
Uniform synthesis of palladium species confined in a small-pore zeolite via full ion-exchange investigated by cryogenic electron microscopy†
Yongwoo
Kim‡
a,
Jongbaek
Sung‡
 ab,
Sungsu
Kang
ab,
Jaeha
Lee
a,
Min-Ho
Kang
ab,
Sungha
Hwang
a,
Hayoung
Park
ab,
Joodeok
Kim
ab,
Younhwa
Kim
ab,
Eunwon
Lee
a,
Gyeong-Su
Park
c,
Do Heui
Kim
*a and
Jungwon
Park
*ab
ab,
Sungsu
Kang
ab,
Jaeha
Lee
a,
Min-Ho
Kang
ab,
Sungha
Hwang
a,
Hayoung
Park
ab,
Joodeok
Kim
ab,
Younhwa
Kim
ab,
Eunwon
Lee
a,
Gyeong-Su
Park
c,
Do Heui
Kim
*a and
Jungwon
Park
*ab
aSchool of Chemical and Biological Engineering, Institute of Chemical Process, Seoul National University, Seoul 08826, Republic of Korea. E-mail: jungwonpark@snu.ac.kr; dohkim@snu.ac.kr
bCenter for Nanoparticle Research, Institute for Basic Science (IBS), Seoul 08826, Republic of Korea
cDepartment of Materials Science and Engineering, Research Institute of Advanced Materials, Seoul National University, Seoul 08826, Republic of Korea
First published on 26th April 2021
Abstract
Finely dispersing noble metal species with high phase homogeneity in zeolites is crucial to develop an efficient catalyst. However, for palladium-impregnated zeolites, fully utilizing active palladium species in small-pore zeolites with high dispersion is challenging despite the establishment of a general synthetic approach of ion-exchange and subsequent thermal treatment to generate encapsulated nanoparticles. Herein, we achieve full dispersion of isolated Pd2+ ions in a small-pore SSZ-13 zeolite via a controlled ion-exchange process, and successfully generate uniformly dispersed nano-sized PdO clusters in SSZ-13 supported by mechanistic understanding of nanoparticle growth. Direct investigation via cryogenic electron microscopy and ultramicrotomy allows the successful artifact-free imaging of electron-beam-sensitive zeolite-based catalysts, and reveals that the formation of nano-sized PdO clusters during thermal treatment is governed by the rapid nucleation and suppressed particle growth in a confined space. Through fully utilizing active Pd species in SSZ-13 by controlled ion-exchange and rationalized thermal treatment, enhanced catalytic performances toward a passive NOx adsorber and CH4 combustion are achieved.
Introduction
Palladium species encapsulated small-pore zeolites have attracted significant attention owing to their distinctive catalytic performances.1–5 Compared to other zeolites with a larger pore size, small-pore zeolites can stabilize nano-sized noble metal species to show comparably high stability, which is crucial for catalytic reactions under realistic conditions.3,6–8 Particularly, applications of Pd-impregnated small-pore zeolite catalysts for after-treatment and hydrogen generation have been successfully demonstrated.3,6,7 In these cases, various Pd phases such as isolated Pd2+ ions, Pd, and PdO can exist in the zeolite, all of which show different catalytic activities depending on the reaction.3–5,7,9 For example, isolated Pd2+ ions are active for passive NOx adsorbers (PNAs),5,9 while most catalytic reactions including catalytic combustion of CH4 require highly dispersed Pd or PdO nanoparticles for high catalytic activity.7,10 Therefore, generating confined Pd species with high dispersion and with a homogeneous phase in small-pore zeolites is necessary to efficiently utilize the expensive Pd for catalytic applications.Among various synthetic methods to locate noble metal species in zeolites,11–14 introducing noble metal precursors by ion-exchanging them with cationic species of zeolites and subsequently generating nanoparticles via thermal treatment is recognized as a practical method.1 However, in general, poor controllability inherent in both ion-exchange and thermal treatment hinders the efficient utilization of Pd in small-pore zeolites. The unwanted adsorption of precursors on the external surface of the zeolite occurs concurrently with ion-exchange at the internal channel of the zeolite, resulting in the formation of large-sized particles.15,16 For small-pore zeolites such as SSZ-13 (channel size of 0.38 × 0.38 nm2), ion-exchange is further suppressed by the limited accessibility of the zeolite channels, causing a serious problem toward achieving high ion-exchange efficiency.2,17 Furthermore, the formation of nanoparticles from isolated Pd2+ ions during thermal treatment is complicated by multiple processes including the nucleation from the isolated Pd2+ ions and the confined growth of nanoparticles in the nano-sized pores of zeolite, limiting the formation of highly dispersed active Pd species with a homogeneous phase. Direct observation of nanoparticles confined in the zeolite, if enabled, can facilitate the optimization of complex particle growth in this system. Transmission electron microscopy (TEM) is a promising method that ensures microscopic imaging with sufficient spatial resolution,18–20 but the zeolites are generally electron-beam sensitive, exhibiting a collapse of microstructures when they are exposed to an electron-beam used in typical TEM.21–23 In particular, small-pore zeolites such as SSZ-13 are vulnerable to electron-beam-induced degradation due to their weak mechanical strength and low framework density.24,25 Furthermore, the deteriorated image resolution of the nanoparticles embedded in the zeolite also hinders the application of conventional TEM in resolving particle formation in zeolites.21,22 Hence, understanding the ion-exchange process and the detailed growth behavior of Pd species in the zeolite, ideally by direct observation with an advanced TEM technique, is required to achieve the efficient synthesis of highly dispersed Pd species with high phase homogeneity for catalytic applications.
Herein, the fully isolated Pd2+ ions in SSZ-13 is synthesized by achieving full ion-exchange efficiency. Thereafter, the generation and growth behavior of the nano-sized PdO clusters in SSZ-13 are directly investigated through a combination of cryogenic scanning transmission electron microscopy (cryo-STEM) and ultramicrotomy with minimized electron-beam induced damage. The obtained mechanistic understanding allows successful preparation of uniformly dispersed nano-sized PdO clusters confined in a small-pore SSZ-13 zeolite from fully isolated Pd2+ ions, by controlling the thermal treatment that induces rapid nucleation and suppressed nanoparticle growth. The enhanced PNA and CH4 combustion performances highlight the importance of our method comprising full ion-exchange efficiency and rationalized nanoparticle growth for Pd-impregnated zeolite catalysts.
Result and discussion
SSZ-13 with a low Si/Al ratio of 4.5, determined by 29Si magic angle spinning nuclear magnetic resonance (Si-MAS-NMR), was synthesized for ensuring a sufficient amount of Al-sites to stabilize isolated Pd2+ ions (Fig. S1 and Table S1†).26,27 The typical CHA-structure of synthesized SSZ-13 is confirmed by X-ray diffraction (XRD) (Fig. S2†). The extent of isolated Pd2+ ions in SSZ-13 is affected in the ion-exchange process between the Pd precursor and SSZ-13 in the aqueous solution. We prepared various Pd-exchanged SSZ-13 samples using different combinations of water-soluble Pd precursors (Pd(NO3)2 and Pd(NH3)4Cl2) and zeolite-forms (H-SSZ-13 and NH4-SSZ-13) (Experimental section and Table S2†). Among the as-prepared samples, only the sample prepared with Pd(NH3)4Cl2 and NH4-SSZ-13 does not show any clustered Pd precursors on the external surface of the zeolite, whereas a large amount of clustered Pd precursors are commonly observed on the external surface of the zeolite for samples prepared with either Pd(NO3)2 or H-SSZ-13, as shown in TEM images in Fig. 1a (Fig. S3†). The state of the Pd-ionic complex in the precursor solution immensely affects the dispersion of Pd precursors in SSZ-13, because the Pd-ionic complex is exchanged with H+ or NH4+ of SSZ-13. The Pd-ionic complex in the Pd(NH3)4Cl2 solution exists as a positively charged form of [Pd(NH3)4]2+,28 whereas Pd(NO3)2 solution contains negatively charged species,29 as confirmed by the UV-vis spectra in Fig. S4.† During ion-exchange in the aqueous solution, the negative charge of the Pd-ionic complex in Pd(NO3)2 solution hinders diffusion into the negatively charged zeolite channel and ion-exchange with positively charged H+ or NH4+.11 In addition, Pd-hydrate complexes can exist in the Pd(NO3)2 solution, and prevent ion-exchange due to its large hydrated radius.30 The XPS analysis of the as-prepared samples before the calcination step also indicate that the surface Pd/Al atomic ratios of samples prepared with Pd(NO3)2 are much higher than those of samples prepared with Pd(NH3)4Cl2, as listed in Table S3.† This indicates that the Pd-ionic complex in Pd(NH3)4Cl2 solution can penetrate into SSZ-13 much better than that in Pd(NO3)2 solution. Furthermore, the similar level between the surface Pd/Al atomic ratios and the bulk Pd/Al atomic ratios of the samples prepared with Pd(NH3)4Cl2 demonstrates that the Pd precursors were well-distributed through SSZ-13, but not concentrated on the surface (Table S3†). The zeolite-form also affects the ion-exchange efficiency. The ion-exchange equilibrium between the Pd2+ ions and H-form (or NH4-form) zeolite is as follows:| Pd2+ + 2Al–OH (or 2Al–ONH4) ↔ 2(Al–O)Pd + 2H+ (or 2NH4+) |
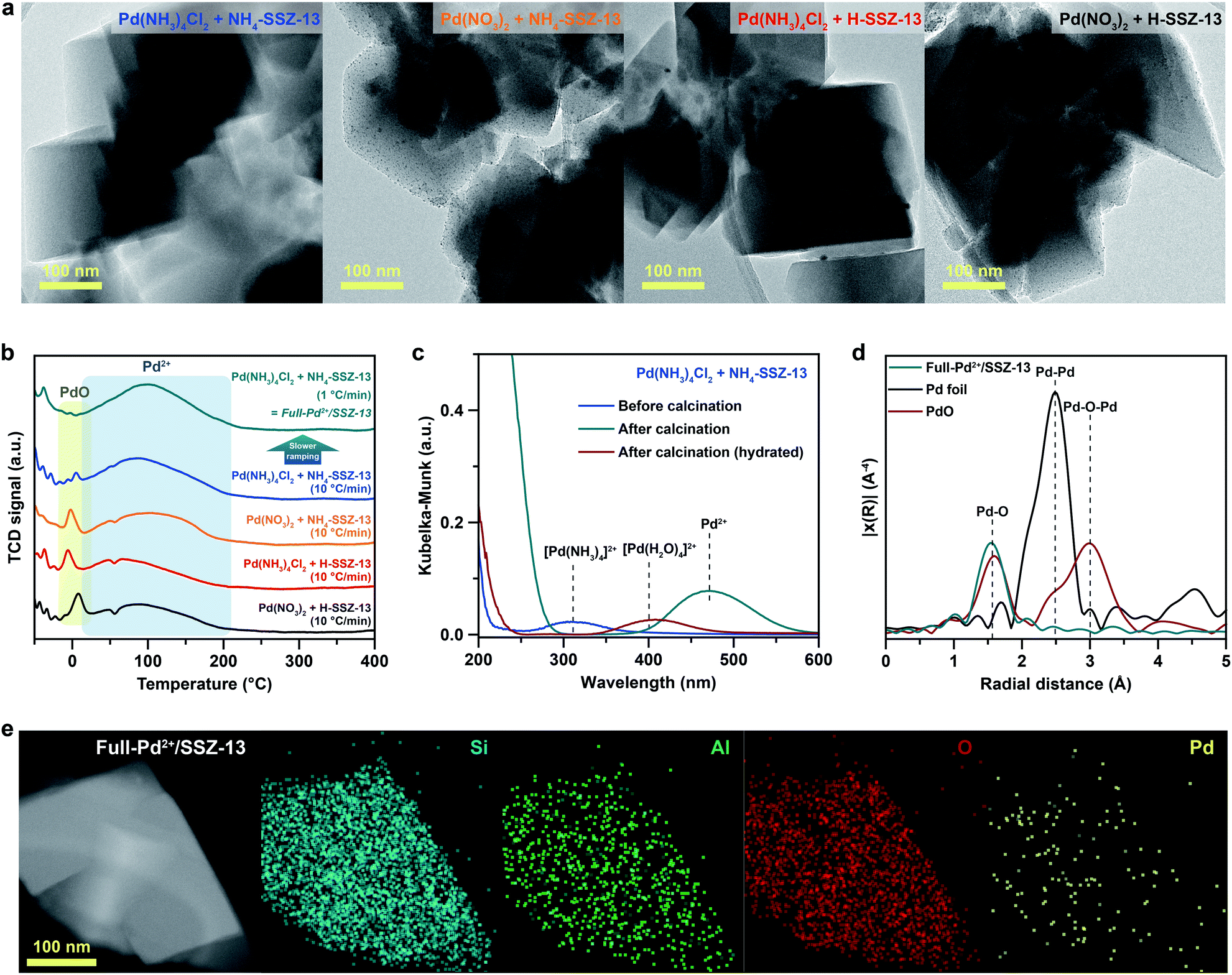 | ||
| Fig. 1 (a) TEM images of Pd-exchanged SSZ-13 samples prepared with different combinations of Pd precursors and zeolite-forms, before calcination. (b) Cryo-H2-TPR profiles of the Pd-exchanged SSZ-13 samples prepared using different combinations of precursors, zeolite-forms, and ramping rates for calcination. (c) Solid-state UV-vis spectra of Pd-exchanged SSZ-13 samples prepared with Pd(NH3)4Cl2 and NH4-SSZ-13, before and after calcination. (d) k3-weighted Fourier-transformed EXAFS spectra of Full-Pd2+/SSZ-13, Pd foil, and PdO reference. (e) Cryogenic HAADF-STEM image and EDS maps of Full-Pd2+/SSZ-13. | ||
Based on Le Chatelier's principle, the acidic environment in the zeolite limits the shift of the equilibrium to the right due to the formation of H+.5 Meanwhile, since the formation of NH4+ is less affected by the acidic environment, the ion-exchange of the NH4-form zeolite is facilitated.31
The calcination step that decomposes the ion-exchanged Pd precursors also affects the extent of isolated Pd2+ ions. The cryogenic H2 temperature programmed reduction (cryo-H2-TPR) profile of Pd-exchanged SSZ-13 samples evaluates the extent of isolated Pd2+ ions in SSZ-13 after calcination with a ramping rate of 10 °C min−1, as shown in Fig. 1b.30,32,33 The broad peak at approximately 100 °C arises from the reduction of the isolated Pd2+ ions, whereas the sharp peak at approximately 0 °C originates from the reduction of PdO that is not ion-exchanged.30,32,33 All Pd-exchanged SSZ-13 samples contain a large peak of isolated Pd2+ ions due to the sufficient amount of Al-sites, whereas the peak intensity of PdO varies according to a combination of precursors and zeolite-forms. Pd-exchanged SSZ-13 prepared using Pd(NH3)4Cl2 and NH4-SSZ-13 (the sample with a negligible amount of clustered Pd precursors in Fig. 1a) contains a trace amount of PdO. Conversely, a notable amount of PdO was observed in samples prepared using either Pd(NO3)2 or H-SSZ-13, indicating that the application of Pd(NH3)4Cl2 and NH4-SSZ-13 provides the highest ion-exchange efficiency.
To maintain the full ion-exchange efficiency of Pd2+ ions even after the calcination step, the Pd-exchanged SSZ-13 sample prepared with Pd(NH3)4Cl2 and NH4-SSZ-13 were calcined with different ramping rates. The cryo-H2-TPR profiles of these samples demonstrate that the intensity of the PdO peak increases with increasing the ramping rate for calcination (Fig. S5†). According to the solid-state UV-vis analysis in Fig. 1c, the Pd species in the Pd-exchanged SSZ-13 prepared using Pd(NH3)4Cl2 and NH4-SSZ-13 exist as Pd(NH3)42+ before the calcination, indicating that the calcination step decomposes Pd(NH3)42+ species into isolated Pd2+ ions. However, when the ramping rate for calcination is too fast, the unstable Pd2+ ions, decomposed from Pd(NH3)42+ species, can assemble to form PdO particles instead of interacting with Al-sites of SSZ-13. Hence, it can be deduced that the slow ramping rate is required to ensure sufficient time for the Pd2+ ions to be stabilized at Al-sites thereby suppressing the agglomeration of the Pd2+ ions. We also note that the redispersion of Pd species during the calcination can contribute to generating isolated Pd2+ ions,34 but for the case of the Pd-exchanged SSZ-13 sample prepared with Pd(NH3)4Cl2 and NH4-SSZ-13, the redispersion of surface Pd species into the internal zeolite was less noticeable since the Pd species had already been well-distributed through the SSZ-13 (Fig. 1b and Table S3†).
Based on the understanding of the formation of isolated Pd2+ ions through ion-exchange in aqueous solution and the subsequent calcination step, Pd-exchanged SSZ-13 with full ion-exchange efficiency (denoted as Full-Pd2+/SSZ-13) was prepared using Pd(NH3)4Cl2 and NH4-SSZ-13, and applying a slow ramping rate of 1 °C min−1 during calcination. The fully isolated Pd2+ ions in the Full-Pd2+/SSZ-13 was confirmed by the absence of the PdO peak in the cryo-H2-TPR profile, as shown in Fig. 1b. In addition, the extended X-ray absorption fine structure (EXAFS) spectrum of Full-Pd2+/SSZ-13 demonstrates one major peak at a radial distance of 1.6 Å corresponding to the Pd–O bond, as displayed in Fig. 1d. The absence of the Pd–Pd and Pd–O–Pd peaks in the EXAFS spectrum indicates that the Pd species thoroughly exists as isolated Pd2+ ions.5,30,35 The TEM image of Full-Pd2+/SSZ-13 demonstrates none of the PdO particles on the external surface of the zeolite, whereas the agglomerated PdO particles are present in other Pd-exchanged SSZ-13 samples with insufficient ion-exchange efficiency, as shown in Fig. S6.† The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image and energy dispersive X-ray spectroscopy (EDS) maps of Full-Pd2+/SSZ-13 confirmed the presence of well-dispersed Pd2+ ions, as illustrated in Fig. 1e. We emphasize that the full ion-exchange efficiency of Pd2+ ions in Full-Pd2+/SSZ-13 can be achieved when the choice of precursor combination and the ramping rate for calcination are optimized (Fig. S7†).
In order to control the generation of nano-sized PdO clusters from isolated Pd2+ ions, Full-Pd2+/SSZ-13 was treated with H2 at 700 °C for different times of 1, 5, 10, and 60 min. Subsequently, the catalysts were gently oxidized to transform the metallic Pd into PdO, an active species for CH4 combustion. Note that the differences in the crystallinity and the Brunauer–Emmett–Teller (BET) surface area between the thermal-treated Pd/SSZ-13 catalysts with different H2 treatment times were negligible (Fig. S8†).
Cryo-STEM coupled with ultramicrotomy was utilized to directly observe the generated Pd species confined in the SSZ-13 zeolite. Microscopic observation of the nano-sized Pd species located at the internal site of the zeolite is usually hindered due to the deteriorated image resolution caused by the surrounding zeolite particle. In addition, both the metal species and the zeolites are prone to electron-beam induced damage, resulting in unwanted agglomeration of metal species and radiolysis of the zeolite during TEM imaging.5,22,25 In particular, since the SSZ-13 used in this study has a relatively low framework density of 15.1 T/1000 Å3,36 and also has high Al content that easily traps water,23,30 the structural degradation of SSZ-13 zeolite during electron-beam irradiation is severe during conventional TEM imaging (Movie S1†).21 To overcome these obstacles, we ultramicrotomed Pd/SSZ-13 samples into ∼50 nm thick sections to reveal the internal Pd species in the zeolite.37,38 Thereafter, to minimize the electron-beam induced damage during imaging while obtaining high Z-contrast, the ultramicrotomed sections were observed under the STEM mode at a cryogenic temperature of −180 °C.25,39–41 The overall process of ultramicrotomy and cryo-STEM imaging is illustrated in Fig. 2a and b. The undesired electron-beam effect is further avoided by applying minimal scanning during cryo-STEM imaging. We emphasize that this method ensures the observation of nano-sized clusters confined in the internal site of the high-Al and small-pore zeolite without structural ambiguity, while preventing electron-beam induced artifacts (Fig. S9 and Movie S2†).
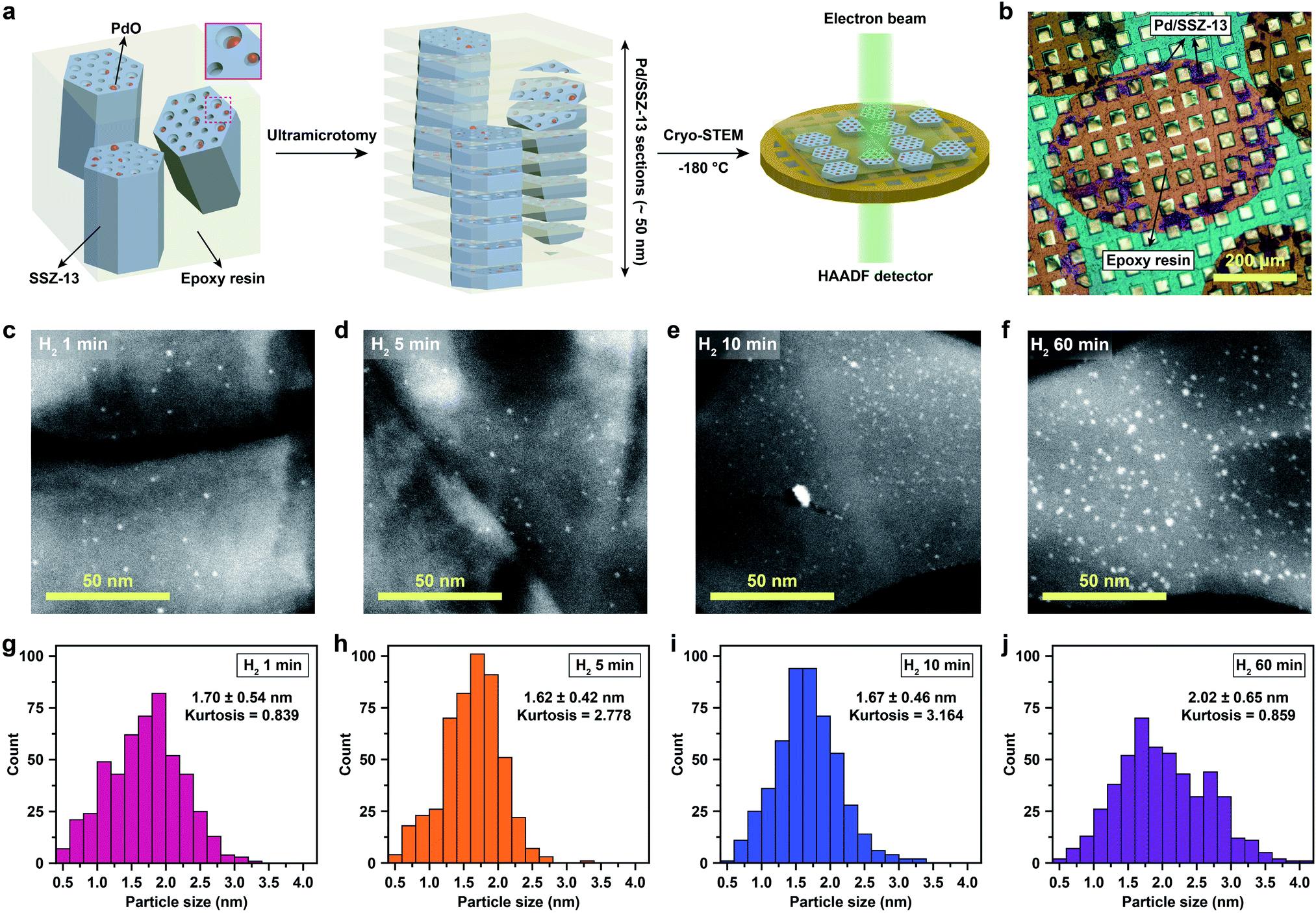 | ||
| Fig. 2 (a) Illustration of ultramicrotomy and cryo-STEM imaging. (b) Optical microscope image of the ultramicrotomed Pd/SSZ-13 catalyst on the TEM grid. (c)–(f) Representative cryo-STEM images of thermal-treated Pd/SSZ-13 catalysts with H2 treatment times of (c) 1 min, (d) 5 min, (e) 10 min, and (f) 60 min. (g)–(j) Size distribution of the nano-sized PdO clusters for the thermal-treated Pd/SSZ-13 catalysts with H2 treatment times of (g) 1 min, (h) 5 min, (i) 10 min, and (j) 60 min. | ||
The cryo-STEM imaging of the thermal-treated Pd/SSZ-13 catalysts with different H2 treatment times elucidates the structural evolution of the nano-sized PdO clusters in the zeolite during thermal treatment. Owing to the full ion-exchange and the successful minimization of the electron-beam induced damage, the Pd species are unobservable in the cryo-STEM images of the ultramicrotomed Full-Pd2+/SSZ-13 before the thermal treatment (Fig. S10 and S11†).5 The observation of the well-dispersed nano-sized PdO clusters in the thermal-treated Pd/SSZ-13 catalysts indicates that thermal treatment successfully generated nano-sized PdO clusters confined in SSZ-13 (Fig. 2c–f). Furthermore, the majority of the nano-sized PdO clusters are observed in the interior site of the ultramicrotomed Pd/SSZ-13 sections, indicating that the nano-sized PdO clusters predominantly exist in the internal pores of the SSZ-13 zeolite.42 However, dispersion of the generated nano-sized PdO clusters varied with the H2 treatment time, as shown in Fig. 2c–f. We obtained the size distribution of nano-sized PdO clusters in each sample by quantifying 500 nanoparticles locating at the internal pores of zeolite without ambiguity of their locations, such as the external surface of the zeolite, as shown in Fig. 2g–j. Notably, the average particle size of a 1 min H2-treated sample is 1.70 ± 0.54 nm and the values are almost stable for 5 min and 10 min of H2 treatment with slightly reduced values of 1.62 ± 0.42 nm and 1.67 ± 0.46 nm, respectively. The average particle size slowly increases to 2.02 ± 0.65 nm after 60 min of H2 treatment owing to particle growth by the sintering process, which is not as significant as expected for the sintering in typical supported catalyst systems.43,44 We note that the average particle sizes in Fig. 2g–j are similar to the average pore diameter of SSZ-13 (1.8 nm), obtained from Ar adsorption–desorption measurements (Fig. S12 and Table S1†). This non-typical growth behavior of the PdO nanoparticles is also depicted in the overall sharpness of size distribution, as indicated by the kurtosis value that statistically represents the heaviness of tail in the size distribution (Fig. 2g–j). Although the size distributions of Pd/SSZ-13 treated with H2 for 1 min and 60 min are broad, indicated by a relatively low kurtosis value of 0.839 and 0.859 respectively, those treated with H2 for 5 min and 10 min show focused size distributions with a high kurtosis value above 2.7.
Nanoparticle growth in supported catalyst systems generally shows an increase in both the average size and the broadness of size distribution, mainly attributed to Ostwald ripening.45,46 However, the nanoparticle growth observed in Fig. 2g–j shows a size-focusing behavior at moderate H2 treatment times of 5 min and 10 min. The cryo-H2-TPR profiles of the thermal-treated Pd/SSZ-13 catalysts indicate that a significant amount of Pd2+ ions remained after 1 min of H2 treatment and disappeared after 5 min, as represented in Fig. 3a. These observations imply that the nucleation of isolated Pd2+ ions rapidly occurs and reaches the full conversion of Pd2+ ions during the first few minutes of H2 treatment. In our study, the observed growth begins from Full-Pd2+/SSZ-13 with a high concentration of well-dispersed isolated Pd2+ ions, and therefore, homogeneous nucleation of isolated Pd2+ ions is likely to occur and the growth pattern deviates from that of typical supported catalyst systems. Furthermore, the subsequent growth of the generated PdO nanoparticles is relatively sluggish by showing only a 0.32 nm increase in the average size from 1 min to 60 min of H2 treatment (Fig. 2g and j). Such suppressed nanoparticle growth behaviour would be attributed to the confined environment by the surrounding zeolite, because our thermal treatment includes harsh reduction conditions that would otherwise cause severe Pd sintering.47–49 Therefore, combining the effect of rapid nucleation and suppressed nanoparticle growth, the generation of nano-sized PdO clusters with high size-uniformity was achieved by a H2 treatment of 5 min. Note that the rapid nucleation and size-focusing behavior is also observed from nanoparticle growth that includes a burst nucleation of monomers after overcoming the critical monomer concentrations.50,51
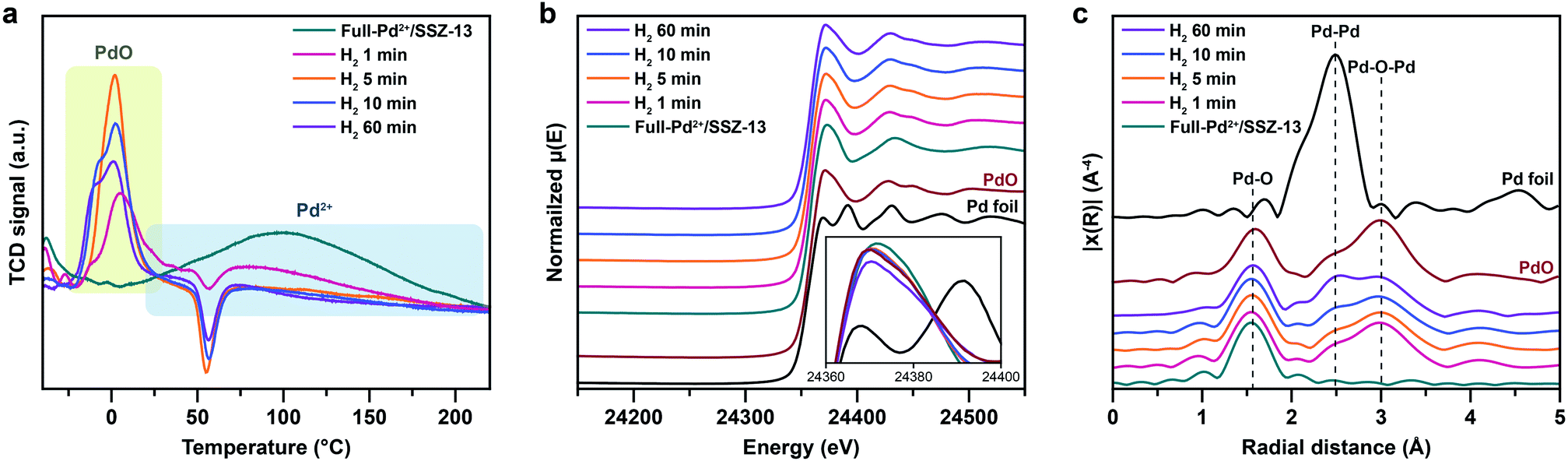 | ||
| Fig. 3 (a) Cryo-H2-TPR profiles, (b) normalized XANES spectra and (c) k3-weighted Fourier-transformed EXAFS spectra of Full-Pd2+/SSZ-13 and thermal-treated Pd/SSZ-13 catalysts with different H2 treatment times. | ||
We further confirmed that the synthesis process with full ion-exchange and an optimized H2 treatment time of 5 min ensures the minimized generation of different Pd phases at unwanted locations other than the internal pore sites of the zeolite, thus resulting in a formation of nano-sized PdO clusters with high phase homogeneity. The inhomogeneity of PdO was observed for the thermal-treated Pd/SSZ-13 catalysts with 10 min and 60 min of H2 treatment, as indicated by the bimodal PdO peak in the cryo-H2-TPR profiles in Fig. 3a. The X-ray absorption near edge structure (XANES) spectra of most of the thermal-treated Pd/SSZ-13 catalysts were similar to those of the PdO reference, whereas the thermal-treated Pd/SSZ-13 catalyst with 60 min of H2 treatment was partially oxidized, as indicated by the reduced white line intensity in Fig. 3b. A consistent result was shown in the EXAFS spectra demonstrated in Fig. 3c. The thermal-treated Pd/SSZ-13 catalysts with 1 min and 5 min of H2 treatment exhibited an atomic structure similar to that of the PdO reference, whereas those with further H2 treatment show development of the Pd–Pd peak at approximately 2.5 Å, particularly for the Pd/SSZ-13 catalyst with 60 min of H2 treatment. In addition, the cryo-STEM images of ultramicrotomed Pd/SSZ-13 indicate the existence of internal voids, as shown in Fig. 4a. Along with the external surface of the zeolite, these internal voids inherently existing in SSZ-13 can serve as agglomeration sites for Pd species during thermal treatment, resulting in the gradual development of large-sized internal Pd species with extended H2 treatment times (Fig. S13†). We suspect that the partially oxidized Pd species are formed at the internal void where full oxidation of Pd can be suppressed due to limited volumetric expansion of large-sized Pd species. Indeed, the aberration-corrected TEM analysis of the internal void species revealed that different phases such as Pd and PdO2, thermodynamically unfavorable phases in an oxidative environment, coexist along with PdO at this site, as illustrated in Fig. 4b and c (Fig. S14 and S15†).52–54 However, a unimodal PdO peak in the cryo-H2-TPR profile and the negligible development of Pd species at internal voids were observed for thermal-treated Pd/SSZ-13 catalyst prepared from Full-Pd2+/SSZ-13 and with 5 min of H2 treatment, as demonstrated in Fig. 3a and S12.† Consequently, only the sample prepared with full ion-exchange efficiency and optimized H2 treatment of 5 min enables the uniform generation of nano-sized PdO clusters with high phase homogeneiety.
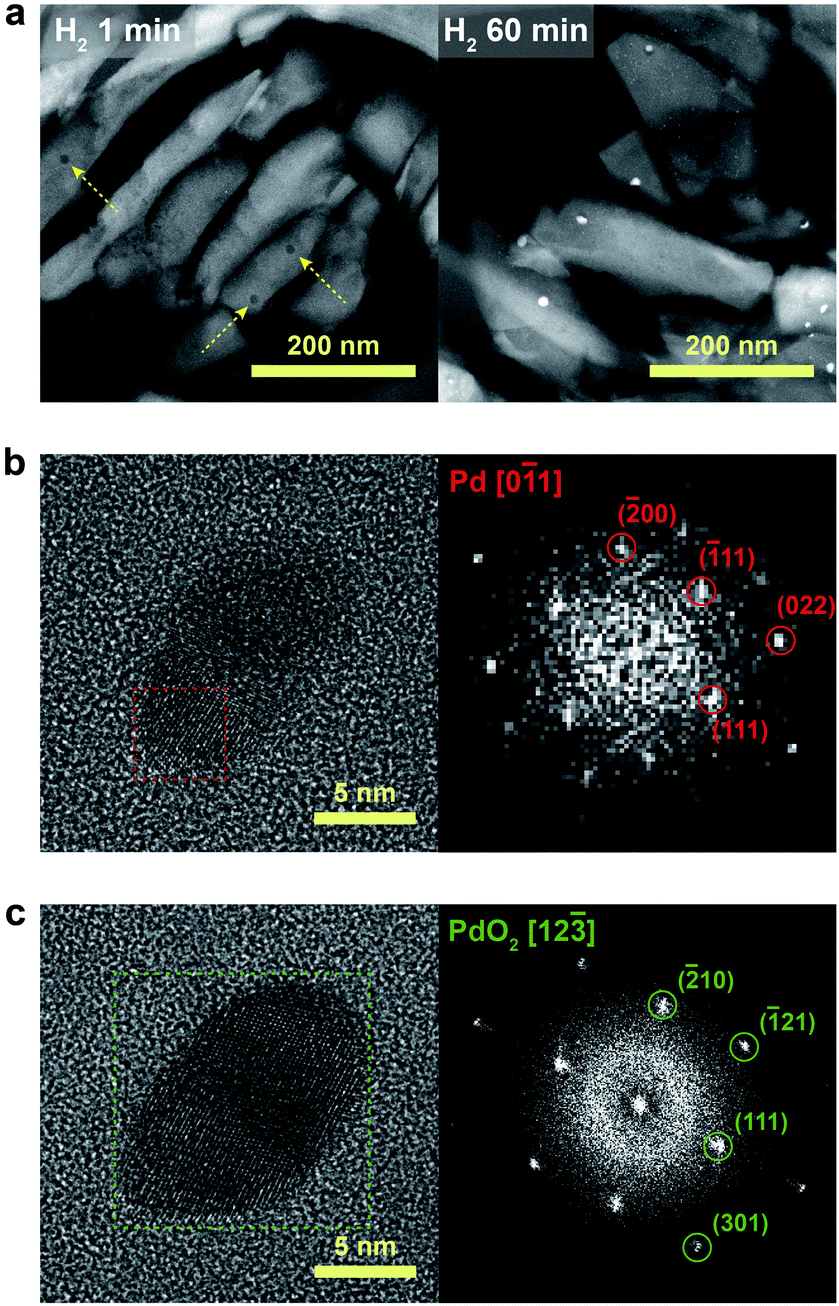 | ||
| Fig. 4 (a) Cryo-STEM images of ultramicrotomed Pd/SSZ-13 catalysts with H2 treatment times of 1 min and 60 min. Yellow arrows indicate the internal void sites. (b) and (c) Aberration-corrected TEM images and the selected-area fast Fourier transform (FFT) patterns of (b) Pd and (c) PdO2 located at the internal void sites of the 60 min H2-treated Pd/SSZ-13 catalyst. | ||
The PNA and CH4 combustion performances of diverse Pd/SSZ-13 catalysts consisting either isolated Pd2+ ions or nano-sized PdO clusters are evaluated to elucidate the advantage of full ion-exchange efficiency and rationalized thermal treatment. Since the isolated Pd2+ ions are active to adsorb NOx, the PNA ability of the Pd/SSZ-13 catalysts before the thermal treatment was tested. Among the various samples prepared with different combinations of precursors, zeolite-forms, and ramping rates for calcination, Full-Pd2+/SSZ-13 demonstrates the highest PNA ability, as this catalyst fully utilizes Pd species as isolated Pd2+ ions (Fig. 5a). Specifically, the amount of NO adsorbed on Full-Pd2+/SSZ-13 is 68.2 μmol gcat−1, corresponding to 0.8 NO/Pd ratio. This is a comparably high value, as demonstrated in our previous research.30 It should be noted that CO was added in a reactant feed since CO facilitates the adsorption of NO on Pd/SSZ-13 by forming NO–Pd–CO complexes41 (Fig. S16†). Meanwhile, CH4 hardly adsorbed on Pd/SSZ-13 (Fig. S16†). Meanwhile, the CH4 combustion activity of Full-Pd2+/SSZ-13 and the thermal-treated Pd/SSZ-13 catalysts prepared with full ion-exchange efficiency and with different H2 treatment times is displayed with light-off curves in Fig. 5b. Full-Pd2+/SSZ-13 exhibits a significantly lower catalytic activity compared to the thermal-treated Pd/SSZ-13 catalysts, indicating that the formation of PdO nanoparticles is required for CH4 combustion. Importantly, the thermal-treated Pd/SSZ-13 catalyst prepared with full ion-exchange efficiency and with 5 min of H2 treatment shows the highest CH4 combustion activity. The CH4 combustion activation barriers of the thermal-treated Pd/SSZ-13 catalysts range from 142 to 148 kJ mol−1, consistent with the reported values of PdO in the Pd-impregnated zeolite catalysts (139–158 kJ mol−1), whereas that of Full-Pd2+/SSZ-13 is 241.7 kJ mol−1, as displayed in Fig. 5c.7,16 The similar activation barriers of the thermal-treated Pd/SSZ-13 catalysts indicate that the different activities between them are not attributed to the thermodynamic barrier. Considering that CH4 combustion over the Pd-impregnated zeolite is reported to be a structure-insensitive reaction,7,55,56 the different activities are ascribed to the phase homogeneity of Pd species as a PdO phase, and the dispersion of generated nano-sized PdO clusters. This explains our observation that the thermal-treated Pd/SSZ-13 catalyst with 5 min of H2 treatment shows the most prominent CH4 combustion activity due to the full conversion of isolated Pd2+ ions and the generation of size-focused nano-sized PdO clusters with high phase homogeneity. The low CH4 combustion activity of the 1 min H2-treated Pd/SSZ-13 catalyst can be explained by the insufficient conversion of isolated Pd2+ ions. Due to the similar size of PdO clusters in 5 min and 10 min of H2 treatment, the CH4 combustion activity of the 10 min H2-treated Pd/SSZ-13 catalyst is similar to that of the 5 min H2-treated Pd/SSZ-13 catalyst, but a minor reduction in 10 min was observed due to some agglomeration of Pd at the void site shown in Fig. S13.† Meanwhile, the increase of particle size and the development of unwanted Pd phases at the internal void site were clearly observed in the 60 min H2-treated Pd/SSZ-13 catalyst, which explains the significantly reduced activity after 60 min of H2 treatment. We also note that the CH4 combustion activity of various Pd-exchanged SSZ-13 catalysts without thermal treatment (i.e. samples right after the calcination) shown in Fig. S17† is significantly low, compared to that of thermal-treated Pd/SSZ-13 catalysts shown in Fig. 5b.
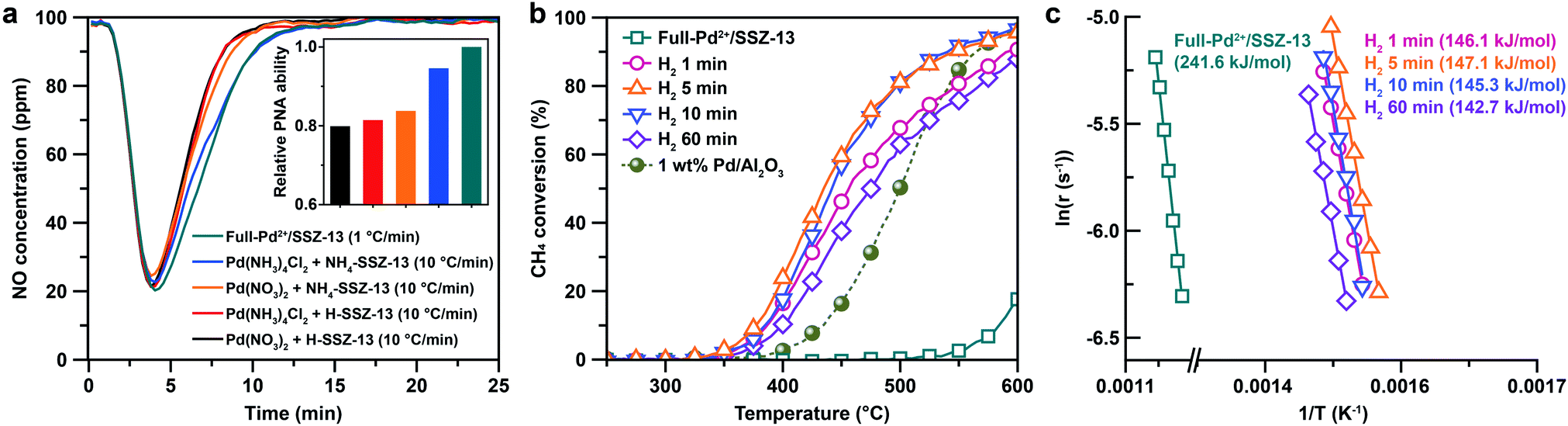 | ||
| Fig. 5 (a) PNA ability of Pd-exchanged SSZ-13 catalysts prepared with different combinations of the Pd precursors, zeolite-forms, and ramping rates. (b) CH4 combustion light-off curves and (c) Arrhenius plots of the Full-Pd2+/SSZ-13 and thermal-treated Pd/SSZ-13 catalysts with different H2 treatment times. The CH4 combustion light-off curves of 1 wt% Pd/Al2O3 is also given in (b) for comparison. | ||
The advantage of full ion-exchange before thermal treatment was further confirmed by comparing the catalytic activities of the thermal-treated Pd/SSZ-13 catalysts with the same H2 treatment time of 5 min, but prepared using different combinations of Pd precursors and zeolite-forms. The light-off curves demonstrated that the thermal-treated Pd/SSZ-13 catalyst prepared from Full-Pd2+/SSZ-13 shows the highest CH4 combustion activity compared to the other thermal-treated Pd/SSZ-13 catalysts prepared with insufficient ion-exchange efficiency, as shown in Fig. S18.† In addition, the CH4 combustion activation barriers of these catalysts were all found to be similar (Fig. S19†). Hence, the lower CH4 combustion activity of the thermal-treated Pd/SSZ-13 catalysts prepared with insufficient ion-exchange efficiency is attributed to the pre-existence of large-sized PdO particles shown in Fig. S6,† which hinders the full conversion of isolated Pd2+ ions into the uniformly nano-sized PdO clusters.
Conclusions
In summary, based on the understanding of the ion-exchange process, the fully isolated Pd2+ ions in a small-pore SSZ-13 zeolite is achieved. Controlled thermal treatment of the fully isolated Pd2+/SSZ-13 catalyst uniformly generates nano-sized PdO clusters with high phase homogeneity by rapid nucleation and suppressed nanoparticle growth, which is directly investigated via advanced microscopic methods of cryo-STEM and ultramicrotomy. The maximized PNA and CH4 combustion performances of the prepared Pd/SSZ-13 catalysts show that our study provides an efficient and practical route to uniformly disperse noble metal species in small-pore zeolites for various catalytic reactions, and introduces microscopic methods that can be widely applied in investigating zeolite-based catalysts.Experimental
Chemicals
Sodium hydroxide (NaOH), sodium silicate (Ni2SiO3) solution, ammonium nitrate (NH4NO3), palladium nitrate dihydrate (Pd(NO3)2·2H2O), and tetraamminepalladium chloride monohydrate (Pd(NH3)4Cl2·H2O) were purchased from Sigma-Aldrich. NH4–Y zeolite (CBV500, Si/Al2 = 5.2) was acquired from Zeolyst, and N,N,N-trimethyl-1-adamantylammonium hydroxide (TMAdaOH) was supplied by Sachem, Inc. Alumina (Al2O3) was obtained from Sasol.Catalyst preparation
Characterization
where ISi(nAl) is the intensity of Si(nAl) that shares n oxygens with Al.
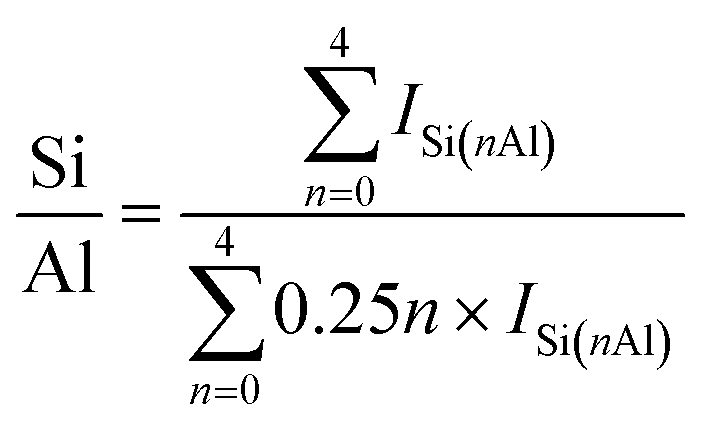
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 350 eV). The step and duration times of the X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) were 1.0 eV and 2 s, and 0.30 nm−1 and 3 s, respectively. The obtained data were processed using the Athena program (Demeter, U.S.A.).
350 eV). The step and duration times of the X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) were 1.0 eV and 2 s, and 0.30 nm−1 and 3 s, respectively. The obtained data were processed using the Athena program (Demeter, U.S.A.).
TEM sample preparation, imaging, and analysis
To image the Pd species in SSZ-13, ultramicrotomy of the synthesized Pd/SSZ-13 catalysts was conducted. First, the synthesized Pd/SSZ-13 catalysts were dispersed in an epoxy resin, and then cured at 70 °C for more than 8 h. Thereafter, the resin-embedded catalysts were sliced into ∼50 nm thick sections using an ultramicrotome (EM UC7, Leica) obtained from the National Instrumentation Center for Environmental Management (NICEM), Seoul National University. The ultramicrotomed sections were loaded onto carbon-deposited copper grids for further investigation.TEM/STEM imaging of the Pd/SSZ-13 catalysts (either whole particle or sliced sections) was performed on a JEOL-2100F (JEOL Ltd) equipped with an UltraScan 1000XP CCP detector (Gatan) and a HAADF detector, under an acceleration voltage of 200 kV. Energy-dispersive X-ray spectroscopy (EDS) was performed and analyzed using the AZtecTEM software equipped in the same microscope. To image the ultramicrotomed Pd/SSZ-13 sections, STEM imaging at a cryogenic temperature of −180 °C was conducted using a cryo-transfer holder (Model 626, Gatan). High-resolution TEM images of Pd/SSZ-13 catalysts were obtained from JEOL-ARM200F (JEOL Ltd) equipped with a spherical aberration corrector in the objective lens and cold field emission gun, with an acceleration voltage of 80 kV. JEOL-ARM200F is installed at the National Center for Inter-university Research Facilities (NCIRF), Seoul National University.
We note that all the STEM images of the ultramicrotomed Pd/SSZ-13 catalysts presented in this study were obtained with a minimal application of scanning mode during the imaging, to further avoid the electron-beam induced damage. That is, the scanning mode was only applied at a low-magnification (typically 1/5 magnification of that of cryo-STEM images in Fig. 2c–f) to quickly search the sample of interest. At high-magnification where the images were collected for size quantification, the focus was adjusted outside the region of interest (ROI), and thus the Pd/SSZ-13 sections located in the ROI were scanned only once during the imaging.
To quantify the size of the nano-sized PdO clusters in cryo-STEM images, a homemade MATLAB algorithm was developed. Particularly, the pixel area of the individual nano-sized PdO clusters located at the internal pores of the zeolite was extracted via manual measurements and converted to nm.2 The diameter of the nano-sized clusters was mathematically calculated by assuming a spherical morphology of the measured nanoparticles. After obtaining the particle size data of 500 nano-sized PdO clusters for each thermal-treated Pd/SSZ-13 catalyst, the kurtosis of size distribution was calculated based on the following equation:
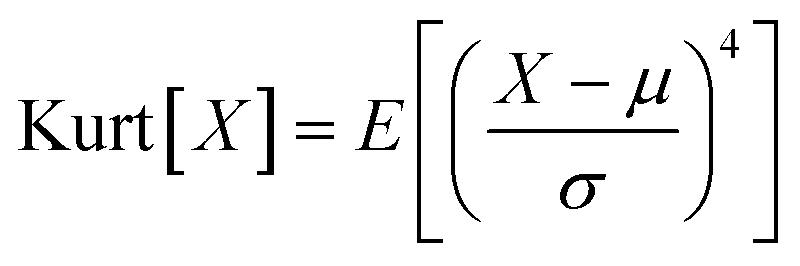
Passive NOx adsorber (PNA) and CH4 combustion
The catalytic performances of Pd/SSZ-13 catalysts for PNA and CH4 combustion were evaluated using a packed bed reactor system. Particularly, 0.05 g of sieved Pd/SSZ-13 catalyst (150–180 μm) was loaded in a 1/2 inch quartz tubular reactor. To control the temperature of the reaction, a thermocouple was placed right above the sample. The feed gas was introduced through a 1/4 inch stainless line, and the line was heated at 120 °C to prevent condensation of water. The loaded catalyst was pretreated under 10 vol% O2 at 500 °C for 30 min. After the pretreatment, the temperature was cooled to 120 °C.Various reactants such as O2, H2O, CO2, CH4, CO, and NO were introduced in the feed gas to simulate the realistic condition. Initially, to expose the catalyst under steam-containing conditions, 10 vol% O2, 5 vol% H2O 5 vol of CO2, and balanced N2 were added to the feed gas at 120 °C. After stabilizing for 30 min, 100 ppm of NO, 250 ppm of CO, and 1000 ppm of CH4 were added to the feed gas. Therefore, the final feed gas contained 100 ppm NO, 250 ppm CO, 1000 ppm CH4, 10 vol% O2, 5 vol% H2O, 5 vol% CO2, and balance N2. Notably, the total flowrate was maintained at 200 sccm in all cases.
After the catalyst was exposed to the final feed gas, the temperature was maintained at 120 °C for 20 min. During this period, to evaluate the PNA performance of the catalyst, the changes in the concentration of NO, NO2, CO, and CH4 were measured using an online FT-IR spectrometer (Nicolet iS-50, Thermo Scientific) with a 2 m gas cell. Specifically, the IR spectra were collected every 10 s via the IR series method to track the continuous changes in the concentrations of the reactants (Fig. S16†). The concentration of NO in the outlet gas was diminished under 100 ppm (the concentration in the feed gas) due to the adsorption of NO on the catalyst, and then slowly reached 100 ppm. The PNA performance of the catalyst was assessed based on the amount of adsorbed NO.
On the other hand, the CH4 combustion activity was measured while ramping the temperature to 600 °C with a ramping rate of 5 °C min−1. The concentrations of NO, NO2, CO, and CH4 in the outlet gas were measured again using an online FT-IR spectrometer.
The conversion and reaction rate of CH4 during the reaction were calculated based on the following equations:
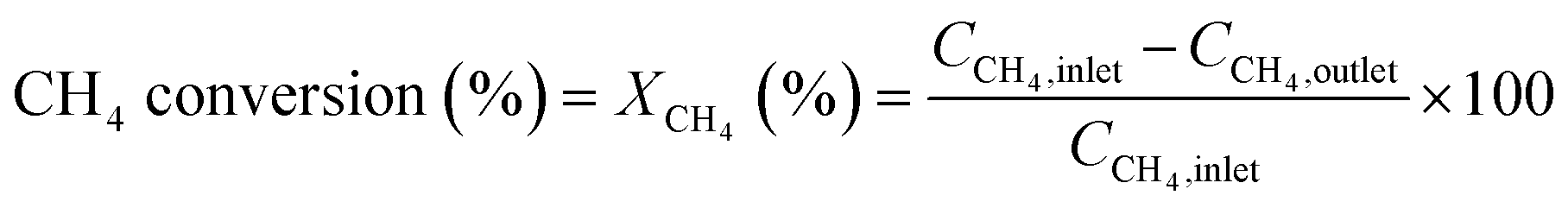
where CCH4,inlet is the inlet concentration of CH4, CCH4,outlet is the outlet concentration of CH4 after the reaction, v is the volumetric flow rate, and XCH4 is the conversion of CH4. ML and MW are the Pd loading in the catalyst and the molecular weight of the Pd, respectively. Only the data where the XCH4 value was below 20% were used for the Arrhenius plot.
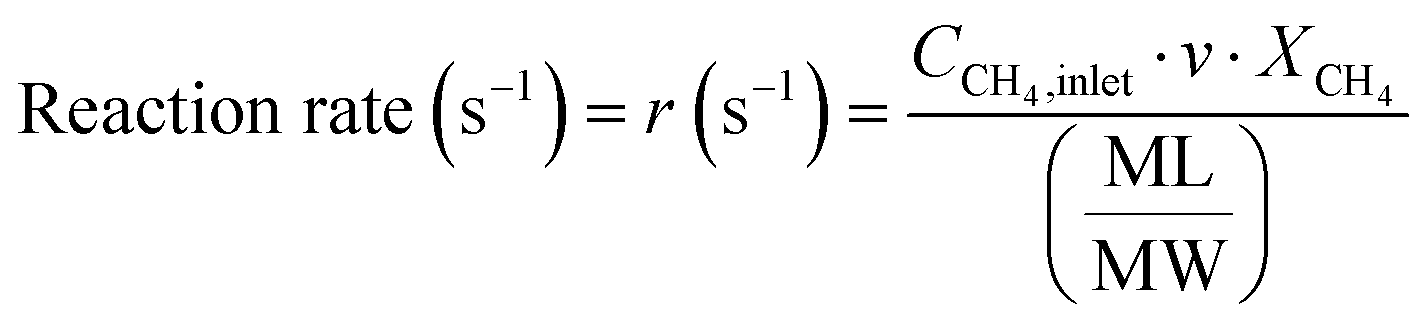
The reliability of the catalyst preparation and reaction system was confirmed via a reproducibility and repeated test, as shown in Fig. S20.†
Author contributions
Yongwoo Kim: conceptualization, investigation, visualization, writing – original draft, and writing – review & editing. Jongbaek Sung: conceptualization, investigation, visualization, writing – original draft, and writing – review & editing. Sungsu Kang: writing – review & editing. Jaeha Lee: writing – review & editing. Min-Ho Kang: writing – review & editing. Sungha Hwang: writing – review & editing. Hayoung Park: writing – review & editing. Joodeok Kim: writing – review & editing. Younhwa Kim: writing – review & editing. Eunwon Lee: writing – review & editing. Gyeong-Su Park: methodology, and writing – review & editing. Do Heui Kim: supervision, conceptualization, funding acquisition, and writing – review & editing. Jungwon Park: supervision, conceptualization, funding acquisition, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. Park acknowledges the support from the Institute for Basic Science (IBS-R006-D1), the National Research Foundation of Korea (NRF) grant funded by the Korea government (MIST) (NRF-2017R1A5A1015365 and NRF-2019M3E6A1064877), Korea Toray Science Foundation, and the LG Chem Open Innovation Fund. D. H. Kim acknowledges the support from the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (MSIP) (NRF-2016R1A5A1009592). We thank Eunjin Kim of the National Instrumentation Center for Environmental Management (NICEM), Seoul National University for providing scientific feedback on ultramicrotomy. The experiments at PLS-II were also supported in part by the Ministry of Science, ICT, and Future Planning (MSIP) and the Pohang University of Science and Technology (POSTECH).References
- Y. Chai, W. Shang, W. Li, G. Wu, W. Dai, N. Guan and L. Li, Adv. Sci., 2019, 6, 1900299 Search PubMed.
- M. Choi, Z. Wu and E. Iglesia, J. Am. Chem. Soc., 2010, 132, 9129–9137 Search PubMed.
- N. Wang, Q. Sun, R. Bai, X. Li, G. Guo and J. Yu, J. Am. Chem. Soc., 2016, 138, 7484–7487 Search PubMed.
- B. Wen, Q. Sun and W. M. H. Sachtler, J. Catal., 2001, 204, 314–323 Search PubMed.
- K. Khivantsev, N. R. Jaegers, L. Kovarik, J. C. Hanson, F. Tao, Y. Tang, X. Zhang, I. Z. Koleva, H. A. Aleksandrov, G. N. Vayssilov, Y. Wang, F. Gao and J. Szanyi, Angew. Chem., Int. Ed., 2018, 57, 16672–16677 Search PubMed.
- H. Xu, W. Chen, Q. Wu, C. Lei, J. Zhang, S. Han, L. Zhang, Q. Zhu, X. Meng, D. Dai, S. Maurer, A.-N. Parvulescu, U. Müller, W. Zhang, T. Yokoi, X. Bao, B. Marler, D. E. De Vos, U. Kolb, A. Zheng and F.-S. Xiao, J. Mater. Chem. A, 2019, 7, 4420–4425 Search PubMed.
- J. B. Lim, D. Jo and S. B. Hong, Appl. Catal., B, 2017, 219, 155–162 Search PubMed.
- Z. Wu, S. Goel, M. Choi and E. Iglesia, J. Catal., 2014, 311, 458–468 Search PubMed.
- Y. Ryou, J. Lee, S. J. Cho, H. Lee, C. H. Kim and D. H. Kim, Appl. Catal., B, 2017, 212, 140–149 Search PubMed.
- J. G. McCarty, Catal. Today, 1995, 26, 283–293 Search PubMed.
- Y.-H. Chen, C.-Y. Mou and B.-Z. Wan, Appl. Catal., B, 2017, 218, 506–514 Search PubMed.
- J. Hwang, G. Kwak, Y.-J. Lee, Y. T. Kim, I. Jeong, S. Kim, K.-W. Jun, K.-S. Ha and J. Lee, J. Mater. Chem. A, 2015, 3, 23725–23731 Search PubMed.
- E. Kang, H. Jung, J.-G. Park, S. Kwon, J. Shim, H. Sai, U. Wiesner, J. K. Kim and J. Lee, ACS Nano, 2011, 5, 1018–1025 Search PubMed.
- P. Losch, W. Huang, O. Vozniuk, E. D. Goodman, W. Schmidt and M. Cargnello, ACS Catal., 2019, 9, 4742–4753 Search PubMed.
- J. de Graaf, A. J. van Dillen, K. P. de Jong and D. C. Koningsberger, J. Catal., 2001, 203, 307–321 Search PubMed.
- A. W. Petrov, D. Ferri, F. Krumeich, M. Nachtegaal, J. A. van Bokhoven and O. Kröcher, Nat. Commun., 2018, 9, 2545 Search PubMed.
- Y. Zheng, L. Kovarik, M. H. Engelhard, Y. Wang, Y. Wang, F. Gao and J. Szanyi, J. Phys. Chem. C, 2017, 121, 15793–15803 Search PubMed.
- K. Manthiram, B. J. Beberwyck and A. P. Alivisatos, J. Am. Chem. Soc., 2014, 136, 13319–13325 Search PubMed.
- K. Manthiram, Y. Surendranath and A. P. Alivisatos, J. Am. Chem. Soc., 2014, 136, 7237–7240 Search PubMed.
- V. Ortalan, A. Uzun, B. C. Gates and N. D. Browning, Nat. Nanotechnol., 2010, 5, 506–510 Search PubMed.
- M. Juneau, R. Liu, Y. Peng, A. Malge, Z. Ma and M. D. Porosoff, ChemCatChem, 2020, 12, 1826–1852 Search PubMed.
- O. Ugurlu, J. Haus, A. A. Gunawan, M. G. Thomas, S. Maheshwari, M. Tsapatsis and K. A. Mkhoyan, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 113408 Search PubMed.
- S. X. Wang, L. M. Wang and R. C. Ewing, J. Nucl. Mater., 2000, 278, 233–241 Search PubMed.
- Z. Li, M. C. Johnson, M. Sun, E. T. Ryan, D. J. Earl, W. Maichen, J. I. Martin, S. Li, C. M. Lew, J. Wang, M. W. Deem, M. E. Davis and Y. Yan, Angew. Chem., Int. Ed., 2006, 45, 6329–6332 Search PubMed.
- Q. Chen, C. Dwyer, G. Sheng, C. Zhu, X. Li, C. Zheng and Y. Zhu, Adv. Mater., 2020, 32, 1907619 Search PubMed.
- J. R. Di Iorio, S. Li, C. B. Jones, C. T. Nimlos, Y. Wang, E. Kunkes, V. Vattipalli, S. Prasad, A. Moini, W. F. Schneider and R. Gounder, J. Am. Chem. Soc., 2020, 142, 4807–4819 Search PubMed.
- J. R. Di Iorio, C. T. Nimlos and R. Gounder, ACS Catal., 2017, 7, 6663–6674 Search PubMed.
- K. Mech, P. Żabiński, R. Kowalik and K. Fitzner, Electrochim. Acta, 2013, 104, 468–473 Search PubMed.
- M. Benkhaled, S. Morin, C. Pichon, C. Thomazeau, C. Verdon and D. Uzio, Appl. Catal., A, 2006, 312, 1–11 Search PubMed.
- J. Lee, J. Kim, Y. Kim, S. Hwang, H. Lee, C. H. Kim and D. H. Kim, Appl. Catal., B, 2020, 277, 119190 Search PubMed.
- A. M. Beale, F. Gao, I. Lezcano-Gonzalez, C. H. F. Peden and J. Szanyi, Chem. Soc. Rev., 2015, 44, 7371–7405 Search PubMed.
- C. Stolz, A. Sauvage, P. Massiani and R. Kramer, Appl. Catal., A, 1998, 167, 113–121 Search PubMed.
- H. Matsumoto and S. Tanabe, J. Mater. Sci. Lett., 1992, 11, 623–626 Search PubMed.
- T. M. Lardinois, J. S. Bates, H. H. Lippie, C. K. Russell, J. T. Miller, H. M. Meyer, K. A. Unocic, V. Prikhodko, X. Wei, C. K. Lambert, A. B. Getsoian and R. Gounder, Chem. Mater., 2021, 33, 1698–1713 Search PubMed.
- H. Jeong, J. Bae, J. W. Han and H. Lee, ACS Catal., 2017, 7, 7097–7105 Search PubMed.
- B. Liu, Y. Zheng, N. Hu, T. Gui, Y. Li, F. Zhang, R. Zhou, X. Chen and H. Kita, Microporous Mesoporous Mater., 2014, 196, 270–276 Search PubMed.
- J. Zecevic, G. Vanbutsele, K. P. de Jong and J. A. Martens, Nature, 2015, 528, 245–248 Search PubMed.
- J.-O. Bovin, V. Alfredsson, G. Karlsson, A. Carlsson, Z. Blum and O. Terasaki, Ultramicroscopy, 1996, 62, 277–281 Search PubMed.
- H. Wang, Y. Li, Y. Li, Y. Liu, D. Lin, C. Zhu, G. Chen, A. Yang, K. Yan, H. Chen, Y. Zhu, J. Li, J. Xie, J. Xu, Z. Zhang, R. Vilá, A. Pei, K. Wang and Y. Cui, Nano Lett., 2019, 19, 1326–1335 Search PubMed.
- Y. Li, Y. Li, A. Pei, K. Yan, Y. Sun, C.-L. Wu, L.-M. Joubert, R. Chin, A. L. Koh, Y. Yu, J. Perrino, B. Butz, S. Chu and Y. Cui, Science, 2017, 358, 506–510 Search PubMed.
- K. Khivantsev, F. Gao, L. Kovarik, Y. Wang and J. Szanyi, J. Phys. Chem. C, 2018, 122, 10820–10827 Search PubMed.
- K. Cheng, L. I. van der Wal, H. Yoshida, J. Oenema, J. Harmel, Z. Zhang, G. Sunley, J. Zečević and K. P. de Jong, Angew. Chem., Int. Ed., 2020, 59, 3592–3600 Search PubMed.
- X. Chen, Y. Cheng, C. Y. Seo, J. W. Schwank and R. W. McCabe, Appl. Catal., B, 2015, 163, 499–509 Search PubMed.
- A. D. Benavidez, L. Kovarik, A. Genc, N. Agrawal, E. M. Larsson, T. W. Hansen, A. M. Karim and A. K. Datye, ACS Catal., 2012, 2, 2349–2356 Search PubMed.
- T. Epicier, S. Koneti, P. Avenier, A. Cabiac, A.-S. Gay and L. Roiban, Catal. Today, 2019, 334, 68–78 Search PubMed.
- T. W. Hansen, A. T. DeLaRiva, S. R. Challa and A. K. Datye, Acc. Chem. Res., 2013, 46, 1720–1730 Search PubMed.
- L. Shang, T. Bian, B. Zhang, D. Zhang, L.-Z. Wu, C.-H. Tung, Y. Yin and T. Zhang, Angew. Chem., Int. Ed., 2014, 53, 250–254 Search PubMed.
- G. Prieto, J. Zečević, H. Friedrich, K. P. de Jong and P. E. de Jongh, Nat. Mater., 2013, 12, 34–39 Search PubMed.
- S. Zhang, M. Cargnello, W. Cai, C. B. Murray, G. W. Graham and X. Pan, J. Catal., 2016, 337, 240–247 Search PubMed.
- S. G. Kwon and T. Hyeon, Small, 2011, 7, 2685–2702 Search PubMed.
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 Search PubMed.
- F. Yin, S. Ji, P. Wu, F. Zhao and C. Li, J. Catal., 2008, 257, 108–116 Search PubMed.
- H. Xiong, K. Lester, T. Ressler, R. Schlögl, L. F. Allard and A. K. Datye, Catal. Lett., 2017, 147, 1095–1103 Search PubMed.
- M. Cargnello, V. V. T. Doan-Nguyen and C. B. Murray, AIChE J., 2016, 62, 392–398 Search PubMed.
- J.-H. Park, B. Kim, C.-H. Shin, G. Seo, S. H. Kim and S. B. Hong, Top. Catal., 2009, 52, 27–34 Search PubMed.
- G. Zhu, J. Han, D. Y. Zemlyanov and F. H. Ribeiro, J. Am. Chem. Soc., 2004, 126, 9896–9897 Search PubMed.
- S. I. Zones, J. Chem. Soc., Faraday Trans., 1991, 87, 3709–3716 Search PubMed.
- Y. Gu, R. P. Zelinsky, Y.-R. Chen and W. S. Epling, Appl. Catal., B, 2019, 258, 118032 Search PubMed.
- J. Lee, Y. Ryou, S. Hwang, Y. Kim, S. J. Cho, H. Lee, C. H. Kim and D. H. Kim, Catal. Sci. Technol., 2019, 9, 163–173 Search PubMed.
- G. Engelhardt, U. Lohse, V. Patzelová, M. Mägi and E. Lippmaa, Zeolites, 1983, 3, 233–238 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta00468a |
| ‡ Y. Kim and J. Sung contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |