
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bimetal–organic framework derived multi-heterostructured TiO2/CuxO/C nanocomposites with superior photocatalytic H2 generation performance†
Mian Zahid
Hussain
ab,
Bart
van der Linden
c,
Zhuxian
Yang
a,
Quanli
Jia
d,
Hong
Chang
a,
Roland A.
Fischer
 b,
Freek
Kapteijn
c,
Yanqiu
Zhu
a and
Yongde
Xia
*a
b,
Freek
Kapteijn
c,
Yanqiu
Zhu
a and
Yongde
Xia
*a
aCollege of Engineering, Mathematics and Physical Sciences, University of Exeter, Exeter EX4 4QF, UK. E-mail: Y.Xia@exeter.ac.uk
bCatalysis Research Center, Technical University of Munich, Garching 85748, Germany
cCatalysis Engineering, Chemical Engineering Department, Delft University of Technology, van der Maasweg, 9, 2629 HZ Delft, Netherlands
dHenan Key Laboratory of High Temperature Functional Ceramics, Zhengzhou University, Zhengzhou, 450052, China
First published on 26th December 2020
Abstract
In situ formation of p–n heterojunctions between TiO2 and CuxO in heteroatom-doped carbon nanocomposites and their applications in photocatalytic H2 evolution were demonstrated. One-step pyrolysis of bimetal–organic-frameworks NH2-MIL-125(Ti/Cu) in steam at 700 °C forms a p–n heterojunction between TiO2 and CuxO nanoparticles. Concurrently, a phasejunction between nitrogen/carbon co-doped anatase and rutile TiO2 is formed, accompanied by the formation of CuxO heterostructures. These multi-heterostructures are embedded in N-containing and hydrophilic carboxyl functionalized carbon matrix. The optimized TiO2/CuxO/C composite multi-heterostructures offer multiple pathways for photoinduced electrons and holes migration, absorb more visible light, and provide an increased number of active sites for photocatalytic reactions. Without loading expensive noble metals, the TiO2/CuxO/C nanocomposite derived at 700 °C in steam exhibited a superior photocatalytic H2 generation activity of 3298 μmol gcat−1 h−1 under UV-Visible light, 40 times higher than that of commercial TiO2. This work offers a simple approach to fabricate novel photocatalytic nanocomposites for efficient H2 generation.
1. Introduction
Depleting fossil fuel reserves, enormous CO2 emissions and consequential environmental pollution are the biggest challenges to our society. To keep the pace of industrial growth and global economies, enormous efforts have been devoted to finding low cost and sustainable clean energy solutions.1 Hydrogen (H2) gas evolution from water splitting by using sunlight is regarded as one of the cheapest and cleanest energy sources.1–5 Commercially, H2 is largely produced by methane steam reforming which is an energy-consuming and environmentally unsustainable process. H2 evolution from water splitting under sunlight mimics natural photosynthesis and it is generally accepted that if achieved at an economically affordable scale, photocatalytically generated H2 can be commercially used as a clean fuel in automobiles, fuel cells and domestic heating.6–11Photocatalytic H2 evolution mainly consists of three steps: (i) generation of electrons and holes by absorbing UV and/or visible light, (ii) separation and transfer of these photogenerated electrons and holes to the surface of the photocatalyst, and finally (iii) photocatalytic reduction and oxidation reactions at the active sites to generate H2 and O2 respectively.3,5 The band gap energy of the photocatalyst must be around or below 3 eV and above 2.3 eV with suitable conduction band (CB) and valence band (VB) edge potentials (vs. NHE) to overcome the over-potential needed for the hydrogen evolution reaction (HER). Besides, the photocatalyst must be physically and chemically stable during the photocatalytic reactions to ensure stable H2 evolution for a longer period.12,13 TiO2 is proved to be the most promising semiconducting material due to its appropriate conduction band position (more negative than 0 eV vs. NHE at pH = 0) and valence band positon (more positive than 1.23 eV vs. NHE at pH = 0). However, due to the large energy band gaps, these photocatalysts can only be activated under UV light. To overcome the challenges of the wide energy band gap, several methods have been reported such as the doping of anionic (like N, C, S, and P) and cationic (like metals atoms) species into semiconducting metal oxides to create new energy levels above the valence and below the conduction band edges to narrow down the energy band gaps respectively.14 Although doping of such species in pure TiO2 results in enhanced visible light absorption, a serious challenge of low performance for such single metal oxide based photocatalysts is the recombination of photogenerated electrons and holes.15–19 Therefore, it is important to improve charge separation and charge migration in the photocatalyst particles for an enhanced quantum yield.
Many strategies have been developed to increase charge separation and charge migration efficiencies of photocatalytic systems, such as constructing a phasejunction between the different phases of the same metal oxide (e.g. anatase and rutile phase TiO2) or a heterojunction at the interfaces of different metal and/or non-metal compounds (e.g. TiO2/CuOx, Cu2O/g-C3N4) to spatially separate the photogenerated electrons and holes.3,4,20–22 Moreover, the number of active sites for the photocatalytic water reduction reaction is increased with the help of noble metal (e.g. Pt, Au and Pd) cocatalysts.3,17,23–26 Although many of these strategies can enhance the photocatalytic H2 evolution performance compared to commercially available pure TiO2 nanoparticles, the synthesis methods are generally complicated, expensive and not commercially affordable. A promising solution to solve these problems is to synthesize metal compound/carbon composite materials as photocatalysts. However, the H2 evolution efficiencies of such metal compound/carbon semiconducting photocatalysts frequently remain low, which can be attributed to surface agglomerations and less accessible active sites.22,27 The conventional approaches such as physical mixing of high surface area graphene or carbon materials and metal oxides hardly form a homogeneous distribution of metal oxide in carbon materials with convincing interfacial contacts for better charge transfer and quenching of surface charge recombination.28,29
Metal–organic frameworks (MOFs) possess exceptionally high BET surface areas and tunable porosities with highly accessible catalytic active sites. They have recently been explored as photocatalysts for H2 evolution.8,30–33 For instance, Karthik et al. recently reported that Cu2+ coordinated Ti-MOF exhibited much higher photocatalytic H2 evolution performance than the pristine Ti-MOF under sunlight due to d–d transition and enhanced absorption of visible light.34 Despite their fascinating morphology, promising textural and chemical properties, these photoactive MOFs suffer from poor charge generation, and low charge transfer as well as structural instability, as the photocatalytic H2 evolution reaction is carried out in aqueous media, which makes the pristine MOFs less favorable materials to be directly used as photocatalysts.9,33,35–37 However, in recent years MOFs have been proven to be excellent precursors and sacrificial templates to derive metal oxide/carbon nanocomposites. Photoactive transition metal based MOFs can be used as sacrificial templates to derive in situ N and/or C doped single/multi-metal oxide nanoparticles embedded in functionalized porous carbon structures with inherited morphologies, high BET surface areas, modifiable textural properties and tunable heterostructures for applications in energy and environment related fields.12,30,35,38–40 For example, Liu et al. reported Fe-MOF/melamine derived α-Fe2O3/g-C3N4 nanocomposites with a 2D nanosheet morphology. With Pt as the cocatalyst, these optimized nanocomposites forming a Z-scheme heterojunction between α-Fe2O3 and g-C3N4 generated 2.07 mmol H2 gcat−1 h−1 under visible light irradiation.41 Xu et al. prepared NH2-UiO-66/melamine derived high performing C–ZrO2/g-C3N4 nanocomposites forming a type II staggered heterojunction. Instead of Pt, they loaded 20 wt% Ni2P as a cocatalyst and a photosensitizer Eosin Y (EY), the resulting material generated 10.04 mmol gcat−1 h−1 H2 under visible light. The formation of a metal oxide/carbon nitride heterojunction and the presence of a cocatalyst facilitate accelerated charge transfer and the surface photocatalytic reactions respectively. Without the Ni2P cocatalyst, C–ZrO2/g-C3N4 could only produce 0.036 mmol gcat−1 h−1 H2 under the same conditions.42 Recently, Hao et al. demonstrated that when decorated with Pt cocatalyst, Cu–TiO2/C hollow nanospheres derived from an external-templated-MOF precursor, SiO2/HKUST-1-Ti can be a high performing photocatalyst.43 Though photocatalytically efficient, to avoid the use of expensive cocatalyst Pt and the complicated multi-step synthetic procedures, the band gap positions of in situ formed heterojunctions, and the physicochemical properties as well as the morphologies of the MOF derived nanocomposites can be effectively adjusted to achieve high performing and stable photocatalysts for H2 evolution.
In this work, we present that a Cu2+ coordinated bimetal–organic framework (bi-MOF), NH2-MIL-125(Ti/Cu), can be employed as an appropriate sacrificial template to simply derive a TiO2/CuxO/C multi-heterojunction for enhanced photocatalytic H2 evolution. Without loading expensive noble metals (Pt, Au or Pd) and additional cocatalysts/photosensitizers, the nanocomposite derived from bi-MOF(Ti/Cu) at optimal 700 °C under water vapor exhibited superior photocatalytic H2 generation performance and produced 3298 μmol gcat−1 h−1 H2 gas under UV-Vis light, which is 40 fold higher than that of the commercial TiO2 (P-25) reference sample. This study demonstrates the successful formation of bi-MOF derived nanocomposite multi-heterostructures with simultaneous phasejunctions between anatase/rutile TiO2 and Cu2O/CuO as well as a p–n heterojunction between TiO2 and CuxO, which exhibited remarkable photocatalytic H2 evolution performance. Based on our findings, this study suggests that by optimizing the relative Cu loading on NH2-MIL-125(Ti/Cu) and the Ti/Cu ratios in derived bi-metal oxide nanocomposites, the photocatalytic H2 evolution activity can be further enhanced manifold.
2. Experimental section
2.1. Synthesis of NH2-MIL-125(Ti)
For synthesis, 60 mmol (10.87 g) of 2-aminoterephthalic acid was dissolved in a solvent mixture of dimethylformamide (DMF) and methanol (MeOH) with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio (75 mL each) in a 500 mL screw jar under stirring for 30 min. Then 20 mmol (6.88 mL) of Ti(OBu)4 was added slowly under constant stirring for 15 minutes before placing it in an oil bath at 130 °C. After 40 hours, a yellow colored product was collected by centrifugation followed by washing twice with DMF to remove all unreacted organic ligand species and then twice with MeOH for solvent exchange. The obtained NH2-MIL-125(Ti) was finally dried at 80 °C in air overnight.32
:1 volume ratio (75 mL each) in a 500 mL screw jar under stirring for 30 min. Then 20 mmol (6.88 mL) of Ti(OBu)4 was added slowly under constant stirring for 15 minutes before placing it in an oil bath at 130 °C. After 40 hours, a yellow colored product was collected by centrifugation followed by washing twice with DMF to remove all unreacted organic ligand species and then twice with MeOH for solvent exchange. The obtained NH2-MIL-125(Ti) was finally dried at 80 °C in air overnight.32
2.2. Synthesis of NH2-MIL-125(Ti/Cu)
To load Cu species into the MOF crystals, first, 2 g of activated NH2-MIL-125(Ti) was dispersed in a mixture of DMF and MeOH with a 1:1 ratio (100 mL each) for 1 hour under ultrasonication. 0.87 g of Cu(NO3)2·3H2O was dissolved in 20 mL methanol and poured into the MOF dispersion before placing it in an oil bath at 120 °C for 12 hours. Dark green color crystals of NH2-MIL-125(Ti/Cu) were collected upon centrifugation followed by washing twice with MeOH for solvent exchange. The obtained NH2-MIL-125(Ti/Cu) was dried at 80 °C in air overnight.
2.3. Synthesis of multi-heterostructured TiO2/CuxO/C nanocomposites
In situ nitrogen/carbon co-doped TiO2/CuxO/C nanocomposites were prepared by one-step direct carbonization of sacrificial template NH2-MIL-125(Ti/Cu) at 550, 700 and 800 °C under water vapor carried by argon gas which is formed via argon gas passing through a water buffer bottle at room temperature. For each sample, 500 mg of the as-prepared NH2-MIL-125(Ti/Cu) was loaded in an alumina boat and placed at the center of a flow-through quartz tube sitting in a tube furnace. Before starting the heating process, the quartz tube was purged with argon gas for 30 minutes to make sure that no oxygen is present in the tube. The heating was started under an argon flow only at a flow rate of 50 mL min−1. When the tube furnace reached the desired temperature, water vapor was introduced along with the argon flow for 2 hours and then it was cooled down to room temperature naturally under an argon flow only. The resulting products were designated as TiCuC550, TiCuC700 and TiCuC800 for the sample derived from the pyrolysis of NH2-MIL-125(Ti/Cu) at temperatures of 550, 700 and 800 °C respectively. For comparison, a sample derived from the pyrolysis of NH2-MIL-125(Ti) without the Cu2+ loading was also synthesized at 700 °C in water vapor for 2 hours and the sample is labelled TiC700.2.4. Materials characterization
Powder X-ray diffraction (PXRD) measurements of the sacrificial template (2θ = 5–50°) and its derived composites (2θ = 5–90°) were performed by employing Bragg–Brentano geometry in a PANalytical Empyrean diffractometer equipped with a PANalytical PIXcel 1D detector. X-ray Cu Kα radiation was λ1 = 1.5406 Å, λ2 = 1.5444 Å, I2/I1 = 0.5. The voltage and current were 45 kV and 40 mA, respectively. The morphologies of the NH2-MIL-125(Ti/Cu) and derived TiO2/Cu2O/C composites were characterized using an xT Nova Nanolab 600 FIB coupled with an in situ SEM all in one unit. Transmission electron microscopy (TEM) (JEM 2100 LeB6 200 kV) coupled with energy dispersive X-ray spectroscopy (EDX) was employed to obtain high-resolution TEM images and elemental mapping. Thermal stability and the weight loss (wt%) of activated MOFs and the derived composites were measured using a Mettler Toledo TGA/STA 409 PC apparatus with a continuous heating ramp of 10 °C min−1 applied under a synthetic air flow. To investigate the nature of carbon and metal oxide species, Raman spectra were recorded employing a Renishaw inVia Reflex Raman System RL532C, Class 3B in the range from 50 to 3000 cm−1. Fourier-transform infrared (FT-IR) spectra of the samples were recorded in the range of 4000–400 cm−1 using a Bruker Optics Tensor-27 FTIR spectrometer. For surface chemical analysis, a Kratos Axis Ultra DLD spectrometer with a monochromated Al Kα X-ray source operating at 168 W (12 mA × 14 kV) was employed. Data were collected with pass energies of 160 eV for survey spectra, and 20 eV for high-resolution scans with step sizes of 1 eV and 0.1 eV respectively over an area approximately 300 × 700 μm2 under a chamber pressure of 1 × 10−9 bar. Data were analyzed using CasaXPS (v2.3.23) after subtraction of a Shirley background and using modified Wagner sensitivity factors as supplied by the manufacturer. XPS depth profiling (DP) experiments were conducted to find out the distribution of each element (atomic concentrations (%)) using a 110 μm diameter spot after etching over a 2 × 2 mm area using 1 × 10−6 Torr argon at 4 kV. The energy band gaps of the derived nanocomposites were estimated from the UV-Vis absorption spectra acquired by employing a ThermoScientific Evolution 200 spectrophotometer. The specific surface areas (SSA) and the pore size distributions (PSD) of the composites were measured by N2 sorption at 77 K on a Quantachrome autosorb iQ2 ASiQwin apparatus equipped with a micropore port (1 × 10−5 bar) via the conventional volumetric technique. Before surface area analysis, the samples were degassed at 180 °C for 6 hours under vacuum. The pore size distribution was determined using the non-local density functional theory (NLDFT) method. A CHI660E electrochemical workstation was employed to measure electrochemical impedance spectra (EIS) at room temperature. In a typical three-electrode electrochemical setup, a carbon rod and Ag/AgCl were used as the counter and reference electrodes respectively. A finely polished mirror-like glassy carbon (GC) electrode (2.8 cm2) was used as a working electrode, prepared to test the EI spectra by depositing a 5 μL solution of the sample (2 mg of photocatalyst dispersed in 1 mL of water–ethanol (4:1 v/v) solution with additional 5 μL of 5 wt% Nafion solution using an ultrasonic bath for 60 min) followed by drying in air overnight. All EIS measurements were performed at the same potential in a 0.5 M H2SO4 solution used as an electrolyte.
2.5. Hydrogen evolution reaction (HER) measurements
Photocatalytic hydrogen evolution reaction (HER) experiments were carried out in a methanol/water solution under UV-Visible light using a 500 W Xe/Hg lamp (66983, Newport) with a 285 nm cut-off optical filter. It involved a custom-made Pyrex-glass reactor, a CP 9001 gas chromatograph (GC, Chrompack) for analysis of the headspace and the light source. Light intensity was measured with an AvaSpec-3648-2-USB2 (Avantes, the Netherlands). The reactor has a total volume of 42.1 mL including 17.1 mL of headspace. The reactor is equipped with a water jacket to precisely control the temperature. The light emitted by the Xe/Hg lamp passes through a lens assembly (77330, Newport) (focusing the beam with a spot size of 2.23 cm2 reaching the reactor window) and a H2O filter (61945, Newport). A probe of the headspace is analyzed every 30 min using the GC. In a typical experiment, 10 mg of the photocatalyst was suspended in 7.5 mL of CH3OH and 17.5 mL of H2O. The suspension was then placed in the reactor and was purged by an argon flow of 30 mL min−1 for 30 min to deoxygenate the system. The temperature of the reactor was fixed at 30 °C. After completely deoxygenating the reactor, illumination was applied followed by GC analysis. The suspension was kept under stirring during the reaction with a small Argon flow through the headspace towards the GC sample loop. The apparent quantum yields (AQY%) of the derived nanocomposites were calculated at 435 nm using the following equation:44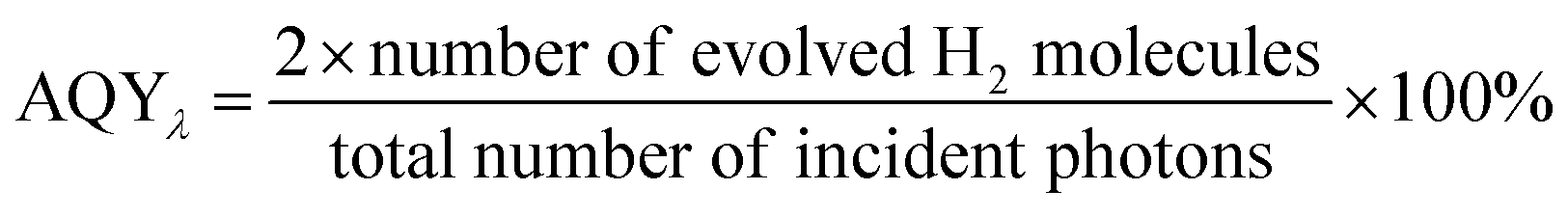 | (1) |
3. Results and discussion
3.1. Synthesis and materials characterization
The powder X-ray diffraction (PXRD) patterns (Fig. S1, ESI†) of activated MOF, NH2-MIL-125(Ti) and bi-MOF, NH2-MIL-125(Ti/Cu) confirm the successful formation and incorporation of Cu2+ into the NH2-MIL-125(Ti/Cu) with no formation of additional phases. PXRD peaks that appeared at 2θ values of 6.8° and 9.8° are in good agreement with the simulated XRD pattern of NH2-MIL-125(Ti). To confirm that the Cu2+ ions are coordinated to the amino (–NH2) group, the XPS spectra of N 1s of NH2-MIL-125(Ti/Cu) were recorded. As shown in Fig. S2 in the ESI,† deconvoluted peaks appeared at 399.3 and 400.1 eV, which can be assigned to the positively charged amino group (–NH2+) due to charge transfer to the coordinated Cu2+ species. The emergence of the shoulder peak can be attributed to the donation of non-bonded lone pair electrons of the –NH2 group to the Cu2+ ions resulting in the formation of quaternary valence –NH2+.34Pyrolysis of bi-MOF NH2-MIL-125(Ti/Cu) at 550, 700 and 800 °C in water vapor, resulted in the decomposition of NH2-MIL-125(Ti/Cu) into TiO2, CuxO and Cu species embedded in a porous carbon matrix. The PXRD patterns (Fig. 1a) show the main peaks of anatase (101) and rutile (110) appearing at 2θ of 25.2° and 36.4° respectively with varying intensity. Depending upon the pyrolysis temperatures, the ratios of the anatase to rutile phase of TiO2 polymorphs were calculated to be 91/9, 45/55 and 10/90 for samples derived at pyrolysis temperatures of 550, 700 and 800 °C under water vapor respectively. In a recent study, we confirmed the role of temperature and the gaseous atmosphere in the crystallinity and phase formation of TiO2 nanoparticles derived from NH2-MIL-125(Ti).45 Using the Scherrer equation, the average particle sizes of anatase and rutile phases of TiO2 were calculated to be 2.6/4.8, 10.3/18.8 and 11.4/23.4 nm for the samples obtained at 550, 700 and 800 °C respectively. The formation of larger TiO2 nanoparticles at higher temperature might be due to the sintering effect.38,45 In addition, along with the formation of optimized TiO2 polymorphs, the TiO2/CuxO heterojunction formed during the pyrolysis of the Cu-loaded bi-MOF NH2-MIL-125(Ti/Cu) at 700 °C which is confirmed by employing several characterization techniques. Das et al. reported that the metals with a reduction potential above −0.27 V such as Cu2+ usually form pure metal nanoparticles (Cu0) upon pyrolysis of the MOFs in an inert atmosphere.46 However, pyrolysis under controlled water vapor (PXRD Fig. 1a) results in the transformation of Cu2+ into Cu2O at 550 °C as a dominant phase with a trace amount of the CuO phase, whereas on pyrolysis at 700 °C, Cu2O and CuO phases co-exist with some residual Cu0 nanoparticles. However, at a pyrolysis temperature of 800 °C, CuO becomes the dominant phase with a small amount of Cu2O and Cu0 nanoparticles in the resulting composite. The highlighted PXRD peaks of CuxO (inset in Fig. 1a) suggest that the pyrolysis temperatures also affect the 2θ values of the crystallites and, therefore, the particle sizes of the formed CuxO. These observations are further confirmed by XPS of Cu 2p as well as Raman spectra.
 | ||
| Fig. 1 (a) Powder X-ray diffraction patterns, (b) TGA in air, (c) SEM images and (d) representative schematic diagrams of bi-MOF NH2-MIL-125(Ti/Cu) derived nanocomposites TiCuC550, TiCuC700 and TiCuC800 obtained at pyrolysis temperatures of 550, 700 and 800 °C respectively. | ||
The thermogravimetric analysis (TGA) profiles of NH2-MIL-125(Ti/Cu) and the derived composites were recorded under a synthetic air flow to quantify the amount of TiO2, CuxO and C species. As shown in Fig. S3 in the ESI,† NH2-MIL-125(Ti) and NH2-MIL-125(Ti/Cu) exhibited around 9% weight loss up to 300 °C due to the release of adsorbed moisture and the residual organic linker. The NH2-MIL-125(Ti) framework starts to decompose at around 350 °C in air releasing primarily CO2 and NOx due to the oxidation of the organic linker and leaving behind 34 wt% TiO2 nanoparticles.47 However, the TGA profile of the bi-MOF, NH2-MIL-125(Ti/Cu) shows the decomposition of the framework starting at around 250 °C due to the breaking of Cu–N bonds resulting in the formation of CuO nanoparticles under an air flow. The weight percentage of Cu species is estimated to be 10.5 wt% with the molar ratio of Ti:Cu being 1:0.30, which is in good agreement with EDX elemental mapping results. Similarly, the TGA profiles of NH2-MIL-125(Ti/Cu) derived nanocomposites presented in Fig. 1b indicate that the amount of carbon for samples obtained at pyrolysis temperatures of 550, 700 and 800 °C is 27, 15 and 5 wt% respectively, which is burned off in the temperature range of 300–400 °C in air. The successive decrease in the amount of carbon in derived samples with increasing pyrolysis temperature is observed because some of the formed carbons can readily react with water vapor at a high pyrolysis temperature and release as CO/CO2.
The representative SEM images suggest that with the increase of the pyrolysis temperature from 550 °C to 800 °C, the thermal decomposition of bi-MOF NH2-MIL-125(Ti/Cu) in water vapor results in the formation of carbon, TiO2 and CuxO nanocomposites with disc-like morphologies similar to their precursor NH2-MIL-125(Ti/Cu) with a particle size up to 1.5 μm (Fig. 1c); however, the particle surface of the nanocomposites turn rougher compared to their precursor. At a pyrolysis temperature of 550 °C in water vapor, the TiO2 and CuxO nanoparticles are buried inside the carbon matrix of sample TiCuC550 with a rougher surface compared to NH2-MIL-125(Ti/Cu). For sample TiCuC700 obtained at 700 °C, more TiO2 and CuxO species are exposed but still embedded homogeneously in a porous carbon matrix. In the case of sample TiCuC800 at a pyrolysis temperature of 800 °C in water vapor, more formed carbon is turned into CO2, which results in the TiO2 and CuxO nanoparticles being fully exposed. Based on the results from TGA and the representative SEM images, a schematic diagram of NH2-MIL-125(Ti/Cu) derived nanocomposites (Fig. 1d) is presented. This observation is also in good agreement with the TEM images of the samples derived at 550, 700 and 800 °C (Fig. S4a–c, ESI†) which show a relative recession in the carbon matrix as well as enlargement of TiO2 nanoparticles with the increase of the pyrolysis temperature.
The textural properties of the bi-MOF precursor together with its derived nanocomposites were evaluated via gas sorption analysis. The N2 sorption isotherms for the precursor NH2-MIL-125(Ti) and NH2-MIL-125(Ti/Cu) (Fig. S5a, ESI†) are typical microporous materials with a narrow pore size distribution (Fig. S5b, ESI†). Their specific surface area and pore volume are summarized in Table S1,† which are in good agreement with the literature reports.34 N2 sorption isotherms for the MOF derived nanocomposites largely exhibited reversible adsorption and desorption branches (Fig. 2a) and their textural properties are summarized in Table 1. In general, a higher pyrolysis temperature results in an increase in the BET surface area of the resulting samples. Despite the higher weight percentage of carbon, the relatively lower BET surface area in sample TiCuC550 might be due to the presence of residual species from the decomposed organic linkers blocking the pores, whereas the relatively lower BET surface area in the sample TiCuC800 compared to TiCuC700 can be due to the evaporation of most of the carbon (lower weight percentage) in the form of CO2. In addition, sample TiCuC700 with the highest BET surface area is dominated with relatively narrow micropores; however, mesopores turn to be dominant in sample TiCuC800 (inset in Fig. 2a). The higher specific surface area of the materials not only provides more exposed photocatalytic active sites, but also is beneficial to support the metal oxides species with better distribution throughout the carbon matrix without agglomerations. Moreover, the higher specific surface area of the micro/mesoporous carbons in the composites decorated with hydrophilic carboxylate functional groups can accommodate more H2O molecules that can readily interact with the catalytic active sites in the composites, and therefore results in the increase of the photocatalytic activity for the redox reaction.
 | ||
| Fig. 2 (a) N2 sorption isotherms (inset shows their corresponding pore size distribution), (b) Raman spectra, (c) Tauc plots (inset shows their corresponding UV-Vis absorption spectra) and (d) XPS valence band positions of TiCuC550 (black), TiCuC700 (red) and TiCuC800 (blue). In (a), the filled and empty symbols represent adsorption and desorption branch of isotherms respectively. | ||
| Sample | BET surface area (m2 g−1) | Total pore volume (cm3 g−1) | Energy band gap (eV) | H2 evolution (μmol gcat−1 h−1) | AQY at 435 nm (%) |
|---|---|---|---|---|---|
| TiCuC550 | 122 | 0.17 | 2.54 | 198 | 0.45 |
| TiCuC700 | 337 | 0.26 | 2.47 | 3298 | 7.79 |
| TiCuC800 | 277 | 0.37 | 2.56 | 1360 | 3.12 |
| TiC700 | 378 | 0.33 | 2.95 | 426 | — |
| Comm. TiO2 | 41 | 0.21 | 3.16 | 82 | — |
Raman spectra were recorded to investigate the vibrational modes of TiO2 and CuxO as well as the nature of carbon present in these samples. In pure TiO2 the prominent vibrational mode Eg(1) of anatase TiO2 appears at 144 cm−1.48 However, in samples TiCuC550 and TiCuC700 this mode is observed at 146 and 154 cm−1 respectively, whereas it is negligibly small in sample TiCuC800. This shift of the Eg(1) mode in TiCuC700 may be ascribed to the dominance of the rutile phase along with the phase change of anatase, and the lattice vibrations as well as the formation of localized defects due to in situ anionic doping of N and C atoms into TiO2 nanoparticles, which will be further confirmed by UV-Vis absorption spectra and XPS. As exhibited in PXRD results, at 550 °C the poorly crystalline anatase phase of TiO2 is dominant while at 700 °C both highly crystalline anatase and rutile phases are formed with a phase ratio of 45/55.45 At 800 °C, the anatase phase almost disappears with the dominant emergence of the rutile phase. This is also in very good agreement with Raman spectra results. At 700 °C, a slightly broader but intense peak of the Eg(1) vibrational mode is observed. The inset in Fig. 2b shows the deconvoluted peaks of the Ti–O symmetric bending mode (B1g) of the anatase phase at 412 cm−1 and the vibrational modes Eg of the rutile phase at 430 and 442 cm−1 in TiCuC700 and TiCuC800 respectively. Moreover, the rutile phase also shows a similar shift in Eg vibrational modes, which suggests that the presence of N and C atoms may cause a dopant induced strain on the TiO2 crystal lattices that result in a shift in the frequencies of Eg modes.49 It is worth noting that for the NH2-MIL-125(Ti) derived TiC700 nanocomposite obtained under identical pyrolysis conditions (in water vapor at 700 °C for 2 hours) in the absence of Cu species, the Eg vibrational modes of the rutile phase appear at 441 cm−1.45 This indicates that the presence of Cu2O and/or Cu nanoparticles at the interfacial contact with TiO2 nanoparticles may modify the band positions and therefore result in a shift in the frequencies of vibrational modes. The vibrational bands of Cu2O (inset in Fig. 2b) can be observed at around 285, 266, 246 and 630 cm−1 for sample TiCuC700, which are also all observed in samples TiCuC550 and TiCuC800 with slightly shifted frequencies and varying intensities. While the peaks at around 246, 285 and around 585 cm−1 can be ascribed to the second-order Raman allowed vibrational modes of Cu2O crystals, the peak at around 630 cm−1 may correspond to the Bg allowed stretching modes of Cu2O/CuO nanoparticles.50–52
As discussed above, the precursor NH2-MIL-125(Ti/Cu) thermally decomposes under water vapor and the organic linker in the precursor transforms into N and O-containing functional porous carbon. Fig. 2b shows the signature peaks of D and G bands in the first-order Raman spectra of MOF derived carbon, appearing at around 1352 and 1593 cm−1 respectively in samples TiCuC550 and TiCuC700. However, these D and G bands are slightly shifted to 1342 and 1598 cm−1 in sample TiCuC800. The D band is due to the breathing modes of sp2 hybridized carbon atoms in hexagonal rings and represents the number of defects and disorders in carbon. The increase in the intensity, as well as the redshift of the D band (Fig. 2b) of carbon present in TiCuC700 and TiCuC800, correspond to the formation of a higher amount of surface defects in these samples. The G band in the Raman spectra of carbon arises from the bond stretching of all sp2 atoms in hexagonal rings and chains of carbon. This band appears due to the formation of nanocrystalline carbon.53,54 The blueshift in the G bands of derived carbon in samples TiCuC550, TiCuC700 and TiCuC800 can be attributed to the oxidation of the organic linker NH2-BDC under water vapor to form N-containing and hydroxyl/carboxyl group functionalities.55 The ID/IG ratios in TiCuC550, TiCuC700 and TiCuC800 are calculated to be 0.63, 0.92 and 0.91 respectively which indicate that the higher pyrolysis temperature results in a higher amount of defects in the formed carbon matrix.56
Pyrolysis of MOFs under water vapor also functionalizes the porous carbon matrix with hydrophilic carboxylic groups.56–58 The presence of surface functional groups in the NH2-MIL-125(Ti/Cu) derived nanocomposites is confirmed by FT-IR spectra. As shown in Fig. S6 in the ESI,† a strong band that appeared at 1630 cm−1 can be assigned to the carboxyl (–COOH) functional groups introduced via water vapor during the pyrolysis process at high temperature.56 While the strong peak at 1385 cm−1 can be ascribed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond stretching, the C–N bond vibration at around 1240 cm−1 arises from the N species decorated on the carbon matrix.34,58 As demonstrated in Fig. S6 in the ESI,† the CHx bond stretching at around 2900 and 1400 cm−1 is negligible,59 and C–O, N–O, OC–OH and C–N bonds are clearly presented between 1700 and 1200 cm−1 as surface functional groups in these as-prepared TiO2/CuxO/C composites. The stretching modes for pure Cu2O and TiO2 usually appear at 630 and 628 cm−1 respectively.60–62 However, in NH2-MIL-125(Ti/Cu) derived nanocomposites, the Ti–O and Cu–O peaks appeared at 633, 628 and 608 cm−1 in samples TiCuC550, TiCuC700 and TiCuC800 respectively. This frequency shift of Ti–O and Cu(I)–O bond stretching modes may be due to the transformation of crystalline phases and the doping of N and C atoms into the TiO2 and Cu2O nanoparticles, as well as the localized surface defects formed during the thermal decomposition of the bi-MOF precursor into the metal oxide species.61
O bond stretching, the C–N bond vibration at around 1240 cm−1 arises from the N species decorated on the carbon matrix.34,58 As demonstrated in Fig. S6 in the ESI,† the CHx bond stretching at around 2900 and 1400 cm−1 is negligible,59 and C–O, N–O, OC–OH and C–N bonds are clearly presented between 1700 and 1200 cm−1 as surface functional groups in these as-prepared TiO2/CuxO/C composites. The stretching modes for pure Cu2O and TiO2 usually appear at 630 and 628 cm−1 respectively.60–62 However, in NH2-MIL-125(Ti/Cu) derived nanocomposites, the Ti–O and Cu–O peaks appeared at 633, 628 and 608 cm−1 in samples TiCuC550, TiCuC700 and TiCuC800 respectively. This frequency shift of Ti–O and Cu(I)–O bond stretching modes may be due to the transformation of crystalline phases and the doping of N and C atoms into the TiO2 and Cu2O nanoparticles, as well as the localized surface defects formed during the thermal decomposition of the bi-MOF precursor into the metal oxide species.61
The UV-Vis absorption spectra of NH2-MIL-125(Ti) and NH2-MIL-125(Ti/Cu) are shown in Fig. S7 in ESI.† The Cu2+ ions coordinated to the –NH2 functional group improves the visible light absorption for NH2-MIL-125(Ti/Cu), which results in a redshift from 470 to 484 nm due to the d–d transitions. The change of color (Fig. S8a and b, ESI†) for NH2-MIL-125(Ti) from yellow to greenish for NH2-MIL-125(Ti/Cu) is consistent with the redshift towards the visible region.34,63 The energy band gaps of NH2-MIL-125(Ti/Cu) derived nanocomposites were estimated from the UV-Vis absorption spectra (inset in Fig. 2c). The Tauc plots (Fig. 2c) show that the energy band gaps of in situ N/C co-doped TiCuC550, TiCuC700 and TiCuC800 are estimated to be 2.54, 2.47 and 2.56 eV respectively. These samples are predominantly black (Fig. S8c–e, ESI†) due to the presence of a significant amount of porous carbons. It is worth noting that the non-linear change in energy band gaps in the bi-MOF (Ti/Cu) derived samples can be attributed to the different types of heterostructures. The relatively narrower energy band gap in sample TiCuC700 can be referred to the formation of multi-heterostructures due to the simultaneous presence of the phasejunction between anatase and rutile TiO2, and the p–n heterojunction between TiO2 and CuxO. To further confirm the shift in energy band positions due to N/C co-doping and the formation of the p–n heterojunction between TiO2 and CuxO, XPS spectra at a lower binding energy were recorded. As shown in Fig. 2d, the valence band spectra exhibit two shoulder-like bends attributed to the valence band edges of TiO2 and Cu2O nanoparticles with respect to the Fermi level. The valence band positions of TiO2 nanoparticles were estimated to be 1.49, 1.36 and 1.48 eV for TiCuC550, TiCuC700 and TiCuC800 respectively. For Cu2O, the valance band positions were estimated to be 0.23, 0.11 and 0.12 eV for samples TiCuC550, TiCuC700 and TiCuC800 respectively. The obvious shift in the valance band positions of Cu species also confirms the transformation of Cu2O into CuO at the higher temperature. As confirmed by PXRD, the anatase to rutile phase ratio in sample TiCuC700 is 0.8:1; therefore, a big shift in the valence band position of TiO2 in TiCuC700 may be due to the band bending at the interfaces of anatase and rutile phases which form a phasejunction. Based on the values of energy band gaps obtained from the Tauc plots and the valence band positions with respect to the core levels of Ti 2p and Cu 2p, a schematic diagram of energy band alignments (Fig. S9, ESI†) for NH2-MIL-125(Ti/Cu) derived nanocomposite TiCuC700 can be drawn between n-type TiO2 and p-type Cu2O forming a p–n heterojunction.64–66 Due to the formation of a p–n heterojunction between N/C co-doped TiO2 and Cu2O nanoparticles and the presence of homogeneously dispersed Cu0 atoms without agglomerations in porous carbon, these black nanocomposites are expected to provide not only better visible light absorption, but also increased photogeneration as well as improved separation of electrons and holes that can result in enhanced photocatalytic H2 evolution.
The high-resolution transmission electron microscopy (HRTEM) images of TiCuC550, TiCuC700 and TiCuC800 (Fig. 3a–f) confirm that the TiO2, Cu2O and Cu0 interfaces are physically in contact forming the heterojunctions. Fig. 3a and d show the predominant anatase phase of TiO2 with an interplanar spacing of 0.32 nm and Cu2O present in TiCuC550 as confirmed by PXRD. However, for sample TiCuC700, well crystalline anatase (101) and rutile (110) TiO2 polymorphs with an interplanar spacing of 0.32 and 0.31 nm respectively can be seen in Fig. 3b and e along with the Cu2O and Cu0 nanoparticles. The crystalline sizes observed in HRTEM images are in good agreement with the particle sizes calculated from the PXRD patterns using the Scherrer equation, which are around 10 nm and 20 nm for the rutile and anatase phases of TiCuC700 respectively. The reported lattice spacing of pure TiO2 is around 0.35 and 0.32 nm for anatase and rutile phases respectively.67 The shift in the interplanar spacing of the NH2-MIL-125(Ti/Cu) derived nanocomposites indicates that the doping of N and C atoms into TiO2 creates the localized energy states above the valence band which may result in the narrowing of the lattice spacing.15,68 For sample TiCuC800 (Fig. 3c and f) the predominant rutile phase of TiO2 forms a heterojunction with CuxO nanoparticles. The interplanar spacing of rutile TiO2 estimated from SAED (inset in Fig. 3c) is 0.31 nm which is consistent with the value calculated from PXRD patterns.
 | ||
| Fig. 3 Representative HRTEM images of (a and d) TiCuC550, (b and e) TiCuC700 and (c and f) TiCuC800 (SAED in the inset); (g) EDX elemental mappings of representative sample TiCuC700. | ||
Energy dispersive X-ray (EDX) elemental mapping was performed to investigate the distribution of elements Ti, O, Cu, C and N in the NH2-MIL-125(Ti/Cu) derived nanocomposites. It is evident from the representative elemental mappings of TiCuC700 (Fig. 3g) that all the elemental species are uniformly distributed throughout the nanocomposite. The EDX elemental mappings of NH2-MIL-125(Ti/Cu) and the derived TiCuC550 and TiCuC800 (Fig. S10a, b and c, ESI,† respectively) also confirm that all the elements are homogeneously present in these samples.
The elemental composition, and the chemical and electronic states of each element in the NH2-MIL-125(Ti/Cu) derived nanocomposites were investigated by X-ray photoelectron spectroscopy (XPS). The element survey spectrum (Fig. 4a) confirms the presence of Ti, Cu, O, C and N species in all three composites, which is also in agreement with EDX elemental analysis. As shown in Fig. 4b, the Ti 2p3/2 peak for samples TiCuC550, TiCuC700 and TiCuC800 appears at 458.3, 458.5 and 458.2 eV respectively, which confirm the Ti4+ oxidation state of O–Ti–O in TiO2 nanoparticles. In pure TiO2 nanoparticles, this peak appears at around 459.3 eV.69 The shift of the Ti 2p3/2 peak towards a lower binding energy compared to the pure TiO2 is caused by the doping of N and C species into TiO2 crystals, which is also confirmed by C 1s and N 1s XPS spectra, as well as the formation of oxygen-related defects due to the substituted N atoms.70,71 Moreover, the formation of a heterojunction at the interfaces of CuxO nanoparticles and TiO2 nanoparticles may also influence the local chemical environment around Ti atoms which can also result in the shift in the binding energy.72 In sample TiCuC700, the presence of anatase (101) and rutile (110) phases with a ratio of 0.8:1 (based on the PXRD pattern) may cause a relatively smaller shift in binding energies compared to the single phase TiO2.
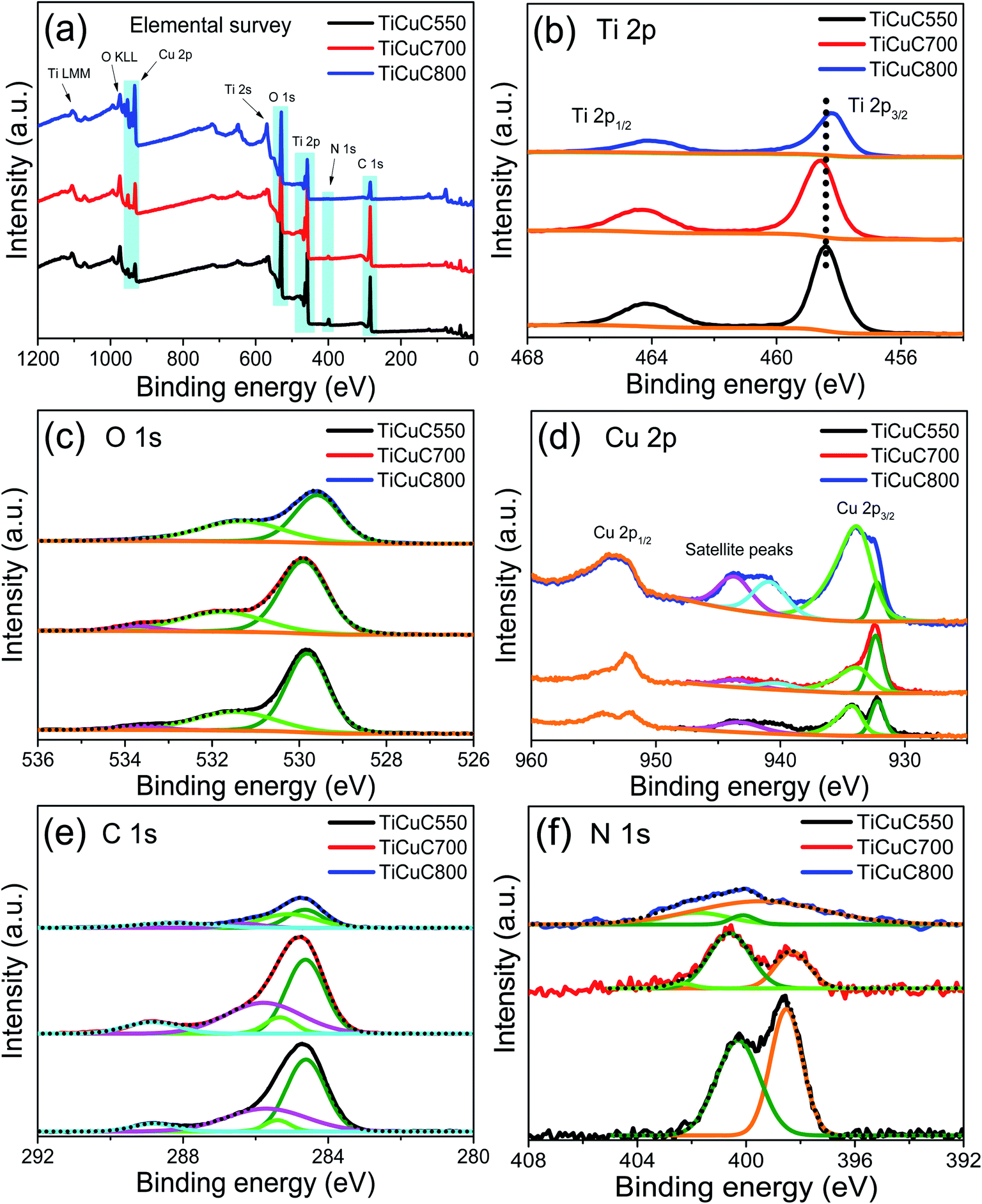 | ||
| Fig. 4 XPS spectra for (a) elemental survey, (b) Ti 2p, (c) O 1s, (d) Cu 2p, (e) C 1s, and (f) N 1s of samples TiCuC550 (black), TiCuC700 (red) and TiCuC800 (blue). | ||
The XPS signals of O 1s (Fig. 4c) can be deconvoluted into three peaks, appearing at 533.6, 531.5 and 529.8 eV for sample TiCuC550. The strongest peak at around 529.8 eV is attributed to the O lattice in Ti–O and Cu–O bonds in the TiO2 and CuxO nanoparticles, whereas the second peak observed at around 531.5 eV can be assigned to the hydroxyl (–OH) groups present on the surface of the TiCuC nanocomposites.21 The third peak with low intensity at around 533.6 eV can be ascribed to the surface functionalized carboxyl groups (OC–OH) attached on the carbon matrix.73 These peaks are slightly shifted for sample TiCuC700 where these peaks are observed at 533.7, 531.8 and 529.9 eV respectively. For sample TiCuC800, these peaks are shifted to 533.7, 531.3 and 529.6 eV respectively which confirms the effect of temperature on the localized chemical environment and atomic distribution of the O 1s species.
The XPS spectra of Cu 2p3/2 (Fig. 4d) for sample TiCuC550 are deconvoluted into two main peaks at around 933.9 and 932.2 eV that can are attributed to the Cu(II) and Cu(I)/Cu(0) chemical states respectively.66,74 Two satellite peaks are observed at 939.9 and 943.3 eV. For sample TiCuC700, the main peaks of Cu(II) and Cu(I)/Cu(0) appear at 933.5 and 932.4 eV respectively. However, for sample TiCuC800, these two peaks of Cu(II) and Cu(I)/Cu(0) are observed at 933.4 and 932.2 eV respectively. Obviously, the binding energies of Cu(II) peaks are shifted for sample TiCuC550 at 933.9 eV to 933.4 eV for composite TiCuC800. The shift in binding energies can be attributed to the transformation of crystalline phases of Cu(II) and Cu(I). It is worth noting that the change in the relative intensities of Cu(I) and Cu(II) peaks confirms the formation of dominant Cu2O nanoparticles with a minor amount of CuO nanoparticles present in samples obtained at lower pyrolysis temperatures (550 °C and/or 700 °C), whereas CuO is the dominant phase with a minor amount of Cu2O and Cu0 nanoparticles in TiCuC800. These observations are consistent with the PXRD (Fig. 1a) and Raman spectra (Fig. 2b) results. The relative atomic concentration of Cu(II) in sample TiCuC800 is much higher compared to that in samples TiCuC550 and TiCuC700, due to the presence of a higher amount of CuO nanoparticles at the outer surface of the MOF-derived TiCuC800 nanocomposite as XPS is a surface-sensitive spectroscopic technique. As confirmed by TGA and EDX elemental mapping, pyrolysis of NH2-MIL-125(Ti/Cu) in a water vapor atmosphere at 800 °C results in some of the carbon species in the MOF turning into CO2, leaving more TiO2 and CuO/Cu2O species exposed to the outer surface of the nanocomposite (schematic diagram Fig. 1d). Moreover, a high pyrolysis temperature is helpful to the fast diffusion of Cu species and results in a high concentration of copper oxides migrating to the outer surface of the nanocomposite. This observation is in very good agreement with the SEM, BET and XPS results. XPS depth profiling can be helpful to identify the atomic distribution of the elemental species. The depth profiles of selected sample TiCuC700 with an etched depth up to 160 nm (Fig. S11 in the ESI†) confirm the atomic concentrations of Ti 2p, O 1s, C 1s, N 1s and Cu 2p averaged to be 16, 33, 44.8, 2.5 and 3.7% respectively, distributed uniformly throughout the matrix. However, the relatively higher amount of C at the surface of the sample is due to the contamination of adventitious carbon.
The XPS results of C 1s for all three samples (Fig. 4e) exhibited the main peak at 284.6 eV which is attributed to the sp2 (CC) bond in hybridized porous carbon, while a low-intensity peak that appeared at 285.4 eV can be assigned to the surface functionalized C–N bond.75–77 A broader peak observed at 285.7 eV is attributed to the C–O bond whereas a small peak at 288.8 eV represents the carboxyl (OC–OH) functional groups attached to the porous carbon matrix due to the pyrolysis of NH2-MIL-125(Ti/Cu) in a water vapor atmosphere.58,73 The peak intensity of C 1s for sample TiCuC800 is, in general, lower than that for samples TiCuC700 and TiCuC550, which is in agreement with the decreased amount of carbon in the sample TiCuC800, as observed by TGA (Fig. 1b). The N 1s XPS spectrum of sample TiCuC550 (Fig. 4f) can be deconvoluted into two peaks at 398.5 and 400.3 eV with the atomic concentrations of 1.92% and 1.85%, which correspond to the pyridinic and highly coordinated quaternary pyrrolic N species respectively.78 However, these pyridinic and pyrrolic N peaks for samples TiCuC700 and TiCuC800 are slightly shifted to 398.3 and 400.6 eV respectively. The change in the relative intensities of the pyridinic and pyrrolic N peaks further confirms the formation of defects in the porous carbon matrix at a higher pyrolysis temperature. In sample TiCuC800, these peaks appeared at around 399.5 and 400.1 eV respectively. It is worth noting that the intensities of these N 1s peaks are reduced from sample TiCuC550 to TiCuC800 because the increased pyrolysis temperature results in the easy breaking of C–N bonds to form NOx and consequently fewer N species remained in the formed composites. In our recent study, we demonstrated that during the pyrolysis of NH2-MIL-125(Ti) under water vapor at 700 °C, the N atoms from the –NH2-BDC organic linker not only functionalize the porous carbon matrix but also cause in situ doping of interstitial N atoms into TiO2. Due to the oxygen-rich gaseous atmosphere (water vapor), the formation of oxygen-rich N like interstitial/intraband states above the valance band can be anticipated.45,71 The N 1s XPS peaks of such interstitial N sites usually appear above 400 eV.71 A low-intensity peak at around 402.4 eV in samples TiCuC700 and TiCuC800 can be assigned to the doped oxygen-rich N species forming interstitial/intraband states above the valance band of TiO2.71,77
3.2. Photocatalytic hydrogen evolution performance and interpretation
The photocatalytic hydrogen evolution reaction (HER) of the NH2-MIL-125(Ti/Cu) derived nanocomposites was evaluated under UV-Visible light filtered using a 285 nm cut-off optical filter in a methanol/water solution without using any noble metals such as Pt, Au or Pd as an electron mediator. The photocatalytic HER reactor setup is shown in Fig. S12 in ESI.† Under UV-Visible light, the HER activities of the NH2-MIL-125(Ti/Cu) derived nanocomposites change with reaction (Fig. 5a) which demonstrate that the pyrolysis temperature of the samples plays a very important role in determining their photocatalytic HER performance. After 6 hours of UV-Vis light exposure, the H2 evolution activities are 556, 18455 and 6880 μmol gcat−1 for samples TiCuC550, TiCuC700 and TiCuC800 respectively, which are much higher than that of commercial TiO2 (P-25) measured to be 493 μmol gcat−1 under identical experimental conditions. It is obvious that the nanocomposite TiCuC700, derived from NH2-MIL-125 (Ti/Cu) under water vapor at 700 °C exhibits much higher H2 evolution activity than other studied nanocomposites under UV-Vis light. As presented in Table S2,† without the loading of noble metals as the co-catalyst, sample TiCuC700 shows one of the highest photocatalytic H2 evolution performances amongst the relevant materials reported in the literature.
 | ||
| Fig. 5 (a) Photocatalytic H2 evolution performance of samples change with the reaction time; (b) comparison of photocatalytic H2 evolution in μmol gcat−1 h−1 of TiCuC550 (black), TiCuC700 (red) and TiCuC800 (blue); (c) the recyclability test of the selected TiCuC700 nanocomposite and (d) EIS Nyquist plots of TiCuC550, TiCuC700 and TiCuC800 at the same potential in 0.5 M H2SO4. | ||
The high photocatalytic H2 evolution activities of NH2-MIL-125(Ti/Cu) derived nanocomposites, particularly nanocomposite TiCuC700 without loading an expensive cocatalyst such as Pt or Au, may be due to the formation of N/C co-doped TiO2 polymorph nanoparticles as well as producing a p–n heterojunction between the TiO2 and CuxO nanoparticles. These nanoparticles are homogeneously distributed in the N and carboxyl group (–COOH) functionalized high specific surface area porous carbon matrix without agglomerations, which result in increased absorption of incident light and more accessible active sites for photocatalytic reactions, consequently leading to significantly enhanced photocatalytic H2 evolution performance.
As presented in Fig. 5b, these nanocomposites TiCuC550, TiCuC700 and TiCuC800 demonstrated H2 evolution activities of 198, 3298 and 1360 μmol gcat−1 h−1 respectively. To elucidate the effect of CuxO/Cu0 species on the H2 evolution performance of the nanocomposites, a sample TiC700 derived from NH2-MIL-125(Ti) without the Cu2+ loading was synthesized under identical experimental conditions for comparison. This sample shows a H2 evolution activity of 426 μmol gcat−1 h−1 under the identical experimental conditions. Obviously, the presence of Cu species in TiCuC700 benefits the formation of the TiO2/CuxO p–n heterojunction that helps to enhance the H2 evolution activity remarkably up to 7.7 fold compared to that of the sample TiC700. The apparent quantum yields (AQY%) of the derived nanocomposites at 435 nm are calculated based on the calculation procedure provided in the ESI† and the results are summarized in Table 1. The AQY value for sample TiCuC700 is the highest amongst the studied nanocomposites and it is 17 and 2.5 times that for samples TiCuC550 and TiCuC800 respectively. The AQY (%) values for these composites are, in general, consistent with their H2 evolution activities.
The stability of the best performing sample TiCuC700 was also evaluated. For this test, UV-Vis light was turned on after 30 min of loading the photocatalyst in MeOH/H2O solution. Then after every 5 hours of reaction, UV-Vis light was turned off and the system was purged with argon for half an hour before turning on UV-Vis light again. As presented in Fig. 5c, sample TiCuC700 shows generally good stability towards the photocatalytic H2 evolution performance in MeOH/H2O solution under UV-Vis light. After 5 consecutive cycles, this nanocomposite exhibited a less than 15% decrease in photocatalytic H2 evolution activities. The decrease in photocatalytic performance may be due to the photocorrosion of the catalysts.
Since the photogenerated charge (e−/h+) density and charge transfer of photocatalysts are widely considered as important issues that can affect the photocatalytic HER activity of the nanomaterials,34 the electric charge transfer resistances (Rct) of the nanocomposites were measured by electrochemical impedance spectroscopy (EIS). The diameter of the arc or semi-circle in the high frequency region of the Nyquist diagram represents the interfacial resistance between the electrode and the electrolyte.78,79 The Nyquist impedance spectra (Fig. 5d) show that the Rct values of samples TiCuC550, TiCuC700 and TiCuC800 are 277, 64 and 187 Ω respectively. The relatively smaller Rct value of TiCuC700 suggests the formation of multi-heterojunctions including the phase junctions between anatase/rutile TiO2 and Cu2O/CuO and the heterojunction between TiO2 and CuxO, which provide multiple pathways for electrons that enhance the electrical conductivity and charge transfer efficiency of the materials.80 These results are in very good agreement with their photocatalytic HER results. Moreover, the Rct value of commercial reference TiO2 (P-25) is 1745 Ω (as shown in Fig. S13, ESI†), which is much higher than those of the NH2-MIL-125(Ti/Cu) derived bimetallic TiO2/CuxO/C nanocomposites. This observation is also consistent with its photocatalytic HER performance.
Based on the characterization and photocatalytic HER application results of the NH2-MIL-125(Ti/Cu) derived nanocomposites, a schematic diagram of the photocatalytic mechanism for the best performing sample TiCuC700, which is obtained at a pyrolysis temperature of 700 °C in water vapor, is proposed in Fig. 6. In sample TiCuC700, the anatase to rutile TiO2 phase percentage is 45% to 55% (based on PXRD) which is close to the optimum ratio 1:1. Since the anatase and rutile phases of pure TiO2 polymorphs formed type II staggered bands where the valence band of the rutile phase is 0.55 eV above that of the anatase phase. Meanwhile, the conduction band of the rutile phase is also positioned 0.35 eV above that of the anatase phase, which results in the formation of a phasejunction.64 In such a type II phasejunction with staggered band alignment, the anatase phase possesses higher electron affinity and the photogenerated electrons tend to migrate from the conduction band of the rutile phase to that of the anatase phase. For NH2-MIL-125(Ti/Cu) derived nanocomposites such as TiCuC700, the valence bands of anatase and rutile phases are further elevated with respect to the Ti 2p core levels due to the doping of N/C atoms into the crystal lattices of TiO2, which is confirmed by the UV-Vis absorption spectra and the XPS valance band position of Ti2p. This further narrows the overall energy band gaps (2.47 eV) of the TiCuC700 nanocomposite compared to the pure TiO2 nanoparticles (3.16 eV), which substantially favors the improved absorption of visible light in an expanded wavelength range. The second important factor that can enhance the photocatalytic activity of the nanocomposite is the formation of the Cu2O/CuO heterojunction. As estimated from XPS, the valance band edges of Cu2O and CuO with respect to the Cu 2p core level lie at 932.3 and 933.5 eV respectively and a heterojunction can be formed between Cu2O and CuO nanoparticles.81 Moreover, the valence band edge of Cu2O/CuO lies at 0.11 eV below the Fermi level which is close to the conduction bands of TiO2. These energy band alignments of the nanoparticles with respect to the Fermi level enable the TiO2 and CuxO nanoparticles to form a p–n heterojunction between them. In addition, the presence of Cu0 nanoparticles may also facilitate faster electron transfer from the conduction bands, and suppress the charge recombination as well as prolong the lifetime of the photogenerated electrons, which result in a better interaction between these photogenerated electrons and the water molecules.21 As a result, multi-heterojunctions formed in the nanocomposite sample TiCuC700 enable multiple pathways to transfer photogenerated electrons to reduce water molecules with significantly improved absorption of visible light due to the remarkable decrease of the overall energy band gap of the nanocomposite, which results in the highest photocatalytic HER performance of the nanocomposite TiCuC700.
 | ||
| Fig. 6 The schematic illustration of the proposed mechanism of photocatalytic H2 evolution over NH2-MIL-125(Ti/Cu) derived multi-heterojunction TiCuC700 under UV-Vis light. | ||
In this proposed mechanism, the electron-rich anatase phase of TiO2 and the CuO act as photocatalytic active sites to reduce the water molecules to H2 species, whereas the oxidation of hole scavenger methanol takes place at the valence bands of the rutile phase of TiO2 and Cu2O. Moreover, the photogenerated electrons can migrate from Cu2O to anatase TiO2. In addition, the high surface area porous carbon matrix that is decorated with N species and hydrophilic hydroxyl/carboxyl functional groups also enhances the accessibility to the photocatalytic active sites.82 Consequently, water molecules can have a significantly improved interaction with the active sites and the photocatalytic H2 evolution activity for sample TiCuC700 increases up to 40 fold compared to that of commercial TiO2 (P-25). On the other hand, the relatively lower HER performance for samples TiCuC550 and TiCuC800 obtained at pyrolysis temperatures of 550 and 800 °C respectively is due to the formation of only a single heterojunction between TiO2 and CuxO nanoparticles with a relatively wider band gap. Only the anatase phase with poorly crystalline TiO2 and Cu2O phases predominantly exists in sample TiCuC550 whereas only the rutile phase of TiO2 and CuO are present in nanocomposite TiCuC800. This current work demonstrates that making use of the binary metal–organic framework as a sacrificial template, nanocomposites with multi-heterojunction and tuneable polymorph components of N/C co-doped TiO2 and CuxO as well as, a high BET surface area porous carbon matrix with hydrophilic functional groups can be readily produced via a simple one-step method. The resulting nanocomposites are excellent photocatalytic materials which can dramatically enhance H2 evolution activities at room temperature. The photocatalytic performance of these MOF derived TiO2/CuxO/C nanocomposites could be further improved by adjusting the amount of Cu loading on NH2-MIL-125(Ti), deciphering the individual role of TiO2, CuxO and C species through in situ/operando techniques and the optimization of the formation of the multi-heterojunction in the MOF derived nanocomposites.
4. Conclusion
The formation of a p–n heterojunction between TiO2 and CuxO nanoparticles is achieved via controlled pyrolysis of a bi-MOF, NH2-MIL-125(Ti/Cu) under a water vapor atmosphere. It is found that pyrolysis of bi-MOF at 550 °C only results in poorly crystalline anatase TiO2 and Cu2O, while at a pyrolysis temperature of 800 °C only rutile TiO2 with predominant CuO nanoparticles forms the heterojunction. However, pyrolysis of bi-MOF at 700 °C under a water vapor atmosphere leads to the in situ formation of optimal well-crystalline anatase and rutile phases of TiO2 with a type-II staggered phasejunction as well as the formation of a heterostructure between Cu2O and CuO. Most importantly, these TiO2 and CuxO form the p–n heterojunction due to the appropriate alignment of energy band gaps. These multi-heterostructured nanocomposites with narrowed energy band gaps embedded in a high surface area porous carbon matrix retain the disc-like morphology inherited from the NH2-MIL-125(Ti/Cu) precursor. The symbiotic structural, textural and semiconducting properties provide easily accessible multiple active sites, which result in higher charge generation for photocatalytic redox reactions. Moreover, these multi-heterojunctions offer multiple pathways to photo-induced electrons and holes for improved charge migration that reduce bulk charge recombination. In addition, the anionic doping of N and C atoms into the TiO2 lattices narrows the energy band gap which enhances the absorption of visible light. Consequently, NH2-MIL-125(Ti/Cu) derived nanocomposite TiCuC700 without loading any noble metals exhibits a superior photocatalytic H2 evolution activity of 3298 μmol gcat−1 h−1 under UV-Vis light, which is 40 fold higher than that of commercial TiO2 (P-25). The one-step formation of the optimized TiO2/CuxO multi-heterojunctions and the inherited textural properties of the MOF precursor make these bi-MOF derived nanocomposites excellent materials for photocatalytic H2 generation. It can be concluded that the formation of multi-heterostructures between N/C co-doped optimal TiO2 and CuxO, functionalization of the porous carbon matrix and tuned textural properties are the key parameters which determine the photocatalytic activities of these nanocomposites. Further efforts to develop experimental approaches that can individually tune the above-mentioned properties of materials may result in even higher photocatalytic H2 evolution performance. This work paves a solid way to develop novel nanocomposites from MOFs with desired semiconducting properties for highly efficient photocatalytic hydrogen production performance.Author contributions
Mian Zahid Hussain: investigation, methodology, and data curation; writing – original draft. Bart van der Linden: methodology, data curation, and validation. Zhuxian Yang: data curation, validation, and writing – review & editing. Quanli Jia: investigation, and data curation. Hong Chang: investigation, validation, and data curation. Roland A. Fischer: supervision, writing – review & editing. Freek Kapteijn: methodology, writing – review & editing. Yanqiu Zhu: supervision, writing – review & editing. Yongde Xia: supervision, conceptualization, methodology, validation, writing – review & editing.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
The authors are thankful to EPSRC CDT in Metamaterials (XM2) at the University of Exeter for financial support.References
- G. U. Fayomi, S. E. Mini, O. S. I. Fayomi, O. Oyeleke, D. O. Omole and I. I. Akinwumi, IOP Conf. Ser.: Mater. Sci. Eng., 2019, 640, 012097 CrossRef CAS
.
- S. Cao, L. Piao and X. Chen, Trends Chem., 2020, 2, 57–70 CrossRef CAS
- J. Low, J. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Adv. Mater., 2017, 29, 1601694 CrossRef
- J. Fu, J. Yu, C. Jiang and B. Cheng, Adv. Energy Mater., 2017, 8, 1701503 CrossRef
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS
- Y. Qi, S. Chen, M. Li, Q. Ding, Z. Li, J. Cui, B. Dong, F. Zhang and C. Li, Chem. Sci., 2017, 8, 437–443 RSC
- J. Qiu, X. Zhang, Y. Feng, X. Zhang, H. Wang and J. Yao, Appl. Catal., B, 2018, 231, 317–342 CrossRef CAS
- J. Zhu, P.-Z. Li, W. Guo, Y. Zhao and R. Zou, Coord. Chem. Rev., 2018, 359, 80–101 CrossRef CAS
- H. Wang, Q.-L. Zhu, R. Zou and Q. Xu, Chem, 2017, 2, 52–80 CAS
- Z. Tong, D. Yang, Y. Sun, Y. Nan and Z. Jiang, Small, 2016, 12, 4093–4101 CrossRef CAS
- H. Zou, B. He, P. Kuang, J. Yu and K. Fan, Adv. Funct. Mater., 2018, 28, 1706917 CrossRef
- H. Luo, Z. Zeng, G. Zeng, C. Zhang, R. Xiao, D. Huang, C. Lai, M. Cheng, W. Wang, W. Xiong, Y. Yang, L. Qin, C. Zhou, H. Wang, Y. Zhou and S. Tian, Chem. Eng. J., 2020, 383, 123196 CrossRef CAS
- N. Serpone, A. V. Emeline, V. K. Ryabchuk, V. N. Kuznetsov, Y. M. Artem'ev and S. Horikoshi, ACS Energy Lett., 2016, 1, 931–948 CrossRef CAS
- D. E. Scaife, Sol. Energy, 1980, 25, 41–54 CrossRef CAS
- G. Yang, Z. Jiang, H. Shi, T. Xiao and Z. Yan, J. Mater. Chem., 2010, 20, 5301–5309 RSC
- G. Yan, M. Zhang, J. Hou and J. Yang, Mater. Chem. Phys., 2011, 129, 553–557 CrossRef CAS
- Q. Wang, T. Hisatomi, S. S. K. Ma, Y. Li and K. Domen, Chem. Mater., 2014, 26, 4144–4150 CrossRef CAS
- D. H. Wang, L. Jia, X. L. Wu, L. Q. Lu and A. W. Xu, Nanoscale, 2012, 4, 576–584 RSC
- C. Moslah, M. Kandyla, G. A. Mousdis, G. Petropoulou and M. Ksibi, Phys. Status Solidi A, 2018, 215, 1800023 CrossRef
- S. Kampouri, C. P. Ireland, B. Valizadeh, E. Oveisi, P. A. Schouwink, M. Mensi and K. C. Stylianou, ACS Appl. Energy Mater., 2018, 1, 6541–6548 CrossRef
- H. Hou, M. Shang, F. Gao, L. Wang, Q. Liu, J. Zheng, Z. Yang and W. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 20128–20137 CrossRef CAS
- Y.-H. Zhang, Y.-L. Li, B.-B. Jiu, F.-L. Gong, J.-L. Chen, S.-M. Fang and H.-L. Zhang, Nanotechnology, 2019, 30, 145401 CrossRef CAS
- C. Han, Y. Wang, Y. Lei, B. Wang, N. Wu, Q. Shi and Q. Li, Nano Res., 2014, 8, 1199–1209 CrossRef
- F. Xu, W. Xiao, B. Cheng and J. Yu, Int. J. Hydrogen Energy, 2014, 39, 15394–15402 CrossRef CAS
- M. E. Aguirre, R. Zhou, A. J. Eugene, M. I. Guzman and M. A. Grela, Appl. Catal., B, 2017, 217, 485–493 CrossRef CAS
- Y. Liao, P. Deng, X. Wang, D. Zhang, F. Li, Q. Yang, H. Zhang and Z. Zhong, Nanoscale Res. Lett., 2018, 13, 221 CrossRef
- J. Fu, S. Cao and J. Yu, J. Materiomics, 2015, 1, 124–133 CrossRef
- H. Hou, F. Gao, L. Wang, M. Shang, Z. Yang, J. Zheng and W. Yang, J. Mater. Chem. A, 2016, 4, 6276–6281 RSC
- K. Qi, B. Cheng, J. Yu and W. Ho, Chin. J. Catal., 2017, 38, 1936–1955 CrossRef CAS
- Y.-Z. Chen, R. Zhang, L. Jiao and H.-L. Jiang, Coord. Chem. Rev., 2018, 362, 1–23 CrossRef CAS
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS
- M. Sohail, Y.-N. Yun, E. Lee, S. K. Kim, K. Cho, J.-N. Kim, T. W. Kim, J.-H. Moon and H. Kim, Cryst. Growth Des., 2017, 17, 1208–1213 CrossRef CAS
- M. A. Nasalevich, R. Becker, E. V. Ramos-Fernandez, S. Castellanos, S. L. Veber, M. V. Fedin, F. Kapteijn, J. N. H. Reek, J. I. van der Vlugt and J. Gascon, Energy Environ. Sci., 2015, 8, 364–375 RSC
- P. Karthik, A. R. M. Shaheer, A. Vinu and B. Neppolian, Small, 2020, 16, 1902990 CrossRef CAS
- W. Wang, X. Xu, W. Zhou and Z. Shao, Adv. Sci., 2017, 4, 1600371 CrossRef
- M. A. Nasalevich, M. van der Veen, F. Kapteijn and J. Gascon, CrystEngComm, 2014, 16, 4919–4926 RSC
- L. Zeng, X. Guo, C. He and C. Duan, ACS Catal., 2016, 6, 7935–7947 CrossRef CAS
- L. Oar-Arteta, T. Wezendonk, X. Sun, F. Kapteijn and J. Gascon, Mater. Chem. Front., 2017, 1, 1709–1745 RSC
- F. Song, W. Li and Y. Sun, Inorganics, 2017, 5, 40 CrossRef
- M. H. Yap, K. L. Fow and G. Z. Chen, Green Energy Environ., 2017, 2, 218–245 CrossRef
- J. Liu, X. Zhao, P. Jing, W. Shi and P. Cheng, Chem. - Eur. J., 2019, 25, 2330–2336 CAS
- J. Xu, J. Gao, Y. Qi, C. Wang and L. Wang, ChemCatChem, 2018, 10, 3327–3335 CrossRef CAS
- H. Chen, Z.-G. Gu, S. Mirza, S.-H. Zhang and J. Zhang, J. Mater. Chem. A, 2018, 6, 7175–7181 RSC
- L. Yuan, C. Han, M.-Q. Yang and Y.-J. Xu, Int. Rev. Phys. Chem., 2016, 35, 1–36 Search PubMed
- M. Z. Hussain, Z. Yang, B. van der Linden, Z. Huang, Q. Jia, E. Cerrato, R. A. Fischer, F. Kapteijn, Y. Zhu and Y. Xia, J. Energy Chem., 2021, 57, 485–495 Search PubMed
- R. Das, P. Pachfule, R. Banerjee and P. Poddar, Nanoscale, 2012, 4, 591–599 RSC
- S. Hu, M. Liu, K. Li, Y. Zuo, A. Zhang, C. Song, G. Zhang and X. Guo, CrystEngComm, 2014, 16, 9645–9650 RSC
- O. Frank, M. Zukalova, B. Laskova, J. Kürti, J. Koltai and L. Kavan, Phys. Chem. Chem. Phys., 2012, 14, 14567–14572 RSC
- W. F. Zhang, Y. L. He, M. S. Zhang, Z. Yin and Q. Chen, J. Phys. D: Appl. Phys., 2000, 33, 912 CrossRef CAS
- S. Chaurasiya, J. Udaya Bhanu and P. Thangadurai, Trans. Indian Inst. Met., 2018, 71, 1185–1191 CrossRef CAS
- N. G. Elfadill, M. R. Hashim, K. M. Chahrour, M. A. Qaeed and M. Bououdina, Superlattices Microstruct., 2015, 85, 908–917 CrossRef CAS
- T. H. Tran and V. T. Nguyen, Mater. Sci. Semicond. Process., 2016, 46, 6–9 CrossRef CAS
- P. K. Chu and L. Li, Mater. Chem. Phys., 2006, 96, 253–277 CrossRef CAS
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS
- L. Bokobza, J.-L. Bruneel and M. Couzi, J. Carbon Res., 2015, 1, 77–94 CrossRef
- M. Z. Hussain, A. Schneemann, R. A. Fischer, Y. Zhu and Y. Xia, ACS Appl. Energy Mater., 2018, 1, 4695–4707 CrossRef CAS
- B. Chen, G. Ma, D. Kong, Y. Zhu and Y. Xia, Carbon, 2015, 95, 113–124 CrossRef CAS
- M. Z. Hussain, G. S. Pawar, Z. Huang, A. A. Tahir, R. A. Fischer, Y. Zhu and Y. Xia, Carbon, 2019, 146, 348–363 CrossRef CAS
- J. Y. Choi, C. K. Lim, B. Park, M. Kim, A. Jamal and H. Song, J. Mater. Chem. A, 2019, 7, 15068–15072 RSC
- W. Ho, Q. Tay, H. Qi, Z. Huang, J. Li and Z. Chen, Molecules, 2017, 22, 677 CrossRef
- B. Qiu, Y. Zhou, Y. Ma, X. Yang, W. Sheng, M. Xing and J. Zhang, Sci. Rep., 2015, 5, 8591 CrossRef CAS
- A. Sahai, N. Goswami, S. D. Kaushik and S. Tripathi, Appl. Surf. Sci., 2016, 390, 974–983 CrossRef CAS
- M. A. Nasalevich, C. H. Hendon, J. G. Santaclara, K. Svane, B. van der Linden, S. L. Veber, M. V. Fedin, A. J. Houtepen, M. A. van der Veen, F. Kapteijn, A. Walsh and J. Gascon, Sci. Rep., 2016, 6, 23676 CrossRef CAS
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. Catlow, M. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh and A. A. Sokol, Nat. Mater., 2013, 12, 798–801 CrossRef CAS
- A. Meng, L. Zhang, B. Cheng and J. Yu, Adv. Mater., 2019, 31, 1807660 CrossRef
- W. Zhen, W. Jiao, Y. Wu, H. Jing and G. Lu, Catal. Sci. Technol., 2017, 7, 5028–5037 RSC
- M. Rehan, X. Lai and G. M. Kale, CrystEngComm, 2011, 13, 3725–3732 RSC
- P. Ribao, M. J. Rivero and I. Ortiz, Environ. Sci. Pollut. Res., 2017, 24, 12628–12637 CrossRef CAS
- U. Diebold and T. E. Madey, Surf. Sci. Spectra, 1996, 4, 227–231 CrossRef CAS
- R. Asahi, T. Morikawa, H. Irie and T. Ohwaki, Chem. Rev., 2014, 114, 9824–9852 CrossRef CAS
- C. Di Valentin, E. Finazzi, G. Pacchioni, A. Selloni, S. Livraghi, M. C. Paganini and E. Giamello, Chem. Phys., 2007, 339, 44–56 CrossRef CAS
- J. Xing, Z. P. Chen, F. Y. Xiao, X. Y. Ma, C. Z. Wen, Z. Li and H. G. Yang, Chem.–Asian J., 2013, 8, 1265–1270 CrossRef CAS
- P. Iamprasertkun, A. Krittayavathananon and M. Sawangphruk, Carbon, 2016, 102, 455–461 CrossRef CAS
- X. Qiu, M. Miyauchi, K. Sunada, M. Minoshima, M. Liu, Y. Lu, D. Li, Y. Shimodaira, Y. Hosogi, Y. Kuroda and K. Hashimoto, ACS Nano, 2012, 6, 1609–1618 CrossRef CAS
- J. Ederer, P. Janoš, P. Ecorchard, J. Tolasz, V. Štengl, H. Beneš, M. Perchacz and O. Pop-Georgievski, RSC Adv., 2017, 7, 12464–12473 RSC
- H. Wang, X. Yuan, Y. Wu, G. Zeng, X. Chen, L. Leng, Z. Wu, L. Jiang and H. Li, J. Hazard. Mater., 2015, 286, 187–194 CrossRef CAS
- Y. Yuan, K. Sheng, S. Zeng, X. Han, L. Sun, I. Lončarić, W. Zhan and D. Sun, Inorg. Chem., 2020, 59, 5456–5462 CrossRef CAS
- Z. Huang, Z. Yang, M. Z. Hussain, B. Chen, Q. Jia, Y. Zhu and Y. Xia, Electrochim. Acta, 2020, 330, 135335 CrossRef CAS
- J. Ângelo, P. Magalhães, L. Andrade and A. Mendes, Appl. Surf. Sci., 2016, 387, 183–189 CrossRef
- D. Barreca, G. Carraro, A. Gasparotto, C. Maccato, M. Cruz-Yusta, J. L. Gómez-Camer, J. Morales, C. Sada and L. Sánchez, ACS Appl. Mater. Interfaces, 2012, 4, 3610–3619 CrossRef CAS
- H. Li, Z. Su, S. Hu and Y. Yan, Appl. Catal., B, 2017, 207, 134–142 CrossRef CAS
- Q. Xu, L. Zhang, J. Yu, S. Wageh, A. A. Al-Ghamdi and M. Jaroniec, Mater. Today, 2018, 21, 1042–1063 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta10853g |
| This journal is © The Royal Society of Chemistry 2021 |