
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organic photoelectrode engineering: accelerating photocurrent generation via donor–acceptor interactions and surface-assisted synthetic approach†
Yaroslav S.
Kochergin
 ab,
Seyyed Mohsen
Beladi-Mousavi
a,
Bahareh
Khezri
a,
Pengbo
Lyu
c,
Michael J.
Bojdys
d and
Martin
Pumera
*aefg
ab,
Seyyed Mohsen
Beladi-Mousavi
a,
Bahareh
Khezri
a,
Pengbo
Lyu
c,
Michael J.
Bojdys
d and
Martin
Pumera
*aefg
aCenter for Advanced Functinal Nanorobots, Department of Inorganic Chemistry, Faculty of Chemical Technology, University of Chemistry and Technology Prague, Technická 5, 166 28 Prague, Czech Republic. E-mail: Martin.Pumera@vscht.cz
bInstitute of Organic Chemistry and Biochemistry of the CAS, Flemingovo nám. 2, 166 10 Prague, Czech Republic
cDepartment of Physical and Macromolecular Chemistry, Faculty of Sciences, Charles University, Hlavova 8, 128 43 Prague 2, Czech Republic
dDepartment of Chemistry, King's College London, Britannia House Guy's Campus, 7 Trinity Street, SE1 1DB, London, UK
eFuture Energy and Innovation Laboratory, Central European Institute of Technology, Brno University of Technology, Pukryňova 123, 61200 Brno, Czech Republic
fDepartment of Medical Research, China Medical University Hospital, China Medical University, No. 91 Hsueh-Shih Road, Taichung, Taiwan
gDepartment of Chemical and Biomolecular Engineering, Yonsei University, 50 Yonsei-ro, Seodaemun-gu, Seoul 03722, Korea
First published on 24th February 2021
Abstract
Conventional photoelectrocatalysts composed of precious metals and inorganic elements have limited synthetic design, hence, hampered modularity of their photophysical properties. Here, we demonstrate a scalable, one-pot synthetic approach to grow organic polymer films on the surface of the conventional copper plate under mild conditions. Molecular precursors, containing electron-rich thiophene and electron-deficient triazine-rings, were combined into a donor–acceptor π-conjugated polymer with a broad visible light adsorption range due to a narrow bandgap of 1.42 eV. The strong charge push–pull effect enabled the fabricated donor–acceptor material to have a marked activity as an electrode in a photoelectrochemical cell, reaching anodic photocurrent density of 6.8 μA cm−2 (at 0.6 V vs. Ag/AgCl, pH 7). This value is 3 times higher than that of the model donor–donor thiophene-only-based polymer and twice as high as that of the analogue synthesized in bulk using the heterogenous CuCl catalyst. In addition, the fabricated photoanode showed a 2-fold increase in the photoelectrocatalytic oxygen evolution from water upon simulated sunlight irradiation with the photocurrent density up to 4.8 mA cm−2 (at 1.0 V vs. Ag/AgCl, pH 14). The proposed engineering strategy opens new pathways toward the fabrication of efficient organic “green” materials for photoelectrocatalytic solar energy conversion.
Introduction
Artificial photosynthesis is a key technology for clean, sustainable, fossil-fuels-free energy generation. The conversion of abundant solar energy into a useful feedstock or chemical fuel using a semiconducting photocatalyst has attracted considerable attention in the scientific community.1 In the light of that, photoelectrochemical (PEC) water splitting into oxygen and hydrogen offers a great potential for clean and secure energy production without dependence on finite and non-renewable resources.2 In this process the harvested light excites charge carriers (electrons and holes) in the semiconducting material, which subsequently promotes the hydrogen evolution (HER) and oxygen evolution (OER) half-reactions at the catalyst–electrolyte interface.3 Therefore, the ideal photocatalyst should possess a broad light absorption range, facilitate fast interfacial charge transfer without rapid charge recombination and be stable under operating conditions.4 Moreover, to meet industrial requirements the active material has to be fabricated and easily scaled up from low-cost and readily available precursors.There is a considerable number of reports about efficient PEC systems based on transition metal oxides,5–7 metal chalcogenides, and dichalcogenides.8–11 Despite their good performance, the majority of these photoactive materials are still suffering from low modularity, narrow light absorption range and require complicated and costly synthetic approaches, such as atomic layer deposition, solvothermal synthesis or solution-based deposition with subsequent high-temperature annealing.12,13 In that respect, organic conjugated semiconducting polymers emerged as a promising alternative for the fabrication of the materials for solar energy harvesting.14–16 The metal-free fully organic molecular backbone enables precise tuneability of photophysical properties, such as bandgap and charge transport, thus, empowering these materials to be utilised as photocatalysts in light-driven H2 production.17 In 2008 graphitic carbon nitride (g-C3N4) became the first example of metal-free synthetic photocatalyst, which was capable to operate under visible light irradiation.18 The wide optical bandgap of 2.7 eV, however, severely limits the light absorption range of g-C3N4 (up to 460 nm), therefore, numerous attempts to modify this material have been made to improve its photocatalytic activity.19–21
Along with g-C3N4, several other classes of organic porous materials, such as covalent organic frameworks (COFs), covalent triazine-based frameworks (CTFs), and conjugated microporous polymers (CMPs) have emerged as promising materials for photocatalytic water splitting reaction.22–24 On one hand, typical synthetic protocols of COFs and CTFs often involve high-temperature/high-pressure conditions, such as solvothermal reaction in pressurised vessels, or ionothermal/superacid-catalysed cyclotrimerization.25,26 Not every molecular precursor can withstand such harsh synthetic conditions. On the other hand, the relatively mild solution-processed bulk polymerisation of CMPs leads to materials with intertwisted molecular backbone struts and, as a consequence, numerous structural defects, which can inhibit the efficient transfer and separation of the photoexcited charge carriers.27 Therefore, it is essential to find a reliable, easy scalable synthetic approach for the fabrication of semiconducting conjugated polymer photocatalyst with improved photophysical properties.
In 2010, Li and co-workers reported a copper-foil catalysed synthesis of a carbon-rich diacetylene-containing network – graphdiyne – a structural analogue to the theoretically predicted graphyne,28 which is believed to exhibit unique optical, electronic and mechanical properties.29 Recently, Schwarz et al. employed a similar synthetic strategy to fabricate photocatalytically active triazine-based analogue of graphdiyne – triazine-based graphdiyne (TzG) framework.30 Here, the copper foil is playing a role of both a catalyst and a surface template, that allows the growing of large-scale and easy-processable polymer films. The inclusion of nitrogen heteroatoms creates an additional charge push–pull effect, thus leading to a comparatively narrow bandgap of 1.91 eV and improved electron transfer processes with the ability to evolve H2 gas from water under light irradiation without the need for a noble metal cocatalyst. However, the use of acetylenic organic polymer frameworks in PEC systems, which combines the benefits of solar energy harvesting and electrochemical water splitting, remains largely unexplored. A recent example from Zhang et al. shows the preparation of poly(1,3,5-triethynylbenzene) (PTEB) with incorporation of thieno[3,2-b]thiophene unit via copper-surface-mediated polycondensation.31 Reported materials have notable activity as photoelectrodes in PEC devices and show promise in light-driven hydrogen evolution reaction.
Herein, we present a fully organic π-conjugated triazine-thiophene (Tz-Th)-based polymer (denoted as Tz-Th@Cu). To synthesise this material, we employed the following engineering strategy: (1) we use a copper foil as both a catalyst and a template to grow a polymer film in a basic solution under mild conditions (60 °C, ambient pressure); (2) we predesign the C3-symmetrical molecular precursor that is able to form a 3D polymer network with an extended π-conjugated backbone; (3) we incorporate into the molecular building block both electron-accepting triazine (C3N3) and electron-donating thiophene repeating units that give the resulting material an additional charge push–pull effect that narrows down the optical band gap to 1.42 eV, and extends the light absorption range up to 845 nm. The D–A polymer concept is believed to improve not only the optical properties, such as the bandgap, but also benefit toward efficient transfer and separation of photoexcited charge carriers, thus leading to better photocatalytic performance of the D–A materials.32 To demonstrate the benefits of D–A polymer architecture, we also synthesised a framework composed of only repeating electron-rich benzotrithiophene core as a material for comparison (denoted as BTT@Cu). As expected, the D–A Tz-Th@Cu network showed better overall performance as a photoelectrode in the PEC cell with marked saturated photocurrent density up to 6.8 μA cm−2 (at 0.6 V vs. Ag/AgCl reference electrode). Given the highest photoelectrochemical activity of the fabricated materials at a potential range between 0–0.8 V (vs. Ag/AgCl, pH = 7), the electrodes can potentially be employed as photoelectrocatalysts for OER. As a proof-of-concept, we demonstrated that D–A Tz-Th@Cu shows a 2-fold increase in the photocurrent density under simulated sunlight illumination (up to 4.8 mA cm−2 at 1.0 V vs. Ag/AgCl), which is more than 10-times higher than that of D–D BTT@Cu (0.46 mA cm−2 at 1.0 V vs. Ag/AgCl). Our results demonstrate that organic conjugated polymers built by the D–A approach and via a robust and easy-scalable Cu-surface-catalysed route can be used to fabricate highly active photoelectrodes for PEC devices.
Results and discussion
Materials synthesis
Molecular precursors 2,4,6-tris(5-ethynylthiophen-2-yl)-1,3,5-triazine (Tz-Th) and 2,5,8-triethynylbenzo[1,2-b:3,4-b′:5,6-b′′]trithiophene (BTT) were prepared by similar reaction protocols (Fig. 1a, Schemes S1 and S2†). In brief, the respective brominated synthon was reacted with trimethylsilyl–acetylene under Negishi cross-coupling reaction conditions. Subsequently, the obtained intermediate was subjected to the deprotection reaction in the presence of a base (aqueous NaOH in the case of Tz-Th, and K2CO3 in the case of BTT, more details can be found in the Experimental section of the ESI†).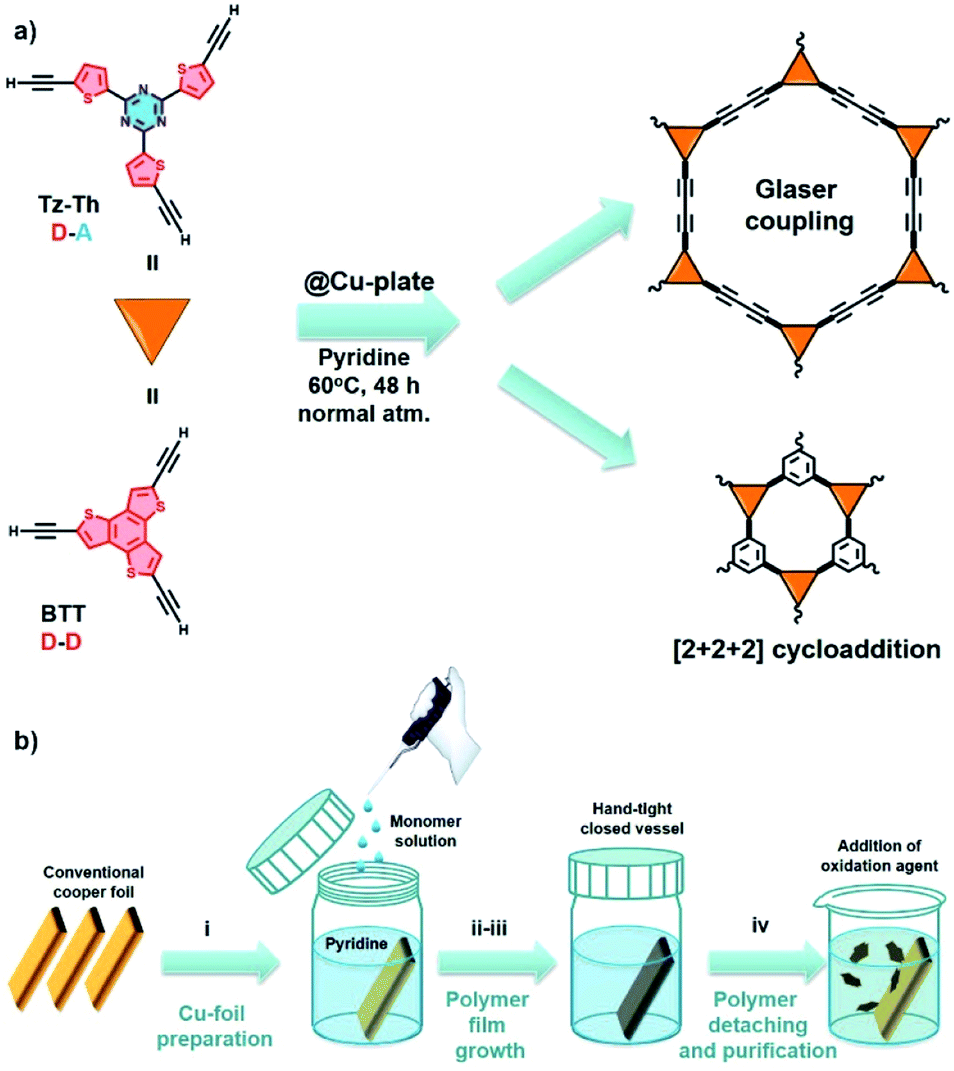 | ||
| Fig. 1 (a) Synthetic pathways toward the preparation of Tz-Th- and BTT-based polymers on the surface of a conventional copper plate (@Cu-plate). There are two possible mechanisms that can occur during the reaction – Glaser-type coupling or [2 + 2 + 2] cyclotrimerization of terminal alkenes; (b) schematic representation of the synthetic procedure that includes: (i) preparation of the Cu-foil, (ii) submerging of the Cu-foil in pyridine and addition of the monomer, (iii) reaction in the hand-tight closed vessel at 60 °C for 48 h; (iv) detaching of the polymer film from Cu-foil with subsequent purification and drying. | ||
We employed “wet” chemistry approach to fabricate polymeric materials by using a conventional copper plate that acts both as a surface template and a catalyst for polymeric network formation. The polymer growth reaction is based on the homocoupling of tris-ethynyl-substituted monomer and can be governed by two possible reaction pathways: (1) Glaser-type (Glaser) polycondensation of terminal alkynes; (2) [2 + 2 + 2] cyclotrimerization (cyclo) of carbon–carbon triple bonds (Fig. 1a).30 At the initial point the synthons have to get absorbed at the catalytically active copper surface, and, due to their increased local concentration at the solid–liquid interface, should form the statistically more preferred [2 + 2 + 2] product. Further during the reaction Cu-ions are leached into the solution due to passivation and solvation effects, and therefore will promote the diffusion-controlled coupling of the terminal alkyne groups via the Glaser mechanism.33 For both Tz-Th@Cu and BTT@Cu we used pyridine that simultaneously acts as a solvent and a base and the reaction mixture was kept under an ambient atmosphere in a hand-tight-closed vessel at 60 °C for 48 h (Fig. 1b). After completion of the reaction, the polymer film can be easily released from the copper surface via the addition of 0.5 M aqueous solution of (NH4)2S2O8 as a mild oxidative agent (Fig. S1†). An average polymer film thickness of ∼1 μm for Tz-Th@Cu and ∼1.5 μm for BTT@Cu was obtained after 48 h, which is evidenced by scanning electron microscopy (SEM, Fig. S2 and S3†). Using atomic force microscopy (AFM) we established that the polymer film growth almost stopped after 24 h of reaction time, which can be explained by the limited diffusion path length of the leached copper species (Fig. S4 and S5†). The reaction was carried out for an additional 24 h to achieve a homogeneous film structure.
Structural characterisation
We probed the chemical make-up of Tz-Th- and BTT-based networks by performing 13C cross-polarization magic-angle-spinning (CP-MAS) solid-state NMR (ssNMR) spectroscopy (Fig. 2a). In the case of Tz-T@Cu we observed characteristic triazine-ring carbon signals at ∼170 ppm. In addition, two distinguishable signals at ∼140 and 130 ppm confirm the presence of other structural motifs – thiophene and phenyl rings. The ssNMR spectrum of BTT@Cu shows a broad feature between 130–150 ppm, which is indicative of the fused benzotrithiophene core.34 Moreover, both materials have a weak signal at ∼80 ppm corresponding to the carbon–carbon triple bonds. This phenomenon points toward the twinned growth of Glaser-type and [2 + 2 + 2] cyclotrimerization heterostructures in both polymers, which was also observed before in the case of TzF/TzG system.30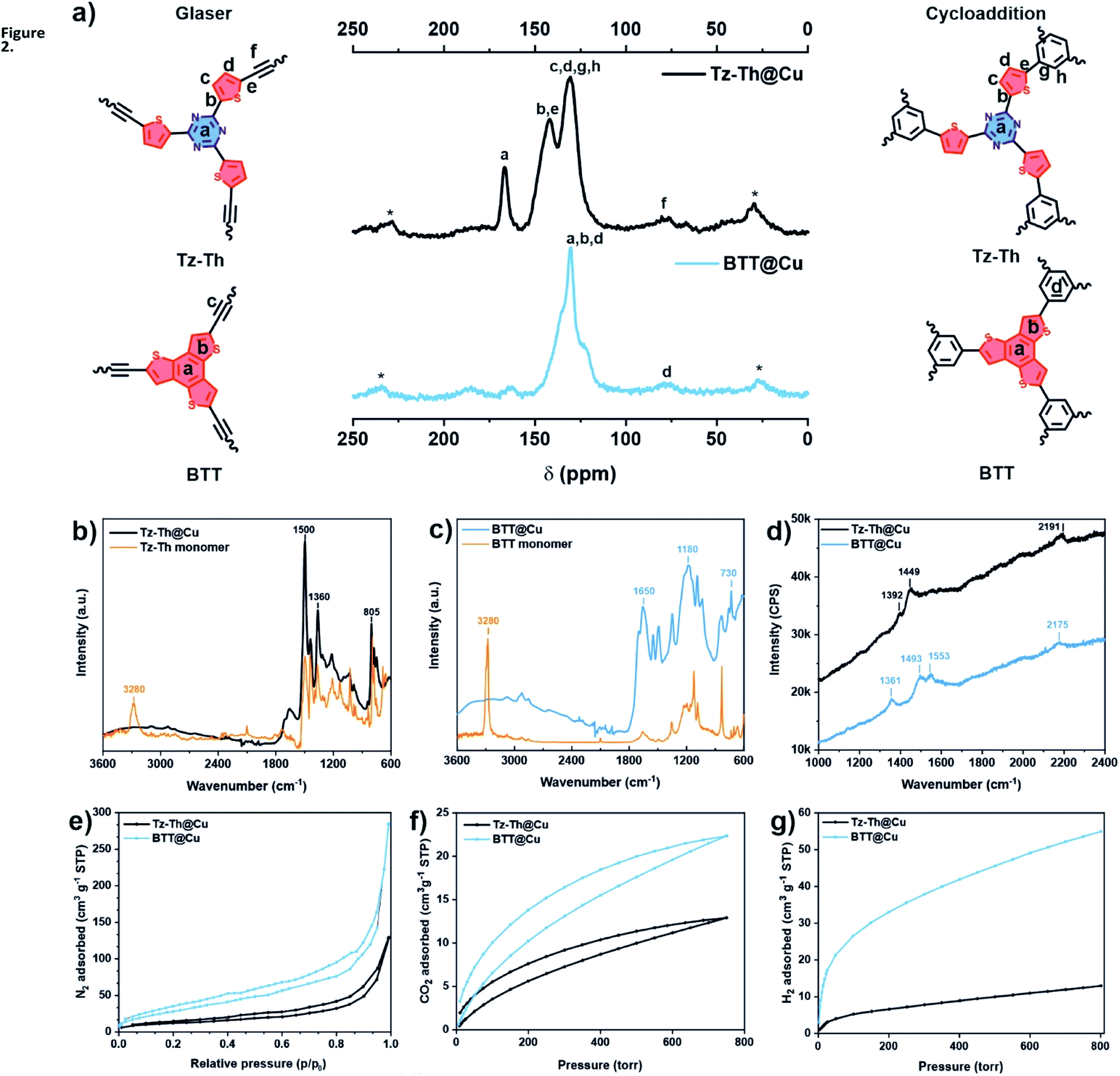 | ||
| Fig. 2 Structural characterisation of the obtained materials. (a) 13C CP-MAS solid-state NMR spectra of Tz-Th@Cu and BTT@Cu. Signals of carbon atoms are labelled in accordance with 2 possible structures – products of Glaser-coupling and cycloaddition reaction (schematic synthons on the left and right of ssNMR spectra. Spinning side bands are marked with an asterisk (*); (b) and (c) FTIR spectra of Tz-Th@Cu and BTT@Cu respectively, showing the comparison with the starting monomers; (d) Raman spectra of both Th–Th@Cu (black) and BTT@Cu (blue); (e) N2 ad-/desorption isotherms measured at 77 K; (f) CO2 ad-/desorption isotherms measured at 298 K and 1 bar; (d) H2 adsorption isotherms recorded at 77 K and 1 bar. | ||
We analysed the elemental composition of the synthesised polymers by combustion elemental analysis (EA). All materials have close to ideal C/N and C/S ratios, which proves the high degree of polymerisation reaction (Tables S1 and S2†). The inductively coupled plasma optical emission spectrometry (ICP-OES) and energy dispersive X-ray spectroscopy (EDS) showed a residual content of P (0.35–0.38 wt%) and Cu (0.3–2.49 wt%), that might arise from the preparation of molecular precursors and Cu-catalysed polycondensation reaction (Tables S1–S4†). We corroborate these findings by performing the thermogravimetric analysis (TGA) under the oxidative atmosphere, which revealed the incombustible inorganic residues from 0.66 to 5.71 wt% (Fig. S6a†). Noteworthy, all prepared materials have excellent structural integrity and, therefore, high thermal stability, which is proved by TGA performed under inert conditions – more than 40 wt% of polymers are remaining after heating up to 1000 °C (Fig. S6b†).
We further analysed the chemical states of the main composition elements (C, N, and S) using high-resolution photoelectron spectroscopy (XPS, Fig. S7 and S8†). Deconvolution of the C 1s region for Tz-Th@Cu resulted in three peaks at 282.6 eV, 284.5 eV, and 286.9 eV, which can be attributed to the sp2 hybridized carbons of the phenyl rings (C–Cphenyl), thiophene ring (C–S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C)/adventitious (Cadv) carbons and triazine ring carbons (C–NC) (Fig. S7b†).35,36 Deconvolution of the N 1s region identified one peak with the binding energy of 397.4 eV, which can be assigned to nitrogen atoms in C3N3 ring (C–NC) (Fig. S7c†). In the S 2p region, we observed two dominant peaks at 162.4 eV and 163.6 eV, which are characteristic for the S 2p3/2 and S 2p1/2 spin–orbit components of sulfur in thiophene units (C–SC) (Fig. S7d†).36 The XPS spectrum of BTT@Cu confirmed the presence of carbon and sulfur as main structural elements with 3 carbon signals in the C 1s region at 282.4 eV, 284.9 eV, and 288.4 eV (C–Cphenyl, C–SC/Cadv, and C–O/CO) and 2 main peaks in the S 2p region at 164.04 and 165.2 eV (2p3/2 and S 2p1/2) (Fig. S8b and c†).
C)/adventitious (Cadv) carbons and triazine ring carbons (C–NC) (Fig. S7b†).35,36 Deconvolution of the N 1s region identified one peak with the binding energy of 397.4 eV, which can be assigned to nitrogen atoms in C3N3 ring (C–NC) (Fig. S7c†). In the S 2p region, we observed two dominant peaks at 162.4 eV and 163.6 eV, which are characteristic for the S 2p3/2 and S 2p1/2 spin–orbit components of sulfur in thiophene units (C–SC) (Fig. S7d†).36 The XPS spectrum of BTT@Cu confirmed the presence of carbon and sulfur as main structural elements with 3 carbon signals in the C 1s region at 282.4 eV, 284.9 eV, and 288.4 eV (C–Cphenyl, C–SC/Cadv, and C–O/CO) and 2 main peaks in the S 2p region at 164.04 and 165.2 eV (2p3/2 and S 2p1/2) (Fig. S8b and c†).
The high degree of network-forming reactions was confirmed via Fourier-transform infrared (FT-IR) spectroscopy – the sharp peak of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H at 3270–3280 cm−1 for both BTT and Tz-Th precursors is absent in the spectra of the respective polymers (Fig. 2b and c). Moreover, there are no strong features which can point toward free OH- and NH-groups of some oxidized or hydrolysed fragments in the region between 3000 and 3600 cm−1. The sharp signals at 1500 cm−1 and 805 cm−1 for Tz-Th@Cu can be attributed to C3N3-ring vibration and breathing modes.37 Typical C–S bond symmetric and asymmetric vibrations in the thiophene ring between 830–730 cm−1 and phenyl ring vibrations in the region from 1400 to 1650 cm−1 can be observed for both Tz-Th@Cu and BTT@Cu.38 Complementary Raman spectra show the presence of CC stretching at 2191 cm−1 for Tz-Th@Cu and at 2175 cm−1 for BTT@Cu, which corroborate the formation of conjugated diyene linkages (Fig. 2d).39 The peaks at ∼1360–1390 cm−1 and ∼1450–1550 cm−1 can be assigned to the aromatic (phenyl, thiophene, triazine) rings breathing and stretching modes, respectively.40
C–H at 3270–3280 cm−1 for both BTT and Tz-Th precursors is absent in the spectra of the respective polymers (Fig. 2b and c). Moreover, there are no strong features which can point toward free OH- and NH-groups of some oxidized or hydrolysed fragments in the region between 3000 and 3600 cm−1. The sharp signals at 1500 cm−1 and 805 cm−1 for Tz-Th@Cu can be attributed to C3N3-ring vibration and breathing modes.37 Typical C–S bond symmetric and asymmetric vibrations in the thiophene ring between 830–730 cm−1 and phenyl ring vibrations in the region from 1400 to 1650 cm−1 can be observed for both Tz-Th@Cu and BTT@Cu.38 Complementary Raman spectra show the presence of CC stretching at 2191 cm−1 for Tz-Th@Cu and at 2175 cm−1 for BTT@Cu, which corroborate the formation of conjugated diyene linkages (Fig. 2d).39 The peaks at ∼1360–1390 cm−1 and ∼1450–1550 cm−1 can be assigned to the aromatic (phenyl, thiophene, triazine) rings breathing and stretching modes, respectively.40
Morphology and porosity investigation
Both Tz-Th and BTT-based polymers have predominantly amorphous morphology in the bulk, which is evidenced by powder X-ray diffraction (PXRD) patterns (Fig. S9†). Broad peaks in the range from 20° to 30° 2θ can be attributed to the stacking distances of individual layers (2.8–3.8 Å), which were also observed for other porous organic networks, such as COFs and CTFs.41 More atomistic structural information for both Tz-Th@Cu and BTT@Cu, however, cannot be deduced from PXRD patterns due to the low scattering contrast of 2D–3D intertwined heterostructures and additional free rotation along the C–C bonds between repeating units, which leads to amorphization of the polymer structures in bulk.The flake-like morphological growth of Tz-Th@Cu and BTT@Cu films is particularly interesting for nanoscale investigation using transmission electron microscopy (TEM). Accordingly, we performed high-resolution TEM imaging of both polymers and observed pronounced moiré fringes with hexagonal repeating patterns, that can be attributed to the crystalline domains formed within the polymeric network (Fig. S10 and S12†).42 The corresponding selected area diffraction (SAED) pattern made from Tz-Th@Cu confirms the hexagonal bonding motif within 2D crystalline lattice of obtained polymer, which is typical for structural analogues, such as CTFs (Fig. S11†).42 The SAED for BTT@Cu shows visible concentric rings, that correspond to regular distances within the principal unit-cells and indicate that this material features polycrystalline morphology (Fig. S13†).43 Both recorded SAED patterns are in a reasonable agreement with simulated monolayer patterns (Fig. S11 and S13†).
We further probed the pore structure of the synthesised polymers using different gas adsorption analyses. The guest accessible surface area of the obtained materials was determined by N2 ad-/desorption experiments at 77 K (Fig. 2e). Both Tz-Th@Cu and BTT@Cu showed moderate nitrogen gas uptake, with respective specific surface areas of 42 and 116 m2 g−1, calculated according to the Brunauer–Emmett–Teller (BET) model (Table 1). The N2 adsorption isotherms show almost no discernible hysteresis loops, which is pointing to the high rigidity of prepared networks. Obtained materials are predominantly mesoporous with the largest pore fraction between 5 and 25 nm, which is proved by pore size distribution (PSD) analysis, performed according to Horvath–Kawazoe (HK) and Barrett–Joyner–Halenda (BJH) models (Fig. S14†) The CO2 adsorption isotherms show steep linear gas uptake without any discernible hysteresis, which is typical for micro-and mesoporous polymers (Fig. 2f).44 In addition, we investigated the hydrogen gas storage capacity of synthesised networks (Fig. 2g). The CO2 and H2 uptake of Tz-Th@Cu have close values of 0.57 and 0.58 mmol g−1, whereas in the case BTT@Cu there is an almost 3-fold difference – 0.99 mmol g−1 for CO2 and 2.45 mmol g−1 for H2 adsorption (Table 1). Higher BET surface area together with the results of CO2 and H2 sorption analyses point to the formation of more uniform pore channels in the case of BTT@Cu, which is the consequence of the preferential AA-stacking structure.
| Material | S BET (m2 g−1) | CO2 uptakeb (mmol g−1) | H2 uptakec (mmol g−1) | E g dir. (eV) | J (μA cm−2) |
|---|---|---|---|---|---|
| a Surface area calculated from N2 adsorption isotherm using BET equation. b CO2 gas uptake measured at 298 K and 1 bar. c H2 gas uptake measured at 77 K and 1 bar. d Optical bandgaps calculated via the Kubelka–Munk function from the diffuse-reflectance UV-Vis adsorption edge. e Saturated photocurrent density obtained at an applied potential of 0.6 V (vs. Ag/AgCl) under simulated sunlight irradiation in 0.01 M Na2SO4 aqueous solution. | |||||
| Tz-Th@Cu | 42 | 0.57 | 0.58 | 1.42 | 6.8 |
| BTT@Cu | 116 | 0.99 | 2.45 | 1.85 | 2.3 |
| Tz-Thb | 868 | 1.04 | 3.42 | 1.48 | 3.7 |
| BTTb | 563 | 1.38 | 4.08 | 1.58 | 2.6 |
Structural arrangement and theoretical modelling
For a better understanding of the polymer structural arrangement, we performed additional density functional theory (DFT) calculations. In the case of [2 + 2 + 2] cycloaddition network, the formation of both AA and AB-stacking structures has similar interaction energies (Eint), hence, similar stability. However, in the case of Glaser-type polymer, the AA-arrangements are more stable than AB-arrangements (Fig. S15–S17 and Table S5†). Considering this, it is difficult to determine the dominant interlayer stacking in the resulting materials solely depending on Eint. To rationalize the difference in BET surface area observed for the prepared materials, we considered the solvation effect of pyridine to explain their structural stability and preferential interlayer stacking. Pyridine as a solvent is also an electron-acceptor and can alter the polarity of the solvated material, thus leading to sliding of the individual polymer layers, and therefore to the formation of a staircase-like AB-stacking arrangement with blocked pore channels and decreased accessible surface area.45 On the one hand, this effect is more pronounced in the case of polar D–A Tz-Th@Cu polymer, where the AB-stacking with the reduced accessible surface area will be favoured. On the other hand, the electron-withdrawing effect of pyridine will have limited influence on the relatively apolar D–D BTT@Cu, therefore causing less interlayer sliding and resulting in the AA-stacked structure with a higher BET surface area.Optoelectronic properties
We further investigated the optical properties of the prepared polymers via diffuse-reflectance UV-Vis performed in solid-state. The UV-Vis spectra confirm that all materials absorb light in the whole visible part of the electromagnetic spectrum with discernible light absorption edges starting around 740 nm for BTT@Cu and 845 nm for Tz-Th@Cu (Fig. 3a). We calculated the optical bandgaps from the UV-Vis adsorption edges by plotting the respective Tauc plots using Kubelka–Munk theory and found them to be equal to 1.42 eV for Tz-Th@Cu and 1.85 eV BTT@Cu (Table 1 and Fig. 3b).46 Theoretical modelling predicts the bandgaps to be in the range from 1.04 to 1.93 eV and 1.30 to 1.95 eV for Tz-Th@Cu and BTT@Cu respectively, depending on the structure and interlayer stacking arrangement (Fig. 3c, S18, S19 and Table S5†). Additionally, the positive slope of the Mott–Schottky curves shows that both prepared materials are n-type semiconductors, and the relative electronic band positions are in broad agreement with theoretical predictions (Fig. S20†).47 | ||
| Fig. 3 Optical properties and bandgap characterisation of the prepared polymer networks. (a) Diffuse reflectance solid-state UV-Vis spectra ND (b) Tauc plots showing an estimation of optical bandgaps of Tz-Th@Cu (black) and BTT@Cu (blue); (c) relative locations of valence band maxima (VBM) and conduction band minima (CBM) for both Tz-Th and BTT-based polymers derived from DFT calculations for different AA and AB-stacking arrangements. The redox potential of water is indicated at pH = 7 and pH = 14. On the bottom – schematic structures of repeating subunits of Glaser- and cycloaddition-types of synthesised frameworks. | ||
According to DFT calculations, the Glaser-type products always have relatively low band gaps, while [2 + 2 + 2] cycloaddition products should have significantly higher energy gaps. Therefore, the high band gap experimentally measured for BTT@Cu (1.85 eV) can be mostly attributed to the [2 + 2 + 2] cycloaddition product, whereas the lower band gap obtained for Tz-Th@Cu (1.42 eV) should be related to the Glaser-type product. However, we cannot completely rule out the existence of [2 + 2 + 2] cycloaddition product in the structure of Tz-Th@Cu. The narrower bandgap value of Tz-Th@Cu polymer is a consequence of its D–A composition – the combination of tectons with different electron affinity (electron-poor triazine and electron-rich thiophene rings) results in a material with a smaller energy gap. We observed a similar phenomenon for S- and N-containing and S-only containing conjugated microporous polymers.34 Moreover, the highly polarised triazine–thiophene couple can also withdraw electron density from electron-rich 1,3-butadyine linkages of the Glaser-coupling component, thus creating an additional charge push–pull effect within the Tz-Th@Cu network. This in turn, can lead to increased mobility of the generated charge carriers under photoelectrochemical conditions.48
Photoelectrodes fabrication and PEC characterisations
Narrow optical bandgaps (1.42–1.85 eV), micro-and mesoporosity, and improved transport of photogenerated electrons and holes make a promise for the synthesised materials to be applied in PEC devices. Accordingly, we conducted photoelectrochemical experiments with Tz-Th@Cu and BTT@Cu polymers spray-coated on an indium tin oxide (ITO) substrate as photoelectrodes in a 0.01 M Na2SO4 solution (pH = 7). The electrochemical measurements were performed in a 3-electrode setup with a Pt-disk counter electrode and applied bias voltage relative to the Ag/AgCl reference electrode. Photoanodes with the area of ∼0.36 cm2 fabricated from both Tz-Th-and BTT-based polymers showed a clear on/off response at an applied bias under intermitted light irradiation (100 mW cm−2) (Fig. 4a). To find the optimal equilibrium between the light adsorption ability and charge-transfer properties, we prepared a series of electrodes with different thicknesses.31 The highest photocurrent for Tz-Th@Cu was achieved for a medium-thick (∼0.9 μm) coating, whereas thinner and thicker polymer films resulted in a lower photocurrent (Fig. S21†). In the case of BTT@Cu the highest photocurrent was achieved at ∼1 μm and attempts to prepare a thicker coating resulted in unstable polymer films, which were easily detached from the ITO substrate when exposed to the electrolyte solution (Fig. S22†).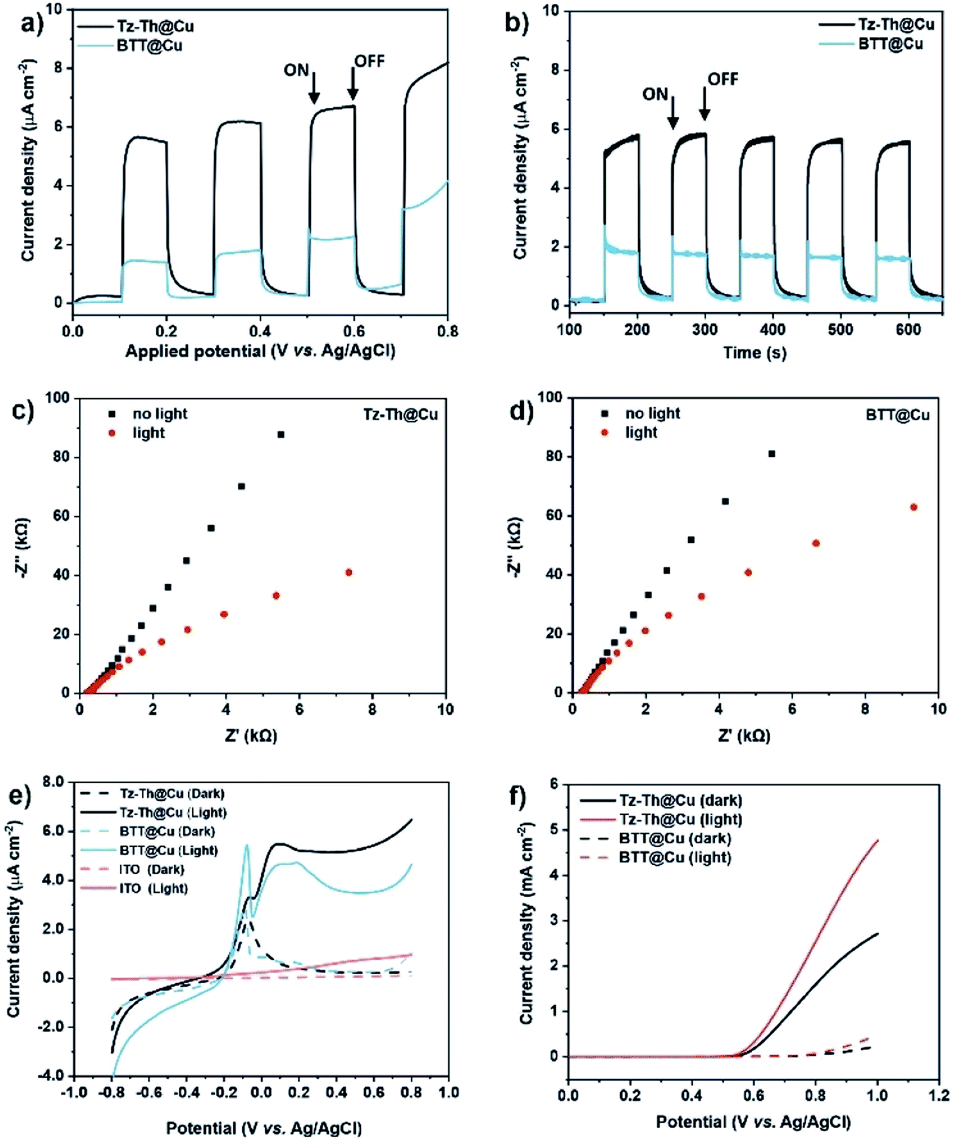 | ||
| Fig. 4 PEC characterization of the Tz-Th- and BTT-based photoanodes. (a) Current density–potential curves vs. applied potential of Tz-Th@Cu (black) and BTT@Cu (blue). Under intermittent irradiation at 1 mV s−1 scan rate; (b) transient current density vs. time at 0.6 V under intermittent light irradiation recorded for both materials; EIS Nyquist plots at applied voltage of 0.6 V under dark (black) and light irradiation (red) of Tz-Th@Cu (c) and BTT@Cu (d); (e) linear sweep voltammetry recorded for obtained polymers at 1 mV s−1 scan rate and applied potential from −0.8 to 0.8 V (a–e measured in 0.01 M Na2SO4, pH = 7 vs. Ag/AgCl reference electrode); (f) photocurrent responses of Tz-Th@Cu (solid lines) and BTT@Cu (dashed lines) under dark (black) and illuminated (red) conditions measured in 1 M NaOH solution (pH = 14) vs. Ag/AgCl reference electrode. | ||
Both materials showed stable switching behaviour at 0.6 V vs. Ag/AgCl with Tz-Th@Cu reaching the saturated photocurrent density of ∼6.8 μA cm−2 and BTT@Cu reaching ∼2.3 μA cm−2 (after subtracting the dark current, Fig. 4b). The appearance of anodic photocurrent confirms that the prepared materials are n-type semiconductors,49 and the obtained values of the photocurrent densities are on par with the highest hitherto reported for organic polymeric photoelectrodes (Table S6†). The benefit of D–A composition of Tz-Th@Cu results in an almost 3-fold increase in photocurrent density, clearly pointing toward improved excited charge transport and separation. The decreased arc radii of the Nyquist plots under light illumination for both Tz-Th@Cu and BTT@Cu suggest the reduced charge transport resistance in comparison to the dark conditions (Fig. 4c and d). This phenomenon confirms the photogeneration of electrons and holes upon simulated sunlight irradiation. Moreover, we compared the charge transfer resistance of both polymers under illuminated conditions, which is significantly lower for Tz-Th@Cu in contrast to BTT@Cu (∼440 kΩ vs. >800 kΩ, Fig. S23†). This fact points toward much better transport of photogenerated charge carriers through the more polarised D–A Tz-Th@Cu network.50 Noteworthy, both synthesised materials show good stability with negligible change in the photocurrent density under constant illumination for more than 2 h, which is imperative for real device fabrication (Fig. S24†).
Unlike with the majority of organic π-conjugated photoelectrodes, both Tz-Th@Cu and BTT@Cu show a more pronounced photocurrent response in the positive potential window from 0 to 0.8 V vs. Ag/AgCl reference electrode (Fig. 4e).51–53 Additionally, calculated band positions for both materials are suitable for water oxidation at the high pH value of the OER electrolyte (1 M NaOH, pH = 14) (Fig. 3c), therefore, as a proof-of-concept, we tested the prepared photoelectrodes in photoelectrochemical OER. The photocurrent density, and hence, the OER activity of Tz-Th@Cu increased almost 2 times under simulated solar irradiation – from 2.6 to 4.8 mA cm−2 at 1.0 V vs. Ag/AgCl reference electrode without the addition of any transition-metal-cocatalyst (Fig. 4f). For BTT@Cu we observed similar behaviour under dark and illuminated conditions – a roughly 2-fold increase in the photocurrent density from 0.26 to 0.46 mA cm−2, which is, however, significantly lower than that of D–A Tz-Th@Cu material (Fig. 4f).
Both synthesised materials have a similar guest-accessible area with a predominance of mesopores, therefore the external surface wettability may play a crucial role in PEC OER (Table 1).54 We estimated the contact angle with water to be slightly less for Tz-Th@Cu compared to BTT@Cu (51.3 vs. 56.5 deg., Fig. S25†). The negligible difference in the surface polarity clearly demonstrates that the affinity to water should not be a detrimental factor for the better OER performance of Tz-Th@Cu. The D–A composition creates a strong charge push–pull effect and improves the photophysical properties of Tz-Th-based network, i.e., narrows down the bandgap and enhances the charge transport in the photoexcited state. We corroborate these findings with GC-MS measurements that show 5.7 increase in the amount of evolved O2 under light illumination for Tz-Th@Cuvs. 4.3 increase in the case of BTT@Cu (Fig. S26 and Table S7†). Moreover, the Tz-Th-based material demonstrated overall better OER efficiency with the ability to produce 2-times and 1.5-times more oxygen with and without light irradiation, respectively. These results are pointing to the accelerated generation and better separation of photoexcited holes that can reach the surface of the photocatalyst and promote the oxygen evolution reaction.
The residual Cu content left in both networks after the synthesis, may influence the performance in electrochemical OER. The EDS showed a similar nonstochiometric amount of Cu left in both materials (2.49 and 2.48 wt% for Tz-Th@Cu and BTT@Cu respectively, Table S2†). Since D–A Tz-Th@Cu performed better in the photoelectrochemical OER, there is no direct correlation between the OER activity and residual Cu content. Moreover, we observed no catalytic activity when conventional Cu foil was used as a working electrode in the OER (Fig. S27†). In addition, we confirmed that increase in the photocurrent is not due to the oxidation of active materials, since their structure remains intact during the OER process. No additional bands appear in the FTIR spectra recorded after the OER measurements under illuminated conditions (Fig. S28†).
Preparation and characterisation of bulk materials
To further demonstrate the benefit of surface-assisted polymerisation for the fabrication of organic photoelectrodes, we prepared Tz-Th- and BTT-based frameworks in bulk using copper(I) chloride as a catalyst (denoted as Tz-Thb and BTTb). We confirmed the similarity of their chemical composition to the Tz-Th@Cu and BTT@Cu by 13C CP-MAS ssNMR (Fig. S29 and S30†), elemental analysis and EDS (Tables S8–S11†), and XPS (Fig. S31 and S32†). Moreover, the completion of the network forming reactions was also evidenced by TGA (Fig. S33†) and FTIR (Fig. S34†). Opposite to the pronounced flake-like morphology of on-copper grown frameworks, Tz-Thb and BTTb show random ‘cauliflower’ aggregation due to kinetically controlled nucleation growth, which is typical for transition-metal-catalysed bulk reactions (Fig. S35†).55 Similarly to their thin-film analogues, bulk Tz-Thb and BTTb have an overall amorphous structure according to PXRD patterns (Fig. S36†). The N2 gas sorption analyses resulted in BET surface areas of 868 and 563 m2 g−1 for Tz-Thb and BTTb, respectively (Fig. S14a†), with the majority of micro- and mesopores having a diameter in the range from 2 to 5 nm according to PSD analysis (Fig. S14b†). The synthetic approach directly influences the morphology, hence the porosity of the prepared materials. Bulk-prepared networks have a higher accessible surface area due to random interdigitation of polymeric struts that formed predominantly under kinetic control. Additionally, Tz-Thb showed CO2 and H2 adsorption capacities of 1.04 and 3.42 mmol g−1, whereas BTTb showed higher capacities of 1.38 and 4.08 mmol g−1 despite lower SBET (Table 1, Fig. S14c and d†). This can be attributed to the larger S-content (Tables S8–S11†), which is believed to be beneficial for CO2 gas capturing.56 Noteworthy, the obtained values for CO2 and H2 adsorption capacities are comparable to the highest of the hitherto reported values for benchmark CMPs and COFs.57,58We affirmed the similarity of optical properties (bandgap and photoluminescence) of bulk materials with their on-surface-grown analogues (Table 1, Fig. S37 and S38†). The PEC measurements showed an almost 2-fold drop in the saturated photocurrent density of Tz-Thb in comparison to the Tz-Th@Cu – from 6.8 to 3.7 μA cm−2, whereas there is almost no difference between BTT@Cu and BTTb – 2.3 and 2.6 μA cm−2 respectively (Table 1 and Fig. S39†). These results indicate that not only the synthetic conditions but also the molecular composition will determine the efficiency of the resulting organic photoelectrode. Here, the strong D–A interactions dramatically improve the intra- and interlayer charge transport within Tz-Th@Cu network, leading to enhanced PEC performance in comparison with the bulk synthesised material.59 In the case of BTT-based frameworks, however, weak D–D interactions do not enable efficient mobility of photoexcited charge carriers, and, consequently, there is no improvement in the photocurrent generation of layered BTT@Cu in comparison to the bulk BTTb.
Conclusions
In conclusion, we demonstrated herein a rational approach toward the design and fabrication of highly efficient photoelectrodes composed of fully organic triazine- and thiophene-based porous polymers. First, the facile and easy-scalable surface-assisted polymerisation using conventional copper foil and mild “wet chemistry” reaction conditions allowed us to produce polymeric heterostructures with pronounced layered morphology and permanent micro- and mesoporosity. Second, the selection of modular molecular precursors enables fine-tuning of the photophysical properties (bandgap, photoluminescence) of the resulting materials. The combination of building blocks with different electron affinity enables strong donor–acceptor interactions, which in turn narrow the bandgap and improve the photoexcited charge transfer within the obtained network. As a result, the photoanode, fabricated from the donor–acceptor Tz-Th@Cu polymer showed a 3-fold increase in PEC performance in comparison to donor–donor BTT@Cu, reaching an exceptional photocurrent density of 6.8 μA cm−2, which is also 3-times higher than that of the bulk synthesised Tz-Thb material (at 0.6 V vs. Ag/AgCl). Moreover, the donor–acceptor Tz-Th@Cu network showed a double increase in activity under simulated sunlight irradiation as OER photoelectrocatalyst, and performed as twice as better in comparison to donor–donor BTT@Cu. The presented strategy, which combines robust and simple copper-surface-assisted synthesis and modular terminal alkyne coupling, may lead to the fabrication of photoelectrochemically active materials for efficient and sustainable utilisation of solar energy.Author contributions
M. P. and Y. S. K. devised and designed the project. Y. S. K. carried out the materials synthesis, SEM, EDS, TEM, TGA, PXRD, FTIR, Raman, AFM, solid-state UV-Vis, and PL measurements, analysed the data and wrote the paper. S. M. B. M. designed and performed the PEC experiments, analysed the data. B. K. performed photoelectrochemical O2 evolution experiments and GC analysis. P. L. performed DFT calculations and analysis the data. M. J. B. contributed reagents and materials. M. P. conceived and designed the project, contributed reagents/materials/analysis tools, and wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the project Advanced Functional Nanorobots (reg. no. CZ.02.1.01/0.0/0.0/15_003/0000444 financed by the EFRR). M. J. B. thanks the European Research Council (ERC) for funding under the Starting Grant Scheme (BEGMAT-678462). Authors acknowledge Dr Jan Plutnar from UCT Prague for XPS measurements, Dr Stanislava Matějková from IOCB Prague for elemental analysis and ICP-OES measurements, Dr Martin Dračínský from IOCB Prague for solid-state NMR measurements, and Arnošt Zukal from J. Heyrovsky Institute of Physical Chemistry for gas adsorption measurements.Notes and references
- F. K. Kessler, Y. Zheng, D. Schwarz, C. Merschjann, W. Schnick, X. Wang and M. J. Bojdys, Nat. Rev. Mater., 2017, 2, 17030 CrossRef CAS.
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- E. Samuel, B. Joshi, M.-W. Kim, M. T. Swihart and S. S. Yoon, Nano Energy, 2020, 72, 104648 CrossRef CAS.
- L. M. Peter, J. Solid State Electrochem., 2013, 17, 315–326 CrossRef CAS.
- K. Sivula, F. Le Formal and M. Grätzel, ChemSusChem, 2011, 4, 432–449 CrossRef CAS.
- P. Luan, M. Xie, D. Liu, X. Fu and L. Jing, Sci. Rep., 2014, 4, 6180 CrossRef CAS.
- Y. Sun, R. Wang and K. Liu, Appl. Phys. Rev., 2017, 4, 011301 Search PubMed.
- Y.-X. Yu, W.-X. Ouyang, Z.-T. Liao, B.-B. Du and W.-D. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 8467–8474 CrossRef CAS.
- J. R. McKone, A. P. Pieterick, H. B. Gray and N. S. Lewis, J. Am. Chem. Soc., 2013, 135, 223–231 CrossRef CAS.
- W. Septina, Gunawan, S. Ikeda, T. Harada, M. Higashi, R. Abe and M. Matsumura, J. Phys. Chem. C, 2015, 119, 8576–8583 CrossRef CAS.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef CAS.
- J. E. Thorne, J.-W. Jang, E. Y. Liu and D. Wang, Chem. Sci., 2016, 7, 3347–3354 RSC.
- C. Wang, L. Wang, J. Jin, J. Liu, Y. Li, M. Wu, L. Chen, B. Wang, X. Yang and B.-L. Su, Appl. Catal., B, 2016, 188, 351–359 CrossRef CAS.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- Q. Yang, M. Luo, K. Liu, H. Cao and H. Yan, Appl. Catal., B, 2020, 276, 119174 CrossRef CAS.
- J. Yu, X. Sun, X. Xu, C. Zhang and X. He, Appl. Catal., B, 2019, 257, 117935 CrossRef CAS.
- G. Zhang, Z.-A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 128, 15940–15996 CrossRef.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2008, 8, 76 CrossRef.
- Y. Zhang, A. Thomas, M. Antonietti and X. Wang, J. Am. Chem. Soc., 2009, 131, 50–51 CrossRef CAS.
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Fu, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 441–444 CrossRef CAS.
- L. Lin, Z. Yu and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 6164–6175 CrossRef CAS.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2015, 55, 1792–1796 CrossRef.
- J. Bi, W. Fang, L. Li, J. Wang, S. Liang, Y. He, M. Liu and L. Wu, Macromol. Rapid Commun., 2015, 36, 1799–1805 CrossRef CAS.
- M. J. Bojdys, Macromol. Chem. Phys., 2016, 217, 232–241 CrossRef CAS.
- Z. Xiang and D. Cao, J. Mater. Chem. A, 2013, 1, 2691–2718 RSC.
- J.-S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS.
- G. Li, Y. Li, H. Liu, Y. Guo, Y. Li and D. Zhu, Chem. Commun., 2010, 46, 3256–3258 RSC.
- F. Diederich and M. Kivala, Adv. Mater., 2010, 22, 803–812 CrossRef CAS.
- D. Schwarz, Y. Noda, J. Klouda, K. Schwarzová-Pecková, J. Tarábek, J. Rybáček, J. Janoušek, F. Simon, M. V. Opanasenko, J. Čejka, A. Acharjya, J. Schmidt, S. Selve, V. Reiter-Scherer, N. Severin, J. P. Rabe, P. Ecorchard, J. He, M. Polozij, P. Nachtigall and M. J. Bojdys, Adv. Mater., 2017, 29, 1703399 CrossRef.
- T. Zhang, Y. Hou, V. Dzhagan, Z. Liao, G. Chai, M. Löffler, D. Olianas, A. Milani, S. Xu, M. Tommasini, D. R. T. Zahn, Z. Zheng, E. Zschech, R. Jordan and X. Feng, Nat. Commun., 2018, 9, 1140 CrossRef.
- Y. S. Kochergin, D. Schwarz, A. Acharjya, A. Ichangi, R. Kulkarni, P. Eliášová, J. Vacek, J. Schmidt, A. Thomas and M. J. Bojdys, Angew. Chem., Int. Ed., 2018, 57, 14188–14192 CrossRef CAS.
- B. F. Straub and C. Gollub, Chem.–Eur. J., 2004, 10, 3081–3090 CrossRef CAS.
- Y. S. Kochergin, Y. Noda, R. Kulkarni, K. Škodáková, J. Tarábek, J. Schmidt and M. J. Bojdys, Macromolecules, 2019, 52, 7696–7703 CrossRef CAS.
- J. P. Paraknowitsch, A. Thomas and M. Antonietti, J. Mater. Chem., 2010, 20, 6746–6758 RSC.
- G. H. Kim, L. Shao, K. Zhang and K. P. Pipe, Nat. Mater., 2013, 12, 719–723 CrossRef CAS.
- P. Katekomol, J. Roeser, M. Bojdys, J. Weber and A. Thomas, Chem. Mater., 2013, 25, 1542–1548 CrossRef CAS.
- Y. S. Kochergin, K. Villa, F. Novotný, J. Plutnar, M. J. Bojdys and M. Pumera, Adv. Funct. Mater., 2020, 30, 2002701 CrossRef CAS.
- J. Wang, S. Zhang, J. Zhou, R. Liu, R. Du, H. Xu, Z. Liu, J. Zhang and Z. Liu, Phys. Chem. Chem. Phys., 2014, 16, 11303–11309 RSC.
- X. Zhang, Q. Zhou, Y. Huang, Z. Li and Z. Zhang, Sensors, 2011, 11, 11510–11515 CrossRef CAS.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS.
- S. Ren, M. J. Bojdys, R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Adv. Mater., 2012, 24, 2357–2361 CrossRef CAS.
- M. Valamanesh, C. Langlois, D. Alloyeau, E. Lacaze and C. Ricolleau, Ultramicroscopy, 2011, 111, 149–154 CrossRef CAS.
- D. Schwarz, A. Acharja, A. Ichangi, P. Lyu, M. V. Opanasenko, F. R. Goßler, T. A. F. König, J. Čejka, P. Nachtigall, A. Thomas and M. J. Bojdys, Chem.–Eur. J., 2018, 24, 11916 CrossRef CAS.
- S. A. Ahmed, Q.-B. Liao, Q. Shen, M. M. F. A. Baig, J. Zhou, C.-F. Shi, P. Muhammad, S. H. Hanif, K. Xi, X.-H. Xia and K. Wang, Chem.–Eur. J., 2011, 17, 2388–2392 CrossRef.
- P. Kubelka and F. Munk, Z. Tech. Phys., 1931, 593–601 Search PubMed.
- X.-J. Wen, C.-G. Niu, L. Zhang and G.-M. Zeng, Dalton Trans., 2017, 46, 4982–4993 RSC.
- H. Sun, I. H. Öner, T. Wang, T. Zhang, O. Selyshchev, C. Neumann, Y. Fu, Z. Liao, S. Xu, Y. Hou, A. Turchanin, D. R. T. Zahn, E. Zschech, I. M. Weidinger, J. Zhang and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 10368–10374 CrossRef CAS.
- P. Bornoz, M. S. Prévot, X. Yu, N. Guijarro and K. Sivula, J. Am. Chem. Soc., 2015, 137, 15338–15341 CrossRef CAS.
- H. E. Katz and Z. Bao, J. Phys. Chem. B, 2000, 104, 671–678 CrossRef CAS.
- S. Otep, T. Michinobu and Q. Zhang, Sol. RRL, 2020, 4, 1900395 CrossRef CAS.
- T. Sick, A. G. Hufnagel, J. Kampmann, I. Kondofersky, M. Calik, J. M. Rotter, A. Evans, M. Döblinger, S. Herbert, K. Peters, D. Böhm, P. Knochel, D. D. Medina, D. Fattakhova-Rohlfing and T. Bein, J. Am. Chem. Soc., 2018, 140, 2085–2092 CrossRef CAS.
- Y. Guo, J. Li, Y. Yuan, L. Li, M. Zhang, C. Zhou and Z. Lin, Angew. Chem., Int. Ed., 2016, 55, 14693–14697 CrossRef CAS.
- J. T. Kirner and R. G. Finke, J. Mater. Chem. A, 2017, 5, 19560–19592 RSC.
- D. Schwarz, Y. S. Kochergin, A. Acharjya, A. Ichangi, M. V. Opanasenko, J. Čejka, U. Lappan, P. Arki, J. He, J. Schmidt, P. Nachtigall, A. Thomas, J. Tarábek and M. J. Bojdys, Chem.–Eur. J., 2017, 23, 13023–13027 CrossRef CAS.
- H. Seema, K. C. Kemp, N. H. Le, S.-W. Park, V. Chandra, J. W. Lee and K. S. Kim, Carbon, 2014, 66, 320–326 CrossRef CAS.
- Y. Xie, T.-T. Wang, X.-H. Liu, K. Zou and W.-Q. Deng, Nat. Commun., 2013, 4, 1960 CrossRef.
- Z. Tan, H. Su, Y. Guo, H. Liu, B. Liao, A. M. Amin and Q. Liu, Polymers, 2020, 12, 719 CrossRef CAS.
- M. Ko, L. Mendecki and K. A. Mirica, Chem. Commun., 2018, 54, 7873–7891 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta11820f |
| This journal is © The Royal Society of Chemistry 2021 |