetrahedral and octahedral coordination (Fig. 4f).61 Compared to Co–O coordination, Co–S coordination generates a scattering pathway at higher radial distance due to the longer average bond length, which ranges from 2.18 to 2.45 in known bulk phases of cobalt sulfide (Table S5 and Fig. S25†). The XANES and EXAFS of these two control samples serve as bookends for the Co–WS2 samples treated with varying amounts of sulfur because we expect the first-shell coordination environment to transform from oxygen-rich to sulfur-rich with increasing K2S loading.
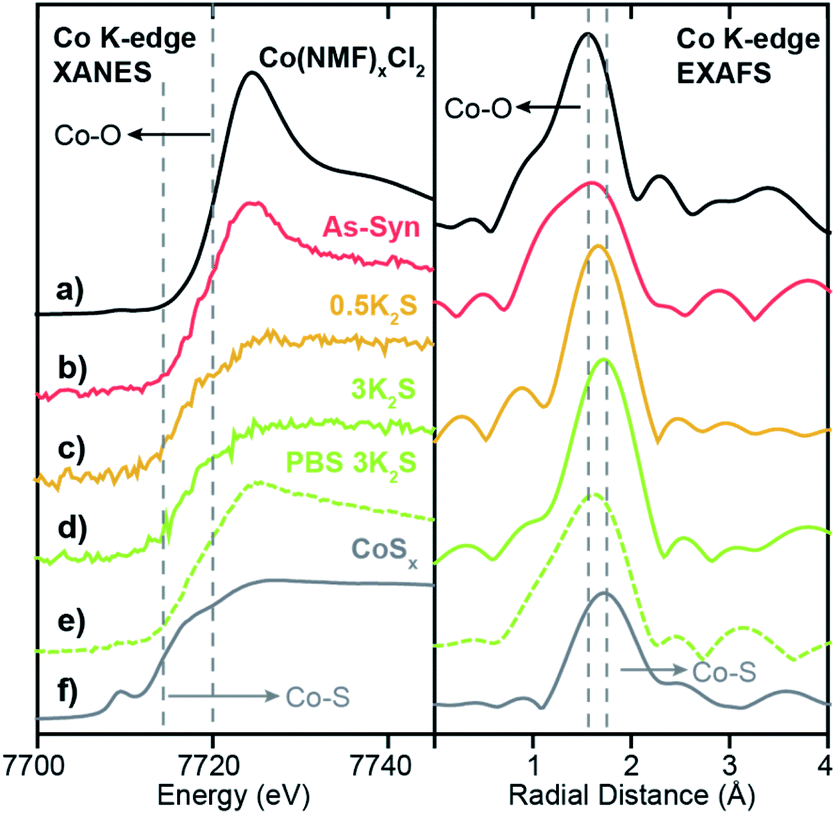 | ||
| Fig. 4 Co K-edge XANES and EXAFS spectra for (a) Co(NMF)xCl2, (b) As-synthesized 0.3Co–WS2, (c) 0.3Co–WS2 annealed with 0.5 equiv. of K2S, (d) 0.3Co–WS2 annealed with 3.0 equiv. of K2S, (e) 0.3Co-WS2 3K2S immersed in 1 M PBS electrolyte, and (f) amorphous CoSx. Dotted lines indicate the edge energies or scattering peaks for Co–O and Co–S coordination. | ||
| Sample | Pair | CN | R (Å) | E 0 (eV) |
|---|---|---|---|---|
| Co(NMF)xCl2 | Co–O | 6.0 ± 0.5 | 2.08 ± 0.007 | −2.3 ± 0.9 |
| 0.3Co–WS2 0.5K2S | Co–O | 3.8 ± 0.2 | 2.10 ± 0.004 | 7.4 ± 0.3 |
| Co–S | 2.5 ± 0.1 | 2.25 ± 0.004 | ||
| 0.3Co–WS2 3K2S | Co–O | 1.7 ± 0.4 | 2.10 ± 0.022 | 3.3 ± 0.6 |
| Co–S | 3.9 ± 0.3 | 2.25 ± 0.006 | ||
| 1.0Co–WS2 As-Syn | Co–O | 4.3 ± 0.4 | 2.08 ± 0.009 | 0.30 ± 0.9 |
| 1.0Co–WS2 0K2S | Co–O | 4.3 ± 0.3 | 2.08 ± 0.007 | −0.29 ± 0.8 |
| 1.0Co–WS2 1K2S | Co–O | 5.5 ± 0.4 | 2.10 ± 0.006 | −0.52 ± 0.7 |
| 1.0Co–WS2 3K2S | Co–O | 4.9 ± 0.2 | 2.10 ± 0.004 | −1.2 ± 0.3 |
| Co–S | 0.5 ± 0.1 | 2.25 ± 0.024 | ||
| 1.0Co–WS2 10K2S | Co–S | 4.4 ± 0.2 | 2.25 ± 0.005 | −3.1 ± 0.5 |
| 1.0Co–WS2 0K2S PBS | Co–O | 4.1 ± 0.1 | 2.04 ± 0.003 | −1.0 ± 0.4 |
| 1.0Co–WS2 1K2S PBS | Co–O | 5.0 ± 0.3 | 2.10 ± 0.006 | −2.1 ± 0.6 |
| Co–S | 0.8 ± 0.2 | 2.25 ± 0.025 | ||
| 1.0Co–WS2 3K2S PBS | Co–S | 4.5 ± 0.2 | 2.25 ± 0.005 | −4.8 ± 0.5 |
| 1.0Co–WS2 10K2S PBS | Co–S | 4.6 ± 0.4 | 2.25 ± 0.007 | −2.8 ± 0.9 |
We begin our XAS analysis with the low loading 0.3Co–WS2nK2S series of samples because their structural homogeneity and lack of aggregation lend themselves to clear structure–activity correlations. In the as-synthesized 0.3Co–WS2 sample, we observe small perturbations to the adsorbed Co coordination environment relative to the free Co(NMF)xCl2 complex. A drop in white line intensity in the XANES and a small shift to longer radial distance for the first Co–X (X = O or S) scattering pathway relative to the free complex are both indicative of an increase in the number of sulfur atoms relative to oxygen atoms in the first coordination sphere (Fig. 4b). However, the persistence of the white line peak indicates that Co atoms remain primarily oxygen-bound in this form, when passively-adsorbed to the Li–WS2 nanosheet in the solution phase. These data are consistent with the observation of Co atoms bound in S-top sites and W-top sites adjacent to vacancies in the high-resolution STEM images of colloidal 0.3Co-WS2. We postulate that the complex likely binds in a mono- or bidentate fashion to dangling sulfur sites on the lithiated WS2 surface, and the remaining coordination sites remain occupied by NMF ligands.
Upon K2S impregnation and annealing, the XAS data for 0.3Co–WS2 shows a significant increase in the Co–S coordination to the WS2 surface. Treatment with 0.5 equiv. or 3 equiv. of K2S completely suppresses the sharp white line feature diagnostic of Co2+–O binding and shifts the Co K-edge energy downwards by 3 eV, approaching the edge energy of the CoSx control sample (Fig. 4c and d). The EXAFS scattering peak also shifts to longer radial distance, and the sample treated with 3 equiv. K2S essentially matches the peak position of CoSx. Fitting of the EXAFS data indicates that the Co–S CN of 0.3Co–WS2 increases steadily from 2.5 at 0.5 equiv. K2S up to 3.9 at 3 equiv. K2S while the Co–O CN drops correspondingly (Table 1). In all of these samples, no significant scattering density is observed beyond 3.0 Å, indicating that Co atoms remain relatively isolated on the WS2 surface even after thermal annealing. No metallic Co–Co scattering or second coordination sphere Co–X–Co (X = O or S) scattering features are observed based on comparisons to Co foil and a variety of known CoOx and CoSx crystalline phases (Fig. S20, S25, Tables S4 and S5†). Together, these data show that Co atomic coordination to the WS2 surface can be readily controlled in the 0.3Co–WS2 system simply by varying the amount of impregnated K2S.
Lastly, we characterized the 0.3Co–WS2 samples after 1 hour immersion in phosphate buffer solution, catalytically equivalent to the material after 5–20 CV scans, to understand how the adsorbed Co sites evolve in aqueous electrolyte (Fig. S26 and Table S6†). XANES data on 0.3Co–WS2 3K2S immersed in PBS shows a similar Co K-edge energy but a slight increase in white line intensity relative to the pre-catalysis material (Fig. 4e). The EXAFS spectrum also shows a small shift in the first-shell scattering peak to lower radial distance. These data indicate that the Co–S bonds that anchor the dopants to the WS2 surface hydrolyze slightly in the presence of aqueous electrolyte, likely causing the decay in catalytic activity observed in the stability tests (Fig. S16 and S17†). However, Co surface sites remain relatively unaggregated on the WS2 surface after the electrolyte treatment, as evidenced by the continued absence of a second coordination sphere Co–S–Co scattering feature between 3–4 Å (Fig. 4e and S24c†). STEM-EDS mapping and elemental analysis after electrolyte treatment show that Co loading and spatial distribution remain unchanged relative to the pre-catalysis samples (Fig. 5a and S27†). Notably, Co atoms are fully retained in the 0.3Co–WS2nK2S samples after immersion in aqueous electrolyte, suggesting that all Co atoms are coordinatively attached to the WS2 surface (Table S7†).
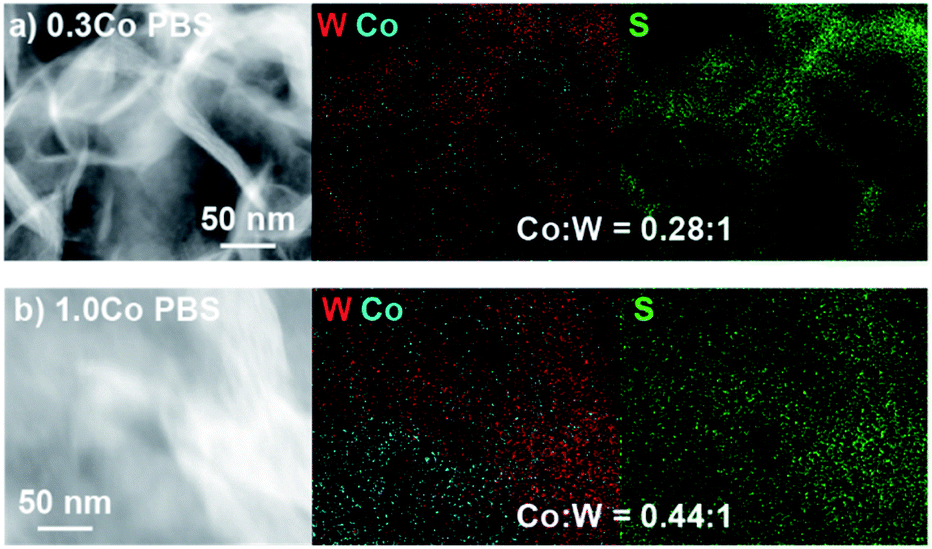 | ||
| Fig. 5 STEM-EDS image and elemental maps of W, Co, and S for (a) 0.3Co–WS2 3K2S and (b) 1.0Co–WS2 3K2S after treatment with PBS electrolyte. | ||
We can now correlate XAS structural characterization on the 0.3Co–WS2 samples to their initial ORR activity because, unlike in bulk or nanoparticle cobalt sulfide materials, the majority of Co atoms are available on the surface for catalysis. We find that 0.3Co–WS2 treated with 3 equiv. K2S, exhibiting three- to four-fold coordination of Co to sulfur atoms on the WS2 surface, provides an optimal electronic and geometric environment for pH 7 ORR catalysis. We hypothesize that the activation of adsorbed Co atoms with K2S up to 3 equiv. stems from the necessity for a more electron-rich metal center, enabled by Co–S coordination, in order to turn over adsorbed OH intermediates in the catalytic cycle.35,62 Further increasing K2S, however, likely causes over-coordination with sulfur and therefore loss of available coordination sites for O2 binding and activation (Fig. S29†). During catalysis, extended exposure to aqueous electrolyte induces partial hydrolysis of Co–S bonds, leading to a slight decay in ORR activity.
As a comparison to our Co–WS2 materials, we also obtained ORR catalytic data on three Co-containing phases with very different bulk composition and local Co coordination environment: Co(OH)2, amorphous CoSx (∼CoS1.2), and CoS2 (Fig. S14 and S15†). The Co(OH)2 nanosheets with pure Co–O coordination show the poorest ORR onset potential of 0.514 V vs. RHE (Fig. 3c and Table S3†). Even the 0.3Co–WS2 sample treated with 0 equiv. K2S is substantially more active due to the interaction with and contribution of the underlying WS2 nanosheets. The amorphous CoSx sample with an intermediate S:Co ratio of ∼1:1 shows the highest Eonset among the bulk phases at 0.767 V vs. RHE, nearly matching the onset potential of the optimal 0.3Co–WS2 3K2S catalyst. The most sulfur-rich sample, comprising CoS2 nanoparticles, exhibits lower ORR activity with an onset potential of 0.660 V vs. RHE, which mirrors the drop in activity of 0.3Co–WS2 10K2S when it becomes over-saturated with sulfur. The similar trends in onset potential as a function of cobalt sulfidation between 0.3Co–WS2 and bulk Co-containing phases provides an initial indication that the local coordination environment identified for Co atoms supported on WS2 may also apply to active sites formed in situ on the surface of bulk cobalt sulfide species during ORR catalysis.
Because XAS spectra represent an average over all Co species in the sample, the high loading 1.0Co–WS2 sample, comprising large Co aggregates and heterogeneous speciation, is much more difficult to directly correlate to catalytic reactivity. Nonetheless, the XAS data at each stage of the synthetic and catalytic process provide useful information about the speciation and evolution of adsorbed Co species at high loading.
In the as-synthesized 1.0Co–WS2, both the XANES and EXAFS at the Co K-edge closely resemble the spectra for the free Co(NMF)xCl2 complex, suggesting that a significant fraction of Co complexes at this high loading are intercalated or weakly physisorbed due to the ionic interaction between the negatively charged WS2 sheets and cationic Co complexes (Fig. 6a). After K2S addition and thermal annealing in the presence of 1 and 3 equiv. K2S, small increases in EXAFS first-shell scattering intensity are observed but with no significant increase in radial distance (Fig. 6b and c). Coupled to the persistently high white line intensity in the XANES, these data indicate that the bulk of the Co atoms in 1.0Co–WS2 remain unsulfidized up to 3 equiv. K2S (Table 1). At 10 equiv. K2S, an abrupt increase in first-shell scattering distance and drop in white line intensity reveals that the majority of Co atoms have been converted to cobalt sulfide species (Fig. 6d). The relatively high equivalents of K2S required to sulfidize the Co species in the 1.0Co–WS2 is consistent with the presence of aggregated and intercalated Co species, clearly indicating that these average XAS spectra are not representative of catalytically active Co species.
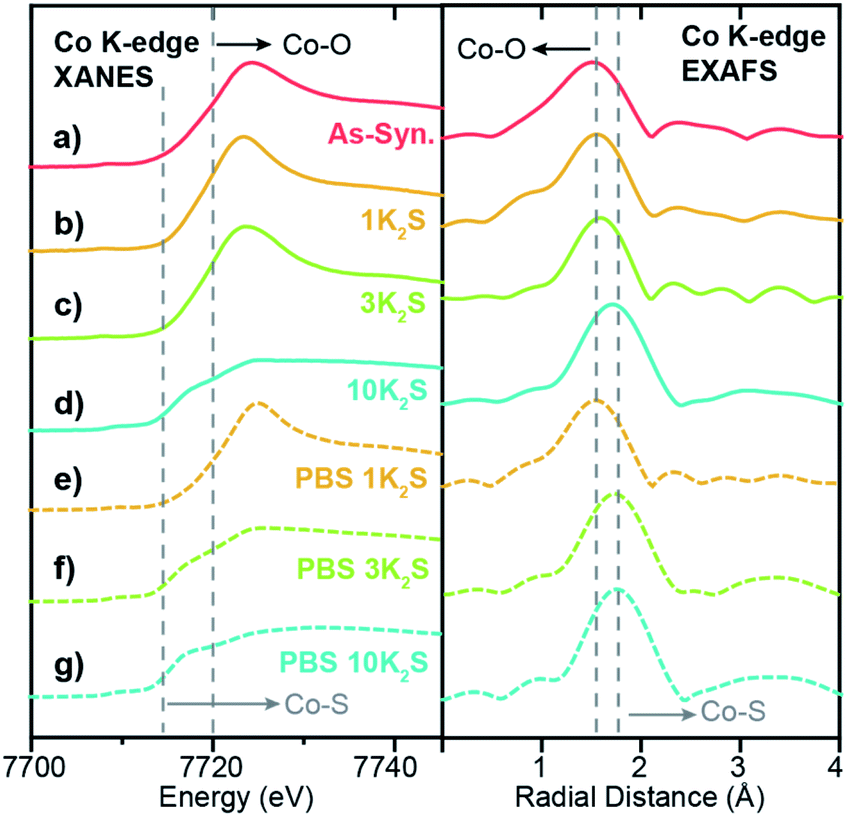 | ||
| Fig. 6 Co K-edge XANES and EXAFS spectra for 1.0Co–WS2 samples at varying stages of synthesis and catalysis. (a) As-synthesized, (b–d) thermally treated with varying equivalents of K2S, and (e–g) immersed in 1 M PBS electrolyte. Dotted lines indicate the edge energies or scattering peaks for Co–O and Co–S coordination. | ||
After the PBS immersion, elemental analysis using XRF reveals that significant amounts of Co are leached out of the 1.0Co–WS2 nanosheets, which is in stark contrast to the full Co retention observed in the 0.3Co–WS2 samples (Table S7†). The Co spatial distribution also becomes more non-uniform and aggregated based on STEM-EDS elemental maps after PBS treatment (Fig. 5b, S28 and Table S8†). The quantity of leached Co is greatest at low K2S loadings, likely because the loosely intercalated Co(NMF)x molecular complexes observed in the as-synthesized sample are not yet converted to insoluble CoSx aggregates and can be readily removed in strongly ionic solutions. As a result, the post-PBS XAS spectra are more representative of the Co species present during catalysis for the high loading 1.0Co–WS2 sample (Fig. 6e–g). The difference in XAS spectra between the pre- and post-electrolyte treatment is clearest in the 1.0Co–WS2 sample at intermediate K2S loading (3 K2S). While the spectrum prior to PBS immersion appears minimally sulfidized, the spectrum after PBS immersion shows the longer radial distance and suppressed white line intensity characteristic of a cobalt sulfide species (Fig. 6c and f). This sample clearly demonstrates that unreacted Co complexes after K2S annealing are dissolved out during the immersion in electrolyte solution, leaving behind only the sulfided Co nanoparticles. Analysing the full series of post-PBS samples, we observe an increase in Co–S CN and decrease in Co–O CN as the K2S loading is increased (Table 1). Full sulfidation is observed by 3 equiv. K2S, which exhibits Co–S CN of 4.5 and no contribution from Co–O. Unlike in the 0.3Co–WS2 samples, however, it is impossible to correlate these average Co–S coordination numbers directly to ORR catalysis because of the presence of large CoSx aggregates and the fact that the majority of Co atoms are not available at the surface for catalysis. As anticipated, all 1.0Co–WS2 samples after PBS treatment display a second coordination sphere Co–X–Co (X = O or S) scattering feature, which corroborates the presence of large aggregates and nanoparticles observed in the STEM-EDS images (Fig. S24†).
Conclusion
In conclusion, we find that relatively isolated Co atoms supported on WS2 show high activity for the pH 7 ORR reaction when partially coordinated to sulfur ligands. Both undercoordination and overcoordination to sulfur prove detrimental to catalysis. The optimal Co active site comprises approximate 3–4-fold coordination to sulfur atoms on the surface of WS2 and two labile coordination sites occupied by oxygen-based solvent ligands. These dilute Co–WS2 catalysts show similar catalytic activity to the most active bulk phases of cobalt sulfide with an onset potential for ORR of 0.798 V vs. RHE in pH 7 phosphate buffer electrolyte. Unlike bulk CoSx systems, however, the majority of Co atoms in dilute Co–WS2 are available for catalysis, and the Co coordination environment obtained by XAS is representative of catalytically active sites. As a result, we postulate that the optimal coordination environment ascertained herein may be representative of the dynamic active sites present in situ on the surfaces of bulk cobalt sulfide catalysts. The activity of dilute Co–WS2 clearly illustrates that the electronic and geometric environment created by Co–S bonds in the first coordination sphere provides a key descriptor for ORR catalysis in cobalt sulfide materials.Author contributions
WH and EM contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Purdue University and the National Science Foundation (CHE-2106450). EM and WH acknowledge support from the Purdue Research Foundation. We acknowledge Dr Rosa E. Diaz for assistance in collecting high resolution HAADF-STEM images. We thank Dr Yujia Ding, Dr Kamil Kucuk, Dr Mark Warren, and Dr Joshua Wright for their help with XAS experiments. Use of the Advanced Photon Source is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-06CH11357. MCRAT operations are supported by the Department of Energy and MRCAT member institutions.References
- C. Santoro, C. Arbizzani, B. Erable and I. Ieropoulos, J. Power Sources, 2017, 356, 225–244 Search PubMed.
- B. E. Logan, B. Hamelers, R. A. Rozendal, U. Schrorder, J. Keller, S. Freguia, P. Aelterman, W. Verstraete and K. Rabaey, Environ. Sci. Technol., 2006, 40, 5181–5192 Search PubMed.
- M. V. Kannan and G. G. Kumar, Biosens. Bioelectron., 2016, 77, 1208–1220 Search PubMed.
- Z. X. Hu, X. X. Zhou, Y. Lu, R. M. Jv, Y. Liu, N. Li and S. W. Chen, Electrochim. Acta, 2019, 296, 214–223 Search PubMed.
- M. Kodali, S. Herrera, S. Kabir, A. Serov, C. Santoro, I. Ieropoulos and P. Atanassov, Electrochim. Acta, 2018, 265, 56–64 Search PubMed.
- L. Z. Liu, G. Zeng, J. X. Chen, L. L. Bi, L. M. Dai and Z. H. Wen, Nano Energy, 2018, 49, 393–402 Search PubMed.
- X. H. Tang and H. Y. Ng, Electrochim. Acta, 2017, 247, 193–199 Search PubMed.
- O. Schaetzle, F. Barriere and U. Schroder, Energy Environ. Sci., 2009, 2, 96–99 Search PubMed.
- B. Yan, N. M. Concannon, J. D. Milshtein, F. R. Brushett and Y. Surendranath, Angew. Chem., Int. Ed., 2017, 56, 7496–7499 Search PubMed.
- J. M. Falkowski, N. M. Concannon, B. Yan and Y. Surendranath, J. Am. Chem. Soc., 2015, 137, 7978–7981 Search PubMed.
- P. W. Cai, J. H. Huang, J. X. Chen and Z. H. Wen, Angew. Chem., Int. Ed., 2017, 56, 4858–4861 Search PubMed.
- W. W. Zhao, P. Bothra, Z. Y. Lu, Y. B. Li, L. P. Mei, K. Liu, Z. H. Zhao, G. X. Chen, S. Back, S. Siahrostami, A. Kulkarni, J. K. Norskov, M. Bajdich and Y. Cui, ACS Appl. Energy Mater., 2019, 2, 8605–8614 Search PubMed.
- H. Behret, H. Binder and G. Sandstede, Electrochim. Acta, 1975, 20, 111–117 Search PubMed.
- H. Behret, H. Binder, W. Clauberg and G. Sandstede, Electrochim. Acta, 1978, 23, 1023–1029 Search PubMed.
- Y. J. Feng, A. Gago, L. Timperman and N. Alonso-Vante, Electrochim. Acta, 2011, 56, 1009–1022 Search PubMed.
- M. R. Gao, J. Jiang and S. H. Yu, Small, 2012, 8, 13–27 Search PubMed.
- V. Vij, S. Sultan, A. M. Harzandi, A. Meena, J. N. Tiwari, W. G. Lee, T. Yoon and K. S. Kim, ACS Catal., 2017, 7, 7196–7225 Search PubMed.
- H. L. Wang, Y. Y. Liang, Y. G. Li and H. J. Dai, Angew. Chem., Int. Ed., 2011, 50, 10969–10972 Search PubMed.
- L. Zhu, D. Susac, M. Teo, K. C. Wong, P. C. Wong, R. R. Parsons, D. Bizzotto, K. A. R. Mitchell and S. A. Campbell, J. Catal., 2008, 258, 235–242 Search PubMed.
- D. Susac, L. Zhu, M. Teo, A. Sode, K. C. Wong, P. C. Wong, R. R. Parsons, D. Bizzotto, K. A. R. Mitchell and S. A. Campbell, J. Phys. Chem. C, 2007, 111, 18715–18723 Search PubMed.
- A. Arunchander, S. G. Peera and A. K. Sahu, ChemElectroChem, 2017, 4, 1544–1553 Search PubMed.
- J. M. Falkowski and Y. Surendranath, ACS Catal., 2015, 5, 3411–3416 Search PubMed.
- Y. F. Cao, Y. Y. Meng, S. C. Huang, S. M. He, X. H. Li, S. F. Tong and M. M. Wu, ACS Sustainable Chem. Eng., 2018, 6, 15582–15590 Search PubMed.
- P. Ganesan, M. Prabu, J. Sanetuntikul and S. Shanmugam, ACS Catal., 2015, 5, 3625–3637 Search PubMed.
- B. L. Chen, R. Li, G. P. Ma, X. L. Gou, Y. Q. Zhu and Y. D. Xia, Nanoscale, 2015, 7, 20674–20684 Search PubMed.
- Q. L. Zhu, W. Xia, T. Akita, R. Q. Zou and Q. Xu, Adv. Mater., 2016, 28, 6391–6398 Search PubMed.
- Z. X. Wu, J. Wang, M. Song, G. M. Zhao, Y. Zhu, G. T. Fu and X. E. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 25415–25421 Search PubMed.
- Y. X. Zhou, H. B. Yao, Y. Wang, H. L. Liu, M. R. Gao, P. K. Shen and S. H. Yu, Chem.–Eur. J., 2010, 16, 12000–12007 Search PubMed.
- Y. P. Tang, F. Jing, Z. X. Xu, F. Zhang, Y. Y. Mai and D. Q. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 12340–12347 Search PubMed.
- S. F. Fu, C. Z. Zhu, J. H. Song, S. Feng, D. Du, M. H. Engelhard, D. D. Xiao, D. S. Li and Y. H. Lin, ACS Appl. Mater. Interfaces, 2017, 9, 36755–36761 Search PubMed.
- Z. Xiao, G. Z. Xiao, M. H. Shi and Y. Zhu, ACS Appl. Mater. Interfaces, 2018, 10, 16436–16448 Search PubMed.
- S. L. Zhang, D. Zhai, T. T. Sun, A. J. Han, Y. L. Zhai, W. C. Cheong, Y. Liu, C. L. Su, D. S. Wang and Y. D. Li, Appl. Catal., B, 2019, 254, 186–193 Search PubMed.
- F. Bai, X. Qu, J. Wang, X. Chen and W. S. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 33740–33750 Search PubMed.
- R. A. Sidik and A. B. Anderson, J. Phys. Chem. B, 2006, 110, 936–941 Search PubMed.
- B. Yan, D. Krishnamurthy, C. H. Hendon, S. Deshpande, Y. Surendranath and V. Viswanathan, Joule, 2017, 1, 600–612 Search PubMed.
- D. Susac, A. Sode, L. Zhu, P. C. Wong, M. Teo, D. Bizzotto, K. A. R. Mitchell, R. R. Parsons and S. A. Campbell, J. Phys. Chem. B, 2006, 110, 10762–10770 Search PubMed.
- X. F. Yang, A. Q. Wang, B. T. Qiao, J. Li, J. Y. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 Search PubMed.
- A. Q. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 Search PubMed.
- G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Acc. Chem. Res., 2019, 52, 237–247 Search PubMed.
- Y. Xin, N. N. Zhang, Q. Li, Z. L. Zhang, X. M. Cao, L. R. Zheng, Y. W. Zeng and J. A. Anderson, ACS Catal., 2018, 8, 1399–1404 Search PubMed.
- G. Q. Gan, X. Y. Li, L. Wang, S. Y. Fan, J. C. Mu, P. L. Wang and G. H. Chen, ACS Nano, 2020, 14, 9929–9937 Search PubMed.
- Y. Xu, M. Y. Chu, F. F. Liu, X. C. Wang, Y. Liu, M. H. Cao, J. Gong, J. Luo, H. P. Lin, Y. Y. Li and Q. Zhang, Nano Lett., 2020, 20, 6865–6872 Search PubMed.
- J. Q. Zhang, Y. F. Zhao, C. Chen, Y. C. Huang, C. L. Dong, C. J. Chen, R. S. Liu, C. Y. Wang, K. Yan, Y. D. Li and G. X. Wang, J. Am. Chem. Soc., 2019, 141, 20118–20126 Search PubMed.
- Q. Wang, X. Huang, Z. L. Zhao, M. Y. Wang, B. Xiang, J. Li, Z. X. Feng, H. Xu and M. Gu, J. Am. Chem. Soc., 2020, 142, 7425–7433 Search PubMed.
- A. Zitolo, V. Goellner, V. Armel, M. T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937–942 Search PubMed.
- L. L. Zhang, A. Q. Wang, W. T. Wang, Y. Q. Huang, X. Y. Liu, S. Miao, J. Y. Liu and T. Zhang, ACS Catal., 2015, 5, 6563–6572 Search PubMed.
- M. Y. Hu, S. N. Li, S. S. Zheng, X. H. Liang, J. X. Zheng and F. Pan, J. Phys. Chem. C, 2020, 124, 13168–13176 Search PubMed.
- W. G. Liu, L. L. Zhang, W. S. Yan, X. Y. Liu, X. F. Yang, S. Miao, W. T. Wang, A. Q. Wang and T. Zhang, Chem. Sci., 2016, 7, 5758–5764 Search PubMed.
- W. G. Liu, L. L. Zhang, X. Liu, X. Y. Liu, X. F. Yang, S. Miao, W. T. Wang, A. Q. Wang and T. Zhang, J. Am. Chem. Soc., 2017, 139, 10790–10798 Search PubMed.
- A. Zitolo, N. Ranjbar-Sahraie, T. Mineva, J. K. Li, Q. Y. Jia, S. Stamatin, G. F. Harrington, S. M. Lyth, P. Krtil, S. Mukerjee, E. Fonda and F. Jaouen, Nat. Commun., 2017, 8, 957 Search PubMed.
- P. Q. Yin, T. Yao, Y. Wu, L. R. Zheng, Y. Lin, W. Liu, H. X. Ju, J. F. Zhu, X. Hong, Z. X. Deng, G. Zhou, S. Q. Wei and Y. D. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 Search PubMed.
- P. Vancso, Z. I. Popov, J. Peto, T. Ollar, G. Dobrik, J. S. Pap, C. Y. Hwang, P. B. Sorokin and L. Tapaszto, ACS Energy Lett., 2019, 4, 1947–1953 Search PubMed.
- L. Ji, P. F. Yan, C. H. Zhu, C. Y. Ma, W. Z. Wu, C. Wei, Y. L. Shen, S. Q. Chu, J. O. Wang, Y. Du, J. Chen, X. A. Yang and Q. Xu, Appl. Catal., B, 2019, 251, 87–93 Search PubMed.
- K. Qi, X. Q. Cui, L. Gu, S. S. Yu, X. F. Fan, M. C. Luo, S. Xu, N. B. Li, L. R. Zheng, Q. H. Zhang, J. Y. Ma, Y. Gong, F. Lv, K. Wang, H. H. Huang, W. Zhang, S. J. Guo, W. T. Zheng and P. Liu, Nat. Commun., 2019, 10, 5231 Search PubMed.
- Q. Wang, Z. L. Zhao, S. Dong, D. S. He, M. J. Lawrence, S. B. Han, C. Cai, S. H. Xiang, P. Rodriguez, B. Xiang, Z. G. Wang, Y. Y. Liang and M. Gu, Nano Energy, 2018, 53, 458–467 Search PubMed.
- B. Mahler, V. Hoepfner, K. Liao and G. A. Ozin, J. Am. Chem. Soc., 2014, 136, 14121–14127 Search PubMed.
- E. Meza, R. E. Diaz and C. W. Li, ACS Nano, 2020, 14, 2238–2247 Search PubMed.
- C. Li, A. P. Tardajos, D. Wang, D. Choukroun, K. Van Daele, T. Breugelmans and S. Bals, Ultramicroscopy, 2021, 221 Search PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 Search PubMed.
- M. Uchikoshi and K. Shinoda, Struct. Chem., 2019, 30, 945–954 Search PubMed.
- L. van Haandel, G. Smolentsev, J. A. van Bokhoven, E. J. M. Hensen and T. Weber, ACS Catal., 2020, 10, 10978–10988 Search PubMed.
- J. K. Norskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, synthetic methods, physical characterization methods, electrochemical methods, and additional tables and figures. See DOI: 10.1039/d1ta02468j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |