
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMelt-quenched porous organic cage glasses†
Michael C.
Brand
 a,
Francesca
Greenwell
a,
Rob
Clowes
a,
Benjamin D.
Egleston
a,
Aiting
Kai
a,
Andrew I.
Cooper
*a,
Thomas D.
Bennett
*b and
Rebecca L.
Greenaway
*ac
a,
Francesca
Greenwell
a,
Rob
Clowes
a,
Benjamin D.
Egleston
a,
Aiting
Kai
a,
Andrew I.
Cooper
*a,
Thomas D.
Bennett
*b and
Rebecca L.
Greenaway
*ac
aDepartment of Chemistry, Materials Innovation Factory, University of Liverpool, 51 Oxford Street, Liverpool, L7 3NY, UK. E-mail: aicooper@liverpool.ac.uk
bDepartment of Materials Science and Metallurgy, University of Cambridge, 27 Charles Babbage Road, Cambridge CB3 0FS, UK. E-mail: tdb35@cam.ac.uk
cDepartment of Chemistry, Molecular Sciences Research Hub, Imperial College London, White City Campus, 82 Wood Lane, W12 0BZ, UK. E-mail: r.greenaway@imperial.ac.uk
First published on 4th May 2021
Abstract
The discrete molecular nature of porous organic cages (POCs) has allowed us to direct the formation of crystalline materials by crystal engineering. It has also been possible to create porous amorphous solids by deliberately disrupting the crystalline packing, either with chemical modification or by processing. More recently, organic cages were used to form isotropic porous liquids. However, the connection between solid and liquid states of POCs, and the glass state, are almost completely unexplored. Here, we investigate the melting and glass-forming behaviour of a range of organic cages, including both shape-persistent POCs formed by imine condensation, and reduced and synthetically post-modified amine POCs that are more flexible and lack shape-persistence. The organic cages exhibited melting and quenching of the resultant liquids provides molecular glasses. One of these molecular glasses exhibited improved gas uptake for both CO2 and CH4 compared to the starting amorphous cage. In addition, foaming of the liquid in one case resulted in a more stable and less soluble glass, which demonstrates the potential for an alternative approach to forming materials such as membranes without solution processing.
Introduction
Porous materials such as metal–organic frameworks (MOFs) and covalent organic frameworks (COFs) have widespread applications such as gas storage, separations, and catalysis. Highly crystalline porous solids with precisely defined pore structures capable of selective host–guest interactions have provoked attention as ‘designable’ materials.1–4 However, long-range order is not a pre-requisite for practical utility. Indeed, amorphous, or disordered solids are employed across the materials spectrum in a diverse range of applications. For example, organic polymers have been studied intensively for separations and catalysis,5 and are used commercially in separation membranes, and inorganic glasses have found uses in display technologies and communications.6Several prototypical reports have recently emerged that connect emerging families of porous materials with topological disorder.7,8 The search for new porous materials9 has led to the study of the liquid states of both metal–organic cages10 and MOFs.11 In the latter case, several zeolitic imidazolate frameworks (ZIFs) were melted at ca. 400 °C, and the resultant liquids cooled to form materials with no long range order, but which nonetheless retain the short range connectivity of the crystalline solid-state; that is, glasses.12,13 The formation of melt-quenched glasses from the liquid state offers some advantages over the crystalline solid state, including the ability to cast materials into transparent, bulk, mechanically-stable morphologies. In theory, a melt-quenched glass, or ‘frozen-liquid’ may be formed by cooling any liquid sufficiently quickly enough to avoid reordering.14,15 Glasses are distinguished from the more general family of amorphous solids by exhibiting a glass transition temperature, Tg; that is, a temperature or temperature range over which material behaviour changes from a solid to a liquid.
In this context, we consider the emerging category of porous materials known as porous organic cages (POCs). POCs are discrete shape-persistent molecules that contain a permanent cavity accessible through windows, which have been investigated as both crystalline and amorphous solids and, more recently, as porous liquids (Fig. 1).16–18 They are often synthesised using analogous dynamic covalent chemistries to those used to assemble COFs, such as imine condensations. However, unlike extended porous frameworks, such as COFs and MOFs, the discrete nature of POCs makes them soluble, meaning that the packing of the individual cages can be directed using solvent to access crystalline solids with different interconnected pore networks; that is, to produce different porous polymorphs. Alternatively, the crystal packing can be disrupted and POCs can be rendered amorphous, which can sometimes result in increased porosity with respect to the crystalline state.19,20 This solution processability also means that POCs can be processed into different physical forms, such as thin films and membranes.21,22 However, this typically requires the use of solvent to cast films onto various supports, or the use of a polymeric additive to form mixed-matrix membranes,21 and not all POCs have good solubility in common organic solvents.
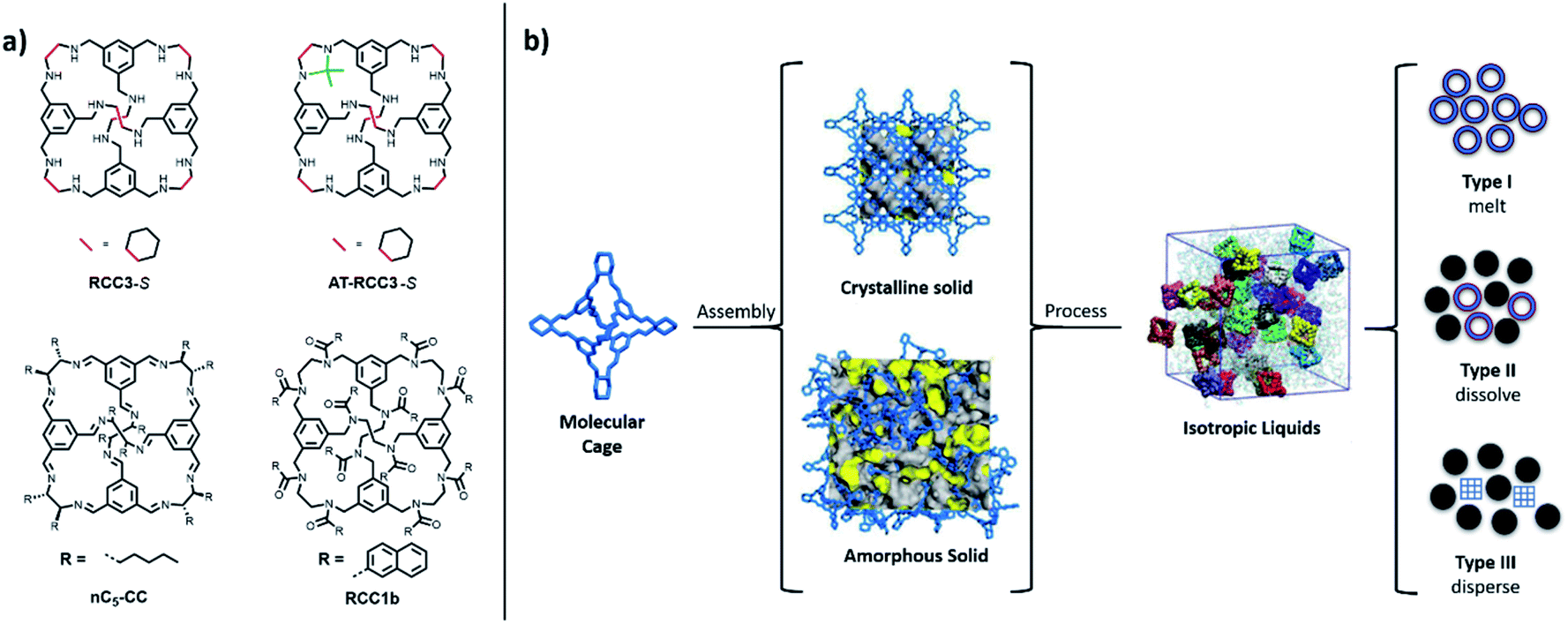 | ||
| Fig. 1 (a) Structures of the organic cages studied for melting and glass formation – all cages are initially formed as a tetrahedral [4+6] cycloimine cage (CC1, CC3-S, nC5-CC) by the reaction between 1,3,5-triformylbenzene (4 equiv.) and a vicinal diamine (6 equiv.); RCC3-S is formed by reducing CC3-S, AT-RCC3-S is formed by subsequent aminal formation between acetone and RCC3-S, and RCC1b is formed by the chemical reduction of CC1 and the subsequent formation of a dodecaamide cage. (b) Crystalline solid, amorphous solid, and isotropic porous liquid states, of porous organic cages are known; the liquid phases can be categorized as Type 1, Type 2, or Type 3; in this study, Type 1 solvent-free POC liquids are investigated. | ||
POCs can also be modified and processed into porous liquids; that is, liquids with permanent intrinsic microporosity. Since the initial conceptualisation of porous liquids in 2007,23 POCs have been the basis of investigations into both Type 1 porous liquids (that is, neat liquids formed from molecules containing permanent cavities) and Type 2 porous liquids (that is, solutions of a porous material in a cavity-excluded solvent).17,24–27 For Type 1 porous liquids, and of particular relevance to this work, POCs that were externally decorated with alkyl groups were synthesised in an attempt to form neat liquid POCs, although a neat Type 1 porous liquid was not fully realised due to interpenetration of the alkyl chains into the cage cavities.28,29 Moreover, while melting points were reported, the potential glass-forming behaviour of these alkylated POCs was not investigated.
Alongside the formation of melt-quenched glasses from porous frameworks, molecular organic glasses have seen increased attention over the past 20 years due to their applications in electronic and optoelectronic devices.30 Bringing together several characteristics of molecular species, including their small size, well-defined structure, and high purity,31 molecular glasses have shown benefits such as higher resolution,32 higher device efficiency, and increased stability.33 While there are many examples of molecular organic glasses, their formation from molecular organic cages with a permanent cavity has not been studied to date. Here, we studied the formation and properties of molecular glasses from melt-quenched organic cage liquids. We suggest that this strategy may be a new, complementary route to functional materials such as membranes.
Experimental
Materials
1,3,5-Triformylbenzene (TFB) and (1S,2S)-cyclohexane-1,2-diamine (S,S-CHDA) were purchased from Manchester Organics; trifluoroacetic acid (TFA) was purchased from TCI UK; sodium borohydride, sodium sulfate, and ethylenediamine (EDA), triethylamine, napthoyl chloride, and formic acid were purchased from Sigma-Aldrich. Dichloromethane (DCM), methanol, chloroform, acetone, and hexane were reagent or HPLC grade and purchased from Fisher Scientific. (6S,7S)-Dodecane-6,7-diamine was synthesised following a literature procedure28via a diaza-Cope rearrangement and hydrolysis using (1R,2R)-1,2-bis(2-hydroxyphenyl)ethylenediamine and hexanal from Sigma-Aldrich, and toluene, 37% hydrochloric acid, and tetrahydrofuran (THF) from Fisher Scientific. All gases (nitrogen, carbon dioxide, methane, xenon, butane) were purchased from BOC at a purity of ≥99.9%. All chemicals and solvents were used as received without any purification.Synthesis of organic cages
Full synthetic protocols and characterisation data can be found in the ESI.†
RCC3-S is synthesised by reducing CC3-S, and AT-RCC3-S is formed by subsequent aminal formation between acetone and RCC3-S, following modified literature procedures.28,34 DCM was added onto TFB (4.0 equiv.) without disturbing the solid, followed by a drop of TFA, and then a solution of S,S-CHDA (6.0 equiv.) in DCM was carefully layered on top. The reaction was left undisturbed at room temperature for 5 days, and the crystals of CC3-S were collected by filtration, washed with methanol, and dried under vacuum. To a solution of CC3-S (1.0 equiv.) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of chloroform:methanol was added sodium borohydride (32.0 equiv.) under nitrogen. The resulting mixture was stirred at room temperature for 1 day, before being quenched with water and concentrated by rotary evaporation. After isolation by aqueous work-up and rotary evaporation, RCC3-S was isolated as a pale yellow foam. To form AT-RCC3-S, RCC3-S was dissolved in acetone and left to stand at room temperature overnight. The colourless crystals of AT-RCC3-S were collected by filtration and washed with acetone, and dried under vacuum.
:1 mixture of chloroform:methanol was added sodium borohydride (32.0 equiv.) under nitrogen. The resulting mixture was stirred at room temperature for 1 day, before being quenched with water and concentrated by rotary evaporation. After isolation by aqueous work-up and rotary evaporation, RCC3-S was isolated as a pale yellow foam. To form AT-RCC3-S, RCC3-S was dissolved in acetone and left to stand at room temperature overnight. The colourless crystals of AT-RCC3-S were collected by filtration and washed with acetone, and dried under vacuum.
nC5-CC is synthesised by refluxing a solution of TFB (4.0 equiv.) and (6S,7S)-dodecane-6,7-diamine28 (7.2 equiv.) in chloroform for 6 days. After concentration on a rotary evaporator, acetone was added to precipitate a solid which was collected by filtration. The solid was then redissolved in a 1:1 mixture of dichloromethane/methanol, filtered, and the dichloromethane carefully removed under reduced pressure until an off-white solid precipitates. The nC5-CC is collected by filtration and dried under vacuum.
RCC1b is synthesised by first reducing CC1 to form RCC1, and RCC1b is then formed by forming a dodecaamide between RCC1 and napthoyl chloride, following modified literature procedures.35 To a cooled solution (0 °C) of TFB (4.0 equiv.) in DCM was added a solution of EDA (6.1 equiv.) in DCM over 48 hours. After filtering and concentration on a rotary evaporator, hexane was added to precipitate CC1 which was collected by filtration. To a solution of CC1 (1.0 equiv.) in a 1:1 mixture of chloroform:methanol was added sodium borohydride (32.0 equiv.) batchwise under nitrogen. The resulting mixture was stirred at room temperature for 18 hours, before being quenched with water and concentrated by rotary evaporation. The resulting solid was sonicated in chloroform, and the filtrate was collected and dried using a rotary evaporator to afford RCC1 as a colourless foamed solid. RCC1b was formed through the addition of triethylamine (15.0 equiv.) to a solution of RCC1 (1.0 equiv.) in chloroform under nitrogen at 0 °C. To this solution napthoyl chloride (12.6 equiv.) was added dropwise and the reaction was warmed to room temperature and stirred for 6 days, before concentration by rotary evaporation. THF was then added to the crude material and the slurry was sonicated before being filtered. The filtrate was concentrated by rotary evaporation and then stirred in MeOH, with the filtrate then decanted off, before the washing was repeated a further 4 times. The washed solid was collected by filtration and dried under vacuum to afford RCC1b.
Preparation and scale-up of glasses
Samples of each cage glass were prepared by subjecting the different as-synthesised organic cages to a heat–cool or heat–cool–heat cycle on a DSC using a ramp rate of 10 °C min−1 up to the required temperatures (20 °C below Td temperature, identified by TGA) before being cooled at the same rate. The glasses were subsequently analysed by PXRD, Raman spectroscopy, 1H NMR spectroscopy, and SEM, before being subjected to high-throughput gas sorption measurements.For the scale-up of the glasses formed from RCC3-S, agRCC3-S and afgRCC3-S, a batchwise approach was used. This was achieved by heating 10 samples of ∼10 mg RCC3-S in separate aluminium DSC pans at 10 °C min−1 to either 250 °C (agRCC3-S) or 300 °C (afgRCC3-S), before cooling at 10 °C min−1 to room temperature. Samples were then removed from the aluminium pans and combined to recover agRCC3-S (42 mg) and afgRCC3-S (84 mg) which were analysed by PXRD and gas sorption.
High-throughput gas sorption
High-throughput gas sorption was performed on a custom platform. A thermal camera is mounted above a sorption chamber and records the temperature change associated with gas adsorption/desorption. Samples were loaded into a 96-well ProxiPlate and degassed under vacuum whilst heating at 80 °C overnight and then left to equilibrate at 25 °C and maintained with a temperature-controlled oil heating unit. Whilst recording, the adsorbate gas is charged into the sorption chamber with approximately a 70 mbar dose. Samples were left to equilibrate for approximately 3 minutes before consecutive dosing. Data interpretation – 7 empty wells were averaged and used as a temperature change reference. This reference value was deducted from the measured temperature change in the sample wells giving temperature change in the samples. The activation temperature of the cages was determined with thermogravimetric analysis.Characterisation methods
Results & discussion
Materials selection
Many POCs undergo thermal decomposition before any solid–liquid transitions occur.28,36,37 To induce melting, a number of design strategies can be used: for example, imine bonds in the cage can be reduced to more stable amines, and optionally post-modified, or alkyl functionalities can be introduced onto the periphery of the cage during synthesis to lower the melting point, Tm. Reduction of the imine bonds tends to yield cages that are more flexible, often leading to the loss of shape-persistence and, hence, loss of porosity. However, porosity can be re-introduced by subsequent reaction of the amines to form more rigidified aminals or amides.34,35 While increasing the alkyl group length was reported to lower Tm in POCs,28 computational modelling suggested that the longer alkyl chains begin to occupy the cavities of neighbouring cages, reducing the porosity. One exception to this was a pentyl substituted cage that was predicted to result in a porous liquid with 30% of the cages empty at any given time, although this was not demonstrated experimentally.29Bearing these design criteria in mind, we synthesized a range of structurally related organic cages, including RCC3, AT-RCC3, nC5-CC (pentyl decorated covalent cage), and RCC1b (Fig. 1a) (see ESI Methods†) and studied them using a combination of powder X-ray diffraction (PXRD), thermogravimetric analysis (TGA), and differential scanning calorimetry (DSC).27,28,35 The presence of different exterior functionalities was explored (that is, no functionalization versus alkyl groups versus aromatic groups), along with the effect of molecular flexibility (shape-persistent imine cages versus reduced, more flexible cages), and the influence of chemical stability (that is, reversible imine bonds versus more stable amine and amide cages). This allowed us to map out the key chemical and structural parameters in the liquid and glass formation for POCs.
Melting and glass behaviour
Characterisation of the various cages by 1H NMR spectroscopy, high resolution mass spectrometry (HRMS), and elemental analysis revealed high purity samples in each case (ESI Fig. 1–10†). All as-synthesised cages were found to pack into amorphous arrangements when isolated by either rapid solvent evaporation or rapid precipitation using solvent exchange, with the exception of AT-RCC3-S, for which the PXRD indicated some crystallinity (ESI Fig. 11–15†). Thermogravimetric analysis (TGA) was then performed after first activating the four samples to remove solvent (see ESI Methods†). Featureless TGA traces were identified, up to decomposition temperatures (Td) in excess of 270 °C (Table 1, ESI Fig. 16–19†), suggesting that essentially all solvent had been removed. DSC was carried out on all samples, revealing complex behaviour upon heating. Specifically, DSC scans of RCC3-S, AT-RCC3-S, nC5-CC, and RCC1b contained unexplained endothermic features prior to thermal decomposition. For example, a broad endotherm was noted for RCC3-S over the temperature range 100–200 °C, before a second, much sharper endotherm at ca. 270 °C occurred, prior to the onset of mass loss (ESI Fig. 20†). The corresponding data for AT-RCC3-S and RCC1b showed endothermic features starting at ca. 130 °C and ca. 230 °C, respectively (ESI Fig. 21 and 22†). The DSC scan for the nC5-CC sample also displays an endotherm, this time at ca. 140–170 °C, immediately followed by a sharp exotherm at 190 °C (ESI Fig. 23†) – alongside being insoluble, subsequent PXRD analysis of the material after this exotherm indicated that the material was still amorphous (ESI Fig. 14†), suggesting that this feature in the DSC may be indicative of conversion of the cage species to an insoluble polymeric network via cross-linking due to the inherent dynamic nature of the imine bonds, prior to the onset of mass loss observed by TGA.| Organic cage | T d/°C | T m/°C | T g/°C | Notes |
|---|---|---|---|---|
| RCC3-S | ca. 300 | 100–200 | 138 | Second sharp endotherm at 270 °C |
| AT-RCC3-S | 350 | 150–250 | 138 | — |
| RCC1b | 300 | 150–250 | 150 | — |
| nC5-CC | 280 | 170–190 | N/A | Sharp exotherm at 190 °C |
To unambiguously identify these endothermic features as melting transitions, Tm, the sample morphology was monitored in melting point apparatus upon heating from room temperature. Melting to macroscale liquid states was observed in each case, at temperatures corresponding to the endothermic features in the DSC scans (Table 1). The melting point ranges were very broad (100 °C range) for the more flexible reduced cages (RCC3-S, AT-RCC3-S, and RCC1b), especially when compared to the more rigid imine cage nC5-CC (20 °C range). This is thought to be due to the amorphous nature of the as-synthesised solids (ESI Fig. 11–14†), with crystalline organic solids traditionally exhibiting much narrower melting points due to the first order transition from crystalline material to glass, especially since the characterisation of the different cages suggested they were of high purity. Since there is no long-range order in the amorphous organic cage solids, and arguably even less order with the more flexible organic cages, the (kinetic) transition from solid to liquid is consistent with the formation of a macroscale liquid state. To illustrate the observed broad melting behaviour, the behaviour of RCC3-S between 100–200 °C is included as a video (ESI Video 1†). Interestingly, continued heating of the liquid formed from RCC3-S led to foaming at 270 °C; that is, at a temperature corresponding to the sharp endothermic feature in the DSC trace (ESI Fig. 20†). As discussed below, this foaming may be due to a thermally induced polymerization reaction.
These observations were then used in a second series of DSC experiments, where the four evacuated samples were heated to temperatures just in excess of their melting point ranges, though below their respective decomposition temperatures and any apparent exotherms. The samples were then cooled back to room temperature, and then reheated once more. The data for RCC3-S show clearly a reversible glass transition at 138 °C (Fig. 2a). Data for AT-RCC3-S and RCC1b were similarly unequivocal, although a glass transition was not observed for nC5-CC (ESI Fig. 24–26). These data are summarized in Table 1.
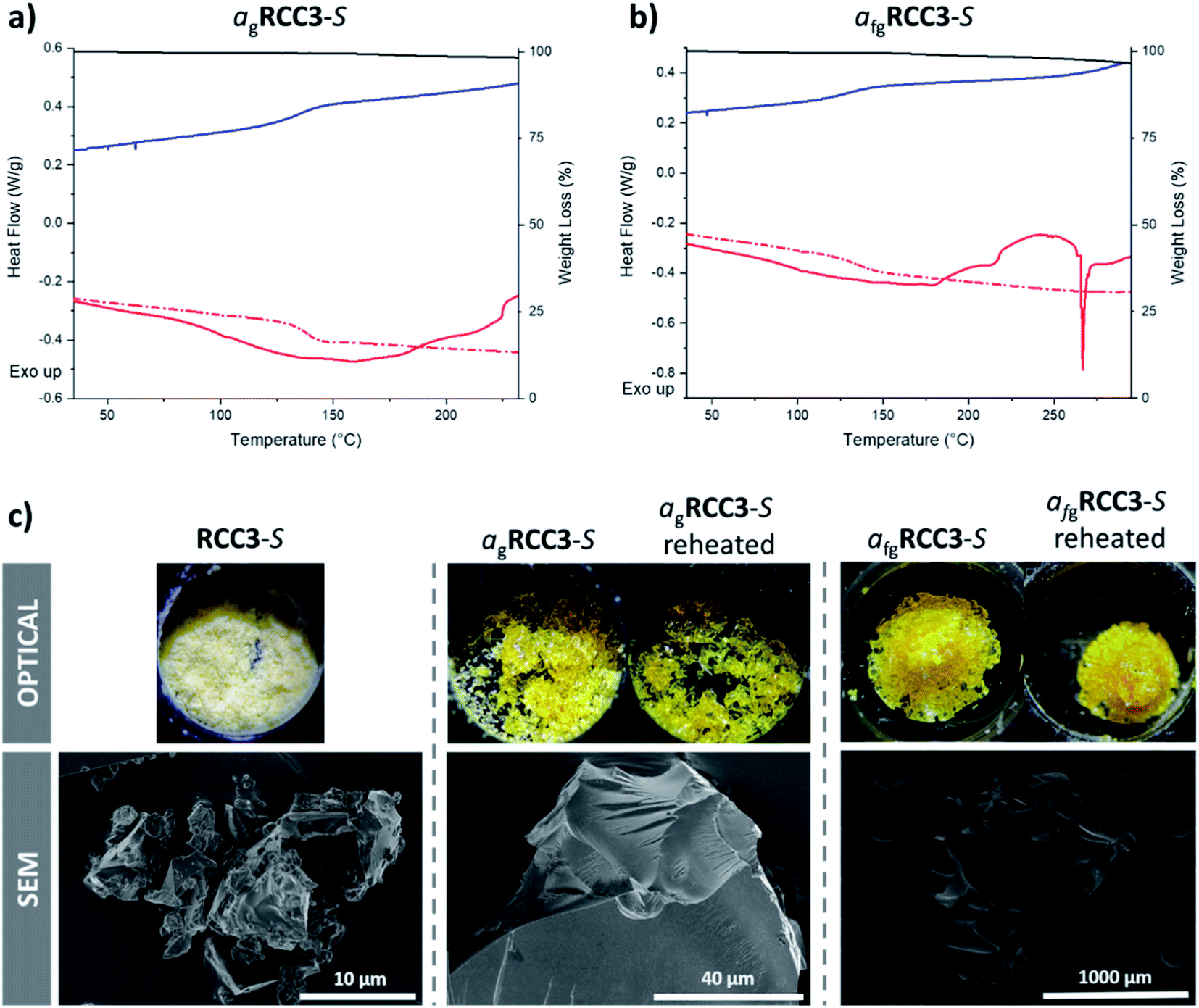 | ||
| Fig. 2 DSC traces and corresponding TGA curve (black) of RCC3-S subjected to heat–cool–heat (HCH) cycles to (a) 250 °C forming agRCC3-S, and (b) 300 °C forming afgRCC3-S. DSC traces: initial upscan shown as a solid red line, downscan as a blue line, and second upscan as a dashed red line. Optical and SEM images of RCC3-S, agRCC3-S, and afgRCC3-S (c). | ||
The curious foaming behaviour of RCC3-S, associated with a sharp endotherm at 270 °C, necessitated investigation of a glass formed from cooling a sample above this temperature (Fig. 2b). A Tg for this foamed glass product was observed at an identical temperature to that of the non-foamed glass; that is, 138 °C.
The higher Tg of RCC1b (150 °C) is ascribed to the sterically bulky external naphthyl-amide functionalisation on the cage (Fig. 1a); the near identical lower Tg values for RCC3-S and AT-RCC3-S (138 °C) can be ascribed to reduced exterior steric bulk. The identical Tg values for RCC3-S and AT-RCC3-S was rationalised by the internal aminal functionalisation in AT-RCC3-S having no effect on phase transitions because it is buried in the cage and not exposed to the surface of the molecule.
Glass structure and properties
The RCC3-S, AT-RCC3-S, and RCC1b samples recovered at room temperature after melting remained amorphous, as did the sample of RCC3-S recovered from the higher temperature (ESI Fig. 11–13†). The nC5-CC sample post-melting, however, displayed Bragg peaks, indicative of crystallisation upon cooling from the liquid state (ESI Fig. 14†). Chemical analysis by 1H NMR spectroscopy (ESI Fig. 27–30†) found the glass state of RCC3-S, and the heated sample of nC5-CC, to be identical to the starting cage materials (ESI Fig. 27 and 30†). While some peak broadening was apparent for the glass state of RCC1b compared to the starting cage material, this may be due to the sterically bulky external naphthyl-amide functionalisation on the cage (ESI Fig. 29†). However, the foamed glass state of RCC3-S was completely insoluble in all solvents tested, suggesting the formation of a polymeric glass (ESI Fig. 27†). Additionally, AT-RCC3-S was also found to be mostly insoluble, but 1H NMR spectroscopy of a small amount of material that dissolved in solution initially suggested that the acetal functionality had been lost and that the material had converted back to RCC3-S (ESI Fig. 28†); however, it has been previously reported that AT-RCC3-S slowly converts back to RCC3-S over time in the same solvents used for 1H NMR analysis.34 Therefore, both the starting (amorphous) and glass phases of AT-RCC3-S were analysed by Raman spectroscopy which indicated that the glass had similar characteristics to the starting cage, confirming that the AT-RCC3-S was still intact and had not converted to RCC3-S (ESI Fig. 31†).Optical images confirmed that all samples underwent appreciable flow during the melting process (Fig. 2c and ESI Fig. 32–34†). RCC3-S, AT-RCC3-S, and RCC1b were all optically transparent after melting, while nC5-CC contained a visible crystalline component, correlating with the observation by PXRD. Appreciable macroscopic differences were noticed between the two samples of RCC3-S cooled from 250 °C and 300 °C: the latter had undergone foaming (Fig. 2c). Scanning electron microscopy (SEM) images were also collected, confirming these observations (Fig. 2c, and ESI Fig. 35–37†). The foaming coincides with conversion of RCC3-S to an insoluble material when it is heated to the higher temperature; this was presumed to be a polymeric network, based on the 1H NMR analysis discussed above. This foaming and the corresponding endotherm observed by DSC is likely due to the loss of a gas, which could potentially be explained by polymerisation occurring with the loss of a gaseous side product such as ethene, compared to exothermic cross-linking to form a polymeric network as observed with nC5-CC. Alternatively, the foaming may be explained by the thermal release of strongly bound gaseous guests, either from within the cage cavities (physisorption) or from a carbamate formed from CO2 binding with the free amines (chemisorption).
In line with previous terminology on the formation of glasses by melt-quenching MOFs,12 the glasses formed here are referred to as agRCC3-S, agAT-RCC3-S, and agRCC1b; the foamed glass is referred to as afgRCC3-S. Given the absence of any observable glass behaviour in nC5-CC, this cage was not investigated further.
High throughput thermal gas adsorption experiments (see Methods†) were carried out on the four glass samples, agRCC3-S, afgRCC3-S, agAT-RCC3-S, and agRCC1b. Initially, CO2 measurements were carried out (Fig. 3a–d, ESI Fig. 38–41†). The quantity of CO2 adsorbed by agRCC3-S, agAT-RCC3-S, and agRCC1b was found to be substantially reduced, albeit not completely lost for agRCC3-S, compared to the amounts adsorbed by the starting amorphous cage materials, demonstrating a decrease in porosity upon glass formation. Subsequent experiments on these glasses, which had been reheated above their respective Tg, indicated that no further reduction in porosity occurred upon crossing the glass transition.
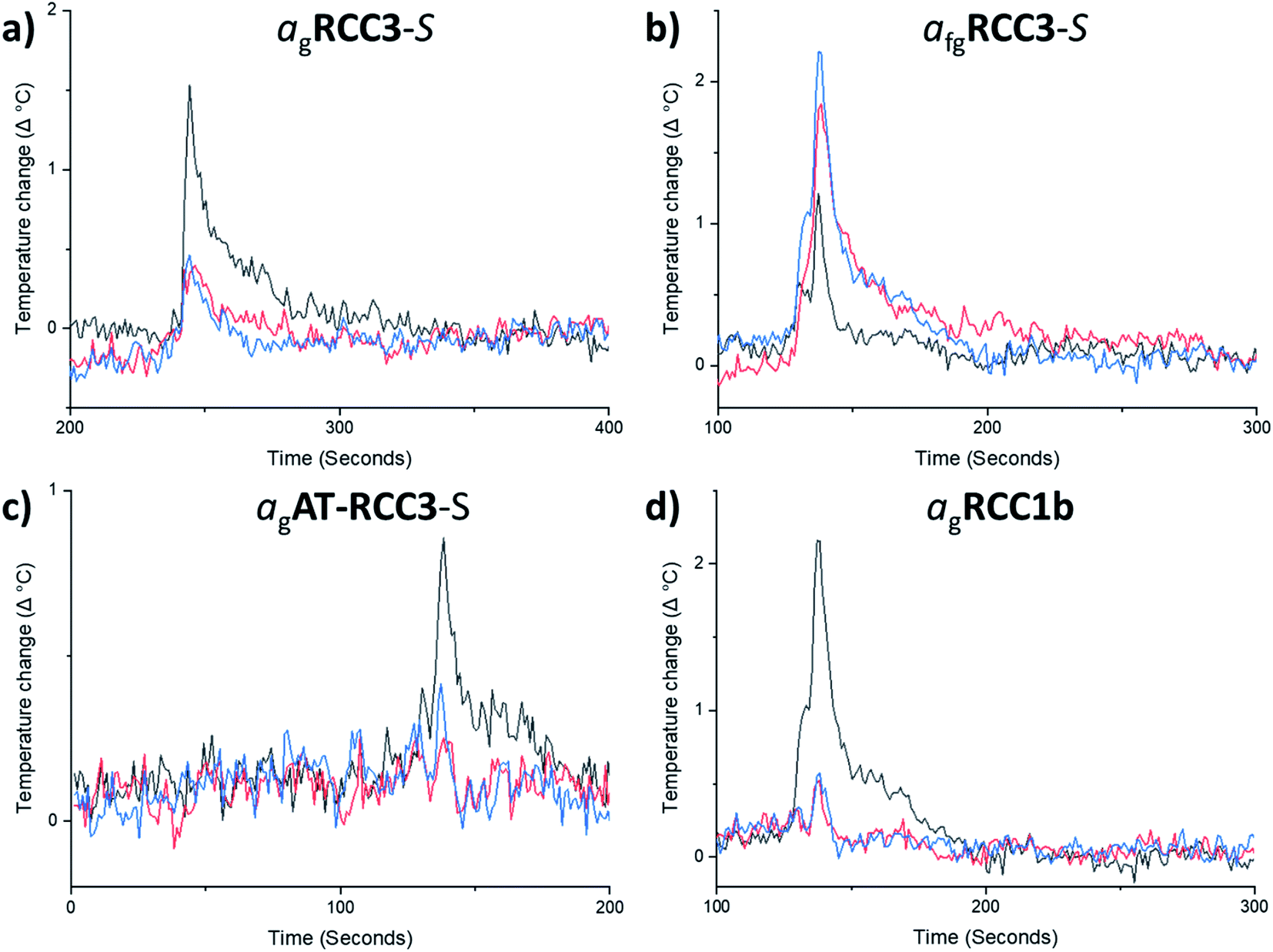 | ||
| Fig. 3 High-throughput thermal CO2 uptake measurements on (a) RCC3-S and agRCC3-S, (b) RCC3-S and afgRCC3-S, (c) AT-RCC3-S and agAT-RCC3-S, (d) RCC1b and agRCC1b. As CO2 is dosed into the evacuated samples of each cage and glass, an increase in temperature indicates the relative porosity in the samples – amorphous solids (black), melt-quenched glasses (red), and reheated glass samples above Tg (blue). The first CO2 addition is shown here for clarity. | ||
By contrast, the quantity of CO2 adsorbed by afgRCC3-S was found to be greater than either RCC3-S or agRCC3-S. In other words, porosity was apparently increased in the foamed glass. This motivated us to probe the uptake of this foamed glass for a range of other gases. CH4, Xe, and nC4H10, were selected because they span a range of kinetic diameters38–40 – CO2 = 3.3 Å, CH4 = 3.8 Å, Xe = 3.96 Å, nC4H10 = 4.3 Å (Fig. 4a–c, ESI Fig. 42–44†). This foamed glass was found to exhibit appreciable uptake of all three gases, although as the kinetic diameter of the gas increased, any difference between the three samples (RCC3-S, afgRCC3-S, and reheated afgRCC3-S) became much less pronounced. Finally, we found that the permanent porosity of the afgRCC3-S sample was retained after 6 weeks, demonstrating the stability of the glass phase (ESI Fig. 45†).
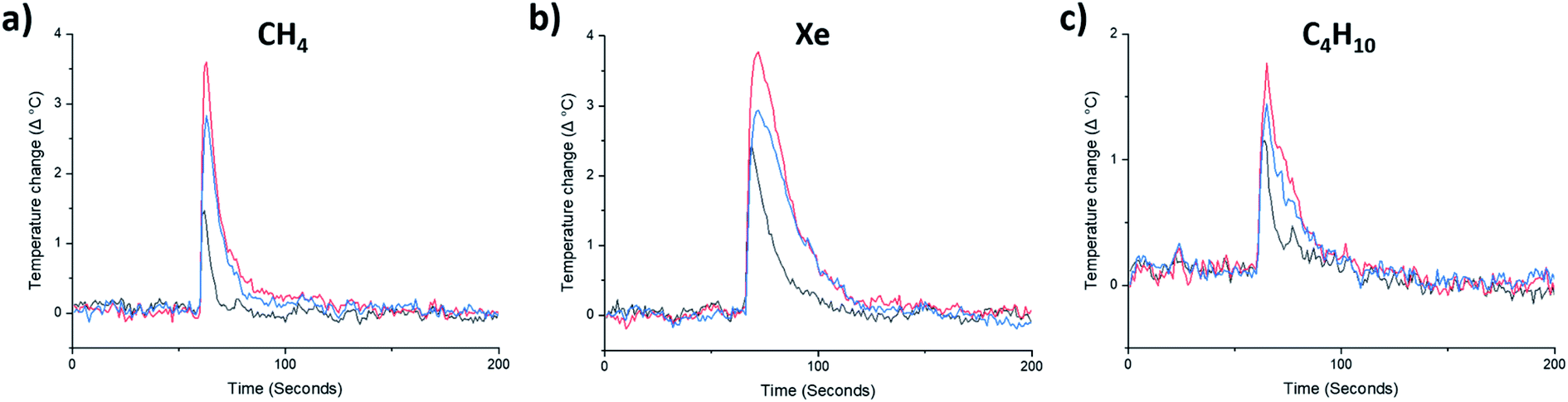 | ||
| Fig. 4 High-throughput gas uptake measurements on RCC3-S and afgRCC3-S: (a) CH4, (b) Xe, and (c) C4H10. As gas is dosed into the evacuated samples of RCC3-S and afgRCC3-S, an increase in temperature indicates the relative porosity of the samples – amorphous solids (RCC3-S, black), melt-quenched glasses (afgRCC3-S, red), and the reheated glass sample above Tg (reheated afgRCC3-S, blue). The first gas addition is shown here for clarity. | ||
Sorption studies
Since the preliminary results of the high-throughput thermal gas uptake screen indicated that afgRCC3-S was porous, the formation of the foamed glass was scaled up for sorption studies. Additionally, for comparison, the non-foamed glass formed from RCC3-S was also scaled up (agRCC3-S) and compared to afgRCC3-S, alongside the parent amorphous solid, RCC3-S. For the scale-up procedure and DSC traces, see ESI Methods and ESI Fig. 46.† Sorption studies were carried out using a range of gases including N2 (77 K), CH4 (273 K), CO2 (273 K), and Xe (273 K) (Fig. 5 and ESI Fig. 47–51†). None of the materials exhibited high BET surfaces areas (RCC3-S 15 m2 g−1, agRCC3-S 25 m2 g−1, afgRCC3-S, 13 m2 g−1), although this is not surprising given the lack of shape-persistence in the RCC3 cage structure, leading to pore collapse as a result of the inherent increased flexibility that occurs on reduction of the imine bonds. Additionally, the pore size distributions were all similar with no major changes observed in the foamed glass, although the total pore volumes indicate that agRCC3-S has the largest pore volume (ESI Fig. 52†), which is also supported by the much larger hysteresis observed in the isotherms for agRCC3-S (Fig. 5). However, while it is difficult to draw quantitative conclusions on the uptake behaviours in these low porosity materials, for CO2 and CH4, both agRCC3-S and afgRCC3-S demonstrated increased uptakes over the starting amorphous cage RCC3-S, albeit to differing degrees (Table 2). In particular, agRCC3-S took up substantially more CH4, even compared to the afgRCC3-S, demonstrating an ∼17 fold increase compared to the starting amorphous cage. This contrasts with the high-throughput gas adsorption screening experiments where that indicated that afgRCC3-S had an apparent increased porosity compared to both RCC3-S and agRCC3-S, at least for CO2. This most likely reflects the semi-quantitative nature of this high-throughput screening approach, since differences in the colour, plate well coverage, and specific heat capacity for each sample can also affect the measured temperature increase. In the case of Xe, both glasses exhibited a reduction in gas uptake compare to RCC3-S: we believe that the Xe uptake in RCC3-S is mostly due to extrinsic porosity that exists between cages in the amorphous material, and this is reduced in the glasses due to the formation of denser amorphous phases.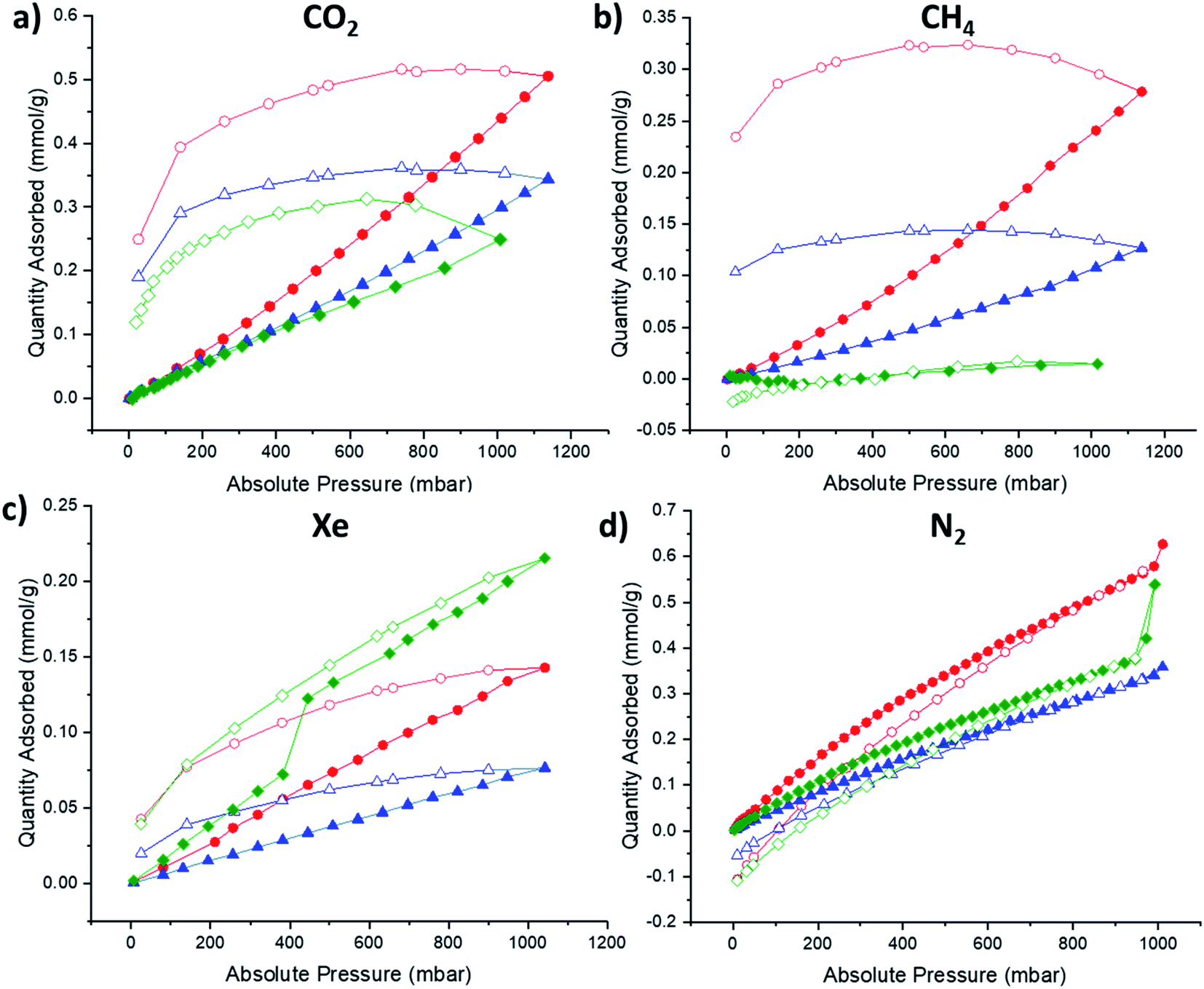 | ||
| Fig. 5 Adsorption (filled) and desorption (empty) isotherms for RCC3-S (green), agRCC3-S (red), afgRCC3-S (blue): (a) CO2, 273 K; (b) CH4, 273 K; (c) Xe, 273 K; (d) N2, 77 K. | ||
| Material | CO2 (mmol g−1) | CH4 (mmol g−1) | Xe (mmol g−1) | N2 (mmol g−1) |
|---|---|---|---|---|
| RCC3-S | 0.25 | 0.014 | 0.22 | 0.54 |
| a g RCC3-S | 0.44 | 0.24 | 0.14 | 0.63 |
| a fg RCC3-S | 0.30 | 0.11 | 0.077 | 0.34 |
While the observed gas uptakes in these melt-quenched organic cage glasses is low, especially when comparing the BET surface areas to crystalline MOFs, COFs, ZIFs, and POCs, which can easily exceed 500 m2 g−1, the uptakes are comparable to previously reported ZIF glasses. For example, agRCC3-S has a CO2 uptake of 9.8 cm3 g−1 (273 K, 1 bar) which is in the same order of magnitude to agZIF-62(Co) (∼17 cm3 g−1, 273 K, 1 bar),41 and a CH4 uptake of 5.4 cm3 g−1 (273 K, 1 bar) which is comparable to that reported for agZIF-76-mbIm (∼6 cm3 g−1, 273 K, 1 bar).42 Therefore the uptake in these materials is promising, especially as future systems could be designed and engineered with the aim of increasing the porosity.
Conclusions
It is possible that most of the observed porosity in the amorphous solid state of these POCs results from extrinsic porosity between cages. In the case of shape-persistent cages, such as nC5-CC, the inherent intrinsic porosity may facilitate connectivity between these extrinsic voids.43 However, for the reduced derivatives RCC3-S, AT-RCC3-S, and RCC1b, the reduction of the imine bonds has previously been reported to increase the flexibility of the structure, thus leading to cavity collapse.34,35 This reduces gas uptake significantly due to the loss of shape-persistence, and any observed porosity is therefore mostly due to the extrinsic cavities.34 The lack of appreciable xenon uptake in the glass samples could therefore be explained by the reduction of extrinsic porosity upon formation of more dense glass phases.The increase in porosity for methane in the two glasses agRCC3-S and afgRCC3-S is encouraging from the point of view of potential separation materials. Polymers of intrinsic microporosity (PIMs) have emerged as an important platform for gas separations,44,45 but to date they have not been processed via the liquid state—they are invariably processed by solution casting, and the solvent used can influence the porosity.46–48 Additionally, while a glass transition for PIM-1 has been reported,49 the glass transition is substantially higher than reported here for these molecular glasses (∼440 °C compared to ∼140 °C for agRCC3-S and afgRCC3-S). The use of meltable organic cages as a glass former could enable casting directly from the liquid state. This has the potential to introduce porosity and also undergo conversion into a more stable and less soluble glass, as in the case of afgRCC3-S. In principle, this could overcome the need for solution processing, both reducing solvent use and providing an alternative approach to the formation of membranes or other separation media—for example, by dip-coating a substrate. In this respect, the lack of high surface areas in these materials may be an advantage, especially for demanding separations of gases with similar kinetic diameters. In addition, if stable liquid porous organic cages can be designed and realised that have a glass transition, as opposed to the flexible reduced organic cages studied here, then melt-quenched porous organic glasses with increased porosity and size-selective pores could be realised. Alternatively, these meltable organic cages could be used to form glass variants of mixed-matrix membranes, where porosity could be induced by introducing a porous material into a non-porous glass to form hybrid materials. In addition to being able to cast directly from the liquid state without the use of solvent, such glasses could also be recyclable and re-castable, opening up a new processing route to molecular glasses with size-selective pores and tuneable porosity. Finally, the formation of POCs, and in particular CC3-S – the precursor to RCC3-S used to form the glasses studied, is highly scalable by both twin-screw extrusion50 and in flow51 from commercially available precursors, which will hopefully enable further research into processing these materials into porous molecular glasses.
Author contributions
M. C. B., F. G., A. K., and R. L. G. carried out the cage synthesis and characterisation. M. C. B. and F. G. carried out the thermal studies, and M. C. B. carried out the property investigation of the solids, liquids, and glasses. B. D. E. carried out the SEM measurements of the solids and glasses. R. C. carried out the sorption experiments. A. I. C., T. D. B., and R. L. G. conceived the project, and the paper was written by R. L. G. and T. D. B. with input from all authors.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank the Engineering and Physical Sciences Research Council (EPSRC) under the grant EP/R005710/1, and for an EPSRC Summer Vacation Bursary at the University of Liverpool. T. D. B. and R. L. G. thank the Royal Society for University Research Fellowships (URF191432 and URF150021). T. D. B. also thanks the Leverhulme Trust for a Philip Leverhulme Prize (2019). We thank Shrey Sharma and Linkam Scientific for the loan of an Optical DSC450 and imaging station. We acknowledge the MicroBioRefinery for assistance with QTOF-MS measurements, and Rachel Kearsey and Alex James (University of Liverpool) for helpful discussions.References
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 Search PubMed.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 Search PubMed.
- A. M. Evans, L. R. Parent, N. C. Flanders, R. P. Bisbey, E. Vitaku, M. S. Kirschner, R. D. Schaller, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Science, 2018, 361, 52–57 Search PubMed.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 Search PubMed.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 Search PubMed.
- J. C. Mauro, C. S. Philip, D. J. Vaughn and M. S. Pambianchi, Int. J. Appl. Glass Sci., 2014, 5, 2–15 Search PubMed.
- A. Carné-Sánchez, G. A. Craig, P. Larpent, T. Hirose, M. Higuchi, S. Kitagawa, K. Matsuda, K. Urayama and S. Furukawa, Nat. Commun., 2018, 9, 2506 Search PubMed.
- S. Kitagawa, Acc. Chem. Res., 2017, 50, 516 Search PubMed.
- A. G. Slater and A. I. Cooper, Science, 2015, 348, 8075 Search PubMed.
- L. Ma, C. J. E. Haynes, A. B. Grommet, A. Walczak, C. C. Parkins, C. M. Doherty, L. Longley, A. Tron, A. R. Stefankiewicz, T. D. Bennett and J. R. Nitschke, Nat. Chem., 2020, 12, 270–275 Search PubMed.
- T. D. Bennett and S. Horike, Nat. Rev. Mater., 2018, 3, 431–440 Search PubMed.
- T. D. Bennett, Y. Yue, P. Li, A. Qiao, H. Tao, N. G. Greaves, T. Richards, G. I. Lampronti, S. A. T. Redfern, F. Blanc, O. K. Farha, J. T. Hupp, A. K. Cheetham and D. A. Keen, J. Am. Chem. Soc., 2016, 138, 3484–3492 Search PubMed.
- T. D. Bennett, J. C. Tan, Y. Yue, E. Baxter, C. Ducati, N. J. Terrill, H. H. M. Yeung, Z. Zhou, W. Chen, S. Henke, A. K. Cheetham and G. N. Greaves, Nat. Commun., 2015, 6, 8079 Search PubMed.
- P. G. Debenedetti and F. H. Stillinger, Nature, 2001, 410, 259–267 Search PubMed.
- C. A. Angell, Science, 1995, 267, 1924–1935 Search PubMed.
- G. Zhang and M. Mastalerz, Chem. Soc. Rev., 2014, 43, 1934–1947 Search PubMed.
- N. Giri, M. G. Del Pópolo, G. Melaugh, R. L. Greenaway, K. Rätzke, T. Koschine, L. Pison, M. F. C. Gomes, A. I. Cooper and S. L. James, Nature, 2015, 527, 216–220 Search PubMed.
- A. I. Cooper, ACS Cent. Sci., 2017, 3, 544–553 Search PubMed.
- T. Hasell, S. Y. Chong, K. E. Jelfs, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2012, 134, 588–598 Search PubMed.
- S. Jiang, K. E. Jelfs, D. Holden, T. Hasell, S. Y. Chong, M. Haranczyk, A. Trewin and A. I. Cooper, J. Am. Chem. Soc., 2013, 135, 17818–17830 Search PubMed.
- A. F. Bushell, P. M. Budd, M. P. Attfield, J. T. A. Jones, T. Hasell, A. I. Cooper, P. Bernardo, F. Bazzarelli, G. Clarizia and J. C. Jansen, Angew. Chem., Int. Ed., 2013, 52, 1253–1256 Search PubMed.
- Q. Song, S. Jiang, T. Hasell, M. Liu, S. Sun, A. K. Cheetham, E. Sivaniah and A. I. Cooper, Adv. Mater., 2016, 28, 2629–2637 Search PubMed.
- N. O'Reilly, N. Giri and S. L. James, Chem.–Eur. J., 2007, 13, 3020–3025 Search PubMed.
- R. L. Greenaway, D. Holden, E. G. B. Eden, A. Stephenson, C. W. Yong, M. J. Bennison, T. Hasell, M. E. Briggs, S. L. James and A. I. Cooper, Chem. Sci., 2017, 8, 2640–2651 Search PubMed.
- R. J. Kearsey, B. M. Alston, M. E. Briggs, R. L. Greenaway and A. I. Cooper, Chem. Sci., 2019, 10, 9454–9465 Search PubMed.
- B. D. Egleston, K. V. Luzyanin, M. C. Brand, R. Clowes, M. E. Briggs, R. L. Greenaway and A. I. Cooper, Angew. Chem., Int. Ed., 2020, 59, 7362–7366 Search PubMed.
- K. Jie, N. Onishi, J. A. Schott, I. Popovs, D. en Jiang, S. Mahurin and S. Dai, Angew. Chem., Int. Ed., 2020, 59, 2268–2272 Search PubMed.
- N. Giri, C. E. Davidson, G. Melaugh, M. G. Del Pópolo, J. T. A. Jones, T. Hasell, A. I. Cooper, P. N. Horton, M. B. Hursthouse and S. L. James, Chem. Sci., 2012, 3, 2153–2157 Search PubMed.
- G. Melaugh, N. Giri, C. E. Davidson, S. L. James and M. G. Del Pópolo, Phys. Chem. Chem. Phys., 2014, 16, 9422–9431 Search PubMed.
- Y. Shirota, J. Mater. Chem., 2000, 10, 1–25 Search PubMed.
- A. Plante, S. Palato, O. Lebel and A. Soldera, J. Mater. Chem. C, 2013, 1, 1037–1042 Search PubMed.
- D. Yang, S. W. Chang and C. K. Ober, J. Mater. Chem., 2006, 16, 1693–1696 Search PubMed.
- S. F. Swallen, K. L. Kearns, M. K. Mapes, Y. S. Kim, R. J. McMahon, M. D. Ediger, T. Wu, L. Yu and S. Satija, Science, 2007, 315, 353–356 Search PubMed.
- M. Liu, M. A. Little, K. E. Jelfs, J. T. A. Jones, M. Schmidtmann, S. Y. Chong, T. Hasell and A. I. Cooper, J. Am. Chem. Soc., 2014, 136, 7583–7586 Search PubMed.
- J. L. Culshaw, G. Cheng, M. Schmidtmann, T. Hasell, M. Liu, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2013, 135, 10007–10010 Search PubMed.
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 Search PubMed.
- M. Mastalerz, Chem. Commun., 2008, 4756–4758 Search PubMed.
- L. M. Robeson, J. Membr. Sci., 1991, 62, 165–185 Search PubMed.
- P. Wollmann, M. Leistner, U. Stoeck, R. Grünker, K. Gedrich, N. Klein, O. Throl, W. Grählert, I. Senkovska, F. Dreisbach and S. Kaskel, Chem. Commun., 2011, 47, 5151–5153 Search PubMed.
- C. G. Saxton, A. Kruth, M. Castro, P. A. Wright and R. F. Howe, Microporous Mesoporous Mater., 2009, 129, 68–73 Search PubMed.
- L. Frentzel-Beyme, M. Kloß, R. Pallach, S. Salamon, H. Moldenhauer, J. Landers, H. Wende, J. Debus and S. Henke, J. Mater. Chem. A, 2019, 7, 985–990 Search PubMed.
- C. Zhou, L. Longley, A. Krajnc, G. J. Smales, A. Qiao, I. Erucar, C. M. Doherty, A. W. Thornton, A. J. Hill, C. W. Ashling, O. T. Qazvini, S. J. Lee, P. A. Chater, N. J. Terrill, A. J. Smith, Y. Yue, G. Mali, D. A. Keen, S. G. Telfer and T. D. Bennett, Nat. Commun., 2018, 9, 5042 Search PubMed.
- E. Berardo, R. L. Greenaway, L. Turcani, B. M. Alston, M. J. Bennison, M. Miklitz, R. Clowes, M. E. Briggs, A. I. Cooper and K. E. Jelfs, Nanoscale, 2018, 10, 22381–22388 Search PubMed.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 Search PubMed.
- P. M. Budd and N. B. McKeown, Polym. Chem., 2010, 1, 63–68 Search PubMed.
- D. Wu, F. Xu, B. Sun, R. Fu, H. He and K. Matyjaszewski, Chem. Rev., 2012, 112, 3959–4015 Search PubMed.
- N. B. McKeown and P. M. Budd, Macromolecules, 2010, 43, 5163–5176 Search PubMed.
- S. Wang, K. Shi, A. Tripathi, U. Chakraborty, G. N. Parsons and S. A. Khan, ACS Appl. Polym. Mater., 2020, 2, 2434–2443 Search PubMed.
- H. Yin, Y. Z. Chua, B. Yang, C. Schick, W. J. Harrison, P. M. Budd, M. Böhning and A. Schönhals, J. Phys. Chem. Lett., 2018, 9, 2003–2008 Search PubMed.
- B. D. Egleston, M. C. Brand, F. Greenwell, M. E. Briggs, S. L. James, A. I. Cooper, D. E. Crawford and R. L. Greenaway, Chem. Sci., 2020, 11, 6582–6589 Search PubMed.
- M. E. Briggs, A. G. Slater, N. Lunt, S. Jiang, M. A. Little, R. L. Greenaway, T. Hasell, C. Battilocchio, S. V. Ley and A. I. Cooper, Chem. Commun., 2015, 51, 17390–17393 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta01906f |
| This journal is © The Royal Society of Chemistry 2021 |