
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWell-defined Cu2O photocatalysts for solar fuels and chemicals
Sourav
Rej†
a,
Matteo
Bisetto†
b,
Alberto
Naldoni
 *ac and
Paolo
Fornasiero
*b
*ac and
Paolo
Fornasiero
*b
aRegional Centre of Advanced Technologies and Materials, Czech Advanced Technology and Research Institute, Palacký University, 779 00 Olomouc, Czech Republic. E-mail: alberto.naldoni@upol.cz
bDepartment of Chemical and Pharmaceutical Sciences, ICCOM-CNR Trieste Research Unit, INSTM-Trieste, University of Trieste, Via L. Giorgieri 1, 34127 Trieste, Italy. E-mail: pfornasiero@units.it
cInstitute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 610054, China
First published on 15th February 2021
Abstract
The shape-controlled synthesis of cuprous oxide (Cu2O) photocatalysts with both low or high index crystal planes has received increasing attention due to their unique facet-dependent properties. Since they are cheap and earth abundant, these well-defined Cu2O nanostructures are extensively used for different photocatalytic reactions, also because of their strong visible light absorption capability. However, further development will still be needed to enhance the efficiency and photostability of Cu2O to expand its industrial application. We start this review by summarizing the synthetic advancement in the facet engineering of Cu2O and other associated hybrid Cu2O-based heterostructures with a special emphasis put on their growth mechanism. We then discuss different facet-dependent properties, which are relevant to photocatalysis. In the subsequent section, we present a critical discussion on the photocatalytic performance of faceted Cu2O nanostructures during organic synthesis, hydrogen production, and carbon dioxide photoreduction. The relation between photocatalytic efficiency and product selectivity with exposed crystal facets or with different compositions of hybrid nanostructures is also discussed. Finally, important strategies are proposed to overcome the photostability issue, while outlining the course of future development to further boost the technological readiness of well-defined Cu2O-based photocatalysts.
 Sourav Rej | Sourav Rej received his PhD degree (2015) from National Tsing Hua University, Taiwan in shape-controlled synthesis of nanocrystals and their catalytic applications under the supervision of Prof. Michael H. Huang. After completing his appointment in the Institute of Bioengineering & Nanotechnology, A*STAR, Singapore; he started his next postdoctoral position in the photoelectrochemistry group at the Regional Centre of Advanced Technologies and Materials, Palacký University, Czech Republic. Now his research interests focus on utilization of plasmonic materials and other associated hybrid nanostructures for solar energy conversion. |
 Matteo Bisetto | Matteo Bisetto obtained his master's degree in materials science (2019) from the University of Padova (Italy). During his thesis, he worked on the development of nanostructured electrodes for PEC Water Splitting in collaboration with the research group of Professor A. Vomiero from LTU of Luleå (Sweden). He began his PhD in Nanotechnology at the University of Trieste in 2019 under the supervision of Prof. P. Fornasiero and T. Montini. Now his research focuses on the development of well-defined shaped nanostructures for catalytic applications. |
 Alberto Naldoni | Alberto Naldoni is deputy head of the photoelectrochemistry group at the Regional Centre of Advanced Technologies and Materials of Palacký University Olomouc. He obtained his PhD (2010) in chemical sciences from the University of Milan before moving to the Italian National Research Council to study photocatalysis and photoelectrochemical water splitting. He spent 3 years as visiting faculty in the Nanophotonics group at the Birck Nanotechnology Center of Purdue University. His research interests focus on solar energy conversion with emphasis on plasmonics, photocatalysis, and photoelectrochemistry. |
 Paolo Fornasiero | Paolo Fornasiero is full professor in inorganic chemistry at the University of Trieste. His research focuses on the application of inorganic chemistry in nanoscience for the preparation of new materials useful in the fields of energy and environmental protection. He is co-author of more than 260 ISI publications, 16 book chapters and 4 patents. He received the 2005 Nasini Gold Medal, the 2013 Chiusoli Gold Medal, the 2016 Heinz Heinemann Award, the 2017 Kramer Award and the 2018 M. T. Messori Roncaglia Award. |
1 Introduction
Recent development of efficient photocatalytic materials was significantly accelerated in order to effectively deploy green, energy efficient, and sustainable technologies.1–6 Generally, these materials are inorganic metal oxide semiconductors and, because of their particular band structure, they allow absorption of a broad part of visible light from the solar spectrum. Furthermore, the electronic structure of these materials provides unique optical behavior and conductivity properties.1,3,4 A typical photocatalytic mechanism consists of three consecutive steps (Fig. 1a): (i) absorption of light with appropriate energy and generation of an electron (e−) hole (h+) pair, which, respectively, (de)localizes in the conduction band (CB) and the valence band (VB) of the semiconductor; (ii) electron and hole separation and migration to the catalyst surface in order to participate in the reduction and oxidation reactions, respectively, at the semiconductor–solution interface; and (iii) if the electron–hole pairs don't take part in the redox reaction, they then undergo the recombination process. The first two steps majorly determine the photocatalytic efficiency of the semiconductor material, whereas the third step is responsible for the performance as such. Thus, to facilitate the solar-to-chemical energy conversion efficiency, different strategies have been developed. They include controlling the morphology, size, and compositions of the photocatalyst for various reactions such as organic reactions, hydrogen production via water splitting (or photoreforming of sacrificial agents), and CO2 reduction.7–14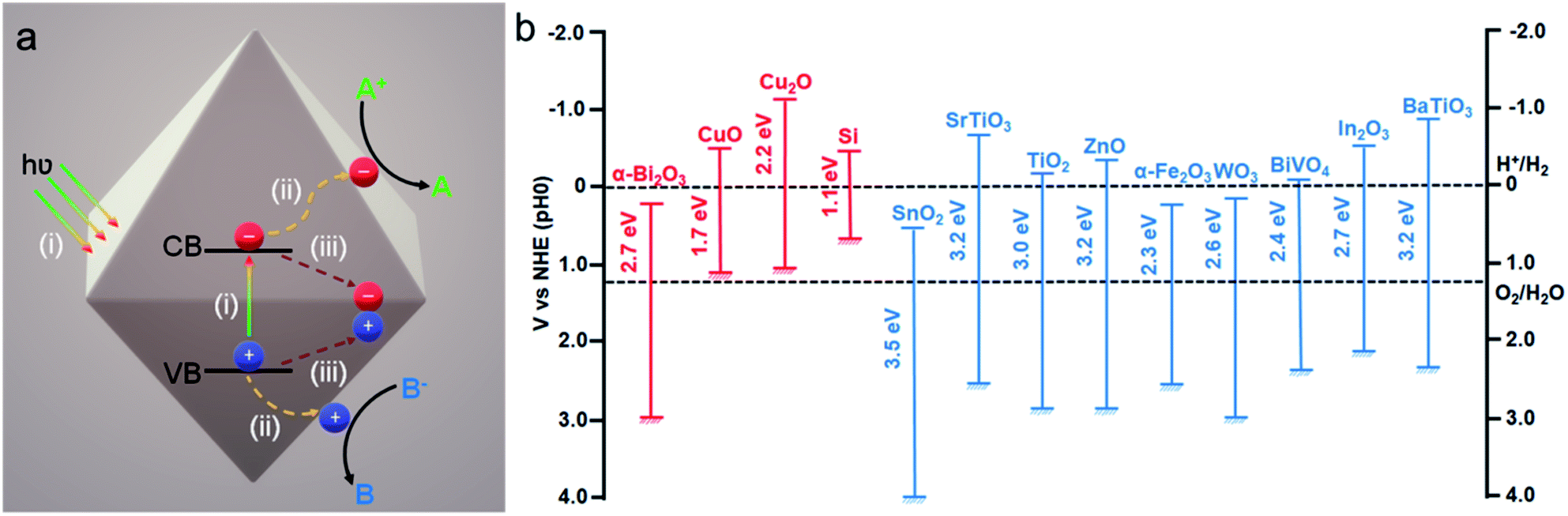 | ||
| Fig. 1 (a) Typical mechanism involved in a photocatalytic process. Adapted with permission from ref. 2, Copyright 2018 Wiley-VCH. (b) Band gap values and band energy alignment with water splitting redox potentials of different common p-type (red) and n-type (blue) semiconductors vs. NHE and at pH 0. Adapted with permission from ref. 1, Copyright 2013 Wiley-VCH. | ||
The synthesis of inorganic metal oxide nanocrystals with a well-defined structure has been extensively studied over the last few decades.15–18 The opportunity to control the morphology and size of nanocrystals will benefit the light absorbing properties and charge carrier management, thus enhancing the photocatalytic activity and product selectivity in the end.19 Moreover, different crystal facets possess different atomic arrangements, which may directly control their physical and catalytic properties.20,21 Therefore, understanding the synthesis and growth mechanism of metal oxide nanocrystals bounded with low or high index facets is really crucial to examining their facet-dependent properties. Common semiconductors like TiO2, ZnO, WO3, and Cu2O attract significant attention due to their surprising photocatalytic activity caused by their suitable band gap energy (Fig. 1b).15–17
Among different photocatalysts, nontoxic, highly abundant cuprous oxide (Cu2O) is a promising photocatalyst for dye degradation, organic reactions, CO2 reduction, and H2 production as it shows a direct band gap structure with a small band gap energy (2.2 eV), which helps this material to absorb efficiently in the visible range of the solar spectrum (Fig. 2).22 Cu2O is the most widely synthesised p-type semiconductor for photocatalysis allowing proper control of its morphology and size obtained under mild synthetic conditions. Therefore, extensive research has been carried out to investigate the Cu2O facet-dependent photocatalytic properties.23–29 However, the photocatalytic activity of Cu2O nanocrystals is still limited by the high recombination rate of the photoexcited electron–hole pairs and also by the photostability issue.30 The photogenerated holes initiate the self-photooxidation of Cu2O, which causes its decreased photostability.30 In order to overcome this issue, different hybrid Cu2O-based nanostructures were developed to enhance the photocatalytic activity by concomitantly increasing the electron–hole separation efficiency. Photoexcited electrons accumulate in the reduction site where they catalyze the reduction reaction; simultaneously, the photogenerated holes are consumed in the oxidation sites by undergoing an oxidation reaction. Such synergistic behavior in the hybrid system not only enhances the photostability, but it may also increase the product selectivity.
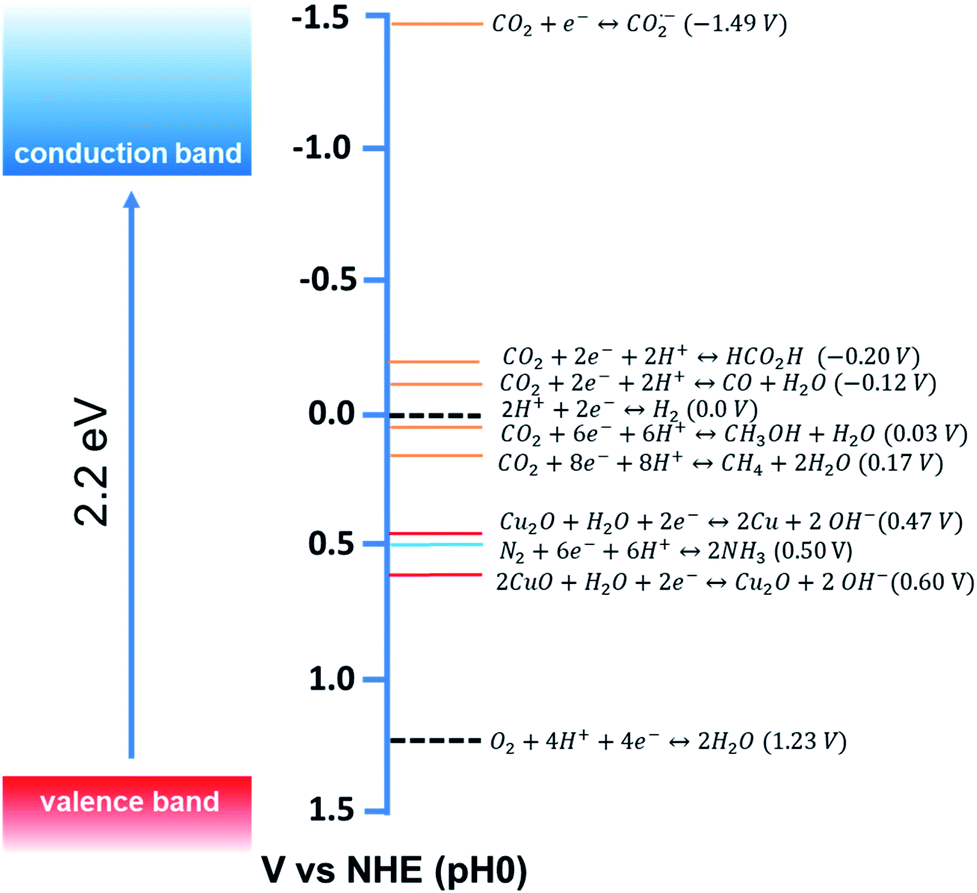 | ||
| Fig. 2 Conduction band and valence band potentials of Cu2O (left side), with the redox potentials of several CO2, N2 and water redox couples at pH = 0 plotted vs. NHE (right side). Adapted with permission from ref. 9, Copyright 2015 American Chemical Society. | ||
Although there are excellent review articles that discuss the synthesis and optical and catalytic properties of well-defined Cu2O nanocrystals, a comprehensive review focusing on the growth mechanism, surfactant removal process, unique facet-dependent properties, and their photocatalytic application in solar energy conversion is still lacking.23,25–29 Hence, in this review, we aim to discuss and summarize all the aspects related to the crystal facet engineering of Cu2O nanocrystals and their photocatalytic application with special emphasis on the stability under different photocatalytic reaction conditions. In the first section, an in-depth discussion on the growth mechanism of well-defined Cu2O nanocrystals and different synthetic procedures for the preparation of diverse nanostructures is presented. The effect of surfactants and additive ions on controlling the morphology of nanocrystals is also highlighted. Successful strategies for the removal of different kinds of surfactants from nanocrystal surfaces have also been discussed. Next, we present the influence of facets on different chemical properties such as selective adsorption behavior, photodeposition of cocatalysts, accumulation of charge carriers on different anisotropic facets, and optical properties. In the last section, we cover the most relevant photocatalytic applications, including H2 production and CO2 reduction, of Cu2O nanocrystals mentioning various strategies that can be employed to improve the photocatalytic activity and stability of Cu2O by forming diverse heterostructures.
2 Shape & size-controlled synthesis of Cu2O nanocrystals
This section starts with discussing the growth mechanism of well-defined nanocrystals from crystalline nuclei in a systematic manner. Then, synthetic strategies for diverse Cu2O nanoarchitectures with precise shape and size control will be presented. The effect of different parameters such as solution pH, concentration of the surfactant, and role of additives will be highlighted. And lastly, different ways of removing the surfactant without altering the shape and size of Cu2O nanocrystals will be presented.2.1 General understanding of the growth direction and exposed crystal facets
Morphology-controlled nanocrystals can be synthesised by different wet chemical synthetic procedures.19 These chemical reactions often appear to be fairly simple, but the exact growth mechanisms inside the glass bottles are extremely complicated. The morphology of the final nanocrystals highly depends on the precise control of temperature, capping agents, different reagent concentrations and also on the sequence of the addition of reagents. Any changes in the aforementioned parameters lead to an unexpected shape and size distribution of the nanocrystals in the reaction mixture. Therefore, scaling up the synthetic processes is not trivial and the industrialization is not easy. The scientific development over the last two decades helped to understand the atomistic details of the evolution pathways in which a precursor compound is converted to atoms, nuclei, and then well-defined nanocrystals. Xia et al. demonstrated that there are three different distinct stages in the growth process: (1) nucleation, (2) evolution of nuclei into seeds, and (3) growth of seeds into nanocrystals.31 During the first step, in a reductive environment, the concentration of reduced metal atoms steadily increases with time. When the concentration of reduced metal atoms reaches a point of supersaturation, the atoms start to aggregate into small clusters, i.e., nuclei formation takes place. Once formed, these nuclei then grow up to a critical size where further structural modification becomes energetically unfavorable, i.e., the seed formation takes place. These single-crystal seeds usually exist as truncated octahedrons (or Wulff polyhedrons) enclosed by a mixture of {111} and {100} facets as they possess a nearly spherical profile, and, thus, the smallest surface area helps to minimize the total interfacial free energy. The density, size distribution, and surface chemistry of these resulting crystalline seeds are highly dependent on the reaction environment of the solution. Once the seeds are formed, the temperature, solvent, reagents, and additives such as surfactant or inorganic ions present in the reaction mixture strongly influence the ultimate growth rate and direction of the seed particles and determine the final morphology of the nanocrystals.The process of a seed growing to a well-defined nanocrystal follows a route to minimizing the total surface energy which is controlled by both thermodynamics and kinetics of the reaction.31 Under optimized synthetic conditions, different surfactants, ions or additives present in the reaction mixture selectively adsorb on a specific facet, as schematically shown in Fig. 3a. During the process of growing a seed, the facets which are not blocked by any additives grow at a higher rate than the unblocked facets.26 Therefore, the fast-growing facets will eventually disappear, resulting in a nanocrystal terminated with blocked facets. This event tailors the shape of the crystal and enables exposure of different facets. According to the Wulff–Kaishew theory, the ratio of the growth rate in the 〈100〉 and 〈111〉 directions, i.e., 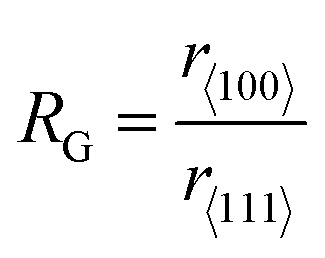 is the deciding factor of the final morphology.32–34 Based on this, when the value of RG = 0.58 for certain reaction conditions is reached, exclusively cube-shaped nanocrystals are formed and bounded with six low-index (100) crystal planes. This kind of environment favors the fast crystal growth along the 〈111〉 direction, compared to the 〈100〉 direction; therefore, the (111) facets disappear from the seed and the cubic shape comes out as the final product. Similarly, the octahedral shape, bounded with eight low-index (111) crystal planes, becomes dominant when RG = 1.73. Three-dimensional geometric shape evolution from cubic to octahedral morphology with different RG values is shown in Fig. 3b. When 0.58 < RG < 1.73, different truncated forms of nanocrystals can be obtained in the reaction medium (Fig. 3c). Although the RG value helps to understand the morphological evolution from the cubic to the octahedral shape, it remains difficult to understand the formation of a rhombic dodecahedral nanocrystal bounded with only {110} low-index facets using this model. However, following a more general approach, it is known that nanocrystals of different shapes can be formed following a change in the reaction cell potential according to the Nernst equation. In fact, it is necessary to have a fine tuning of the pH of the solution and of the reagent concentrations, which determine the electrochemical potential of the Cu2O surface, to produce nanocrystals of different shapes. Hence, formation of different nanocrystals shapes can be understood as resulting from different changes in free energy (ΔG = −nFE) inside the reaction mixture.35,36 Therefore, it can be concluded that the final shape of the nanocrystal and the exposed facets are the result of the interplay between thermodynamics and kinetics of the reaction. A further development in multi-scale materials science simulations on nanoparticles may bring quantitative evaluation of physical parameters that regulate the selective growth of specific crystal facets, thus introducing more reliable design strategies.
is the deciding factor of the final morphology.32–34 Based on this, when the value of RG = 0.58 for certain reaction conditions is reached, exclusively cube-shaped nanocrystals are formed and bounded with six low-index (100) crystal planes. This kind of environment favors the fast crystal growth along the 〈111〉 direction, compared to the 〈100〉 direction; therefore, the (111) facets disappear from the seed and the cubic shape comes out as the final product. Similarly, the octahedral shape, bounded with eight low-index (111) crystal planes, becomes dominant when RG = 1.73. Three-dimensional geometric shape evolution from cubic to octahedral morphology with different RG values is shown in Fig. 3b. When 0.58 < RG < 1.73, different truncated forms of nanocrystals can be obtained in the reaction medium (Fig. 3c). Although the RG value helps to understand the morphological evolution from the cubic to the octahedral shape, it remains difficult to understand the formation of a rhombic dodecahedral nanocrystal bounded with only {110} low-index facets using this model. However, following a more general approach, it is known that nanocrystals of different shapes can be formed following a change in the reaction cell potential according to the Nernst equation. In fact, it is necessary to have a fine tuning of the pH of the solution and of the reagent concentrations, which determine the electrochemical potential of the Cu2O surface, to produce nanocrystals of different shapes. Hence, formation of different nanocrystals shapes can be understood as resulting from different changes in free energy (ΔG = −nFE) inside the reaction mixture.35,36 Therefore, it can be concluded that the final shape of the nanocrystal and the exposed facets are the result of the interplay between thermodynamics and kinetics of the reaction. A further development in multi-scale materials science simulations on nanoparticles may bring quantitative evaluation of physical parameters that regulate the selective growth of specific crystal facets, thus introducing more reliable design strategies.
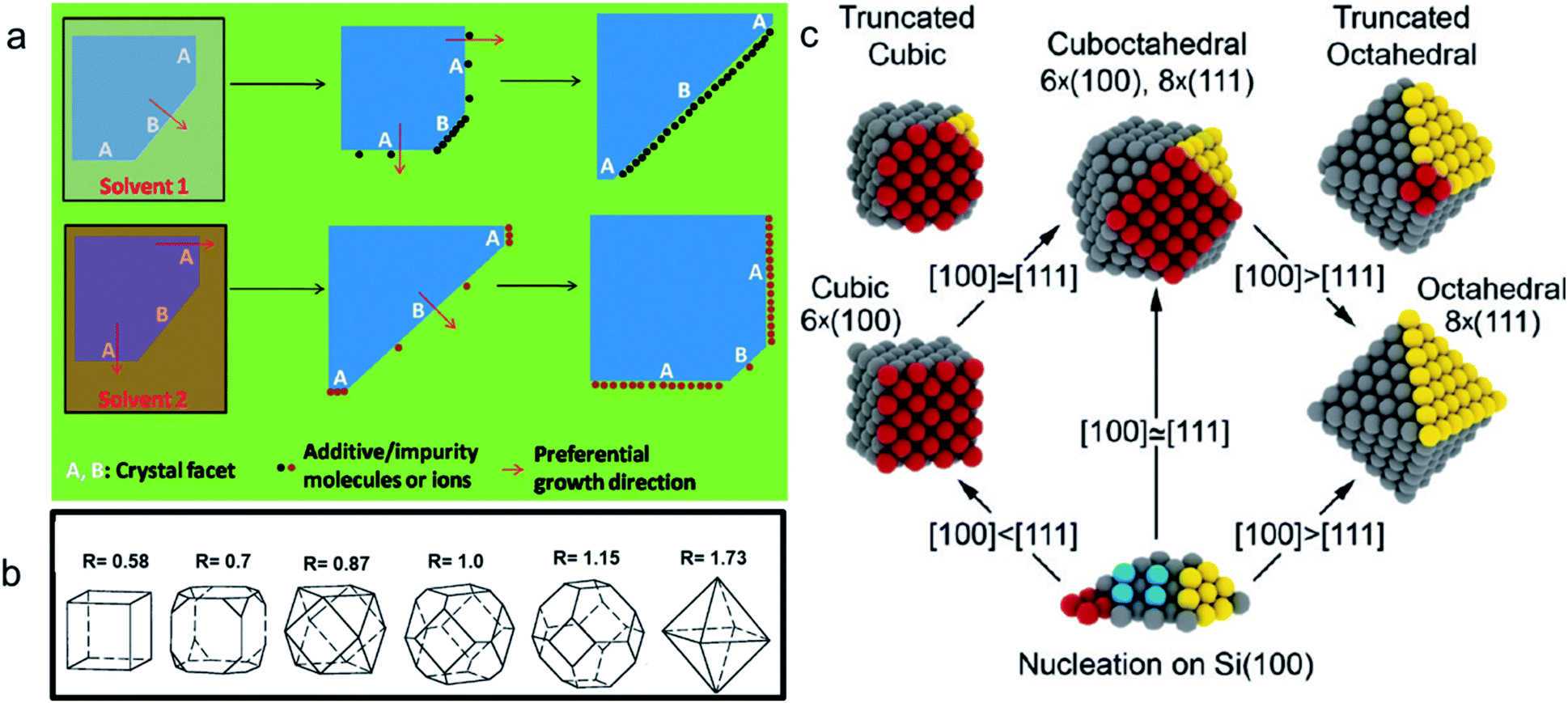 | ||
| Fig. 3 (a) Schematic diagram of the growth mechanism of a seed into a well-defined nanocrystal. Reproduced with permission from ref. 26, Copyright 2011 Royal Society of Chemistry. (b) Value of RG for different morphologies. Reproduced with permission from ref. 32, Copyright 2000 American Chemical Society. (c) Scheme for the shape evolution at different RG values. Red and yellow colored spheres represent the atoms present on (100) and (111) crystal planes in a seed, respectively. Reproduced with permission from ref. 33, Copyright 2010 American Chemical Society. | ||
Although there are enough theory and experimental routes to synthesizing well-defined nanocrystals, the atomic pathways of nanocrystal facet development have been mostly unknown to date because of the lack of direct microscopic observations inside the reaction mixture. Recently, in situ liquid cell transmission electron microscopy (LCTEM) allowed observing single-nanoparticle growth trajectories.36 A liquid cell containing a small amount of liquid reaction mixture inside the high-vacuum microscope can be used to initiate the nanocrystal growth by the introduction of an electron beam that often acts as a reductant. Wu et al. observed the growth process of a Cu2O nanocube by reducing an alkaline copper precursor solution with an electron beam.37 The time-dependent growth process of a Cu2O nanocube observed via an in situ LCTEM is shown in Fig. 4. When the electron beam is introduced into the precursor solution, the Cu2+ ions are reduced to Cu1+ ions and initiate the Cu2O nucleation in the presence of hydroxide anions, as shown by the darker region in Fig. 4a, which then eventually is converted to a nanocube via a complex mechanism. Further development will be needed in this field to gain knowledge of the detailed mechanism underlying morphology-controlled nanocrystal synthesis.38
 | ||
| Fig. 4 Time-dependent in situ TEM images to track the growth of a single Cu2O nanocube at (a) 0, (b) 20, (c) 60, and (d) 120 seconds of reaction time. Reproduced with permission from ref. 37, Copyright 2019 American Chemical Society. | ||
2.2 Synthesis of diverse Cu2O nanostructures
A large number of synthetic wet-chemical routes to producing a wide variety of well-defined Cu2O nanocrystals have been developed over the last two decades. To better understand the effect of the role of different reagents in controlling the morphology, different relevant case studies are presented and discussed in this section.Murphy et al. developed a synthetic procedure of Cu2O nanocubes using an aqueous solution of copper sulfate (CuSO4), NaOH, and cetyltrimethylammonium bromide (CTAB) as the surfactant and sodium ascorbate as the reducing agent.39 The shape and size of the nanocrystals can be tuned by varying the concentration of the surfactant. At low surfactant concentrations, the nanocrystals are ineffectively capped and grow randomly with a spherical morphology. When an optimum concentration is reached, it leads to adequate surface capping, and uniform cubic nanoparticles are formed. TEM images confirm that these crystals appear to be hollow from inside, which is the major drawback for this synthetic condition. At that time, the Wang group also developed a procedure to synthesize Cu2O nanocubes and octahedra.40 Typically, an NH3 solution is first added to an aqueous CuCl2 solution followed by adding NaOH, which leads to the blue precipitation of Cu(OH)2. After that, an aqueous solution of N2H4 is added as the reducing agent to the above-described solution to start the formation of Cu2O nanocrystals. The molar ratios of reagents, i.e.,  and
and 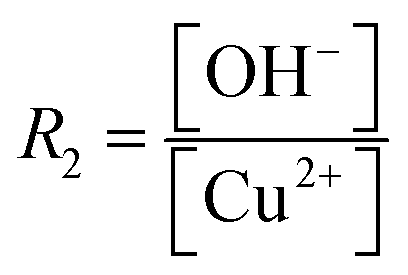 determine the morphology of the corresponding products by affecting the coordination between NH3 and Cu2+ ions, which makes this report interesting. When R1 = 0, R2 = 2, i.e., there is no ammonia present in the system, porous Cu2O spheres were obtained as this morphology has the lowest surface energy. Keeping R2 = 2 fixed, cube-like and octahedral Cu2O nanocrystals were formed when R1 = 4 and 7, respectively. This proved that ammonia plays a critical role in this reaction to control the final morphology of the Cu2O nanocrystals. In the beginning, Cu2+ ions undergo complexation with NH3 molecules forming [Cu(NH3)4]2+; when NaOH is added, NH3 molecules are replaced by OH− ions to form a blue Cu(OH)2 precipitate. Under a fixed R2 value, the maximum number of NH3 molecules are replaced by OH− ions when R1 = 4, whereas the minimum number of NH3 molecules are replaced when R1 = 7. This phenomenon leads to distinct pH values under the two different synthetic conditions. Therefore, RG values (i.e., the ratio of growth rates along the 〈100〉 and 〈111〉 direction) become 0.58 and 1.73 when R1 = 4 and R2 = 7, resulting in cube-like and octahedral nanocrystals, respectively, in the reaction medium. These synthetic conditions provide a stimulating idea on how we can control the shape of the nanocrystals by the fine pH tuning of an inorganic reaction without adding any organic surfactants. However, this approach mainly suffers from inhomogeneity in the size and shape of the nanocrystals, and, therefore, it requires further development.
determine the morphology of the corresponding products by affecting the coordination between NH3 and Cu2+ ions, which makes this report interesting. When R1 = 0, R2 = 2, i.e., there is no ammonia present in the system, porous Cu2O spheres were obtained as this morphology has the lowest surface energy. Keeping R2 = 2 fixed, cube-like and octahedral Cu2O nanocrystals were formed when R1 = 4 and 7, respectively. This proved that ammonia plays a critical role in this reaction to control the final morphology of the Cu2O nanocrystals. In the beginning, Cu2+ ions undergo complexation with NH3 molecules forming [Cu(NH3)4]2+; when NaOH is added, NH3 molecules are replaced by OH− ions to form a blue Cu(OH)2 precipitate. Under a fixed R2 value, the maximum number of NH3 molecules are replaced by OH− ions when R1 = 4, whereas the minimum number of NH3 molecules are replaced when R1 = 7. This phenomenon leads to distinct pH values under the two different synthetic conditions. Therefore, RG values (i.e., the ratio of growth rates along the 〈100〉 and 〈111〉 direction) become 0.58 and 1.73 when R1 = 4 and R2 = 7, resulting in cube-like and octahedral nanocrystals, respectively, in the reaction medium. These synthetic conditions provide a stimulating idea on how we can control the shape of the nanocrystals by the fine pH tuning of an inorganic reaction without adding any organic surfactants. However, this approach mainly suffers from inhomogeneity in the size and shape of the nanocrystals, and, therefore, it requires further development.
To address this problem, Huang et al. developed a strategy to synthesize Cu2O nanocrystals with systematic shape evolution by a single chemical reduction method at room temperature.41 Cubic, truncated cubic, cuboctahedral, truncated octahedral, octahedral, short and extended hexapod structures of Cu2O nanocrystals with systematic morphological evolution have been synthesized by sequential addition of sodium dodecyl sulfate (SDS), an aqueous solution of CuCl2, NaOH, and hydroxylamine hydrochloride (NH2OH·HCl) as reductant. The mixture was aged for 2 h to obtain the products. The systematic variation in the product morphology can be achieved by changing the amount of added NH2OH·HCl, as shown in Fig. 5a–h. Different pH values obtained for different morphologies suggest a clear correlation between the pH and the RG, which proportionally controls the final shape of the nanocrystals, as shown in Fig. 5i. Keeping other parameters fixed, increasing the amount of NH2OH·HCl can make the solution pH decrease from 12 for the nanocube sample to 8.7 for the short hexapod sample. Apart from SEM images, the high morphological uniformity of these Cu2O nanocrystals can be associated with their PXRD patterns, as shown in Fig. 5j. The PXRD patterns evidently show a change in the relative intensities of the (111) and the (200) peaks, which also goes along with a morphology change. Nanocubes show only a strong (200) reflection peak with an extremely weak (111) reflection peak. The intensity of the (111) peak increases progressively as nanocrystals with more {111} surfaces are formed. The two peaks become more comparable in intensity for the type I truncated octahedra. The (111) peak then dominates for octahedra and hexapods.
 | ||
| Fig. 5 SEM images of the Cu2O nanocrystals with various morphologies: (a) cubes, (b) truncated cubes, (c) cuboctahedra, (d) type I truncated octahedra, (e) type II truncated octahedra, (f) octahedra, (g) short hexapods, and (h) extended hexapods. Scale bar = 1 μm. (i) Solution pH values for samples a–h. (j) PXRD patterns of the different shaped Cu2O nanocrystals as shown in a–h. Reproduced with permission from ref. 41, Copyright 2009 American Chemical Society. | ||
Apart from controlling the pH of the solution, the surfactant concentration can also play a crucial role in controlling the systematic shape evolution of the nanocrystals. Polyvinylpyrrolidone [PVP] is one of those surfactants that not only acts as a stabilizer to prevent the aggregation of nanocrystals but also assists the formation of well-defined Cu2O nanocrystals.42 PVP molecules with long chains can be adsorbed on the Cu2O surfaces via both physical and chemical bonding. In particular, PVP preferentially interacts more strongly with the {111} facets than the {100} facets, suppressing the growth of the {111} planes more efficiently than that of the {100} planes. Thus, as shown in Fig. 6, when the PVP concentration is low (0.5 mM), or the PVP is absent in the reaction mixture, the capping effect of PVP toward Cu2O nanocrystals is the weakest or absent, which leads to the formation of cubic shapes. By contrast, when the PVP concentration is high (4.5 mM), it strongly coordinates with the {111} facets and efficiently lowers the surface energy of the corresponding crystal planes. This enhanced coordinating effect of PVP completely blocks the growth on the {111} facets and facilitates the growth on the {100} facets, as shown previously in Fig. 3a. As the growth process takes place rapidly on the {100} facets, they disappear completely, and perfect octahedral morphology appears. Furthermore, different truncated octahedra as intermediate products can be achieved through precise control of PVP concentrations in the 0.5–4.5 mM range. Interestingly, when the PVP concentration becomes 9 mM, spherical nanocrystals are formed due to the high coverage of PVP on all the planes of Cu2O nanocrystals as there is no preferential growth of any particular crystal plane over another.
 | ||
| Fig. 6 Effect of PVP concentration on the Cu2O morphology. Reproduced with permission from ref. 42, Copyright 2010 American Chemical Society. | ||
Similar to PVP, oleic acid also shows facet-selective adsorption properties, which provides control over the final morphology of Cu2O microcrystals.43 Guo et al. demonstrated the reduction of CuSO4 at 100 °C for 1 h with D-(+)-glucose in an aqueous NaOH/ethanol/oleic acid system to synthesize Cu2O microcrystals. In the presence of 1 ml, 2.5 ml, and 4 ml of oleic acid, uniform cubic, octahedral, and rhombic dodecahedral (RD) Cu2O microcrystals were obtained, respectively. This was the first report showing the synthesis of Cu2O RD microcrystals fully bounded by {110} planes, which had a higher surface energy than the corresponding {100} and {111} planes. Later on, Zeng et al. obtained highly uniform ∼70 nm Cu2O RD nanocrystals by the reaction of copper(II) acetate, hexadecylamine, and undecane at 200 °C for 90 min.44 Hexadecylamine played multiple roles in this study, serving as: (1) a chelating ligand to form the [Cu(NH2C16H33)4]2+ complex precursor with Cu2+ ions, (2) a phase-transferring agent to transfer divalent Cu2+ ions into the organic phase, (3) a reducing agent to generate Cu1+ ions, and (4) a surface passivating adsorbate to control the crystal morphology. In addition, ∼9 μm Cu2O RD microcrystals were also synthesized by mixing Cu(NO3)2, formic acid, and NH3 in an ethanol–water solution followed by hydrothermal treatment at 145 °C for 90 min.45
The major drawback of these reaction conditions is that they need high temperature and strong surface binding ligands, and, therefore, further development will be needed to synthesize the RD shape at room temperature using an easily removable surfactant of a considerably small size. Huang et al. then proposed synthetic conditions that would overcome all the above-mentioned problems.46 A systematic shape evolution from cubic to RD nanocrystals was achieved at room temperature by mixing an aqueous solution of CuCl2, SDS surfactant, NaOH, and NH2OH·HCl as the reductant. The systematic shape evolution was achieved only by adjusting the volume of the reducing agent, as shown in Fig. 7. The shape evolution is directly related to the final solution pH and overall reduction rate of the copper precursor. It was demonstrated that cubes are formed at a much faster rate than rhombic dodecahedra, thus linking the formation of different particle morphologies to their different growth rates. This protocol is highly important as it allows achieving a diverse nanocrystal morphology with anisotropic facets.
 | ||
| Fig. 7 Shape evolution from cubic to rhombic dodecahedral morphology with respect to the added amount of hydroxylamine hydrochloride (NH2OH·HCl). Reproduced with permission from ref. 46, Copyright 2012 American Chemical Society. | ||
In addition to the role of the concentration of the surfactant and the reducing agent, different inorganic ions are also crucial to controlling the morphology of the nanocrystal. Xia et al. demonstrated a high temperature ethylene glycol (EG) reduction method for synthesizing Cu2O cubic nanostructures in the presence of chloride ions as the shape directing agent (Fig. 8).47 The in situ formed CuCl intermediate serves as a reservoir to control the supersaturation concentration of the Cu1+ ions in the reaction mixture. Therefore, the Cu2O formation rate significantly slowed down, allowing the seeds to grow into small individual nanocrystals without significant aggregation at the early stages of the reaction. Chloride ions also stabilize the {100} planes of the Cu2O nanocrystals and favors the formation of single-crystalline nanocubes as seeds, which can further grow in size via the Ostwald ripening process. When no chloride ions were added to the reaction mixture, the nucleation and growth of Cu2O nanocrystals took place instantaneously. This uncontrolled growth rate results in the formation of nanoparticles of small size that agglomerated to become polycrystalline colloidal spheres of large sizes in order to reduce the surface energy. Similarly, by tuning the ratio of the concentration of hydroxide and citrate anions, cubes to octahedral-morphological evolution for Cu2O nanocrystals were obtained on conductive substrates by a method of simple chemical deposition.34 Feng et al. also used cyclic scanning electrodeposition (CSE) to convert a thin Cu film into different Cu2O structures with the help of different ions such as NO2−, NO3−, SO42−, and CH3COO− used in the electrolyte.48 Other noticeable examples in this regard can also be seen in the literature.49–55
 | ||
| Fig. 8 Effect of Cl− ions on controlling the morphology of Cu2O nanocrystals. Reproduced with permission from ref. 47, Copyright 2008 Royal Society of Chemistry. | ||
After an in-depth discussion on the development of Cu2O polyhedral structures containing three different low-index facet such as {100}, {111} and {110}, it is necessary to understand the formation mechanism of high-index {hkl} facets (where at least one of the h, k, and l is equal to two or above). Surface energies (γ) corresponding to different Cu2O crystallographic facets usually increase in the order of γ {100} < γ {111} < γ {110} < γ {hkl}. As these high-index facets possess higher surface energy than the corresponding low-index facet, they disappear faster during the crystal growth and are difficult to preserve on the surface of the final nanocrystal.16 The unique feature of these facets is that they consist of a high density of low-coordinated atoms such as steps, edges, and kinks, which show higher catalytic activity by quickly coordinating with reagent molecules when compared to the low-index facets.16 Therefore, it is useful to understand the synthetic recipes and the growth mechanism for the preparation of Cu2O nanocrystals with high-index facets.56–62
Yang et al. synthesised highly symmetric multi-facet polyhedral Cu2O microcrystals partially enclosed with high-index facets (i.e., including low-index {110}, {100}, {111} facets and high-index {544}, {522}, and {211} facets) in the system of Cu2+/NaOH/glucose or ascorbic acid solution at high temperature (Fig. 9a1–c2).56 There are also other methods of synthesizing Cu2O microcrystals with high index facets.57–61 The major drawback of the aforementioned procedures is that the size of Cu2O crystals is on the order of several microns, which means a very low specific surface area and eventually lower photocatalytic activity. To solve this problem, Li et al. synthesized Cu2O truncated concave octahedral nanocrystals of an average edge length of 158 nm, which enclosed {511} high-index facets together with {110} and {100} facets through a system of oil in a water emulsion, as shown in Fig. 9d1–d4.62 Both the solution pH and the oleic acid are critical for the formation of truncated concave octahedral Cu2O nanocrystals.
 | ||
| Fig. 9 Typical FESEM images of polyhedral Cu2O microcrystals: (a1–c1) 50-facet, 74-facet, and 50-facet with high-index {522}, {544} and {211} facets respectively; (a2–c2) representative graphical structure. Reproduced with permission from ref. 56, Copyright 2011 Royal Society of Chemistry. Truncated concave octahedral nanocrystals and their (d1) SEM images, (d2) TEM images, (d3) selective area electron diffraction (SAED) patterns, and (d4) representative models. Reproduced with permission from ref. 62, Copyright 2018 Royal Society of Chemistry. | ||
All the above-mentioned well-defined Cu2O nanocrystals with both low- and high-index facets have one common drawback, i.e., their size is larger than 100 nm or 1000 nm, depending upon different synthetic conditions. That means they possess a low surface area for the given weight of the catalyst, which is related to a lower catalytic activity. Therefore, it is a challenging task to develop a scalable synthetic method for ultrasmall and highly uniform Cu2O polyhedra, keeping the facet intact. Several early trials were made to achieve this goal.63–66 By the reaction of an aqueous mixture of CuSO4, trisodium citrate, and NaOH, 40 nm Cu2O nanocubes were synthesized, but the size of the particles was still not sufficiently small and therefore the synthetic procedure provided low yields.63 Huang's group has recently developed a simple method to produce Cu2O nanocubes and octahedra with a varying size from 16–86 nm and 34–49 nm, respectively (Fig. 10).66 The reaction was carried out at room temperature and lasted 20 min. In a typical reaction, CuSO4, NaOH, and sodium ascorbate are added stepwise to an aqueous SDS surfactant solution. A gradual increase in the amount of sodium ascorbate helped to achieve Cu2O nanocubes of smaller sizes; this is due to the formation of a higher number of uniform nucleation centers at higher concentrations of the reducing agent. There are still a few challenges that need to be overcome in order to achieve RD and other nanostructures with a high-index facet within the sub-50 nm range, which can enhance the catalytic activity by increasing the reactive surface area. Further, the sub-50 nm nanocrystals will have more light absorption capability than scattering, which will increases their photocatalytic performances.
 | ||
| Fig. 10 Size controlled synthesis of uniform Cu2O nanocubes with average edge lengths of (a) 16, (b) 25, (c) 29, (d) 36, (e) 51, (f) 63, (g) 72, and (h) 86 nm. Reproduced with permission from ref. 66, Copyright 2019 American Chemical Society. | ||
It is difficult to control the morphology of Cu2O nanocrystals if the size drops below 10 nm because they try to achieve a spherical structure to minimize the surface energy.67–74 Also, unless a suitable organic surfactant is added, the sub-10 nm nanocrystals undergo aggregation, forming bigger spherical nanocrystals.47 O'Brien et al. synthesized highly uniform monodisperse Cu nanocrystals by reacting copper acetate with oleic acid and trioctylamine, while heating at 180 °C.67 Upon further oxidation in the presence of air and ligand protection, those Cu nanocrystals were systematically converted to highly crystalline 6 nm Cu2O nanocrystals (Fig. 11a). A thin layer of CuO formed at the nanocrystal–ligand interface because of over-oxidation. The corresponding selected area electron diffraction patterns (Fig. 11b) confirmed the cubic crystal structure of Cu2O. Oleic acid stabilized these nanocrystals in solution, which prevented aggregation. A similar procedure was also reported to synthesize hollow and solid Cu2O nanocrystals of 8 nm and 14 nm in size, respectively.72 Further expansion of this idea may lead to many reversible catalytic interconversions of the ligands protecting Cu and Cu2O nanocrystals in the presence of O2 as an oxidant and H2 as a reductant (Fig. 11d).69 During these interconversions, the used bidentate ligands strongly coordinate both oxidized Cu and Cu2O NP surfaces by forming strong covalent bonds, which prevents them from aggregation and size distortion. One-dimensional (1D) nanomaterials such as nanowires are also highly attractive building blocks for photocatalysis because of the inherent anisotropy, which improves charge separation and photocatalytic efficiency.7,70 Cu2O nanowires were also developed by different wet-chemical methods such as the decomposition of the metal–ligand complex route or the use of polyethylene glycol (PEG; Mw 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and hydrazine as the reducing agent at room temperature.73 Most interestingly, Li et al. developed an easy approach to synthesizing bulk quantities of single crystalline Cu2O nanowires with tunable diameter and length. Uniform Cu2O nanowires were prepared through the reduction of cupric acetate with o-anisidine, pyrrole, or 2,5-dimethoxyaniline as the reductant in dilute aqueous solutions under hydrothermal conditions, as shown in Fig. 11c.68 The length of the Cu2O nanowires varied from tens of micrometers to more than one hundred micrometers.
000) and hydrazine as the reducing agent at room temperature.73 Most interestingly, Li et al. developed an easy approach to synthesizing bulk quantities of single crystalline Cu2O nanowires with tunable diameter and length. Uniform Cu2O nanowires were prepared through the reduction of cupric acetate with o-anisidine, pyrrole, or 2,5-dimethoxyaniline as the reductant in dilute aqueous solutions under hydrothermal conditions, as shown in Fig. 11c.68 The length of the Cu2O nanowires varied from tens of micrometers to more than one hundred micrometers.
 | ||
| Fig. 11 Highly single crystalline Cu2O nanocrystals: (a) HRTEM image and (b) SAED pattern. Reproduced with permission from ref. 67, Copyright 2005 American Chemical Society. (c) SEM image of Cu2O nanowires. Reproduced with permission from ref. 68, Copyright 2007 American Chemical Society. (d) Reversible synthesis of ultrasmall Cu NPs and Cu2O NPs from organo-copper(I) reagent. Reproduced with permission from ref. 69, Copyright 2017 American Chemical Society. (e) TEM and (f) AFM images of the ultrathin Cu2O nanosheets with 4 atomic thicknesses and the yellow line in f corresponds to the height profile in (g). Reproduced with permission from ref. 70, Copyright 2014 Elsevier. | ||
Two-dimensional (2D) materials such as nanosheets are also very interesting due to the quick separation of electron–hole pairs from the bulk to the surface as well as their large surface area and efficient light harvesting properties.70 Xie et al. synthesized an ultrathin Cu2O nanosheet with 4 atomic thicknesses by decomposing lamellar Cu2O–oleate complex intermediate microplates, which are formed by periodically stacking organic molecules and inorganic layers, at a temperature of 300 °C for 8 min in air. The TEM image in Fig. 11e shows an almost transparent feature, and the clear Tyndall effect indicates the ultrathin thickness of such Cu2O nanosheets, further verified by their atomic force microscopy (AFM) images, presented in Fig. 11f. The thickness of the nanosheets was around 0.62 nm (Fig. 11g). In addition to the powder well-defined nanocrystals, it is possible to prepare 3D Cu2O arrays on a fluorine doped tin oxide (FTO) thin film or on a Cu mesh using sputtering or anodization and subsequent thermal annealing procedures. Such arrays mainly contain nanowire structures organized over thin films and are less employed in photocatalysis.75–78 All the above-mentioned case studies provide not only a clear stepwise development of the synthesis of diverse well-defined Cu2O nanostructures but also a good opportunity to understand the growth mechanism and role of different ions, solution pH, and surfactant concentration. The remaining synthetic challenges in this field can also be overcome on the basis of these findings.
2.3 Methodologies for removing capping agents
In the previous section, we discussed the role of the different surfactants and their concentrations in controlling both the shape and size of Cu2O nanocrystals. Common surfactants such as SDS, trisodium citrate, CTAB, oleic acid, oleic amine, hexadecylamine, PVP and PEG are majorly used for the synthesis of well-defined Cu2O nanocrystals with the control of size. These surfactants not only stabilize the crystal plane but also prevent any agglomeration during the synthesis and the isolation process of the nanocrystals. After the isolation, the nanocrystals are employed in different catalytic processes in order to evaluate their facet-dependent properties. Different crystal facets have different atomic arrangements of Cu1+ and O2− ions, resulting in different Cu1+ ion surface atomic densities, which may lead to different catalytic activities.46 If the surfactant molecules are still present on these facets, they can block the catalytically active sites and hinder the access of reactant molecules to the reactive sites.79–81 Moreover, under particular reaction conditions, surfactants may decompose and contribute to the formation of hydrogen or carbon-based products, thus affecting the proper evaluation of the photocatalytic activity and product selectivity. This aspect is particularly delicate for CO2 photoreduction and will be discussed in more detail in Section 4.4. Therefore, the removal of the capping agents from the crystal facets must precede any comparison of the facet-dependent catalytic activity. Thus, the surfactant molecules present on the crystal facet can act as poison in the catalytic reaction.It is important to understand the interface between the capping agent and the crystal facet on a nanoscale level before moving on to the removal section.79 There are mainly two different interfaces namely (1) the metal–ligand interface and (2) the ligand–solution interface, as shown in Fig. 12. The intermediate transition zone known as the metal–ligand interphase can have more versatile complex structures depending upon the molecular weight, structure, and functional groups of the surfactant molecules. There is a lack of literature that would evaluate the impact of different surfactant molecules on the activity and selectivity of Cu2O nanocrystals of different shapes. Most studies have been done on Pt, Au, Pd, and Ag nanocrystals.81 At the metal–ligand interface, two major phenomena can occur, both benefiting the catalytic reaction carried out on a metal nanocrystal. Firstly, the discrete molecular orbitals of the functional groups of the ligands can strongly interact with the metal atoms present on the nanocrystal surface and induce charge transfer from the ligand to the metal atom. This effect can modify the local charge density and facilitate any of the steps involved in the catalytic reaction such as adsorption of the reactants, bond-breaking, bond-formation, and desorption of the product molecules, making the reaction more or less selective.79–81 For example, electron donation from an electron-rich capping agent to the surface metal atoms has been used to control the product selectivity of the reaction, by favoring the adsorption of electron deficient substrates and promoting the desorption of electron-rich product molecules from the surface. A second most important effect is the formation of a self-assembled monolayer (SAM) around the nanocrystal surface. Due to the strong affinity of the thiol group present in an alkane thiol, the surfactant adsorbs more strongly with the head group to the metal atoms, while the hydrocarbon tail remains oriented at a well-defined angle with respect to the surface forming the SAM structure. Such an ordered morphology of the SAM can induce a steric hindrance, which can limit the mobility of differently sized reactants and force them to rearrange the adsorption geometry close to the active site, which enables controlling the selectivity or more importantly the stereoselectivity of the products.
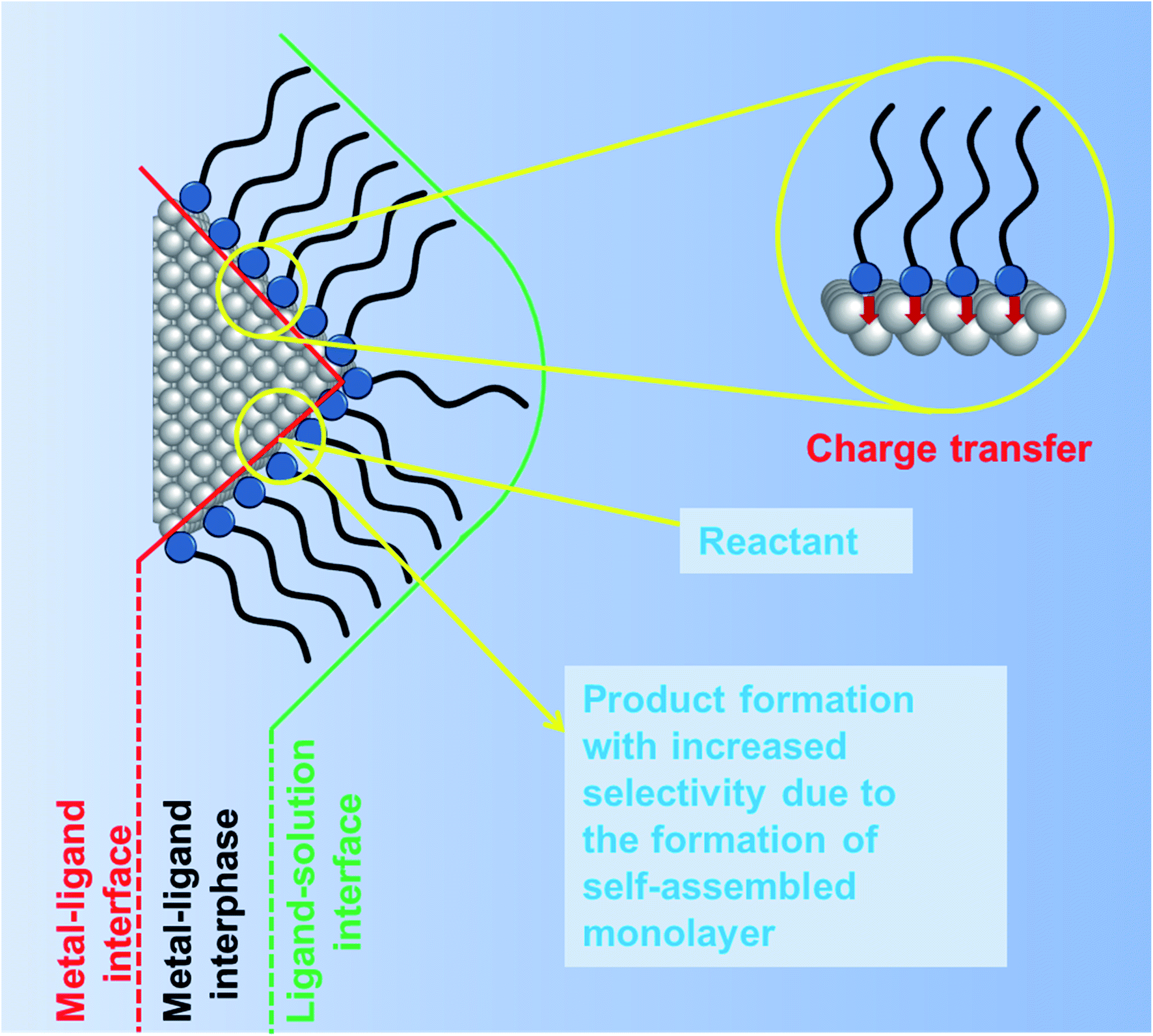 | ||
| Fig. 12 Schematic diagram showing the ligand orientation around a nanocrystal surface from a microscopic point of view. Influence of ligands on the nanocrystal surface and on product selectivity. Adapted with permission from ref. 79, Copyright 2016 MDPI. | ||
Apart from the above-mentioned promotional effects of the capping agent, it is still necessary to completely remove the surfactant molecules from the Cu2O nanocrystal surface in order to compare their facet-dependent catalytic activity in different photocatalytic reactions as these capping agents can reduce the photocatalytic activity by neutralizing the photogenerated electrons or holes via charge transfer. A completely “clean surface” in the solution of nanocrystals cannot be truly obtained, but it can be considered to be free from long-chain sticky surfactant molecules.80,81 After the removal of capping agents, the clean surface is stabilized by small molecules such as solvent molecules or solute ions, which are easily substituted by reactants during catalytic reactions.
There are different, well-established methods to remove the capping agents from the nanocrystal surface.79–81 The most common procedure consists of consecutive sonication steps involving large amounts of solvents and centrifugation to collect the nanoparticles. For example, surfactants such as SDS and trisodium citrate can be easily removed by washing with a 1:1 water/ethanol mixture several times followed by centrifugation.82,83 Therefore, synthetic procedures that use SDS or trisodium citrate as a surfactant for the synthesis of Cu2O nanocrystals are very promising since a clean-surface can be obtained by a simple washing procedure. Other surfactants such as CTAB, oleic acid, oleic amine, hexadecylamine, PVP, and PEG can also be removed from the nanoparticle surface, partially using extensive washing steps, which can lead to the deformation of crystal facets or to leaching. Different strategies have been developed to deal with this problem. One of the them decomposes the capping agents into small molecular fragments by heat or light, followed by their removal by means of a gas flow or solvent.84 This method includes high-temperature thermal annealing and UV-ozone (UVO) irradiation. Surfactants with higher molecular weight such as PVP and PEG can also be removed following this procedure.79–81 One major drawback of this procedure is the partial decomposition of the organic surfactant during the thermal treatment or UVO irradiation with the consequent generation of coke and other species, which can deactivate the catalyst facet. Another strategy makes use of the addition of pure acetic acid to amine-capped nanoparticles causing the protonation of amino groups, which weakens their interaction with the nanocrystal surface.79–81 This accelerates their detachment during the solvent-washing procedure. Surfactants such as oleylamine, hexadecylamine, and oleic acid can be washed off this way. Other strategies are based on adding excessive small molecules (such as 1-butylamine) to displace long-chain hydrocarbon organic surfactants (such as oleylamine, PVP), which also have a stronger capping ability. Due to the much lower boiling point and mass, smaller molecules can be easily removed by extensive washing or vacuum evaporation.79–81
After discussing general approaches to removing the surfactant from the surface of different inorganic nanocrystals, we will now shift the focus onto those procedures that are specifically suitable for Cu2O nanocrystals. It is highly desirable that during the surfactant-removal process, the shape, size, composition, crystallinity, and crystal facet of a Cu2O nanocrystal are preserved. If we change any one of these parameters, the results of studying the facet-dependent properties will be erroneous. It is also crucial to remember that Cu1+ ions in Cu2O can undergo a disproportionation reaction under different chemical environments forming Cu2+ or Cu0 states, which may lead to the chemical decomposition of the crystal or structural modification. Different inorganic ions or molecules can also cause a serious damage to well-defined Cu2O nanocrystals, and these must be identified before conducting the surfactant removal process or pursuing any photocatalytic reactions.
When Cu2O microcrystals are exposed to an aqueous solution of ammonia (pH ∼ 11.4) for 3 or 40 min under stirring conditions, it will bring a certain change to the shape, size and composition, as demonstrated in Fig. 13a1–a3.85 After 3 min, rhombic dodecahedral Cu2O microcrystals undergo an etching process which transforms a smooth {110} surface into a rough and stepped square facet. HRTEM images confirmed that the stepped square facets belong to the Cu2O (100) crystal plane. When the reaction time reaches 40 min, Cu2O rhombic dodecahedral shapes were completely converted into Cu(OH)2 nanobelts (Fig. 13a3), as confirmed by PXRD and XPS. It is important to note that in an aqueous NaOH solution with the same pH value such etching processes of Cu2O nanocrystals cannot be observed.85 Therefore, it can be postulated that the etching process is initiated by the coordination of surface Cu1+ ions present in Cu2O microcrystals with NH3 to form [Cu(NH3)4]1+ species. They undergo an oxidation process by dissolved O2 to produce [Cu(NH3)4]2+, which then slowly precipitates in an aqueous ammonia solution to produce Cu(OH)2. Control experiments proved that the stability of different low index Cu2O crystal planes in the aqueous ammonia solution followed the order {100} > {111} > {110}, suggesting that the cubic is more stable than rhombic dodecahedral morphology in aqueous NH3 solution.85 A similar stability trend can also be observed when Cu2O microcrystals are exposed to an acetic acid solution (pH = 3.5) under stirring conditions.86 Oxidative etching of octahedral and rhombic dodecahedral Cu2O microcrystals in the weak acid solution leads to the formation of stepped layers of square facets exposing {100} planes (Fig. 13b1–b6), evidencing that the Cu2O {100} planes are more stable than the Cu2O {111} and {110} planes. But, in this case, surface chemical composition remains similar to that of Cu2O.
 | ||
| Fig. 13 SEM image of as-synthesized (a1) Cu2O rhombic dodecahedral (RD) nanocrystals. After exposing these microcrystals in aqueous ammonia solution for (a2) 3 min and (a3) 40 min. Reproduced with permission from ref. 85, Copyright 2011 American Chemical Society. (b1) SEM and (b3) TEM images of as-synthesized Cu2O RD. After exposing these microcrystals in aqueous acetic acid solution for 150 min: (b2) SEM, (b4) TEM and (b5 and b6) corresponding HRTEM images with the image of lattice fringes. Reproduced with permission from ref. 86, Copyright 2011 American Chemical Society. (c) High-magnification FESEM image of a Cu2O nanoframe obtained after exposure in air at 20 °C for 16 days. Reproduced with permission from ref. 87, Copyright 2010 Wiley-VCH. TEM images of a single cubic and octahedral Cu2S nanocage viewed over its (d1 and d3) {100} facet and (d2 and d4) {110} edge respectively. Reproduced with permission from ref. 88, Copyright 2011 Wiley-VCH. | ||
Further, a truncated octahedral Cu2O microcrystal has eight hexagonal {111} faces and six {100} faces. When these microcrystals are exposed to air in the presence of PVP, selective etching leads to the formation of a single crystalline Cu2O nanoframe surrounded only by {111} facets, whereas all {100} facets are etched away completely (Fig. 13c).87 This contradictory event can be explained as follows: PVP acts as a capping agent, and preferentially adsorbs on the {111} facets of the Cu2O crystals, which prevents the {111} planes from etching; therefore, {100} facets completely vanish, leading to the formation of hollow structures. Any sulfide anions are also detrimental to Cu2O as they easily replace the oxide anions from the crystal lattice, which leads to the formation of Cu2S crystals.88 A single crystalline cubic and a truncated octahedral Cu2S nanocage have been prepared with ultrathin walls through controlled sulfidation of the corresponding Cu2O crystals, and the acid etching was applied to remove the interior Cu2O portions (Fig. 13d1–d4). This suggests that any sulfide species should always keep away from Cu2O nanocrystals during the surfactant removal, surface modification or photocatalytic reaction.
Not only sulfide but also thiosulfate anions (S2O32−) can induce a strong coordinating etching process on Cu2O nanocrystals. Different metal (M = Mn, Fe, Co, Ni, Zn) hydroxide nanocages can be obtained by reacting a certain amount of Cu2O templates with MCl2·yH2O in an ethanol/water mixed solvent in the presence of PVP (Mw = 30000) and Na2S2O3.89 The formation mechanism can be explained as follows: as per Pearson's hard and soft acid–base (HSAB) principle “soft base form strong complexes with soft acid whereas hard base prefer hard acid”.89 Thus the thiosulfate anion (soft base) forms a stronger bond with Cu1+ cations (soft base) in Cu2O and replaces the lattice oxide anions (hard base). As a result, coordinating etching of Cu2O occurs by forming a soluble [Cu2(S2O3)x]2−2x complex, which is followed by a synchronous complex precipitation process of M(OH)2 around the Cu2O template surface, leading to the formation of metal hydroxide nanocages. This process is termed “coordinating etching and precipitating” (CEP).89 Similar to acetic acid, dilute HCl can also induce facet-selective etching of Cu2O nanocrystals, leading to the formation of diverse single crystalline cubic Cu2O nanoframes.90,91 Therefore, it is essential to remember that acetic acid, hydrochloric acid, ammonia, aqueous solution exposed to air, sulfide, and thiosulfate anions can cause serious damage to the shape and composition of the Cu2O nanocrystal. They should not be used during the removal of capping agents or even in photocatalytic reactions. It should be noted that SDS or trisodium citrate would be a better choice during the synthesis of Cu2O nanocrystals as they can be easily removed by consecutive washing with the water/ethanol mixture.
Regarding PVP, PEG, oleylamine, oleic acid, and hexadecylamine, extra care is needed during the removal steps. The successful removal of the organic surfactant can be then confirmed by FTIR, XPS, and a CO-chemisorption study. Huang et al. used PVP and oleic acid (OA) as capping ligands to prepare octahedral and rhombic-dodecahedral Cu2O microcrystals, respectively, whereas no capping ligands were used for the cubic shape.92 PVP and OA were completely removed from the Cu2O crystal surface by placing them in a U-shaped quartz microreactor, followed by purging in a stream of C3H6, O2 and N2 and heating to the desirable temperature to carry out a controlled oxidation treatment. Oxygen led to the oxidation of capping ligands at low temperature, whereas the coexistence of C3H6, O2 and N2 in the atmosphere cooperatively prevented the Cu2O surface from over-oxidation. A clear CO chemisorption peak could be observed after the removal of the surfactant but not on the capped Cu2O microcrystals (Fig. 14a–c). FTIR (Fig. 14d), PXRD, and XPS also confirmed the successful removal of PVP and OA from the Cu2O microcrystal surface, without changing the morphologies, surface compositions, and structures. In conclusion, with respect to the different types of surfactants used in the synthesis of Cu2O nanocrystals, we must conduct the surfactant-removal steps very carefully, keeping in mind all the previous discussion, so that we do not damage the well-defined nanocrystals before using them in photocatalytic applications. Either multiple washing with a solvent or heat and light treatment under appropriate conditions can be more suitable for achieving this target.
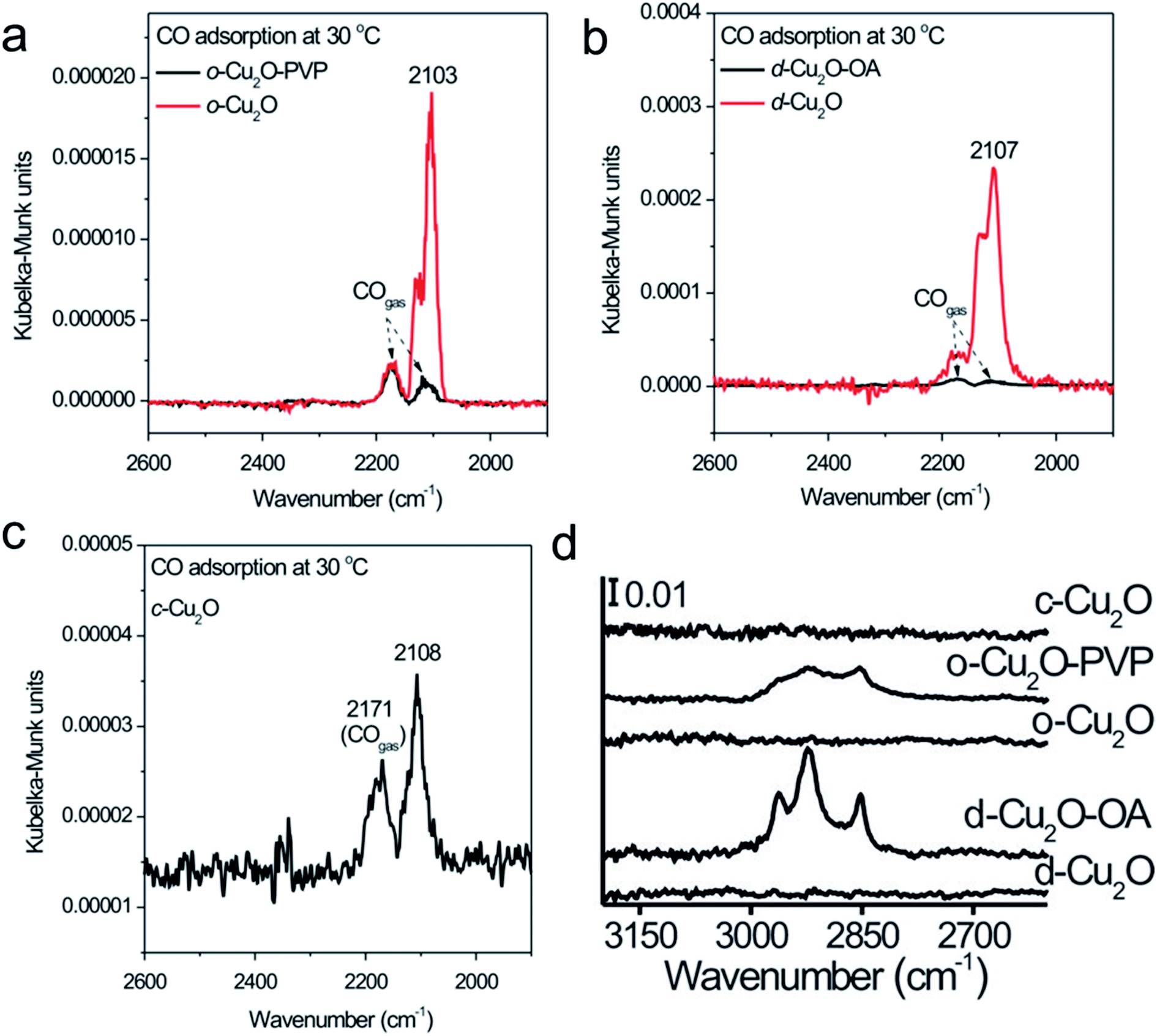 | ||
| Fig. 14 DRIFTS spectra of CO chemisorption at 30 °C on (a) Cu2O octahedra capped with PVP (o-Cu2O-PVP) and capping ligand-free Cu2O octahedra (o-Cu2O), (b) Cu2O rhombic dodecahedra capped with OA (d-Cu2O-OA) and capping ligand-free Cu2O rhombic dodecahedra (d-Cu2O), and (c) capping ligand-free Cu2O cubes (c-Cu2O). (d) FTIR spectra of the corresponding nanocrystals. Reproduced with permission from ref. 92, Copyright 2014 Wiley-VCH. | ||
3 Facet-dependent properties
Well-defined nanocrystals with different crystal facets (i.e. anisotropic facets) show different atomic arrangements. For this reason, they exhibit unique facet-dependent properties such as molecular adsorption, anisotropic redox reaction sites, surface electronic structures, and optical properties. All these facet-dependent properties can have a profound impact on photocatalytic properties; therefore, they will be discussed in detail in the following section.17 We also remark that other fundamental properties that are not covered in depth in this review such as charge mobility, work function, conductivity, and formation energy of point defects are facet-dependent, and may affect photocatalytic processes at the Cu2O surface.3.1 Selective deposition of a co-catalyst
Semiconductor nanocrystals enclosed by anisotropic facets have different electronic band structures (i.e., surface states), which promote the accumulation of the photogenerated electrons and holes on different facets for the reduction and oxidation reaction, respectively.15,17 The presence of a suitable co-catalyst on these redox sites can further enhance the electron–hole separation process, resulting in an enhanced photocatalytic activity. The facet-selective adsorption behavior of surfactants has been deployed to achieve selective deposition of the co-catalyst on Cu2O nanocrystals with anisotropic facets.To probe this phenomenon, Choi et al. electrochemically deposited micrometer-sized cubic, octahedral, and truncated octahedral Cu2O crystals on indium tin oxide (ITO) substrates.93 In the absence of a SDS surfactant, after 15 s, the electrochemical gold deposition showed that gold nanoparticles formed on both {100} and {111} planes (Fig. 15a–c) regardless of the crystal shape, i.e., lack of selective growth was observed. When SDS was introduced into the reaction medium, gold nanocrystals deposition took place on all of the {100} planes belonging to the Cu2O cubic crystals (Fig. 15d), which confirmed that SDS did not adsorb strongly on the {100} planes. However, no gold nanocrystals were found on octahedral Cu2O crystals (Fig. 15e), which indicated that SDS molecules strongly adsorbed on the {111} planes, resulting in the effective inhibition of the nucleation of gold nanocrystals. More surprisingly, gold nanocrystals were selectively deposited only on the {100} planes of truncated octahedral Cu2O crystals, which contained both the {100} and the {111} planes (Fig. 15f), thus confirming the SDS molecule preferential adsorption on the {111} planes of the Cu2O crystal. Therefore, we can choose different surfactant molecules to be used in combination with diverse Cu2O morphologies in order to investigate their preferential adsorption and obtain facet-selective deposition of co-catalysts, which will help to improve the activity and selectivity of the investigated photocatalytic reactions. Even in the absence of any reducing agents, HAuCl4 can be reduced to Au nanocrystals in the presence of Cu2O nanocrystals at room temperature; the driving force for this in situ reduction can be explained in terms of the standard reduction potential difference between AuCl4−/Au [0.93 V vs. the standard hydrogen electrode (SHE)] and Cu2+/Cu1+ [0.15 V vs. SHE] pairs.94,95 A similar galvanic displacement reaction can also be seen for H2PdCl4 and H2PtCl6 precursors. During this process, a certain degree of surface etching can occur in Cu2O crystals, which will be spontaneously covered by newly-formed metal nanocrystals. Du et al. showed that in the absence of any surfactants, Au nanocrystals were first selectively deposited only on all tips of an octahedral shape via in situ reduction, and then they covered the edges when using an increased amount of a gold precursor solution. Further, an increase of the amount of Au caused random coverage of all facets, edges, and tips of the octahedral Cu2O nanocrystals, indicating the loss of selective deposition. The main reason behind the observed behavior is that different facets possess different surface energies that follow the order as: γ(facets) < γ(edges) < γ(tips). Thus, the selective deposition of metal nanocrystals on Cu2O nanocrystals with anisotropic facets can be controlled by utilizing preferential adsorption of additives or controlling the amount of added metal precursors under suitable reaction conditions. Such hybrid nanocatalysts efficiently reduce the photogenerated electron–hole recombination, which results in an enhanced photocatalytic reaction rate.15
 | ||
| Fig. 15 Surfactant assisted facet selective Au deposited on Cu2O crystals without (a–c) and with (d–f) sodium dodecyl sulfate. Reproduced with permission from ref. 93, Copyright 2009 American Chemical Society. | ||
3.2 Distribution of electrons and holes on different facets upon illumination
Efficient separation of photogenerated electrons and holes is one of the bottlenecks in achieving very high photocatalytic activity when using a semiconductor nanocrystal.15,17,26 Photogenerated electrons should move to a reductive site to perform the reduction reaction, whereas holes need to reach oxidative sites to perform the oxidation reaction (Fig. 1a). For instance, nanospheres suffer from a high electron–hole recombination rate due to their isotropic geometry. Li et al. extensively used surface photovoltage (SPV) techniques based on Kelvin probe force microscopy (KPFM) and showed that the photogenerated charges can be separated effectively in a high-symmetry Cu2O photocatalyst by asymmetric light irradiation.96–100 The holes and electrons were transferred to the illuminated and shadow regions, respectively, of a single photocatalytic particle. Further investigation in this regard will be needed to identify the catalytic sites on Cu2O nanocrystals with anisotropic facets. Single-particle single-molecule fluorescence photocatalysis could be a suitable tool for on-site observation of interfacial chemical reactions involving charge carriers and reactive oxygen species (ROS), such as singlet oxygen and the hydroxyl radicals, generated by the photoexcitation of semiconductor nanocrystals.101,102 Such studies confirmed that the presence of anisotropic facets can be beneficial to obtaining efficient spatial charge separation. Using the small sized Cu2O crystals with anisotropic facets remains a challenge. It could help to achieve a higher photocatalytic performance in different reactions, which results from their increased high reactive surface area.3.3 Optical properties
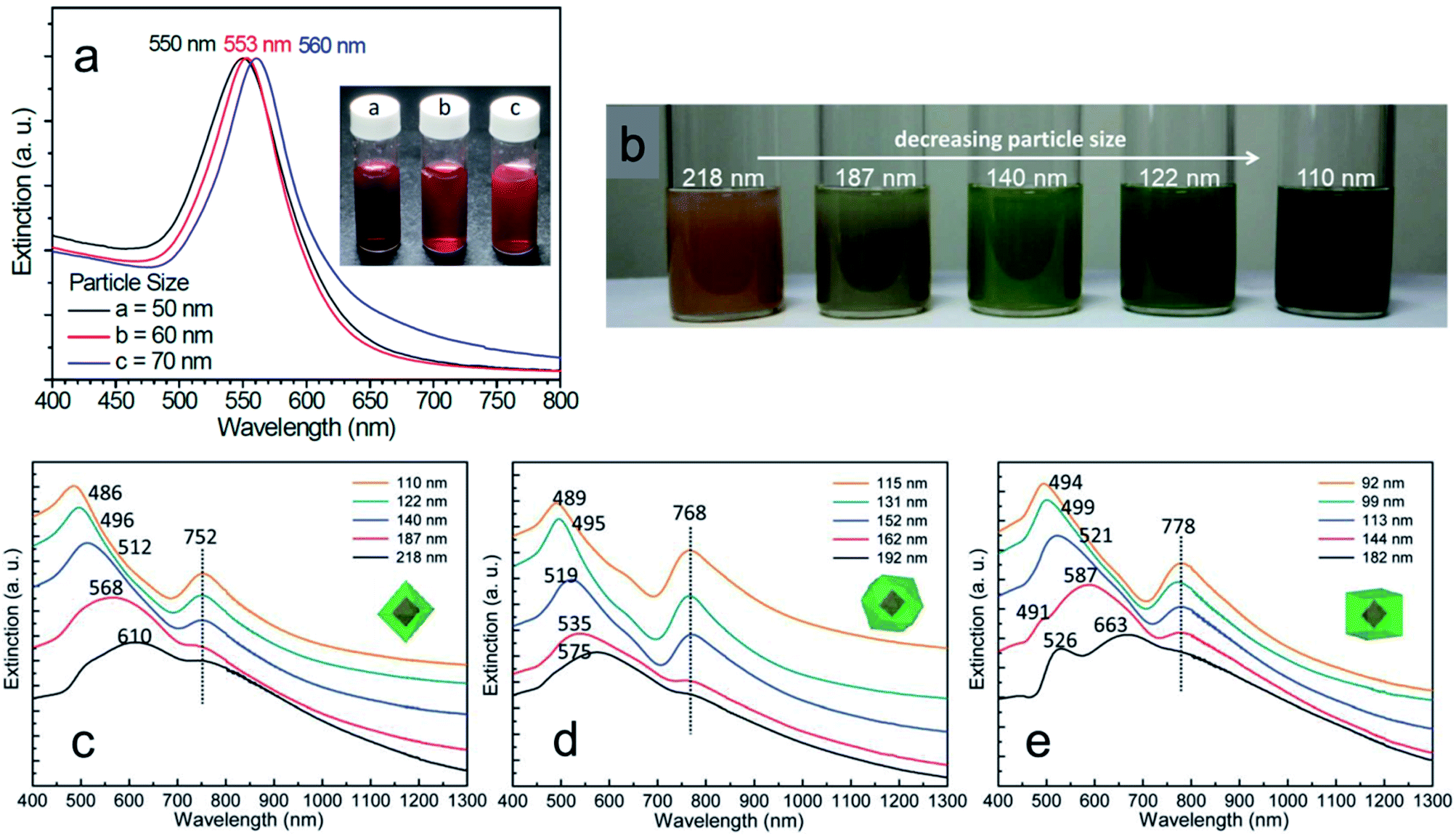
Before moving on to the photocatalytic applications, it is very important to understand how different sizes and shapes of Cu2O nanocrystals affect their optical properties. Diverse synthetic strategies give us an opportunity to better understand the existence of facet-dependent optical properties in Cu2O nanocrystals, which is barely studied.103,104 The absorption peak for Cu2O nanocrystals varies from 450 nm to 700 nm, depending upon the shape and size.105 Generally, while increasing the volume of a spherical Cu2O nanocrystal, the absorption peak will red-shift and broaden systematically, along with the enhanced light scattering.105 However, surprisingly, Cu2O nanocrystals with different facets of similar volume can have different absorption peak positions in the same solvent, which is a purely facet-dependent optical property.106–109Fig. 16a compares the absorption peak position of five different comparable volumes of octahedral and cubic Cu2O samples in absolute ethanol. Interestingly, nanocubes show a more significant absorption band redshift than octahedra of similar volumes. Moreover, the absorption band separation between the corresponding cubes and octahedra is 13–16 nm, and is independent of particle volume.106 This phenomenon is directly related to the exposed crystal facet of the Cu2O nanocrystals. The observed facet-dependent optical effect is due to a thin Cu2O surface layer (∼less than 1.5 nm) that has different band structures and different degrees of band bending with respect to the individual facets of Cu2O.109 The thicknesses of these surface layers were determined through density functional theory (DFT) calculations, which were approximately 6.2, 11.7, and 4.5 Å for the {111}, {100}, and {110} facets of Cu2O respectively. For the three surface atomic layers of (111), (100), and (110) planes, the density of state (DOS) plots resembled those of a metal, a semimetal, and a semiconductor, respectively. Thus, one can treat these thin surface layers of Cu2O as being composed of different materials with different refractive indices. As the refractive index (n) of the medium is directly related to its dielectric constant via εm = n2, a thin shell material with a high dielectric constant causes large redshifts. Fig. 16b present the photoluminescence spectra of three differently sized Cu2O RD nanocrystals showing that the intensities are highest for the smallest nanocrystal.108 Also Cu2O nanocrystals with three different shapes show different colors (Fig. 16c) due to their different band gap energy.108 Thus, it can be concluded that crystal facet engineering can help to tune the band gap of a semiconductor.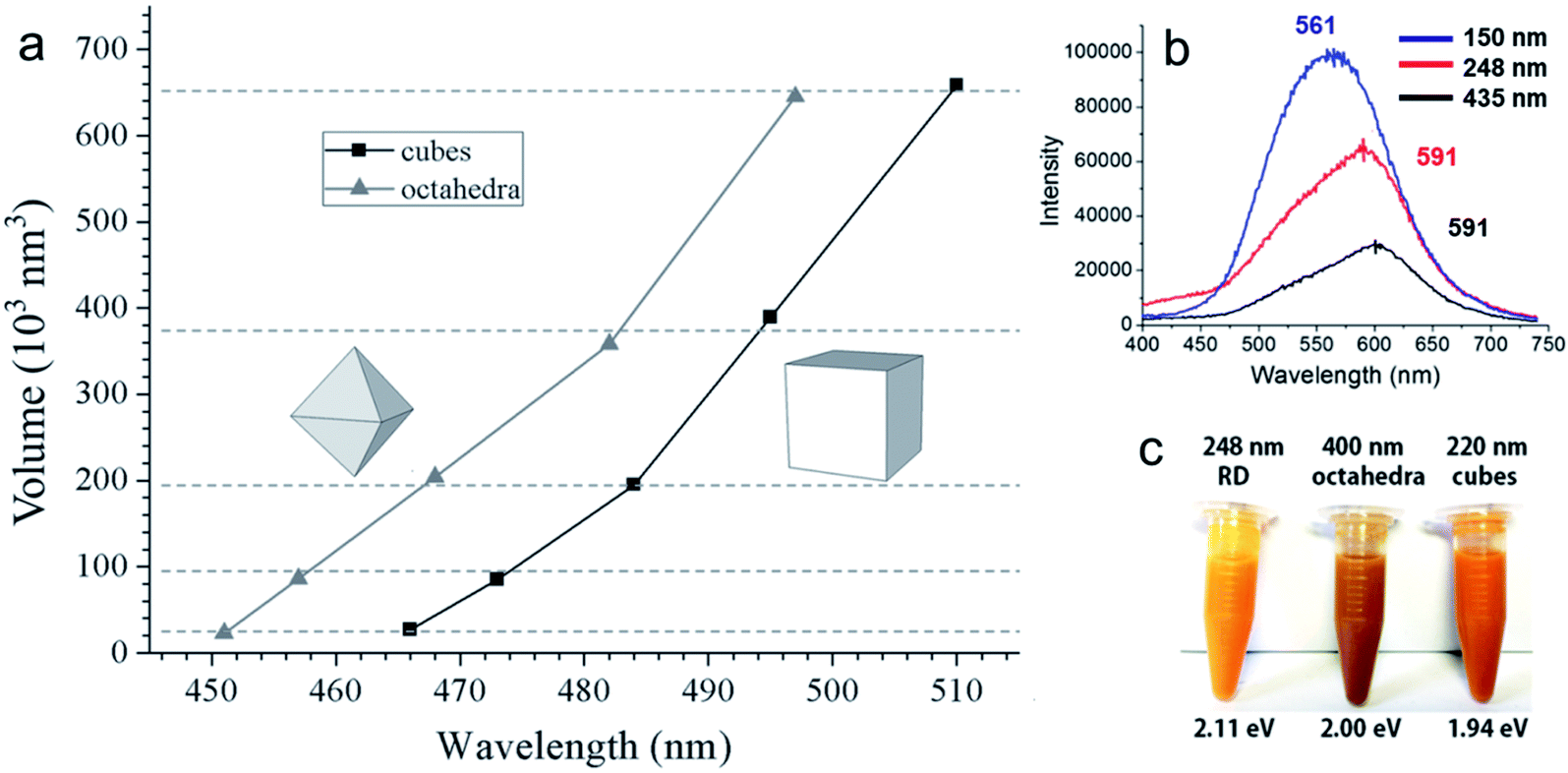 | ||
| Fig. 16 (a) A comparison plot for the variation of volume for Cu2O octahedra and cubes with respect to their absorption peak positions. Reproduced with permission from ref. 106, Copyright 2016 Wiley-VCH. (b) Photoluminescence spectra of different sized Cu2O RD nanocrystals. Excitation wavelength was 380 nm. Reproduced with permission from ref. 108, Copyright 2018 American Chemical Society. (c) Color comparison of three different shaped Cu2O nanocrystals, confirming visually observable optical facet effects. Reproduced with permission from ref. 108, Copyright 2018 American Chemical Society. | ||
Most interestingly, these thin Cu2O surface layers exposing the (111), (100), and (110) planes with different dielectric constants not only influence the absorbance peak position of pure Cu2O nanocrystals but can also cause a large redshift in the localized surface plasmon resonance (LSPR) peak of noble metals.110–113 Huang's group synthesized Au–Cu2O core–shell nanocrystals with octahedral, cuboctahedral, and cubic morphology with high uniformity in size using 50 nm octahedral Au nanocrystals as a core, which showed a LSPR peak at 550 nm (Fig. 17a).110 The colloidal solution color gradually changed from a dark orange color to greenish blue, while the average particle size decreased (Fig. 17b). Surprisingly, the UV-vis absorption spectra of the Au–Cu2O core–shell nanocubes, cuboctahedra, and octahedra showed a fixed LSPR peak position respectively at 752, 768, and 778 nm, though there was a systematic change in the Cu2O shell thickness for each shape. It should be noted that the SPR peak positions are also dependent on the exposed surface facets of the Cu2O shell. Despite the different shell thicknesses and overall sizes, the LSPR peak of the Au core was more redshifted for the cubic morphology (778 nm) than for the octahedral shape (752 nm), whereas cuboctahedral felt in the middle (768 nm) as they both had the {100} and {111} facets. This can be due to the different Cu2O crystal facets having a thin surface layer with different dielectric constants. Therefore, when a plasmonic wave propagates from at the interface between the metal core and the Cu2O shell, it sees the surface layer with different dielectric constants and responds differently to this environmental change, shifting the LSPR peak position accordingly. To further verify this rare optical phenomenon, diverse types of core–shell nanocrystals were also synthesised and studied.111–114 Interestingly, different surface oxidation states or incorporation of various amino acids into the crystal lattice of Cu2O nanocrystals can also lead to different optical and band gap energies.115,116 The unique optical properties of such hybrid nanostructures may also be beneficial for different photocatalytic and photothermal applications as they strongly absorb in the whole solar spectrum.
 | ||
| Fig. 17 (a) UV-vis absorption spectra of the different sized octahedral Au nanocrystals. (b) Color comparison of different sized octahedral Au–Cu2O core–shell nanocrystals. UV-vis absorption spectra of the five different sized Au–Cu2O core–shell nanocrystals with (c) octahedral, (d) cuboctahedral, and (e) cubic shape having a 50 nm octahedral Au core inside them. Reproduced with permission from ref. 110, Copyright 2014 Royal Society of Chemistry. | ||
4 Photocatalysis with well-defined Cu2O nanocrystals
In this section, we will discuss the proper ways of representing common parameters that are directly related to photocatalysis. Photocatalytic applications of well-defined hybrid Cu2O nanocrystals have been studied for different organic reactions, H2 evolution, and CO2 reduction. The photo-instability of Cu2O nanocrystals remains a key challenge to overcome during catalysis. Therefore, different strategies to solve this problem have been emphasized and accompanied with a proper case study.4.1 Figures of merit in photocatalysis
Before starting the discussion about well-defined Cu2O nanocrystal-mediated photocatalytic processes, it is necessary to introduce the figures of merit commonly used to assess the photocatalytic performance of a newly designed photocatalyst with other reported benchmark photocatalysts. It is very common to note that the absence of this crucial information in research papers makes it difficult to compare the photocatalytic efficiency, and also reproduce it, if necessary. Therefore, careful calculation and proper use of the most important figures of merit in research papers should be provided in order to help the research community in this field to facilitate the development of a highly efficient photocatalyst for different photocatalytic reactions.117–124 In this chapter, we will cover the following figures of merit: (a) turnover frequency, (b) specific surface area, and (c) apparent quantum yield and solar quantum yield.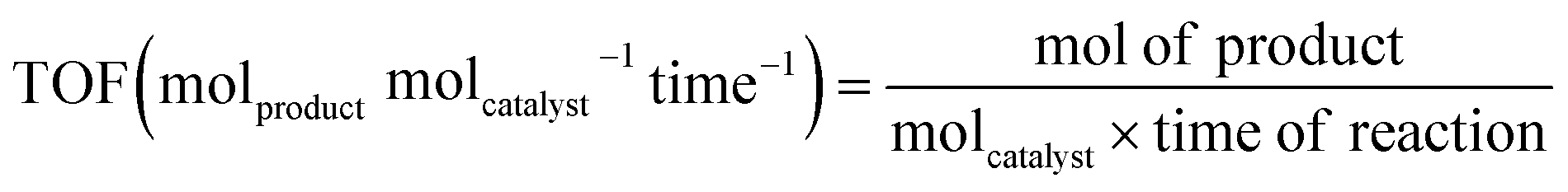
If a hybrid photocatalyst is made of two or more different components, it will be difficult to calculate the molar mass of such a material. In that situation, for simplicity, molcatalyst is often replaced with gcatalyst and the TOF will be expressed as molproduct gcatalyst−1 time−1. This formula is very often used to compare the performance for photocatalytic CO2 reduction and H2 evolution, even if the data provide essential technical information and do not allow a proper comparison of materials with different surface areas.117,123
If we can identify and quantify the fully exposed active sites in a hybrid catalyst that solely carry out the catalytic reaction, then TOF can be expressed as follows
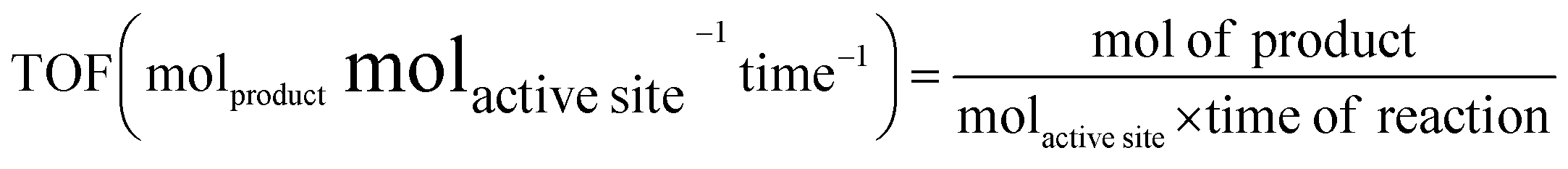
Generally, the specific surface area is defined as the total surface area of a nanocatalyst per unit of mass. Its unit is expressed as m2 g−1 and it must be measured before making any comparison of the facet-dependent photocatalytic reactivity of differently shaped nanocrystals. In a photocatalytic reaction, the amount of differently shaped nanocatalysts must have the same surface area in order to make a fare comparison of facet-dependent photocatalytic reactions. This condition is not often met, and, therefore, one must report the photocatalytic activity normalized over the surface area, which can be termed specific activity per surface area.7
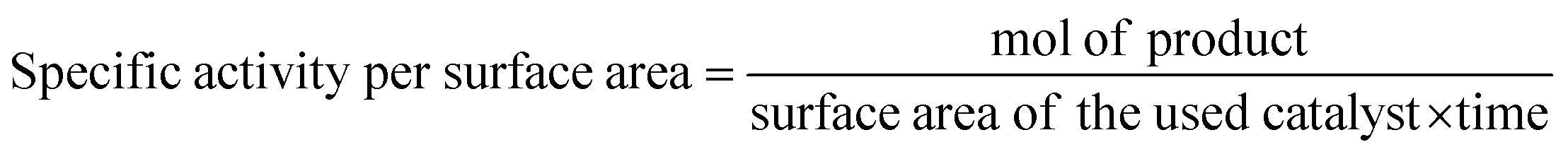
Such practice provides greater insight into the photocatalytic activity of the investigated well-defined nanocrystals. If the synthesized nanocrystals possess a highly uniform shape and size, the surface area can be simply calculated from a particle size distribution plot and also with respect to the geometry of that particular nanostructure. However, if the nanocrystals are not uniform, the specific surface area can be measured by the adsorption–desorption isotherm adopting the Brunauer–Emmett–Teller (BET) method. This method is a more precise way to determine the specific surface area of a nanocrystal as photocatalysts often have a certain degree of mesoporosity, which may increase the final surface area of the material, but this cannot be considered when using mere geometrical calculation. Finally, determination of the surface area of supported nanostructure is far from being trivial.
First, the reaction rate (k) needs to be expressed in (molecules per s) unit for each reaction.
Then, the incident photon flux (Φ) can be calculated as
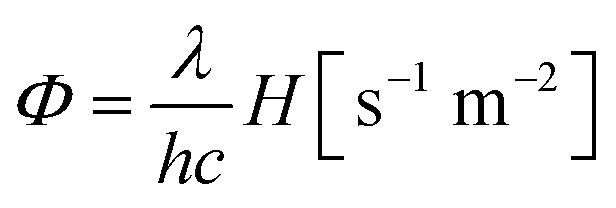 | (1) |
 | (2) |
Consistently, AQY must be reported with indication of the wavelength at which it was calculated and, obviously, comparison between AQY samples must be conducted at an identical wavelength.
Similarly, the solar AQY, i.e., under full solar spectrum irradiation, is calculated as
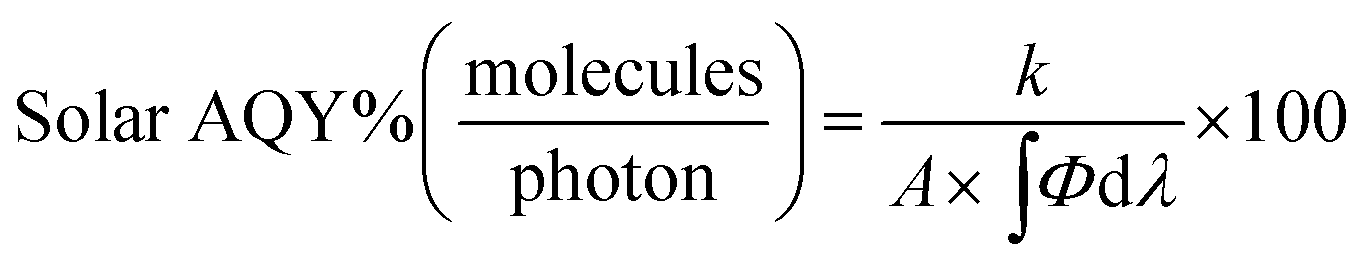
4.2 Photocatalytic organic reactions
Well-defined Cu2O nanocrystals have been employed in many organocatalytic applications under dark conditions because of the versatile catalytic properties of the Cu1+ oxidation state.82,83,126 In addition, Cu2O nanocrystals have also been applied as photocatalysts for different photocatalytic processes due to the suitable light absorption properties in the visible region of the solar spectrum.15,19,26 Therefore, a wide variety of photocatalytic reactions have been studied using Cu2O nanocrystals to understand the effect of different crystal facets. Photocatalytic dye degradation of industrial dye pollutants such as methyl orange (MO) and methylene blue (MB) are used as a common probe reaction to compare the photocatalytic properties of differently exposed crystals facets for Cu2O nanocrystals.19,41,46,127 The rate of photocatalytic dye degradation is directly related to two distinct properties of a semiconductor as follows: (1) strong adsorption of the reactant molecules on the catalytic site present in a crystal facet; (2) efficient electron–hole separation upon exposure of light and their fast migration to the active catalytic sites to generate radical species, which then carry out the oxidation of the dye. From a mechanistic point of view, photoexcited electrons reduce the surface adsorbed molecular oxygen (O2) to form the superoxide anion radical (˙O2−), which upon protonation generates ˙OOH, H2O2, and hydroxyl radicals (˙OH).107 These radicals may then oxidize the solvated MO molecules (via homogeneous catalysis). The photogenerated holes can either directly oxidize the surface-adsorbed dye molecules (via heterogeneous catalysis) or, more frequently, react with OH− and H2O to form the ˙OH radical, which further carries out the reaction. In addition, the complex interfacial charge transfer between the surface adsorbed dye molecules and the photoexcited charge carrier can also lead to efficient dye degradation.128–130 However, this kind of mechanism is generally overlooked and needs future investigation.Huang et al. showed that the rhombic dodecahedral Cu2O nanocrystal has a higher photocatalytic activity than octahedral nanocrystals, whereas the cubic morphology is completely inactive during the photocatalytic MO degradation.41,46,107 It is important to note that the amount of catalysts used in the photocatalytic dye degradation process was normalized against a fixed surface area, which is an important parameter allowing us to compare the catalytic activity of different well-defined nanostructures. The photocatalytic inactivity of Cu2O cubic morphology suggested that photogenerated electrons and holes experience very high energy barriers (Fig. 18a) to reach the active catalytic sites at the exposed {100} surface.41,46,107,127
 | ||
| Fig. 18 (a) Band diagram and charge migration energy barriers for three shaped Cu2O nanocrystals. (b) A schematic diagram showing the migration of photoexcited electrons and holes to the three different Cu2O crystal facets and their corresponding photocatalytic activity. Reproduced with permission from ref. 107, Copyright 2017 Royal Society of Chemistry. (c) EPR spectra of DMPO–OH present in photoirradiated Cu2O cubes, octahedra, and rhombic dodecahedra. Reproduced with permission from ref. 131, Copyright 2016 Wiley-VCH. | ||
To explain the higher photocatalytic activity of RD compared to octahedral nanocrystals, a systematic study has been carried out by adding electron and hole scavengers during the photodegradation of MO.107 The results indicated that in rhombic dodecahedral nanocrystals bounded with {110} faces, both photoexcited electrons and holes reach the surface active sites more efficiently and accelerate the formation of a larger number of active ˙OH radical species, justifying the higher photodegradation rate (Fig. 18b). This {110} facet showed the lowest degree of band bending, which resulted in improved electron and hole transport to the surface active sites. Octahedral nanocrystals enclosed with the {111} facet had moderate band bending, and the photoexcited electrons were able to reach the surface active sites to produce ˙OH radical species, as shown in Fig. 18b.107 The photoexcited holes were blocked by the {111} facets of octahedral nanocrystals, and they did not contribute to the production of ˙OH radicals. The presence of a lower amount of active ˙OH radical species in the reaction medium was the main reason for the lower photocatalytic activity of octahedral nanocrystals, compared to the rhombic dodecahedral nanocrystals. To further verify this fact, the spin trapping agent 5,5-dimethyl-1-pyrroline N-oxide (DMPO) was introduced into a solution containing three differently shaped Cu2O nanocrystals, and the electron paramagnetic resonance (EPR) spectra were recorded after the irradiation of light in the presence of oxygen.131 As shown in Fig. 18c, rhombic dodecahedral nanocrystals produced stronger EPR signals than octahedral nanocrystals, which supported the formation of a higher number of ˙OH radicals in the presence of rhombic dodecahedral nanocrystals, as discussed above.131 Most interestingly, photoexcitation of cubic nanocrystals produced no EPR signals, justifying their photocatalytic inactivity. This study strongly demonstrated that the photocatalytic activity of a semiconductor material can be tuned by controlling the exposed facet of a nanocrystal. Most surprisingly, a recent study demonstrated that a dense decoration of 4-ethynylaniline around the cubic Cu2O nanocrystals altered its band gap and modified the charge carrier transport mechanism through the {100} facet, which led to an enhanced activity.132 Therefore, the surface-bound ligands can have a profound impact on the photocatalytic activity of a semiconductor nanocrystal. The photocatalytic dye degradation efficiency of pristine Cu2O nanocrystals has further been improved by forming heterostructures.133–144 Among them, Cu2O nanocrystals decorated with Au, TiO2, and ZnO nanocrystals are the most widely studied solutions. Such hybrid architectures have also been employed to investigate their peroxidase-like activities toward photocatalytic decomposition of different bacteria under visible light illumination.134,145 All these strategies reduce the photogenerated electron and hole recombination rate and therefore enhance the photocatalytic MO degradation rate by forming a higher number of ˙OH radicals in the reaction medium. The application of highly oxidizing ˙OH radicals can be further extended to other industrially important reactions such as photocatalytic alcohol oxidations or alkene epoxidations.146,147 Thus, well-defined and cheap Cu2O nanocrystals can show product selectivity in such reactions that need to be studied in the future.
Furthermore, different hybrid nanocatalysts have been developed to provide enhanced activity and stability toward tandem photocatalytic catalytic reactions.148–154 Knecht et al. deposited Pd nanocrystals on micron sized cubic Cu2O nanocrystals via a galvanic displacement reaction where surface Cu1+ ions became oxidized to Cu2+ ions and Pd2+ ions were simultaneously reduced to Pd0 nanocrystals.148 Overnight, the galvanic displacement reaction led to the deformation of the surface {100} planes, followed by the formation of a minor amount of CuO and Cu species at the surface of Cu2O nanocrystals. The obtained Cu2O–Pd hybrid nanostructures were then employed in a light-driven tandem hydrodechlorination reaction (Fig. 19a). In this process, first, the Cu2O compartment produced H2via a photocatalytic water splitting reaction, while in the second step, in situ produced H2 was activated by the Pd compartment, where a hydrodechlorination reaction was able to proceed. Conversion of 3-chlorobiphenyl to nontoxic biphenyl in the presence of light is highly important for both organic synthesis and environmental pollution control. No product was obtained when individual Cu2O nanocubes and Pd nanoparticles were used, suggesting the synergistic effect due to the coupling of these two materials. Decoration of Pd nanocrystals on the cubic Cu2O nanocrystals reduced the electron–hole recombination as the photoexcited electrons were transferred from the conduction band of the Cu2O nanocrystals to the Fermi level of the Pd metal. It is interesting to note that the electron transfer becomes possible through the Cu2O {100} plane to Pd nanocrystals due to the deformation in the exposed crystal plane and change in the surface chemical composition, as stated above.
 | ||
| Fig. 19 (a) Scheme of the synthesis of the Cu2O–Pd hybrid photocatalyst for tandem hydrodehalogenation. Reproduced with permission from ref. 148, Copyright 2014, American Chemical Society. (b) Tandem photocatalytic click reaction over the semiconductor–CuO nanohybrid. Reproduced with permission from ref. 149, Copyright 2016 American Chemical Society. | ||
Most surprisingly, Scaiano et al. further extended the concept of heterostructure by decorating CuOx nanoclusters on TiO2 or Nb2O5 semiconductors whose band gaps fall within the 3–3.5 eV range.149 These hybrid nanostructures have been used as efficient catalysts for photocatalytic click reactions, especially Huisgen cycloaddition of azides and terminal alkynes to form triazoles, which has many pharmaceutical applications.82 The click reaction was mainly catalyzed by a Cu1+ oxidation state, not by Cu2+. Therefore, when UV-light impinged the TiO2 or Nb2O5 semiconductors, both of them (Fig. 19b) promoted a valence band electron to the conduction band, which was then trapped by surface CuOx nanoclusters, thus increasing the photoexcited electron–hole separation. After accumulating electrons, CuOx nanoclusters underwent in situ reduction, yielding a Cu1+ oxidation state, which then efficiently catalysed the click reaction. Once the reaction was completed, the Cu1+ oxidation state was regenerated back to a Cu2+ state in the presence of oxygen, demonstrating a reversible regeneration of the photocatalyst. Thus, it can be concluded that a suitable support plays an important role in achieving a high photocatalytic activity and stability of the material during the course of reaction.155
Recently cubic Cu2O nanocrystals were grown on layered molybdenum disulfide (MoS2) and graphene hybrids, which then were used as photocatalysts for a C–C bond formation reaction by oxidative coupling in the presence of oxygen (O2).151 In this case, a question arose: how photoexcited electrons and holes are able to pass via the {100} crystal plane of Cu2O nanocubes? It is due to the deformation of the {100} crystal plane during the course of the reaction as previously illustrated in Fig. 19a. The Cu2O nanocubes present in the hybrid nanocomposite have hollow structures and other distorted shapes in minor concentrations. In addition, a longer reaction time, in the presence of O2, can produce the leaching of Cu1+ ions from the surface and also considerable changes in the chemical composition and atomic orientation of the {100} crystal plane, which create this discrepancy. Therefore, researchers must address these crucial points before claiming that Cu2O nanocubes are a highly active photocatalyst for different reactions.156–161 A recent study showed that when Cu2O nanocrystals with truncated nanocube morphology were used for the photocatalytic Sonogashira reaction in the presence of the K2CO3 and CO2 atmosphere, a CuO layer was formed in the exposed crystal surface.161 Another fascinating hybrid photocatalyst where the Cu2O nanocube was decorated with SnO2 nanoparticles showed a unique post-illumination photocatalytic “memory” effect, which means it can still be catalytically active in the dark after the illumination was switched off for a certain period of time.154 This “memory effect” was possible due to the presence of reversible chemical states (Sn2+vs. Sn4+) in SnO2. In this section, we have discussed common Cu2O photocatalysed organic reactions like dye degradation, click reaction, alcohol oxidation and epoxidation reaction, which are important for environmental applications and industrial organic synthesis. Notably, the application of Cu2O in organic solvents remains of great interest due to the superior chemical and photostability of Cu2O in these media. In the following sections we will turn our attention to photocatalytic reactions that may enable a sustainable energy transition such as H2 evolution and CO2 reduction reaction.
4.3 Photocatalytic H2 evolution reaction
Hybrid Cu2O-based nanocrystals are widely investigated for photocatalytic hydrogen evolution.162–184 In most literature reports, H2 evolution is expressed by μmol gcatalyst−1 h−1. As we know catalytic reactions take place mainly on the catalyst surface; therefore, it would be more correct to report H2 evolution rates after normalization with surface area i.e. μmol m−2 h−1 in addition to μmol gcatalyst−1 h−1 unit. Such a complete picture will be more useful for comparing the H2 evolution rate with those of other catalysts. However, in reality, there is an almost negligible number of articles that report the H2 production for powder photocatalysts in terms of the surface area. In the future, it is recommended to apply such practice during the demonstration of H2 evolution experiments. Similarly, it is also applicable for photocatalytic CO2 reduction reactions, which will be discussed in the following Section 4.4.Lee et al. showed that Cu2O rhombic dodecahedral nanocrystals have higher photocatalytic activity than octahedral shapes whereas cubes are completely inactive for the water splitting reaction (Fig. 20a).162 However, RD nanocrystals lost their photocatalytic activity after 24 h of the photocatalytic reaction in pure water (Fig. 20b). A SEM image of these three different morphologies (Fig. 20c–e) proved the different structure distortion as a consequence of the photocatalytic activity. The RD morphology showed the highest degree of surface distortion and, after use, the highest density of thorns. These thorns are made of CuO as confirmed by XPS (Fig. 20f) and XRD. The presence of any sacrificial reagent is essential to consuming the generated photoexcited holes, with the nature and concentration of these or sacrificial electron donor being relevant for the final performance. In the absence of these sacrificial agents, RD Cu2O nanocrystals are not able to oxidize water to oxygen; instead, they oxidize Cu2O to CuO, producing these thorns. While the amount of CuO in the system increases, there is a change in the chemical composition and in the light absorbing properties with a progressive and dramatic decrease in the photocatalytic hydrogen evolution.162 This observation is very important to understand the photocorrosion properties of Cu2O, which is its weakest point. Increasing the photostability remains a big challenge for the development of low cost efficient photocatalysts for solar to fuel conversion. Different strategies for overcoming the stability issue in Cu2O systems have been developed, and will be discussed as follows.
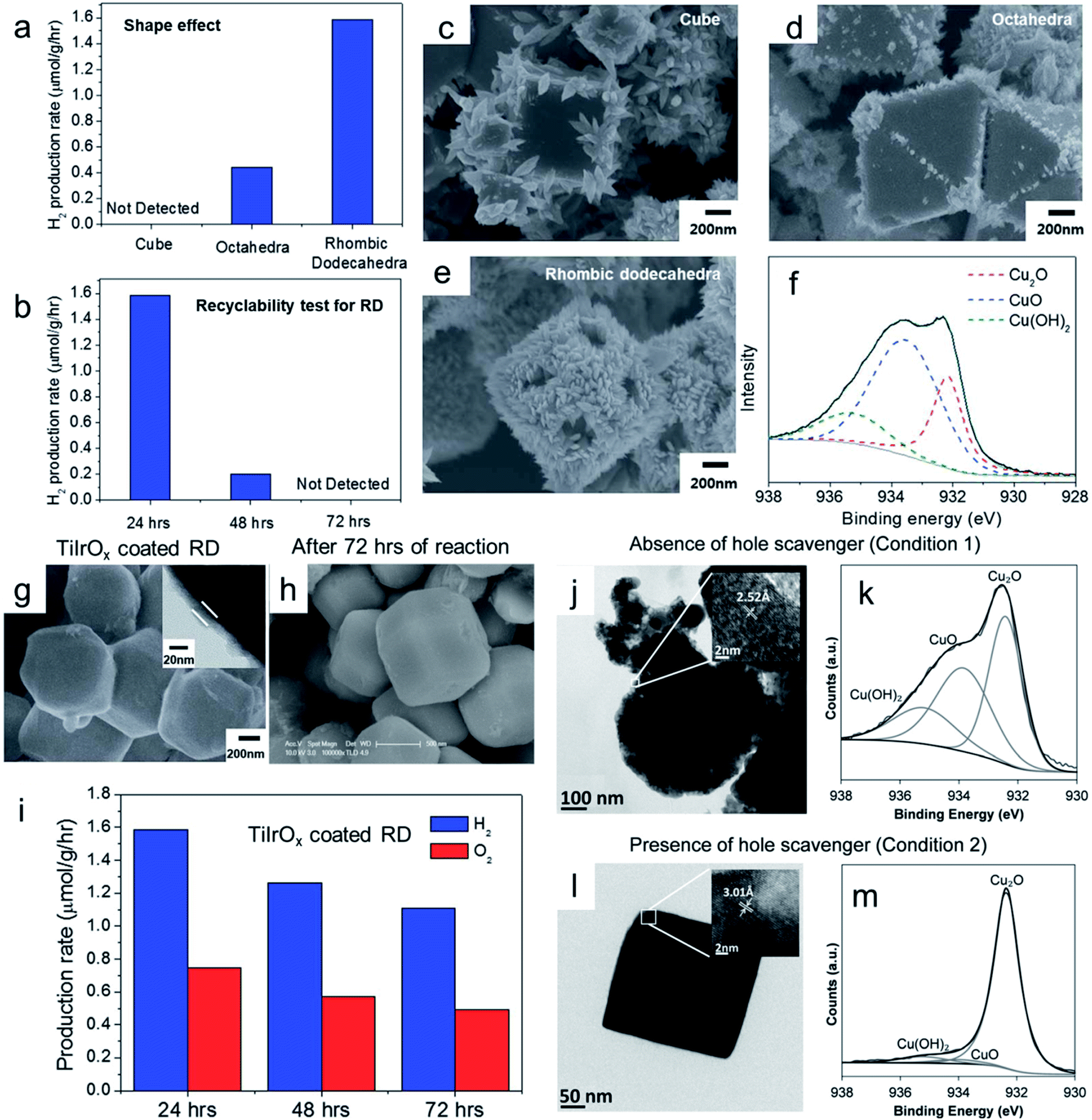 | ||
| Fig. 20 (a) Comparison of H2 production rate from overall water splitting using three different Cu2O morphologies. (b) Recyclable test for Cu2O RD. (c–e) After 9 h of light irradiation in DI–water without bubbling Ar, SEM images of the three different shapes. (f) Corresponding XPS spectrum of RD. SEM image of TiIrOx coated Cu2O RD (g) before, (h) after 72 h of reaction, and (i) their recyclability test. Reproduced with permission from ref. 162, Copyright 2015 Royal Society of Chemistry. (j and l) TEM images and (k and m) XPS of Cu2O nanocubes under two different conditions. Reproduced with permission from ref. 163, Copyright 2018 Wiley-VCH. | ||
The first strategy consists of the uniform ultrathin coating of the Cu2O nanocrystals with a conducting and transparent layer, which allows both electron and hole transfer to its surface active sites, thus minimizing the direct contact between the Cu2O surface and water while keeping the same photocatalytic activity.162 Generally, this strategy is more common for Cu2O based photoelectrode constructions where atomic layer deposition helps to achieve this conformal coating.17,185 One of such attempts is shown in Fig. 20g, where Cu2O RD nanocrystals were uniformly coated with a TiIrOx surface passivation layer (x < 2) with a thickness of 20 nm (inset Fig. 20g). Such a layer may consist of a TiOx layer and IrOx ultrasmall nanocrystals.162 The overall water splitting was achieved with a H2:O2 ratio of 2.13 from the TiIrOx layer coated Cu2O RD (Fig. 20i) nanocrystals. The H2 production rate was comparable with that of pure rhombic dodecahedral Cu2O, indicating efficient transfer of electrons to its surface active sites. In the same way, IrOx facilitated hole separation, thus preventing self-photocorrosion of Cu2O, while enabling water oxidation to produce oxygen. Such techniques also stabilized the shape and morphology of the Cu2O nanocrystals (Fig. 20h) for a longer time during the photocatalytic overall water splitting reaction with a moderate decrease in the catalytic efficiency (Fig. 20i). Further investigation will be needed to find more efficient materials which will be easy to deposit and can have a profound impact on the photocatalytic efficiency of the Cu2O nanocrystals with enhanced photostability.
The second strategy includes the introduction of suitable hole scavengers in the reaction medium. The hole scavengers are efficiently oxidized by the photogenerated holes in Cu2O nanocrystals, which in turn prevents the self-photooxidation process. An interesting study showed that when no hole scavengers were added and Cu2O nanocubes were exposed to pure water in the presence of air and light for a certain period of time, an overall destruction of morphology occurred, as shown in Fig. 20j.163 The XPS analysis confirmed the Cu2O self-photooxidation by holes with the formation of CuO and Cu(OH)2 species (Fig. 20k). Most surprisingly, when ethanol was introduced as a hole scavenger into the system, under the same photocatalytic conditions, the morphology and chemical composition remained almost unchanged (Fig. 20l and m). Due to the favorable redox potential of Na2SO3, it acts as a more effective hole scavenger than ethanol, resulting in a high photocatalytic hydrogen evolution rate in the presence of Cu2O nanocrystals with considerable photostability.163
The third strategy is the successful establishment of a Z-scheme mechanism that efficiently neutralizes the photogenerated hole accumulation in Cu2O, thus preventing its photooxidation.168–173 The Liu group developed a hybrid photocatalyst where 2 nm Cu2O nanocrystals were uniformly photodeposited on oxygen vacancy containing 101-faceted octahedral TiO2 nanocrystals (referred to as Cu2O/T1-VO; Fig. 21a and b).170 Under AM 1.5G irradiation, this hybrid catalyst showed 32.6 mmol g−1 h−1 hydrogen production rate from 10 vol% aqueous methanol solution, which is still the highest reported rate for Cu2O/TiO2 hybrid systems to date (Fig. 21c). It also ensured high photostability over the long-term recyclable experiments (Fig. 21d). No oxidized CuO species were detected in the recycled photocatalyst, which confirmed the efficient neutralization of holes in the Cu2O counterpart via a successful Z-scheme mechanism, as shown in Fig. 21e. An optimum number of surface defects and homogenous distribution of 2 nm Cu2O nanoclusters, along with 101-faceted TiO2 octahedra, acted synergistically, which resulted in high photoactivity and enhanced photostability of Cu2O nanoclusters. Based on these findings, further development will be needed to obtain a highly efficient, low cost and stable Cu2O-based photocatalyst for solar H2 production.
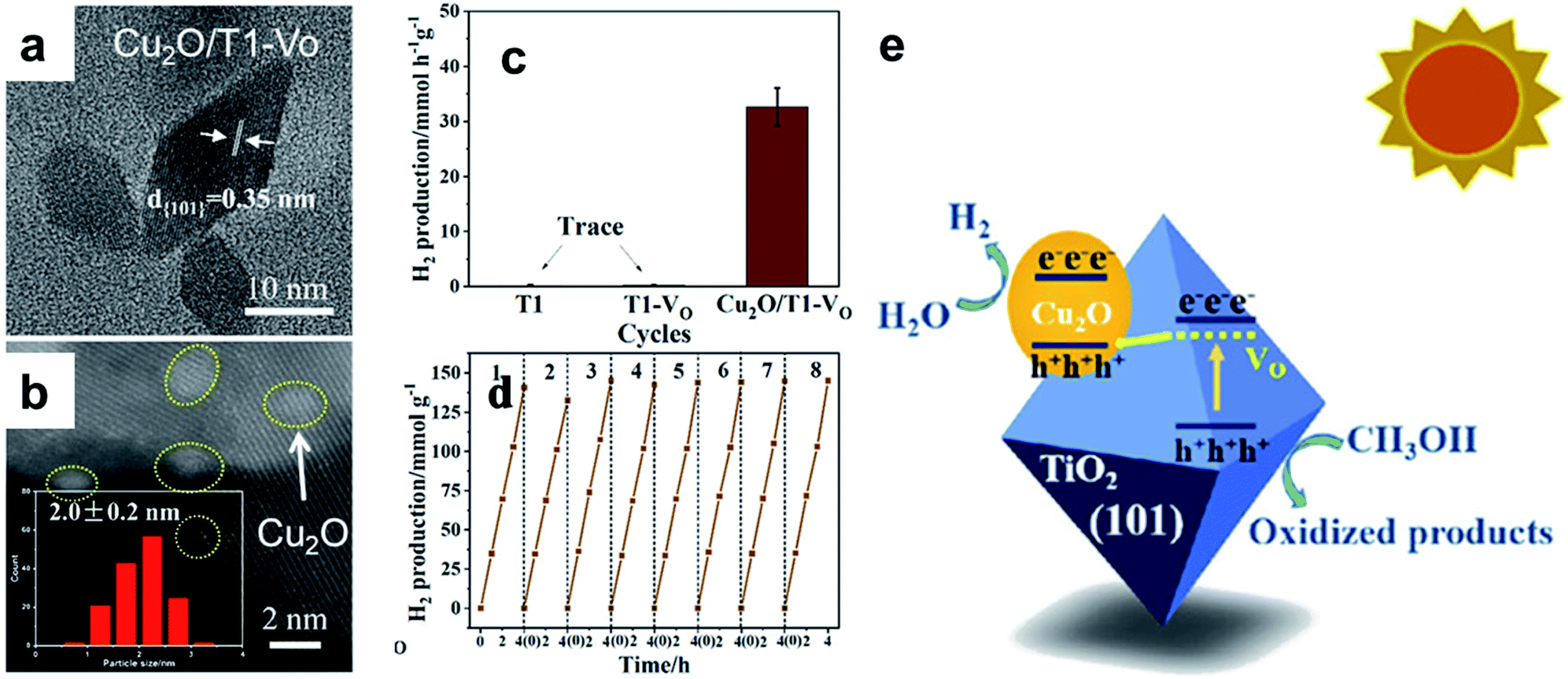 | ||
| Fig. 21 (a) HRTEM and (b) HAADF-STEM image of Cu2O decorated oxygen vacancy (VO) containing 101-faceted TiO2 (referred to as Cu2O/T1-VO). (c) Comparison of H2 production rates of 101-faceted TiO2 (T1), 101-faceted TiO2 having oxygen vacancies (T1-VO), and Cu2O/T1-VO. (d) Recyclability test for Cu2O/T1-VO. (e) Graphical presentation of the interfacial charge transfer mechanism in Cu2O/T1-VO. Reproduced with permission from ref. 170, Copyright 2019 American Chemical Society. | ||
The fourth strategy is the modification of the Cu2O nanocrystal surface by carbon-based materials.165,167,174,179,180 Carbon quantum dots (CQDs),186 nanodiamonds (NDs),165 reduced graphene oxide (rGO),180 and nitrogen-doped carbons174 are widely used to enhance the photocatalytic activity and photostability of Cu2O nanocrystals. Yang et al. showed that the decoration of 50 nm spherical Cu2O nanocrystals with 3 nm NDs enabled a H2 production rate of 1597 μmol g−1 h−1 under simulated solar light irradiation (AM 1.5G, 100 mW cm−2, 1 Sun) with a solar-to-hydrogen conversion efficiency of 0.85%.165 This composite also showed considerable photocatalytic stability. Due to the highly conductive nature of ND and its strong light absorbing properties from UV to NIR regions, the photoexcited electrons from the ND surface were efficiently injected into the Cu2O and simultaneously, the photogenerated holes from Cu2O were injected to ND. The excess holes on the ND surface carried out the photooxidation of ethanol, whereas the excess electrons in Cu2O carried out the photoreduction of H+ to H2. Such effective separation and consumption of photoexcited electrons and holes were the main reasons for the enhanced photostability of ND decorated Cu2O hybrid systems. In another study, glucose was used as the carbon precursor to form a protective 20 nm thick carbon layer coated on a Cu2O nanowire, which resulted in enhanced photostability and water splitting performance.179 The photoexcited electrons were easily transported to the reaction medium via this carbon layer, whereas, because of its hydrophobic nature, it protected Cu2O from H2O. However, organic molecules such as ethanol or methanol make their passage to the Cu2O surface through this carbon layer where they become easily oxidized by photogenerated holes, resulting in enhanced photostability. Such an approach is highly cost-effective and easily scalable; the only thing that needs to be taken into account is how to achieve the optimum thickness of the carbon layer on the Cu2O nanocrystals.
A fifth strategy foresees the formation of heterostructures with plasmonic nanocrystals including sharp vertices and edges.175–178 In this regard, the Han group used hexoctahedral (HOH) Au nanocrystals enclosed by 48 triangular high-index {321} facets as plasmonic antenna and selective overgrowth of Cu2O (reactive site) was achieved on the vertices of the HOH Au NCs with the help of facet-selective adsorption behavior of PVP surfactant (Fig. 22a).176 Such hetero-nanocrystals showed a higher photocatalytic H2 production rate than core–shell nanostructures (Fig. 22b). Such an enhanced rate for Auvertex–Cu2O HNCs could be attributed to the LSPR-induced hot electron transfer from HOH Au nanocrystals to Cu2O. FDTD simulations confirmed that strong electromagnetic field enhancements were mainly localized at the vertices of HNCs where Cu2O nanoclusters were present, and efficient hot electron transfer took place, facilitating the proton reduction process. Due to the formation of a suitable Schottky junction between the Au and Cu2O, the photogenerated holes from the Cu2O nanoclusters efficiently migrated to Au nanocrystals where methanol scavenged the holes due to the availability of the exposed Au surfaces in Auvertex–Cu2O HNCs.176 These kinds of HNCs also contribute to increasing the photostability of Cu2O. Different exotic structures such as hot-dog like Au nanorod@Cu2O@TiO2 (Fig. 22c) nanocrystals and other hybrid systems showed enhanced photocatalytic activity with increased photostability.177,178,187–189
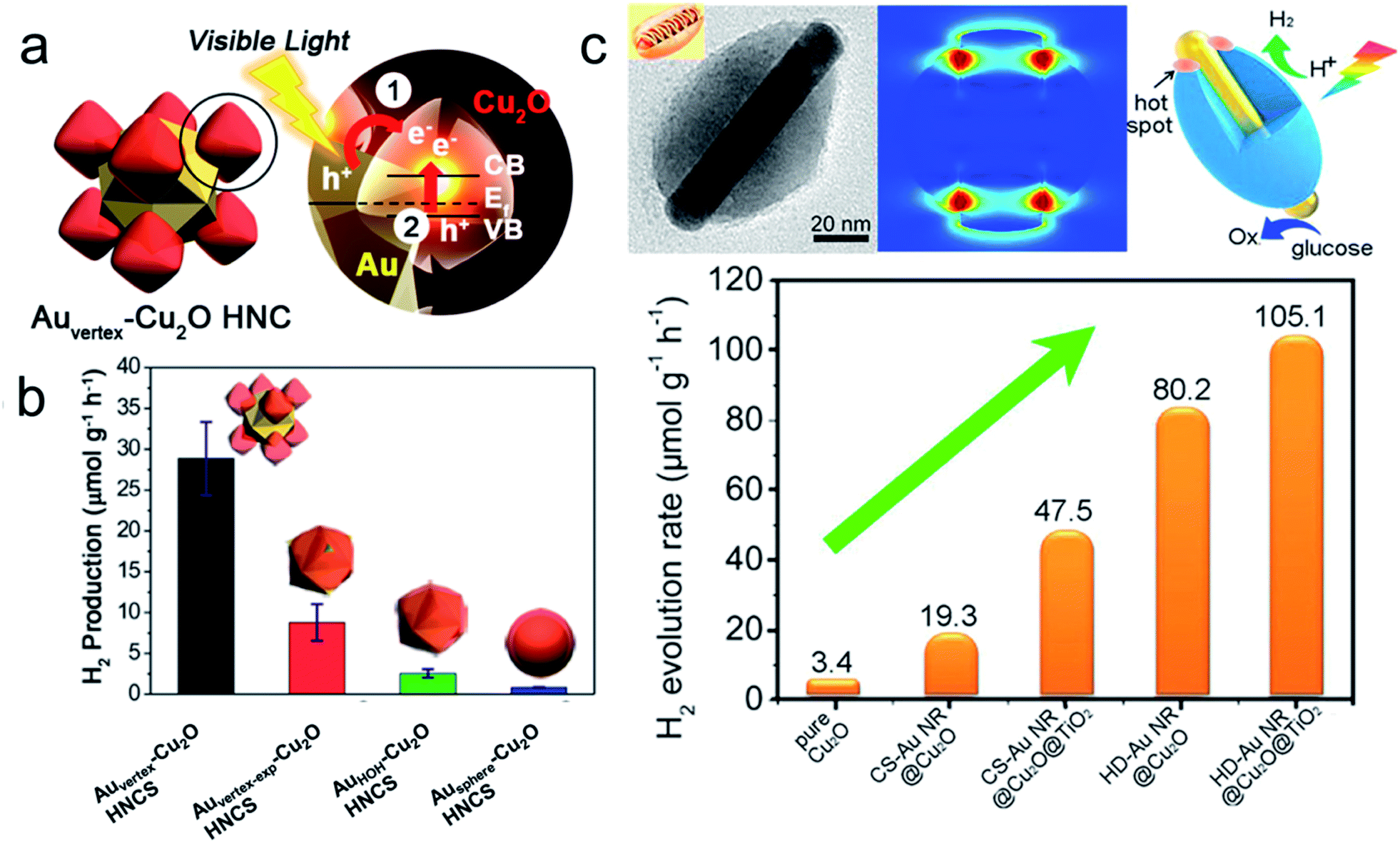 | ||
| Fig. 22 (a) Schematic model for Auvertex–Cu2O heteronanocrystals (HNC) and their SPR-induced hot electron transfer mechanism. (b) Comparison of photocatalytic H2 evolution over different HNCs. Reproduced with permission from ref. 176, Copyright 2016 American Chemical Society. (c) An overall schematic diagram of a hot-dog like Au nanorod@Cu2O@TiO2 heteronanostructure with comparison to hydrogen production activity. Reproduced with permission from ref. 177, Copyright 2017 Elsevier. | ||
Finally, the sixth strategy is based on the careful selection of reagents used during the photocatalytic process. As we have discussed earlier in Section 2.3, the presence of HCl, NH3, or other strong bases such as S2− or S2O32− anions, and dissolved O2, can seriously cause distortion or etching of the Cu2O nanocrystals. Therefore, they should be avoided along with the use of short reaction times and low temperatures in order to increase the photostability of Cu2O nanocrystals. Dang et al. recently showed that in the presence of Na2SO3 as a hole scavenger, Cu2O@Cu7S4 core@shell nanocubes showed efficient photocatalytic hydrogen production with enhanced photostability.178 To summarize, studies of different hybrid photocatalysts and their electron transfer mechanisms provide a fundamental understanding of how to enhance the photocatalytic efficiency and photostability of Cu2O by suppressing its self-photooxidation under light illumination.
4.4 Photocatalytic CO2 reduction reaction
The photocatalytic reduction of CO2 with H2O to solar fuels is considered an artificial photosynthesis reaction. It attracted huge attention due to the recent global warming problems, and motivated the attempt to reduce our dependence on fossil fuels.190–193 Using water as the source of H2 instead of pure H2 gas during this photoreduction process is the most desirable and challenging task. If we can develop such an energy-efficient system, it may be the most sustainable and environmentally green process to complete the carbon cycle.194–198 Major difficulties associated with this photoreduction process are the complex reaction mechanism, which involves different modes of adsorption of CO2 molecules on the catalyst surface and the multi electron-multi protonation coupling–decoupling steps with high activation energy barriers for different intermediates.190 These reaction steps make this reaction photocatalytically less efficient and also lead to generation of a wide variety of products such as CO, CH3OH, HCHO, HCOOH, and CH4 (generally termed C1 products). Complex in situ C–C coupling and further modification of C1 products generate compounds with two carbons (C2 products) such as C2H6, C2H4, CH3CH2OH, CH3CHO, and CH3COOH.190–193 Due to the high activation energy barrier, C2 products are more difficult to form, as compared to C1 products, during the photocatalytic reduction reaction. However, C2 products are of higher importance in the fields of chemical, fuel, and polymer industries. Further coupling of C1 and C2 products can lead to the formation of C3 products, which is rarely formed via a photocatalysed reaction over Cu-based catalysts, but it can be achieved via an electroreduction reaction.199,200 Therefore, a better understanding of the reaction mechanism is necessary in order to improve the product selectivity along with achieving high conversion rates. In this regard, Mul et al. showed that photocatalysts prepared with high molecular weight carbon containing precursors or solvents were contaminated with small quantities of carbon residues even after high temperature calcination.12 These carbon impurities have a profound impact on the photocatalytic water activation and CO2 reduction reaction. Fig. 23a shows in situ DRIFT spectra of the cuprous oxide/TiO2 catalyst after 80 min of photoirradiation under different gaseous atmospheres. When isotopically labeled 13CO2 and H2O were used, the ratio 12CO:13CO was surprisingly 6:1, which indicated that the carbon residues on the catalyst surface were involved in the reactions with 13CO and H2O molecules in the presence of light, and produced 12CO in excess. This is not coming from the artificial photosynthesis reaction, as shown in the following equation:| 13CO2 + 12C → 13CO + 12CO | (3) |
| H2O + 12C → 12CO + H2 | (4) |
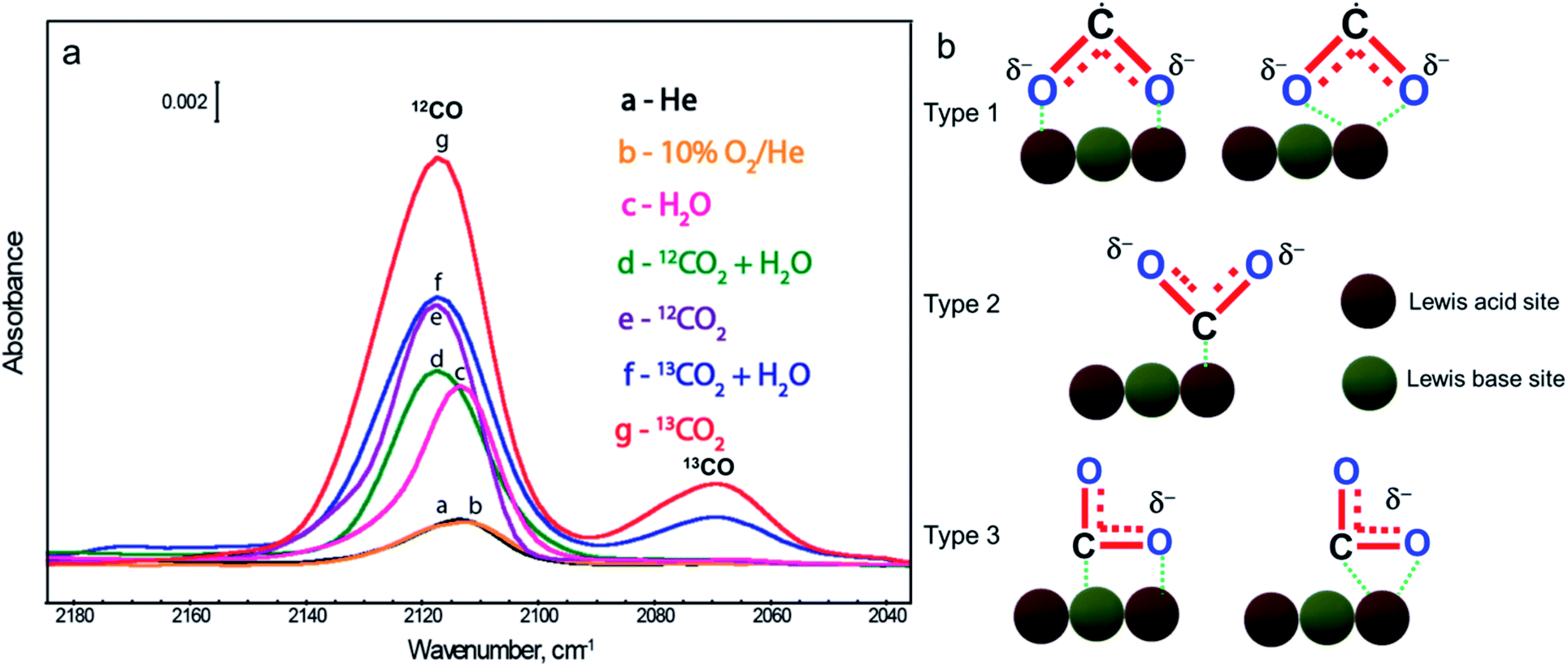 | ||
| Fig. 23 (a) Role of carbon residues in the catalyst: DRIFT spectra showing the distribution of products under different photoreduction conditions. Reproduced with permission from ref. 12, Copyright 2010 American Chemical Society. (b) Different binding mode of CO2˙− radicals on the catalytic site. Reproduced with permission from ref. 190, Copyright 2019 American Chemical Society. | ||
Many recent studies report a high activity with product selectivity for different photocatalysts. Nevertheless, in the future, authors should pay extra attention, before publishing their data, to ruling out any overestimation of the reaction rate coming from carbon contamination present in the photocatalyst surface. This overestimation can be caught by simply carrying out a blank photocatalytic test in the absence of CO2 but in the presence of H2O.12 In addition, photoreduction should also need to be carried out using isotope labeled 13CO2 for further confirmation of the product selectivity and reaction rate. And lastly, it is also necessary to follow the previously described surfactant removal steps (Section 2.3) during the synthesis of the photocatalyst in order to get rid of any unwanted carbon contamination.
The photoreduction of CO2 with H2O to these products is a thermodynamically uphill conversion with a positive Gibbs free energy, ΔG, which makes it difficult to proceed at room temperature and without any supply of high external energies. Therefore, it is of great importance to develop efficient semiconductor-based photocatalysts to achieve a desirable photocatalytic conversion rate with high product selectivity. Different ways have been considered to modify these semiconductors by the fine tuning of the band gap, morphology, and size, and with addition of suitable co-catalysts.190–198 Under suitable light illumination, photoexcited electrons present on the reduction site in a hybrid catalyst typically reduce CO2 to solar fuels, whereas the photoexcited holes present on oxidation sites react with water to produce O2. The determination of a precise mass balance between reagents and products is a critical tool to understand the mechanism of reaction and the intermediates forming over Cu2O photocatalysts. The formation of O2, during CO2 photoreduction in the presence of H2O, represents the first of many redox steps, which ultimately result in H2, CO, CH4, CH3OH, or multi-carbon products. The quantification of O2 therefore may be useful to understand the reaction mechanism, but should be supported by a precise quantification of reaction intermediates/products and by isotopic labelling experiments, which are crucial to unveil the origin of atomic building blocks forming the final molecular products. For an optimum performance, the CB position of the hybrid catalyst must be much higher than the proton-assisted multielectron reduction potentials of CO2 to different products, whereas the VB edge must be much more positive than the water oxidation potential (Fig. 2).190–193 Apart from optimum control of the semiconductor band gap and energy alignment, a right choice of the co-catalyst is also necessary for enhancing the adsorption and activation of CO2 molecules on the active sites, which in turn accelerates the photoreduction of CO2 with high product selectivity.190 As shown in Fig. 2, the photoreduction of CO2 to the CO2˙− radical has a large reduction potential (−1.49 V versus NHE, pH = 0) and also its geometry changes from linear to a bent shape; an increase in steric hindrance will make this preliminary one electron reduction step highly unfavorable. The formation of a destabilized bent CO2˙− radical is the most important step for initiating multistep photoreduction of CO2 and it can be stabilized on the surface active sites via three different coordination modes, as shown in Fig. 23b.190 Different chemical compositions, morphologies, electronic band structures, and the exposed crystal planes have a significant impact on these different kinds of coordination modes, which in turn have a direct effect on the CO2 photoreduction rate and product selectivity. Extensive theoretical work by the Carter group shows that Cu2O is a promising photocatalyst for photoconversion of CO2 to methanol.201–203 Leeuw et al. performed in-depth DFT calculations for different binding modes of CO2 molecules on three low-index Cu2O crystal planes and their corresponding adsorption energy.204 As shown in Fig. 24, the Cu2O (110):Cu–O crystal plane shows the strongest chemisorption of the CO2 molecule, releasing ∼170 kJ mol−1 energy, which supports the formation of the bent CO2˙− radical most efficiently. On the other hand, the Cu2O (111):O surface shows the weakest adsorption, and, therefore, the CO2 molecule remains linear.204 The activation of the CO2 molecule is directly proportional to the adsorption energy. The more negative is the adsorption energy of a CO2 molecule on a particular facet of the Cu2O nanocrystal, the more favorite will be the formation of the bent CO2˙− radical anion. These different adsorption energies and their conformation can directly influence the reaction rate and product selectivity.
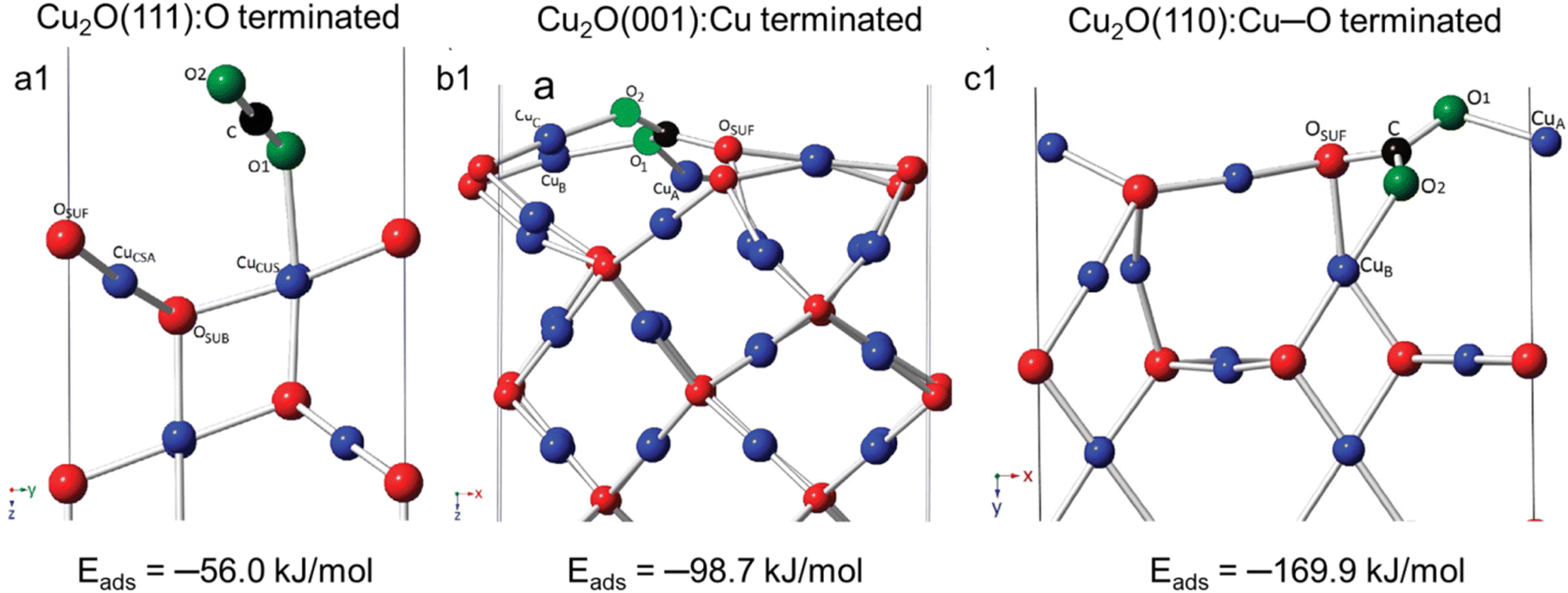 | ||
| Fig. 24 Crystal structures of three low-index surface planes of Cu2O in their most stable configuration along with different binding modes of CO2 molecules and corresponding adsorption energy: (a1) {111}, (b1) {001}, and (c1) {110}. Black and green spheres represent carbon and oxygen atoms in a CO2 molecule. Reproduced with permission from ref. 204, Copyright 2016 American Institute of Physics. | ||
Cubic, octahedral, and rhombic dodecahedral Cu2O crystals decorated with reduced graphene oxide (rGO) nanosheets were recently synthesized, and their photoreduction of CO2 in aqueous solution was compared.205 It is interesting to note that all of these hybrid photocatalysts exclusively produced CH3OH, and other possible products such as HCHO and HCOOH were not detected in the system. The methanol production rate followed the order: cube-Cu2O/rGO < octahedral-Cu2O/rGO < rhombic dodecahedral-Cu2O/rGO. Considering Fig. 18 and 24, the highest photo-conversion of CO2 to MeOH for RD could be explained by: (1) an improved accumulation of photoexcited electrons on the {110} surface and (2) facilitated formation of the bent CO2˙− radical anion. Wrapping with rGO provided higher stability for the Cu2O nanocrystals during the course of the reaction and also better electron–hole separation. Recently, Rajh et al. used in-depth characterization techniques such as correlated scanning fluorescence X-ray microscopy, EPR, and environmental TEM at atmospheric pressure to prove that the (110) facet of Cu2O was more active in the photoreduction of CO2 with H2O to methanol, while the (100) facet was inert during this process.206 Methanol was produced exclusively as the main product, which could be correlated with the fast desorption of methanol from the (110) facet. No other oxidized products such as HCHO and HCOOH were found in the system. This further emphasizes that the facet-dependent photoreduction of CO2 requires precise control of the shape and size of the nanocrystal to achieve a high photoreduction rate and product selectivity, even more than in the case of H2 production.
In addition to pristine Cu2O nanocrystals, different hybrid systems have also been developed in order to enhance their better charge separation and product selectivity during the photoreduction of CO2.186,207–218 A novel carbon quantum dot (CQD) decorated Cu2O heterostructure was synthesized via a one-pot procedure, where ∼5 nm CQDs were homogeneously decorated on the surface of an ∼2 μm spherical crystalline Cu2O particle.186 This hybrid nanostructure showed strong absorption in the complete solar spectrum range with a band gap 1.96 eV, much lower than that of pure bulk Cu2O (2.2 eV). The photoexcited electrons in the Cu2O microsphere selectively reduced CO2 into CH3OH, whereas the holes were transferred to CQDs and oxidized H2O to O2. Thus, the CQD co-catalyst enhanced both the photocatalytic performance and the stability of Cu2O microspheres. Recently, 4.4 nm Cu2O nanocrystals supported on defective graphene were used as an efficient photocatalyst for gas-phase methanation of CO2 in the presence of H2 at high temperature, also known as the Sabatier reaction.207 This hybrid photocatalyst showed exclusive CH4 production with an AQY at 250 °C of 7.84% in the 250–360 nm wavelength range. In the third cycle of the reaction, the photocatalytic activity decreased significantly because of the complete conversion of Cu2O–graphene to Cu–graphene. This conversion was not probably due to the photocatalytic process itself but it was related to the high temperature reductive environment. Doping has also been used as an important tool to tune the band gap of a semiconductor and to enhance the photocatalytic performance and product selectivity. For instance, Cl-doping of Cu2O nanorods has been used to shift the band structure of Cu2O to a more positive valence-band position, which facilitates water oxidation.208 The Cl-doped Cu2O nanorods showed excellent photoreduction of CO2 in the presence of H2O under visible-light irradiation with selective formation of CO.
Another possible way to increase the photoreduction performance of cuprous oxide is the suitable formation of Z scheme-based hybrids by coupling the p-type Cu2O semiconductor with n-type wide bandgap semiconductors such as TiO2, ZnO and W18O49.210,211,219–222 Precise control of the morphology and size can efficiently increase the photoreduction rate and product selectivity. A Z-scheme heterojunction can limit the issue of recombination of the charge carriers, improving the stability of the photocatalyst.
Grela et al. used octahedral Cu2O microcrystals and decorated their 8 {111} crystal planes with TiO2 nanocrystals uniformly.210 When this octahedral Cu2O–TiO2 hybrid photocatalyst was irradiated with UV-visible light (λ ≥ 305 nm) in the presence of water vapor and CO2, the photoreduction reaction proceeded with selective formation of CO. The role of carbon contamination in the catalyst surface was not properly investigated in this study. In addition to this, the octahedral Cu2O–TiO2 hybrid system also showed enhanced photo-stability in comparison with pure Cu2O, which was due to the efficient transfer of photoexcited CB electrons to the photoexcited VB holes of Cu2O. Thus, TiO2 also protected Cu2O from photocorrosion during the course of reaction. These results provided direct evidence of an efficient Z-scheme, which was further confirmed by an in situ analysis of the photocatalyst using XPS and EPR techniques. In addition, Song et al. also developed a hybrid photocatalyst where 40 nm ZnO nanospheres were decorated with multiple 18 nm cubic Cu2O nanocrystals, as shown in Fig. 25c.211 These ZnO–Cu2O hybrid photocatalysts showed a selective CH4 formation with an estimated quantum efficiency (QE) of 1.5% using the 200 to 540 nm wavelength region. A synergistic charge transfer between the ZnO and Cu2O counterparts led to an enhanced CO2 photoreduction rate (Fig. 25g). To understand the enhanced photocatalytic activity, an identical reaction performed under visible light (UV cutoff filter λ > 420 nm) was performed, and a negligible amount of CH4 was obtained. When the cutoff filter was removed, the formation rate for CH4 was similar to that observed for the original reaction, which proved that the operational Z-scheme mechanism under UV-visible light illumination (as shown in Fig. 25h) had caused the hybrid photocatalyst to outperform its individual components. More diverse combinations of hybrid catalysts with different shapes and sizes need to be developed so that they can operate via the Z-scheme mechanism, thus improving the photostability of the Cu2O counterpart with enhanced photoreduction rate and product selectivity. Chen et al. used a simple self-assembly strategy to decorate Ti3C2 QDs onto Cu2O nanowires to achieve selective formation of methanol.212 The photoexcited CB electrons of Cu2O were readily transferred to the attached Ti3C2 QDs, as the latter is a highly conductive material having a EF less negative than the CB of Cu2O, which enhances the charge separation. Similarly, as the EF of Ti3C2 QD is more negative than the redox potential of CO2 to methanol, the accumulated electrons efficiently performed the photoreduction reaction.
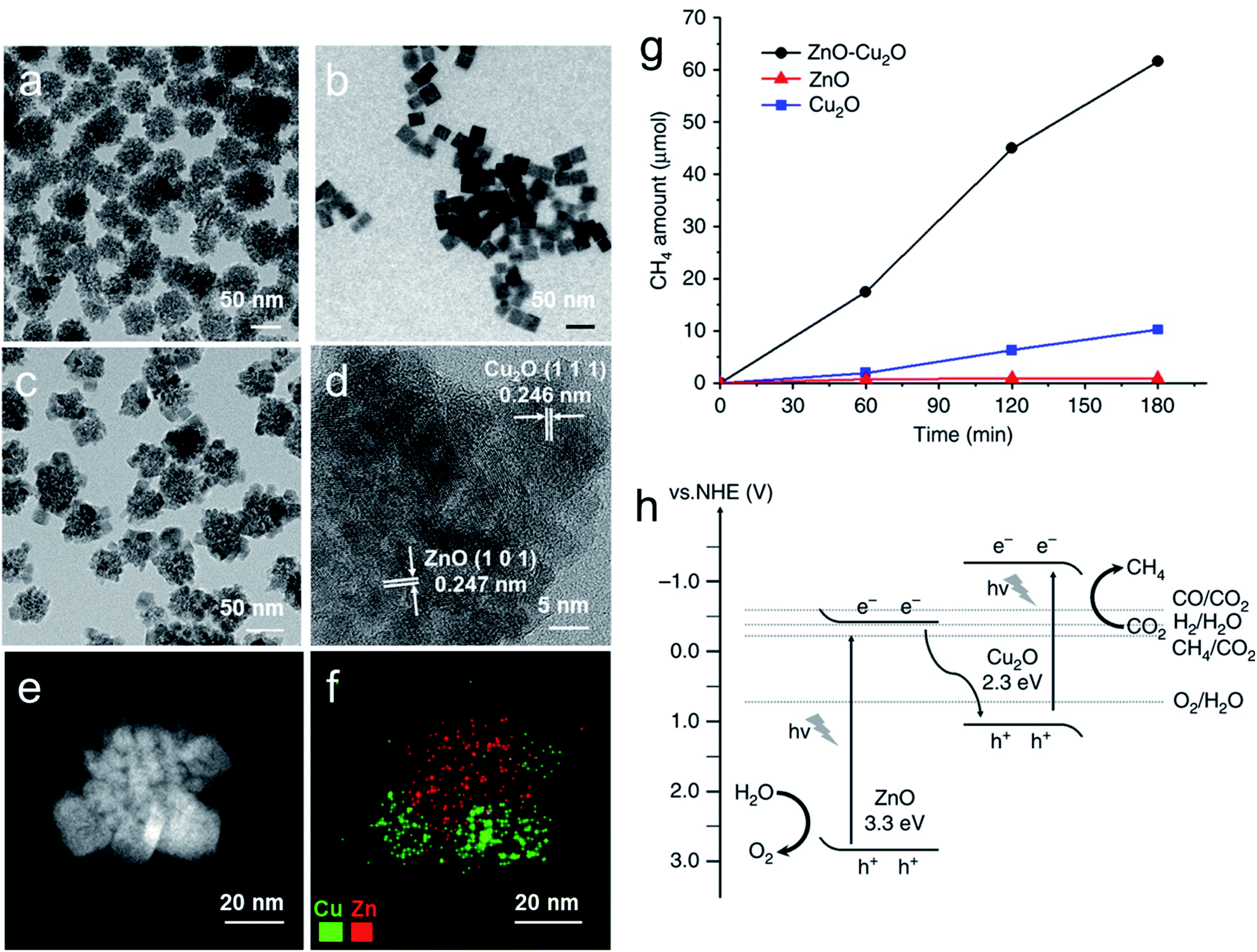 | ||
| Fig. 25 TEM images of (a) ZnO spheres, (b) Cu2O nanocubes, (c) ZnO–Cu2O hybrid nanoparticles, (d) HRTEM, (e) STEM image, and (f) elemental map of an individual ZnO–Cu2O nanoparticle. (g) Comparison of photoreduction of CO2 using the three different catalysts. (h) Band alignment and proposed electron transfer mechanism of the ZnO–Cu2O hybrid catalyst. Reproduced with permission from ref. 211, Copyright 2017 Springer Nature. | ||
Apart from the Z-scheme mechanism, the construction of hybrid ‘antenna-reactor’ photocatalysts also attracted huge attraction due to their enhanced photocatalytic performance compared to their individual components.125,213,214,223 In this system, generally plasmonic materials (such as Au, Ag, Cu, TiN, and Al) act as an ‘antenna’, which mainly absorbs the light and generates hot electrons (and eventually heat). These hot electrons are then transferred to the ‘reactor’ site (usually formed by transition metals such as Pt, Pd, Rh, Ru), which is mostly responsible for performing the photocatalytic reaction.
Halas et al. developed a Al–Cu2O core–shell hybrid photocatalyst where Al was used as a low cost and earth abundant plasmonic core, while the polycrystalline Cu2O shell acted as a catalyst for the reverse water-gas shift (rWGS) reaction (CO2 + H2 ↔ CO + H2O).213 Interestingly, at higher illumination intensities, the Al–Cu2O core–shell hybrid photocatalyst showed a much higher CO formation rate than the individual Cu2O and the Al counterparts. Most surprisingly, under a light driven rWGS reaction, a highly selective formation of CO took place, whereas a purely thermal-driven reaction lost the product selectivity as both CH4 and CO were found in the product mixture. Zhang et al. used a three hetero-compartment hybrid photocatalytic system where the Cu2O hemisphere selectively covered the CdSe part of a plasmonic Au/CdSe nanodumbbell, as shown in Fig. 26a–c.214 It is interesting to note that the use of PEG, methyl ether, and hydrazine as reducing agents led to the formation of these exotic kinds of hybrid nanostructures. Fig. 26d demonstrates that when the photoreduction of CO2 was carried out in a mixed organic solvent (CH3CN:TEOA = 9:1) without adding any water under visible light illumination, the Au/CdSe nanodumbbell generated only H2 without any detectable CO or CH4. The protons for hydrogen production might come from a trace of water in CH3CN. However, when Cu2O fully covered the CdSe hemisphere, a 100% CO selectivity was found without any H2. The addition of different amounts of H2O to the catalytic system systematically increased the production of both CO and CH4 (Fig. 26d). The addition of H2O to the mixture of the CH3CN and TEOA solvents catalyzed the formation of CH4 (a 8e−/8H+ pathway) and CO (a 2e−/2H+ pathway), by providing more protons and electrons into the reaction mixture. The exact electron transfer mechanism for this enhanced photocatalytic preformation is still under investigation.
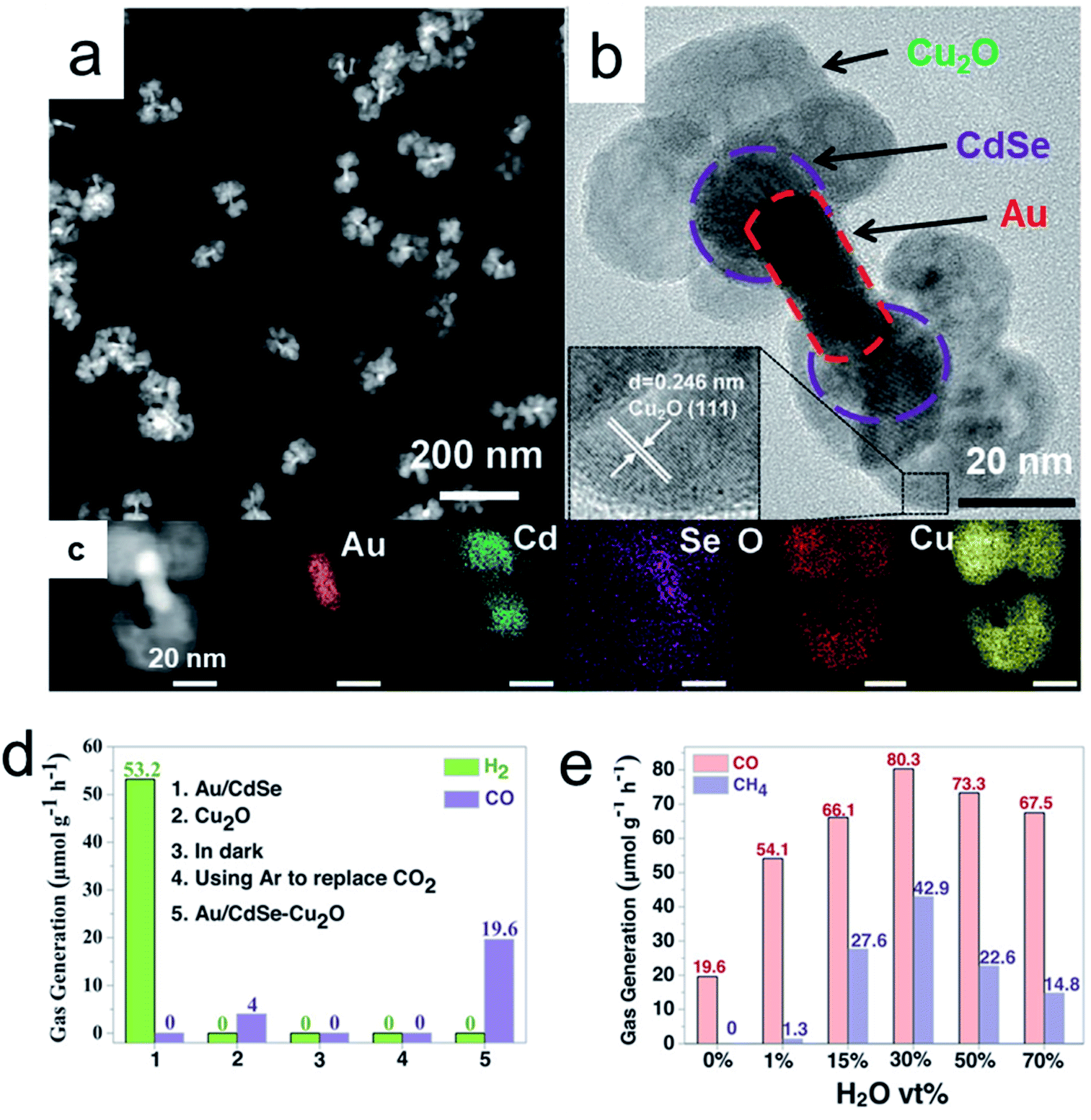 | ||
| Fig. 26 Au/CdSe–Cu2O hybrid nanostructures: (a) STEM image, (b) HRTEM image, (c) STEM image of one nanostructure, and its corresponding EDS elemental map. Control photocatalytic CO2RR experiment for (d) different samples without water (e) with different volumes of water. Reproduced with permission from ref. 214, Copyright 2020 Wiley-VCH. | ||
As discussed above, hydrogen evolution appears as a competitive reduction reaction during the photoreduction of CO2. Thus, is it necessary to suppress the H2 evolution so that we can use the photoexcited electrons more efficiently for the generation of other liquid or gaseous carbon-based solar fuels. The most studied TiO2 (Degussa P25), loaded with Pt, leads to hydrogen evolution during the photoreduction of CO2 in the presence of water, with small fractions of CO and CH4.215 Interestingly, Wang and his co-workers found that when a Cu2O shell was selectively photodeposited on the Pt nanocrystals, it enhanced the production of CH4 and CO over H2 (Fig. 27a). By systematically increasing the Cu content in the catalyst, the selectivity for CO2 reduction increased from 30% to 85% (Fig. 27b). As no high-molecular-weight organic surfactant or solvent was used during the photodeposition process, no carbon contamination was detected on the surface of the final catalyst, and the product selectivity purely came from the photocatalytic conversion of CO2.12
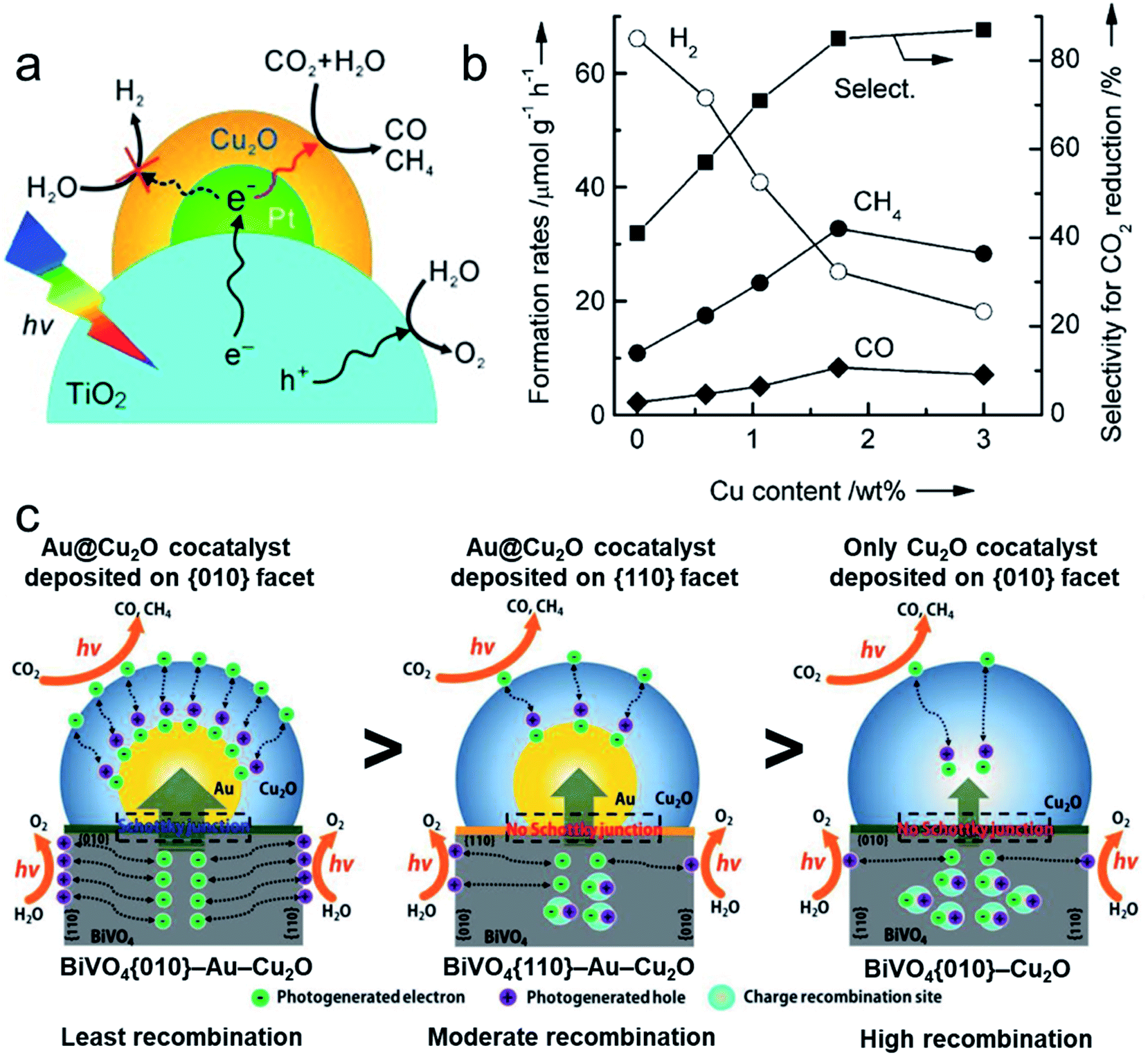 | ||
| Fig. 27 (a) Schematic mechanism of the Cu2O/Pt/TiO2-xh hybrid system and (b) its product selectivity with respect to the Cu content in the hybrid catalysts for the photoreduction of CO2 with H2O. Reproduced with permission from ref. 215, Copyright 2013 Wiley-VCH. (c) Scheme to describe the carrier migration behavior from BiVO4 to Cu2O with and without Au particles deposited on the different facets of BiVO4. Reproduced with permission from ref. 217, Copyright 2018 Wiley-VCH. | ||
Inspired by this work, Yan's group demonstrated a facet-dependent Schottky heterojunction by coupling a BiVO4 truncated octahedron with a hemispherical Au@Cu2O core–shell cocatalyst.217 The truncated octahedron consists of anisotropic facets such as {010} and {110}. Photoexcited electrons can be selectively accumulated on the {010} facets, whereas holes accumulate on the {110} facets. Therefore, Au nanocrystals can be selectively deposited on the {010} facets by photoreduction. By contrast, before Au nanocrystal deposition only on the {110} facets, PVP surfactant is needed to block the {010} facets by preferential adsorption. The Cu2O shell layer was subsequently deposited on Au via a simple chemical reduction, forming three different heterostructures namely BiVO4{010}–Au–Cu2O, BiVO4{110}–Au–Cu2O and BiVO4{010}–Cu2O (as shown in Fig. 27c). Such synthetic strategies are very challenging and need special attention.
An efficient Schottky junction was formed in the BiVO4{010}–Au interface, facilitating the extraction of the photogenerated electrons from BiVO4 and their successful transfer to Cu2O, thus improving the charge separation of the system (Fig. 27c). BiVO4{010}–Au–Cu2O showed a CH4 production rate equal to 3.14 μmol gcat−1 h−1 (1 μmol m−2 h−1 in terms of surface area), which was three and five times higher than that of BiVO4{110}–Au–Cu2O and BiVO4{010}–Cu2O, respectively. The Schottky junction between BiVO4 and Au not only facilitated better charge separation but also prevented the photocorrosion of Cu2O by neutralizing the holes, which resulted in a high photostability of the entire system. Despite the good results, it is worth mentioning that such low CH4 and CO production rates can be associated with surface carbon contamination. Isotope labeled 13CO2 photoreduction was not reported either in this study, and thus some doubts remain on the origin of the detected products. Recently, Lee et al. showed that different surface ligands on the Cu2O/TiO2 photocatalyst surface alter the binding strength of reaction intermediates resulting in different product selectivities during the gas-phase photocatalytic CO2 reduction.224 Therefore, authors should consider any overestimation of the activity for photoreduction of CO2. As discussed previously in Section 4.3, the photostability of Cu2O during photoreduction of CO2 in the presence of H2O can be enhanced following the same approaches employed for H2 production from aqueous solutions: (i) by coating Cu2O nanostructures with an ultrathin conducting and transparent layer, (ii) by forming a successful Z-scheme system, and lastly (iii) by forming a thin carbon layer.75,220,225,226 In general, previous case studies provided an alternative strategy for developing hybrid photocatalytic systems, which can reduce the H2 evolution reduction while improving the formation of solar fuels with suppressed photocorrosion.
5 Conclusion and remarks
Substantial progress in the understanding of synthetic procedures and growth mechanisms of faceted Cu2O crystals opens the door to studying their facet-dependent properties. New physicochemical properties have been discovered by studying the facet-dependent properties of Cu2O crystals, which are different from traditional viewpoints. In particular, facet-engineering of Cu2O nanocrystals provides the opportunity to tune their surface atomic arrangements and light absorbing properties and induce an increased separation of charge carriers on different anisotropic facets, which will result in an enhanced photocatalytic activity.Comparison of the surface energies of Cu2O crystallographic facets reveals that the order is as follows: γ {1 0 0} < γ {1 1 1} < γ {1 1 0} < γ {h k l}. Preparation of 1D, 2D, and 3D Cu2O nanostructures has been reviewed while highlighting the role of a surfactant, solution pH, and different inorganic ions in various synthetic chemical methods. Although obtaining such well-defined morphologies requires the use of templating and capping agents, their careful removal is crucial for guaranteeing the subsequent photocatalytic activity. Clean surfaces must be obtained, and they proved to not only maximize the activity but also ensure the absence of contaminants which can be released during the reaction and may alter the product distribution or can be involved as sacrificial electron donors leading to artefacts. Afterward, suitable steps have been suggested for efficiently removing the surfactant without distorting the facet and chemical composition of the Cu2O nanocrystals.
This precise control of the facet enables unique properties such as facet-selective adsorption of capping agents and facet-selective photodeposition of oxidation and reduction co-catalysts. It was also identified that pure Cu2O nanocrystals showed facet-dependent optical properties. Furthermore, when core–shell nanocrystals were formed with a plasmonic core and Cu2O shell, a large red-shift for the SPR peak of the plasmonic core was observed, which was due to the differently exposed facets of the shell and their corresponding refractive indices and dielectric constants. Such interesting tuning of the optical properties of hybrid core–shell nanocrystals can provide a highly efficient photocatalyst, absorbing from the UV-vis to NIR region of the solar spectrum.
Discussions on the necessary figures of merit in photocatalysis were also reviewed in order to avoid any mistakes during the demonstration of the photocatalytic performance. We discussed the direct relation of the enhanced photocatalytic activity to: (1) efficient adsorption and activation of reactants on the catalyst surface, (2) upon exposure of light, efficient separation of photogenerated electron–hole pairs and their migration to the active catalytic sites via the exposed crystal facet to carry out the photo-redox reaction. Then special emphasis was put on well-defined hybrid Cu2O-based nanostructures, which showed enhanced photocatalytic performance for dye degradation, organic reactions, CO2 reduction, and H2 production, compared to the pristine Cu2O nanostructures. The possible charge transfer mechanisms and the product selectivity were also illustrated for different cases. We particularly emphasized the potential strategies for efficient consumption of photogenerated holes for oxidation reactions, thus reducing the self-photooxidation of Cu2O counterparts, which, in turn, increased the photostability. We hope that the presented in-depth discussion and suggestions will contribute to the discovery of new synthetic strategies to enable the preparation of diverse hybrid Cu2O nanostructures and the fundamental investigation of facet-dependent properties on the nanoscale. This may finally enable the design of low cost and more efficient Cu2O-nanostructures for solar to fuel conversion technologies.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
M. B. and P. F. acknowledge financial support from the European Community (projects H2020 – RIA-CE-NMBP-25 Program – Grant No. 862030 – and H2020-LC-SC3-2019-NZE-RES-CC – Grant No. 884444), INSTM consortium and ICCOM-CNR. S. R. and A. N. gratefully acknowledge the support of the Czech Science Foundation (GACR) through project no. 20-17636S, the Ministry of Education, Youth and Sports of the Czech Republic through project ERC CZ no. LL1903, and the Operational Programme Research, Development and Education – European Regional Development Fund, project no. CZ.02.1.01/0.0/0.0/15_003/0000416.References
- R. Marschall, Adv. Funct. Mater., 2014, 24, 2421–2440 CrossRef CAS.
- M. Xiao, Z. Wang, M. Lyu, B. Luo, S. Wang, G. Liu, H. Cheng and L. Wang, Adv. Mater., 2019, 31, 1801369 CrossRef.
- S. Y. Tee, K. Y. Win, W. S. Teo, L.-D. Koh, S. Liu, C. P. Teng and M.-Y. Han, Adv. Sci., 2017, 4, 1600337 CrossRef.
- W. A. Smith, I. D. Sharp, N. C. Strandwitz and J. Bisquert, Energy Environ. Sci., 2015, 8, 2851–2862 RSC.
- K. C. Christoforidis and P. Fornasiero, ChemCatChem, 2017, 9, 1523–1544 CrossRef CAS.
- M. Cargnello, P. Fornasiero and R. J. Gorte, ChemPhysChem, 2013, 14, 3869–3877 CrossRef CAS.
- M. Cargnello, T. Montini, S. Y. Smolin, J. B. Priebe, J. J. Delgado Jaén, V. V. T. Doan-Nguyen, I. S. McKay, J. A. Schwalbe, M.-M. Pohl, T. R. Gordon, Y. Lu, J. B. Baxter, A. Brückner, P. Fornasiero and C. B. Murray, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3966–3971 CrossRef CAS.
- T. Paik, M. Cargnello, T. R. Gordon, S. Zhang, H. Yun, J. D. Lee, H. Y. Woo, S. J. Oh, C. R. Kagan, P. Fornasiero and C. B. Murray, ACS Energy Lett., 2018, 3, 1904–1910 CrossRef CAS.
- J. L. White, M. F. Baruch, J. E. Pander, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS.
- K. C. Christoforidis and P. Fornasiero, ChemCatChem, 2019, 11, 368–382 CrossRef CAS.
- D. Barreca, P. Fornasiero, A. Gasparotto, V. Gombac, C. Maccato, T. Montini and E. Tondello, ChemSusChem, 2009, 2, 230–233 CrossRef CAS.
- C.-C. Yang, Y.-H. Yu, B. van der Linden, J. C. S. Wu and G. Mul, J. Am. Chem. Soc., 2010, 132, 8398–8406 CrossRef CAS.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS.
- L. Mascaretti, A. Dutta, Š. Kment, V. M. Shalaev, A. Boltasseva, R. Zbořil and A. Naldoni, Adv. Mater., 2019, 31, 1805513 CrossRef.
- S. Bai, L. Wang, Z. Li and Y. Xiong, Adv. Sci., 2017, 4, 1600216 CrossRef.
- S. Sun, X. Zhang, J. Cui, Q. Yang and S. Liang, Nanoscale, 2019, 11, 15739–15762 RSC.
- S. Wang, G. Liu and L. Wang, Chem. Rev., 2019, 119, 5192–5247 CrossRef CAS.
- M. Cargnello, T. R. Gordon and C. B. Murray, Chem. Rev., 2014, 114, 9319–9345 CrossRef CAS.
- M. H. Huang and M. Madasu, Nano Today, 2019, 28, 100768 CrossRef CAS.
- M. H. Huang, S. Rej and S.-C. Hsu, Chem. Commun., 2014, 50, 1634–1644 RSC.
- W. Huang, Acc. Chem. Res., 2016, 49, 520–527 CrossRef CAS.
- Y. Shang and L. Guo, Adv. Sci., 2015, 2, 1500140 CrossRef.
- M. H. Huang and P.-H. Lin, Adv. Funct. Mater., 2012, 22, 14–24 CrossRef CAS.
- M. H. Huang, S. Rej and C.-Y. Chiu, Small, 2015, 11, 2716–2726 CrossRef CAS.
- C.-H. Kuo and M. H. Huang, Nano Today, 2010, 5, 106–116 CrossRef CAS.
- G. Liu, J. C. Yu, G. Q. Lu and H.-M. Cheng, Chem. Commun., 2011, 47, 6763 RSC.
- S. Sun, Nanoscale, 2015, 7, 10850–10882 RSC.
- S. Sun and Z. Yang, RSC Adv., 2014, 4, 3804–3822 RSC.
- S. Sun, X. Zhang, Q. Yang, S. Liang, X. Zhang and Z. Yang, Prog. Mater. Sci., 2018, 96, 111–173 CrossRef CAS.
- C. Y. Toe, J. Scott, R. Amal and Y. H. Ng, J. Photochem. Photobiol., C, 2019, 40, 191–211 CrossRef CAS.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS.
- Z. L. Wang, J. Phys. Chem. B, 2000, 104, 1153–1175 CrossRef CAS.
- A. Radi, D. Pradhan, Y. Sohn and K. T. Leung, ACS Nano, 2010, 4, 1553–1560 CrossRef CAS.
- M. D. Susman, Y. Feldman, A. Vaskevich and I. Rubinstein, ACS Nano, 2014, 8, 162–174 CrossRef CAS.
- B.-H. Kuo, C.-F. Hsia, T.-N. Chen and M. H. Huang, J. Phys. Chem. C, 2018, 122, 25118–25126 CrossRef CAS.
- H.-G. Liao, D. Zherebetskyy, H. Xin, C. Czarnik, P. Ercius, H. Elmlund, M. Pan, L.-W. Wang and H. Zheng, Science, 2014, 345, 916–919 CrossRef CAS.
- Y.-H. Lin, J.-Y. Chen, F.-C. Chen, M.-Y. Kuo, Y.-J. Hsu and W.-W. Wu, Anal. Chem., 2019, 91, 9665–9672 CrossRef CAS.
- Z. Dong, L. Zhang, S. Wang and L. Luo, Nano Energy, 2020, 70, 104527 CrossRef CAS.
- L. Gou and C. J. Murphy, Nano Lett, 2003, 3, 231–234 CrossRef CAS.
- H. Xu, W. Wang and W. Zhu, J. Phys. Chem. B, 2006, 110, 13829–13834 CrossRef CAS.
- J.-Y. Ho and M. H. Huang, J. Phys. Chem. C, 2009, 113, 14159–14164 CrossRef CAS.
- Y. Sui, W. Fu, H. Yang, Y. Zeng, Y. Zhang, Q. Zhao, Y. Li, X. Zhou, Y. Leng, M. Li and G. Zou, Cryst. Growth Des., 2010, 10, 99–108 CrossRef CAS.
- X. Liang, L. Gao, S. Yang and J. Sun, Adv. Mater., 2009, 21, 2068–2071 CrossRef CAS.
- K. X. Yao, X. M. Yin, T. H. Wang and H. C. Zeng, J. Am. Chem. Soc., 2010, 132, 6131–6144 CrossRef CAS.
- X. Lan, J. Zhang, H. Gao and T. Wang, CrystEngComm, 2011, 13, 633–636 RSC.
- W.-C. Huang, L.-M. Lyu, Y.-C. Yang and M. H. Huang, J. Am. Chem. Soc., 2012, 134, 1261–1267 CrossRef CAS.
- M. H. Kim, B. Lim, E. P. Lee and Y. Xia, J. Mater. Chem., 2008, 18, 4069–4073 RSC.
- C. Liu, Y.-H. Chang, J. Chen and S.-P. Feng, ACS Appl. Mater. Interfaces, 2017, 9, 39027–39033 CrossRef CAS.
- M. J. Siegfried and K.-S. Choi, J. Am. Chem. Soc., 2006, 128, 10356–10357 CrossRef CAS.
- M. J. Siegfried and K.-S. Choi, Adv. Mater., 2004, 16, 1743–1746 CrossRef CAS.
- C. J. Munro, E. C. Bell, M. O. Olagunju, J. L. Cohn, E. M. Zahran, L. G. Bachas and M. R. Knecht, ACS Sustain. Chem. Eng., 2019, 7, 17055–17064 CrossRef CAS.
- K. Self and W. Zhou, Cryst. Growth Des., 2016, 16, 5377–5384 CrossRef CAS.
- D. Y. Kim, C. W. Kim, J. H. Sohn, K. J. Lee, M. H. Jung, M. G. Kim and Y. S. Kang, J. Phys. Chem. C, 2015, 119, 13350–13356 CrossRef.
- H. Y. Zhao, Y. F. Wang and J. H. Zeng, Cryst. Growth Des., 2008, 8, 3731–3734 CrossRef CAS.
- Y.-Y. Ma, Z.-Y. Jiang, Q. Kuang, S.-H. Zhang, Z.-X. Xie, R.-B. Huang and L.-S. Zheng, J. Phys. Chem. C, 2008, 112, 13405–13409 CrossRef CAS.
- S. Sun, C. Kong, S. Yang, L. Wang, X. Song, B. Ding and Z. Yang, CrystEngComm, 2011, 13, 2217–2221 RSC.
- Y. Liang, L. Shang, T. Bian, C. Zhou, D. Zhang, H. Yu, H. Xu, Z. Shi, T. Zhang, L.-Z. Wu and C.-H. Tung, CrystEngComm, 2012, 14, 4431–4436 RSC.
- M. Leng, M. Liu, Y. Zhang, Z. Wang, C. Yu, X. Yang, H. Zhang and C. Wang, J. Am. Chem. Soc., 2010, 132, 17084–17087 CrossRef CAS.
- S. Sun, X. Song, Y. Sun, D. Deng and Z. Yang, Catal. Sci. Technol., 2012, 2, 925–930 RSC.
- X. Wang, S. Jiao, D. Wu, Q. Li, J. Zhou, K. Jiang and D. Xu, CrystEngComm, 2013, 15, 1849–1852 RSC.
- X. Wang, C. Liu, B. Zheng, Y. Jiang, L. Zhang, Z. Xie and L. Zheng, J. Mater. Chem. A, 2013, 1, 282–287 RSC.
- Z. Zhao, X. Wang, J. Si, C. Yue, C. Xia and F. Li, Green Chem., 2018, 20, 832–837 RSC.
- I.-C. Chang, P.-C. Chen, M.-C. Tsai, T.-T. Chen, M.-H. Yang, H.-T. Chiu and C.-Y. Lee, CrystEngComm, 2013, 15, 2363–2366 RSC.
- Y.-H. Tsai, K. Chanda, Y.-T. Chu, C.-Y. Chiu and M. H. Huang, Nanoscale, 2014, 6, 8704–8709 RSC.
- Y. Cao, Y. Xu, H. Hao and G. Zhang, Mater. Lett., 2014, 114, 88–91 CrossRef CAS.
- S. Thoka, A.-T. Lee and M. H. Huang, ACS Sustainable Chem. Eng., 2019, 7, 10467–10476 CrossRef CAS.
- M. Yin, C.-K. Wu, Y. Lou, C. Burda, J. T. Koberstein, Y. Zhu and S. O'Brien, J. Am. Chem. Soc., 2005, 127, 9506–9511 CrossRef CAS.
- Y. Tan, X. Xue, Q. Peng, H. Zhao, T. Wang and Y. Li, Nano Lett., 2007, 7, 3723–3728 CrossRef CAS.
- S. D. Pike, E. R. White, A. Regoutz, N. Sammy, D. J. Payne, C. K. Williams and M. S. P. Shaffer, ACS Nano, 2017, 11, 2714–2723 CrossRef CAS.
- S. Gao, Y. Sun, F. Lei, J. Liu, L. Liang, T. Li, B. Pan, J. Zhou and Y. Xie, Nano Energy, 2014, 8, 205–213 CrossRef CAS.
- Y. Xiong, Z. Li, R. Zhang, Y. Xie, J. Yang and C. Wu, J. Phys. Chem. B, 2003, 107, 3697–3702 CrossRef CAS.
- L.-I. Hung, C.-K. Tsung, W. Huang and P. Yang, Adv. Mater., 2010, 22, 1910–1914 CrossRef CAS.
- W. Wang, G. Wang, X. Wang, Y. Zhan, Y. Liu and C. Zheng, Adv. Mater., 2002, 14, 67–69 CrossRef CAS.
- T. Zhou, Z. Zang, J. Wei, J. Zheng, J. Hao, F. Ling, X. Tang, L. Fang and M. Zhou, Nano Energy, 2018, 50, 118–125 CrossRef CAS.
- J.-Y. Li, L. Yuan, S.-H. Li, Z.-R. Tang and Y.-J. Xu, J. Mater. Chem. A, 2019, 7, 8676–8689 RSC.
- J. Hou, H. Cheng, O. Takeda and H. Zhu, Angew. Chem., Int. Ed., 2015, 54, 8480–8484 CrossRef CAS.
- C. Kim, K. M. Cho, A. Al-Saggaf, I. Gereige and H.-T. Jung, ACS Catal., 2018, 8, 4170–4177 CrossRef CAS.
- X. Zhao, Y. Fan, W. Zhang, X. Zhang, D. Han, L. Niu and A. Ivaska, ACS Catal., 2020, 10, 6367–6376 CrossRef CAS.
- S. Campisi, M. Schiavoni, C. Chan-Thaw and A. Villa, Catalysts, 2016, 6, 185 CrossRef.
- Z. Niu and Y. Li, Chem. Mater., 2014, 26, 72–83 CrossRef CAS.
- L. M. Rossi, J. L. Fiorio, M. A. S. Garcia and C. P. Ferraz, Dalton Trans., 2018, 47, 5889–5915 RSC.
- K. Chanda, S. Rej and M. H. Huang, Chem.–Eur. J., 2013, 19, 16036–16043 CrossRef CAS.
- K. Chanda, S. Rej and M. H. Huang, Nanoscale, 2013, 5, 12494–12501 RSC.
- M. Cargnello, C. Chen, B. T. Diroll, V. V. T. Doan-Nguyen, R. J. Gorte and C. B. Murray, J. Am. Chem. Soc., 2015, 137, 6906–6911 CrossRef CAS.
- Q. Hua, K. Chen, S. Chang, Y. Ma and W. Huang, J. Phys. Chem. C, 2011, 115, 20618–20627 CrossRef CAS.
- Q. Hua, D. Shang, W. Zhang, K. Chen, S. Chang, Y. Ma, Z. Jiang, J. Yang and W. Huang, Langmuir, 2011, 27, 665–671 CrossRef CAS.
- Y. Sui, W. Fu, Y. Zeng, H. Yang, Y. Zhang, H. Chen, Y. Li, M. Li and G. Zou, Angew. Chem., Int. Ed., 2010, 49, 4282–4285 CrossRef CAS.
- C.-H. Kuo, Y.-T. Chu, Y.-F. Song and M. H. Huang, Adv. Funct. Mater., 2011, 21, 792–797 CrossRef CAS.
- J. Nai, Y. Tian, X. Guan and L. Guo, J. Am. Chem. Soc., 2013, 135, 16082–16091 CrossRef CAS.
- C.-H. Kuo and M. H. Huang, J. Am. Chem. Soc., 2008, 130, 12815–12820 CrossRef CAS.
- Y.-H. Tsai, C.-Y. Chiu and M. H. Huang, J. Phys. Chem. C, 2013, 117, 24611–24617 CrossRef CAS.
- Q. Hua, T. Cao, X.-K. Gu, J. Lu, Z. Jiang, X. Pan, L. Luo, W.-X. Li and W. Huang, Angew. Chem., Int. Ed., 2014, 53, 4856–4861 CrossRef CAS.
- C. G. Read, E. M. P. Steinmiller and K.-S. Choi, J. Am. Chem. Soc., 2009, 131, 12040–12041 CrossRef CAS.
- H. Zhu, M. Du, D. Yu, Y. Wang, M. Zou, C. Xu and Y. Fu, Dalton Trans., 2012, 41, 13795–13799 RSC.
- X.-W. Liu, Langmuir, 2011, 27, 9100–9104 CrossRef CAS.
- R. Chen, S. Pang, H. An, J. Zhu, S. Ye, Y. Gao, F. Fan and C. Li, Nat. Energy, 2018, 3, 655–663 CrossRef CAS.
- R. Chen, F. Fan, T. Dittrich and C. Li, Chem. Soc. Rev., 2018, 47, 8238–8262 RSC.
- R. Chen, S. Pang, H. An, T. Dittrich, F. Fan and C. Li, Nano Lett., 2019, 19, 426–432 CrossRef CAS.
- R. Chen, J. Zhu, H. An, F. Fan and C. Li, Faraday Discuss., 2017, 198, 473–479 RSC.
- R. Li, X. Tao, R. Chen, F. Fan and C. Li, Chem.–Eur. J., 2015, 21, 14337–14341 CrossRef CAS.
- T. Tachikawa and T. Majima, Chem. Soc. Rev., 2010, 39, 4802 RSC.
- W.-K. Wang, J.-J. Chen, Z.-Z. Lou, S. Kim, M. Fujitsuka, H.-Q. Yu and T. Majima, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18827–18833 CrossRef CAS.
- M. H. Huang, G. Naresh and H.-S. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 4–15 CrossRef CAS.
- M. H. Huang, Small, 2019, 15, 1804726 CrossRef.
- Q. Ruan, N. Li, H. Yin, X. Cui, J. Wang and H.-Q. Lin, ACS Photonics, 2018, 5, 3838–3848 CrossRef CAS.
- W.-H. Ke, C.-F. Hsia, Y.-J. Chen and M. H. Huang, Small, 2016, 12, 3530–3534 CrossRef CAS.
- C.-Y. Chu and M. H. Huang, J. Mater. Chem. A, 2017, 5, 15116–15123 RSC.
- J.-Y. Huang, M. Madasu and M. H. Huang, J. Phys. Chem. C, 2018, 122, 13027–13033 CrossRef CAS.
- C.-S. Tan, S.-C. Hsu, W.-H. Ke, L.-J. Chen and M. H. Huang, Nano Lett., 2015, 15, 2155–2160 CrossRef CAS.
- Y.-C. Yang, H.-J. Wang, J. Whang, J.-S. Huang, L.-M. Lyu, P.-H. Lin, S. Gwo and M. H. Huang, Nanoscale, 2014, 6, 4316–4324 RSC.
- H.-J. Wang, K.-H. Yang, S.-C. Hsu and M. H. Huang, Nanoscale, 2016, 8, 965–972 RSC.
- S. Rej, H.-J. Wang, M.-X. Huang, S.-C. Hsu, C.-S. Tan, F.-C. Lin, J.-S. Huang and M. H. Huang, Nanoscale, 2015, 7, 11135–11141 RSC.
- K.-H. Yang, S.-C. Hsu and M. H. Huang, Chem. Mater., 2016, 28, 5140–5146 CrossRef CAS.
- C.-F. Hsia, C.-H. Chang and M. H. Huang, Part. Part. Syst. Charact., 2018, 35, 1800112 CrossRef.
- M. Tariq, M. D. Koch, J. W. Andrews and K. E. Knowles, J. Phys. Chem. C, 2020, 124, 4810–4819 CrossRef CAS.
- I. Polishchuk, N. Bianco-Stein, A. Lang, M. Kurashvili, M. Caspary Toroker, A. Katsman, J. Feldmann and B. Pokroy, Adv. Funct. Mater., 2020, 30, 1910405 CrossRef CAS.
- M. Qureshi and K. Takanabe, Chem. Mater., 2017, 29, 158–167 CrossRef CAS.
- K. Takanabe, J. Catal., 2019, 370, 480–484 CrossRef CAS.
- P. V. Kamat and S. Jin, ACS Energy Lett., 2018, 3, 622–623 CrossRef CAS.
- K. Takanabe, ACS Catal., 2017, 7, 8006–8022 CrossRef CAS.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS.
- Md. Hoque and M. Guzman, Materials, 2018, 11, 1990 CrossRef.
- M. Melchionna and P. Fornasiero, ACS Catal., 2020, 10, 5493–5501 CrossRef CAS.
- L. Mascaretti and A. Naldoni, J. Appl. Phys., 2020, 128, 041101 CrossRef CAS.
- S. Rej, L. Mascaretti, E. Y. Santiago, O. Tomanec, Š. Kment, Z. Wang, R. Zbořil, P. Fornasiero, A. O. Govorov and A. Naldoni, ACS Catal., 2020, 10, 5261–5271 CrossRef CAS.
- L. Li, C. Nan, Q. Peng and Y. Li, Chem.–Eur. J., 2012, 18, 10491–10496 CrossRef CAS.
- Y. Su, H. Li, H. Ma, J. Robertson and A. Nathan, ACS Appl. Mater. Interfaces, 2017, 9, 8100–8106 CrossRef CAS.
- N. Manfredi, M. Monai, T. Montini, F. Peri, F. De Angelis, P. Fornasiero and A. Abbotto, ACS Energy Lett., 2018, 3, 85–91 CrossRef CAS.
- A. Dessì, M. Monai, M. Bessi, T. Montini, M. Calamante, A. Mordini, G. Reginato, C. Trono, P. Fornasiero and L. Zani, ChemSusChem, 2018, 11, 793–805 CrossRef.
- O. Bettucci, T. Skaltsas, M. Calamante, A. Dessì, M. Bartolini, A. Sinicropi, J. Filippi, G. Reginato, A. Mordini, P. Fornasiero and L. Zani, ACS Appl. Energy Mater., 2019, 2, 5600–5612 CrossRef CAS.
- G.-Z. Yuan, C.-F. Hsia, Z.-W. Lin, C. Chiang, Y.-W. Chiang and M. H. Huang, Chem.–Eur. J., 2016, 22, 12548–12556 CrossRef CAS.
- T.-N. Chen, J.-C. Kao, X.-Y. Zhong, S.-J. Chan, A. S. Patra, Y.-C. Lo and M. H. Huang, ACS Cent. Sci., 2020, 6, 984–994 CrossRef CAS.
- Q. Wei, Y. Wang, H. Qin, J. Wu, Y. Lu, H. Chi, F. Yang, B. Zhou, H. Yu and J. Liu, Appl. Catal., B, 2018, 227, 132–144 CrossRef CAS.
- M.-Y. Kuo, C.-F. Hsiao, Y.-H. Chiu, T.-H. Lai, M.-J. Fang, J.-Y. Wu, J.-W. Chen, C.-L. Wu, K.-H. Wei, H.-C. Lin and Y.-J. Hsu, Appl. Catal., B, 2019, 242, 499–506 CrossRef CAS.
- L. Liu, W. Yang, W. Sun, Q. Li and J. K. Shang, ACS Appl. Mater. Interfaces, 2015, 7, 1465–1476 CrossRef CAS.
- X. Xu, Z. Gao, Z. Cui, Y. Liang, Z. Li, S. Zhu, X. Yang and J. Ma, ACS Appl. Mater. Interfaces, 2016, 8, 91–101 CrossRef CAS.
- S.-C. Wu, C.-S. Tan and M. H. Huang, Adv. Funct. Mater., 2017, 27, 1604635 CrossRef.
- Y. Ma, X. Zhu, S. Xu, G. He, L. Yao, N. Hu, Y. Su, J. Feng, Y. Zhang and Z. Yang, Appl. Catal., B, 2018, 234, 26–36 CrossRef CAS.
- S. K. Cushing, J. Li, F. Meng, T. R. Senty, S. Suri, M. Zhi, M. Li, A. D. Bristow and N. Wu, J. Am. Chem. Soc., 2012, 134, 15033–15041 CrossRef CAS.
- L. Liu, X. Gu, C. Sun, H. Li, Y. Deng, F. Gao and L. Dong, Nanoscale, 2012, 4, 6351–6359 RSC.
- Y. Lu, X. Zhang, Y. Chu, H. Yu, M. Huo, J. Qu, J. C. Crittenden, H. Huo and X. Yuan, Appl. Catal., B, 2018, 224, 239–248 CrossRef CAS.
- F. Ye, Y. Su, X. Quan, S. Chen, H. Yu and H. Li, Appl. Catal., B, 2019, 244, 347–355 CrossRef CAS.
- J. He, D. W. Shao, L. C. Zheng, L. J. Zheng, D. Q. Feng, J. P. Xu, X. H. Zhang, W. C. Wang, W.-H. Wang, F. Lu, H. Dong, Y. H. Cheng, H. Liu and R. K. Zheng, Appl. Catal., B, 2017, 203, 917–926 CrossRef CAS.
- D. Jiang, J. Xue, L. Wu, W. Zhou, Y. Zhang and X. Li, Appl. Catal., B, 2017, 211, 199–204 CrossRef CAS.
- W. Wang, H. Feng, J. Liu, M. Zhang, S. Liu, C. Feng and S. Chen, Chem. Eng. J., 2020, 386, 124116 CrossRef CAS.
- Y. Chen, M. Wang, Y. Ma, Y. Li, J. Cai and Z. Li, Catal. Sci. Technol., 2018, 8, 2218–2223 RSC.
- C. Zhan, Q. Wang, L. Zhou, X. Han, Y. Wanyan, J. Chen, Y. Zheng, Y. Wang, G. Fu, Z. Xie and Z. Tian, J. Am. Chem. Soc., 2020, 142, 14134–14141 CrossRef CAS.
- E. M. Zahran, N. M. Bedford, M. A. Nguyen, Y.-J. Chang, B. S. Guiton, R. R. Naik, L. G. Bachas and M. R. Knecht, J. Am. Chem. Soc., 2014, 136, 32–35 CrossRef CAS.
- B. Wang, J. Durantini, J. Nie, A. E. Lanterna and J. C. Scaiano, J. Am. Chem. Soc., 2016, 138, 13127–13130 CrossRef CAS.
- Y. You, F. Cao, Y. Zhao, Q. Deng, Y. Sang, Y. Li, K. Dong, J. Ren and X. Qu, ACS Nano, 2020, 14, 4178–4187 CrossRef CAS.
- Z. Li, Y. Pi, D. Xu, Y. Li, W. Peng, G. Zhang, F. Zhang and X. Fan, Appl. Catal., B, 2017, 213, 1–8 CrossRef CAS.
- X. Han, X. He, F. Wang, J. Chen, J. Xu, X. Wang and X. Han, J. Mater. Chem. A, 2017, 5, 10220–10226 RSC.
- X. Han, X. He, L. Sun, X. Han, W. Zhan, J. Xu, X. Wang and J. Chen, ACS Catal., 2018, 8, 3348–3356 CrossRef CAS.
- L. Liu, W. Sun, W. Yang, Q. Li and J. K. Shang, Sci. Rep., 2016, 6, 20878 CrossRef CAS.
- W. Huang, Q. Liu, Z. Zhou, Y. Li, Y. Ling, Y. Wang, Y. Tu, B. Wang, X. Zhou, D. Deng, B. Yang, Y. Yang, Z. Liu, X. Bao and F. Yang, Nat. Commun., 2020, 11, 2312 CrossRef CAS.
- S. Liu, Z. Yang, X. Liu, R. Liu, G. Wang, Q. Wang, H. Li and P. Guo, ACS Appl. Nano Mater., 2018, 1, 6038–6045 CrossRef CAS.
- Y. Su, H. Li, H. Ma, H. Wang, J. Robertson and A. Nathan, ACS Omega, 2018, 3, 1939–1945 CrossRef CAS.
- D.-F. Zhang, H. Zhang, L. Guo, K. Zheng, X.-D. Han and Z. Zhang, J. Mater. Chem., 2009, 19, 5220–5225 RSC.
- Y.-C. Pu, H.-Y. Chou, W.-S. Kuo, K.-H. Wei and Y.-J. Hsu, Appl. Catal., B, 2017, 204, 21–32 CrossRef CAS.
- A. E. Kandjani, Y. M. Sabri, S. R. Periasamy, N. Zohora, M. H. Amin, A. Nafady and S. K. Bhargava, Langmuir, 2015, 31, 10922–10930 CrossRef CAS.
- M. Shanmugam, A. Sagadevan, V. P. Charpe, V. K. K. Pampana and K. C. Hwang, ChemSusChem, 2020, 13, 287–292 CrossRef CAS.
- Y. Kwon, A. Soon, H. Han and H. Lee, J. Mater. Chem. A, 2015, 3, 156–162 RSC.
- C. Y. Toe, Z. Zheng, H. Wu, J. Scott, R. Amal and Y. H. Ng, Angew. Chem., Int. Ed., 2018, 57, 13613–13617 CrossRef CAS.
- S. Kumar, C. M. A. Parlett, M. A. Isaacs, D. V. Jowett, R. E. Douthwaite, M. C. R. Cockett and A. F. Lee, Appl. Catal., B, 2016, 189, 226–232 CrossRef CAS.
- Z. Lin, J. Xiao, L. Li, P. Liu, C. Wang and G. Yang, Adv. Energy Mater., 2016, 6, 1501865 CrossRef.
- L. Wang, J. Ge, A. Wang, M. Deng, X. Wang, S. Bai, R. Li, J. Jiang, Q. Zhang, Y. Luo and Y. Xiong, Angew. Chem., Int. Ed., 2014, 53, 5107–5111 CAS.
- S. Bai, J. Ge, L. Wang, M. Gong, M. Deng, Q. Kong, L. Song, J. Jiang, Q. Zhang, Y. Luo, Y. Xie and Y. Xiong, Adv. Mater., 2014, 26, 5689–5695 CrossRef CAS.
- Y. Liu, B. Zhang, L. Luo, X. Chen, Z. Wang, E. Wu, D. Su and W. Huang, Angew. Chem., Int. Ed., 2015, 54, 15260–15265 CrossRef CAS.
- Y. Liu, Z. Ye, D. Li, M. Wang, Y. Zhang and W. Huang, Appl. Surf. Sci., 2019, 473, 500–510 CrossRef CAS.
- T. Wei, Y.-N. Zhu, X. An, L.-M. Liu, X. Cao, H. Liu and J. Qu, ACS Catal., 2019, 9, 8346–8354 CrossRef CAS.
- G. Li, J. Huang, J. Chen, Z. Deng, Q. Huang, Z. Liu, W. Guo and R. Cao, ACS Omega, 2019, 4, 3392–3397 CrossRef CAS.
- X.-Y. Liu, W.-D. Wei, S.-C. Cui and J.-G. Liu, Catal. Lett., 2016, 146, 1655–1662 CrossRef CAS.
- C. Peng, P. Wei, X. Li, Y. Liu, Y. Cao, H. Wang, H. Yu, F. Peng, L. Zhang, B. Zhang and K. Lv, Nano Energy, 2018, 53, 97–107 CrossRef CAS.
- L. Lu, X. Xu, J. Yan, F.-N. Shi and Y. Huo, Dalton Trans., 2018, 47, 2031–2038 RSC.
- Y. Lou, Y. Zhang, L. Cheng, J. Chen and Y. Zhao, ChemSusChem, 2018, 11, 1505–1511 CrossRef CAS.
- J. W. Hong, D. H. Wi, S.-U. Lee and S. W. Han, J. Am. Chem. Soc., 2016, 138, 15766–15773 CrossRef CAS.
- X. Yu, F. Liu, J. Bi, B. Wang and S. Yang, Nano Energy, 2017, 33, 469–475 CrossRef CAS.
- M. Zhang, Z. Chen, Y. Wang, J. Zhang, X. Zheng, D. Rao, X. Han, C. Zhong, W. Hu and Y. Deng, Appl. Catal., B, 2019, 246, 202–210 CrossRef CAS.
- Z. Zhang, R. Dua, L. Zhang, H. Zhu, H. Zhang and P. Wang, ACS Nano, 2013, 7, 1709–1717 CrossRef CAS.
- E. Kecsenovity, B. Endrődi, P. S. Tóth, Y. Zou, R. A. W. Dryfe, K. Rajeshwar and C. Janáky, J. Am. Chem. Soc., 2017, 139, 6682–6692 CrossRef CAS.
- M. Singh, D. Jampaiah, A. E. Kandjani, Y. M. Sabri, E. Della Gaspera, P. Reineck, M. Judd, J. Langley, N. Cox, J. van Embden, E. L. H. Mayes, B. C. Gibson, S. K. Bhargava, R. Ramanathan and V. Bansal, Nanoscale, 2018, 10, 6039–6050 RSC.
- X. Yu, X. Liu, B. Wang, Q. Meng, S. Sun, Y. Tang and K. Zhao, Nanoscale, 2020, 12, 1912–1920 RSC.
- G. Wang, R. van den Berg, C. de Mello Donega, K. P. de Jong and P. E. de Jongh, Appl. Catal., B, 2016, 192, 199–207 CrossRef CAS.
- C. Wang, B. Ma, S. Xu, D. Li, S. He, Y. Zhao, J. Han, M. Wei, D. G. Evans and X. Duan, Nano Energy, 2017, 32, 463–469 CrossRef CAS.
- X. Mao, A. C. Foucher, T. Montini, E. A. Stach, P. Fornasiero and R. J. Gorte, J. Am. Chem. Soc., 2020, 142, 10373–10382 CrossRef CAS.
- H. Li, X. Zhang and D. R. MacFarlane, Adv. Energy Mater., 2015, 5, 1401077 CrossRef.
- H. Li, W. Ali, Z. Wang, M. F. Mideksa, F. Wang, X. Wang, L. Wang and Z. Tang, Nano Energy, 2019, 63, 103873 CrossRef CAS.
- Z. Hu, Y. Mi, Y. Ji, R. Wang, W. Zhou, X. Qiu, X. Liu, Z. Fang and X. Wu, Nanoscale, 2019, 11, 16445–16454 RSC.
- Y.-H. Chiu, S. B. Naghadeh, S. A. Lindley, T.-H. Lai, M.-Y. Kuo, K.-D. Chang, J. Z. Zhang and Y.-J. Hsu, Nano Energy, 2019, 62, 289–298 CrossRef CAS.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS.
- J.-Y. Li, L. Yuan, S.-H. Li, Z.-R. Tang and Y.-J. Xu, J. Mater. Chem. A, 2019, 7, 8676–8689 RSC.
- S. Sun, X. Zhang, J. Cui, Q. Yang and S. Liang, Chem.–Asian J., 2019, 14, 2912–2924 CrossRef CAS.
- L. Wan, Q. Zhou, X. Wang, T. E. Wood, L. Wang, P. N. Duchesne, J. Guo, X. Yan, M. Xia, Y. F. Li, A. A. Jelle, U. Ulmer, J. Jia, T. Li, W. Sun and G. A. Ozin, Nat. Catal., 2019, 2, 889–898 CrossRef CAS.
- J. K. Stolarczyk, S. Bhattacharyya, L. Polavarapu and J. Feldmann, ACS Catal., 2018, 8, 3602–3635 CrossRef CAS.
- T. K. Todorova, M. W. Schreiber and M. Fontecave, ACS Catal., 2020, 10, 1754–1768 CrossRef CAS.
- J. Albero, Y. Peng and H. García, ACS Catal., 2020, 10, 5734–5749 CrossRef CAS.
- X. Jiao, K. Zheng, Z. Hu, Y. Sun and Y. Xie, ACS Cent. Sci., 2020, 6, 653–660 CrossRef CAS.
- L. Liu, S. Wang, H. Huang, Y. Zhang and T. Ma, Nano Energy, 2020, 75, 104959 CrossRef CAS.
- T.-T. Zhuang, Y. Pang, Z.-Q. Liang, Z. Wang, Y. Li, C.-S. Tan, J. Li, C. T. Dinh, P. De Luna, P.-L. Hsieh, T. Burdyny, H.-H. Li, M. Liu, Y. Wang, F. Li, A. Proppe, A. Johnston, D.-H. Nam, Z.-Y. Wu, Y.-R. Zheng, A. H. Ip, H. Tan, L.-J. Chen, S.-H. Yu, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Catal., 2018, 1, 946–951 CrossRef CAS.
- X. Wang, Z. Wang, T.-T. Zhuang, C.-T. Dinh, J. Li, D.-H. Nam, F. Li, C.-W. Huang, C.-S. Tan, Z. Chen, M. Chi, C. M. Gabardo, A. Seifitokaldani, P. Todorović, A. Proppe, Y. Pang, A. R. Kirmani, Y. Wang, A. H. Ip, L. J. Richter, B. Scheffel, A. Xu, S.-C. Lo, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Commun., 2019, 10, 5186 CrossRef.
- L. I. Bendavid and E. A. Carter, J. Phys. Chem. B, 2013, 117, 15750–15760 CrossRef CAS.
- L. I. Bendavid and E. A. Carter, J. Phys. Chem. C, 2013, 117, 26048–26059 CrossRef CAS.
- L. I. Bendavid and E. A. Carter, Surf. Sci., 2013, 618, 62–71 CrossRef CAS.
- A. K. Mishra, A. Roldan and N. H. de Leeuw, J. Chem. Phys., 2016, 145, 044709 CrossRef.
- S.-H. Liu, J.-S. Lu, Y.-C. Pu and H.-C. Fan, J. CO2 Util., 2019, 33, 171–178 CrossRef CAS.
- Y. A. Wu, I. McNulty, C. Liu, K. C. Lau, Q. Liu, A. P. Paulikas, C.-J. Sun, Z. Cai, J. R. Guest, Y. Ren, V. Stamenkovic, L. A. Curtiss, Y. Liu and T. Rajh, Nat. Energy, 2019, 4, 957–968 CrossRef CAS.
- D. Mateo, J. Albero and H. García, Energy Environ. Sci., 2017, 10, 2392–2400 RSC.
- L. Yu, X. Ba, M. Qiu, Y. Li, L. Shuai, W. Zhang, Z. Ren and Y. Yu, Nano Energy, 2019, 60, 576–582 CrossRef CAS.
- Y. Pu, Y. Luo, X. Wei, J. Sun, L. Li, W. Zou and L. Dong, Appl. Catal., B, 2019, 254, 580–586 CrossRef CAS.
- M. E. Aguirre, R. Zhou, A. J. Eugene, M. I. Guzman and M. A. Grela, Appl. Catal., B, 2017, 217, 485–493 CrossRef CAS.
- K.-L. Bae, J. Kim, C. K. Lim, K. M. Nam and H. Song, Nat. Commun., 2017, 8, 1156 CrossRef.
- Z. Zeng, Y. Yan, J. Chen, P. Zan, Q. Tian and P. Chen, Adv. Funct. Mater., 2019, 29, 1806500 CrossRef.
- H. Robatjazi, H. Zhao, D. F. Swearer, N. J. Hogan, L. Zhou, A. Alabastri, M. J. McClain, P. Nordlander and N. J. Halas, Nat. Commun., 2017, 8, 27 CrossRef.
- H. Wang, H. Rong, D. Wang, X. Li, E. Zhang, X. Wan, B. Bai, M. Xu, J. Liu, J. Liu, W. Chen and J. Zhang, Small, 2020, 16, 2000426 CrossRef CAS.
- Q. Zhai, S. Xie, W. Fan, Q. Zhang, Y. Wang, W. Deng and Y. Wang, Angew. Chem., Int. Ed., 2013, 52, 5776–5779 CrossRef CAS.
- Z. Xiong, Z. Lei, C.-C. Kuang, X. Chen, B. Gong, Y. Zhao, J. Zhang, C. Zheng and J. C. S. Wu, Appl. Catal., B, 2017, 202, 695–703 CrossRef CAS.
- C. Zhou, S. Wang, Z. Zhao, Z. Shi, S. Yan and Z. Zou, Adv. Funct. Mater., 2018, 28, 1801214 CrossRef.
- L. D. M. Torquato, F. A. C. Pastrian, J. A. L. Perini, K. Irikura, A. P. d. L. Batista, A. G. S. de Oliveira-Filho, S. I. Córdoba de Torresi and M. V. B. Zanoni, Appl. Catal., B, 2020, 261, 118221 CrossRef CAS.
- M. Jiang, C. Li, K. Huang, Y. Wang, J.-H. Liu, Z. Geng, X. Hou, J. Shi and S. Feng, ACS Appl. Mater. Interfaces, 2020, 12, 35113–35119 CrossRef CAS.
- F. Zhang, Y.-H. Li, M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, Appl. Catal., B, 2020, 268, 118380 CrossRef CAS.
- S. Ali, J. Lee, H. Kim, Y. Hwang, A. Razzaq, J.-W. Jung, C.-H. Cho and S.-I. In, Appl. Catal., B, 2020, 279, 119344 CrossRef CAS.
- X. Zhao, Y. Fan, W. Zhang, X. Zhang, D. Han, L. Niu and A. Ivaska, ACS Catal., 2020, 10, 6367–6376 CrossRef CAS.
- M. Sayed, L. Zhang and J. Yu, Chem. Eng. J., 2020, 397, 125390 CrossRef CAS.
- S. Jeong, G.-M. Kim, G.-S. Kang, C. Kim, H. Lee, W.-J. Kim, Y. K. Lee, S. Lee, H. Kim, H. K. Lim and D. C. Lee, J. Phys. Chem. C, 2019, 123, 29184–29191 CrossRef CAS.
- L. Yuan and Y.-J. Xu, Appl. Surf. Sci., 2015, 342, 154–167 CrossRef CAS.
- M.-Q. Yang and Y.-J. Xu, Nanoscale Horiz., 2016, 1, 185–200 RSC.
Footnote |
| † Both of these authors contribute equally in this work. |
| This journal is © The Royal Society of Chemistry 2021 |