
Sustainable S cathodes with synergic electrocatalysis for room-temperature Na–S batteries†
Hanwen
Liu
 a,
Wei-Hong
Lai
a,
Yaru
Liang
b,
Xin
Liang
c,
Zi-Chao
Yan
a,
Hui-Ling
Yang
a,
Yao-Jie
Lei
a,
Pei
Wei
d,
Si
Zhou
ad,
Qin-Fen
Gu
e,
Shu-Lei
Chou
a,
Hua Kun
Liu
a,
Shi Xue
Dou
a and
Yun-Xiao
Wang
*a
a,
Wei-Hong
Lai
a,
Yaru
Liang
b,
Xin
Liang
c,
Zi-Chao
Yan
a,
Hui-Ling
Yang
a,
Yao-Jie
Lei
a,
Pei
Wei
d,
Si
Zhou
ad,
Qin-Fen
Gu
e,
Shu-Lei
Chou
a,
Hua Kun
Liu
a,
Shi Xue
Dou
a and
Yun-Xiao
Wang
*a
aInstitute for Superconducting and Electronic Materials, Australian Institute of Innovative Materials, University of Wollongong, Innovation Campus, Squires Way, North Wollongong, NSW 2500, Australia. E-mail: yunxiao@uow.edu.au
bPowder Metallurgy Research Institute, State Key Laboratory of Powder Metallurgy, Central South University, Lushan South Road, Changsha, 410083, China
cSchool of Materials Science and Engineering, Engineering Research Center of High Performance Copper Alloy Materials and Processing, Ministry of Education, Hefei University of Technology, Hefei 230009, Anhui, P. R. China
dKey Laboratory of Materials Modification by Laser, Ion and Electron Beams, Dalian University of Technology, Ministry of Education, Dalian 116024, China
eAustralian Synchrotron, 800 Blackburn Road, Clayton, VIC 3168, Australia
First published on 20th November 2020
Abstract
A novel sulfiphilic host is reported with single Co atoms (Co1) and ZnS quantum dots (ZnS-QDs) (∼10 nm) grown in N-doped carbon microparticles. The introduction of Co cations into the precursor is crucial, because it leads to the dispersion of single-atom Co on the carbon matrix, decreasing the size and improving the dispersion of the resultant ZnS-QDs as well. Benefitting from the resilient and stable carbon framework, the single-atom Co and small ZnS-QDs synergically electro-catalyze the S redox process, which can greatly enhance sodium polysulfide conversion and reduction of non-conductive Na2S. The refined S cathode achieves remarkable cycling capability (640 mA h g−1 after 500 cycles at 0.1 A g−1), enhanced capacity retention, and outstanding energy density (541 W h kg−1), giving it great promise for scientific research and practical applications.
Introduction
Na, as a low-cost and abundant alternative to Li, has attracted tremendous research attention towards rechargeable sodium-storage technologies beyond the cell chemistry of the lithium-ion counterparts. The impressive electrochemical characteristics as well as inexpensive and abundant resources of S and Na enable room-temperature Na–S (RT Na–S) batteries to be promising for large-scale energy storage. Compared to Li–S batteries, RT Na–S batteries show comparable theoretical capacity (1675 mA h g−1) and energy density (1274 W h kg−1).1 However, studies on RT Na–S batteries are still in an infancy stage and many issues on cathodes, anodes, and electrolytes are still unsolved. There is still a long way for Na–S batteries to catch up with the Li counterparts. The essential electrochemical performance, including specific energy, rate capability, cycling life, and safety issues, is even not satisfactory in coin-cells for RT Na–S batteries.2 In comparison, recent studies on Li–S batteries have made significant progress toward practical applications with the pouch-type scale.3–5 Learning from successful experience of Li–S batteries, current research on RT Na–S batteries mainly focuses on solving the slow redox kinetics of sulfur and the dissolution of sodium polysulfide intermediates (NaPSs).6–9 Three principles can be adopted: (1) a host with good conductivity is important to improve the sluggish kinetics of sulfur; (2) a porous structure with a medium specific area and good volume stability can effectively increase the volumetric energy density and cycling life; (3) suitable catalysts with high adsorption toward polysulfides can limit the shuttle effect, thereby achieving high energy density and cycling life. In contrast to Li–S batteries, the S hosts in RT Na–S batteries are required to be multifunctional, which are expected not only to improve the conductivity of S and Na2S, but also to be enclosed to confine the highly solvable NaPSs. More importantly, the S redox reaction in RT NaS batteries is much kinetically sluggish, and the addition of electrocatalysts is essential. One of the most effective strategies is to capture NaPSs in carbon matrices with the addition of catalysts.10 Porous carbon can store sulfur and improve its conductivity, while catalysts are important for the adsorption and conversion of NaPSs, thus ensuring stable reversible capacities.11 Compared with the different types of catalysts such as transition metals12 and metal oxides,13 metal sulfide catalysts have achieved outstanding performance due to their relatively high bonding energies with NaPSs.14 This has been proven by many studies. Yan et al. applied FeS2 in the S host and successfully improved the slow kinetics of sulfur.15 NiS2 was doped into porous carbon nanotubes and effectively limited the dissolution of NaPSs.16 The strong adsorption capability of metal sulfide toward NaPSs significantly alleviates the loss of active materials in RT Na–S batteries. Nevertheless, the electrocatalytic activity of metal sulfide is greatly hindered by their intrinsic poor conductivity.17,18 To improve their conductivity, reducing the size of metal sulfide is one of the best tactics.19 Introducing single-atom metals is also a promising strategy to improve the overall conductivity and accelerate the charge transfer of S cathodes.20 Moreover, single-atom metals can also catalyse the NaPSs conversion in RT Na–S batteries.21 Recently, Chen et al. utilized Ni atoms to modify the separator and found that its Ni–N4 structure could reversibly catalyse the conversion of NaPSs.22 Zhang and co-workers applied atomic Co in the S cathode, in which the Co atoms accelerated the reduction reaction from Na2S4 to Na2S.23 Thus, it is a promising approach to synergistically combine metal sulfides and single-atom metal on the same carbon support. Combined with the high conductivity and electrocatalysis of a single atom metal, a polar sulfide metal with strong bonding ability toward NaPSs is expected to make much enhanced Na-storage properties of S cathodes for RT Na–S batteries.24Herein, we present a novel sulfiphilic host consisting of atomic Co and ZnS quantum dots grown on a hierarchical carbon support. The doping of Co atoms into the C matrix not only can improve the conductivity of the electrode, but also is responsible for the decreased particle size and improved dispersion of ZnS quantum dots (∼10 nm). Overall, the hierarchical carbon support with high surface area (1012 m2 g−1) serves as a container for S species; the entangling catalysis of polar ZnS QDs and single-atom Co guarantees efficient NaPSs conversion via enhanced polar–polar interactions. In situ synchrotron X-ray diffraction (in situ XRD) and theoretical calculations were employed to understand the electrocatalytic functions of the constructed host for the S cathode. In contrast to Co single atoms alone, the ZnS catalyst is more effective for optimizing the S cathode. This is because it shows strong chemical bonding and fast redox kinetics for the conversion reaction, leading to high utilization of NaPSs. Furthermore, when observed in situ via transmission electron microscopy (TEM), the S host shows reversible volume changes during the sodiation/desodiation processes, indicating the prolonged cycling stability of the S cathode. As a result, the constructed Co1–ZnS/C@S cathode can exhibit outstanding cycling performance with a capacity of 640 mA h g−1 after 500 cycles (0.1 A g−1) and a remarkable energy density of 541 mA h g−1 at 0.1 A g−1, rendering this composite great promise for scientific research and practical applications.
Results and discussion
To understand the size variations of ZnS-QDs due to the modification of single-atom Co1, a ZnS/C@S sample without sing-atom Co1 doping was prepared as illustrated in ESI.† As displayed in Fig. 1a and b, the scanning electron microscopic (SEM) and scanning transmission electron microscopic (STEM) images show the general structure of ZnS/C@S: a homogeneous distribution of ZnS coupled with nanoparticles is embedded on the microspheric skeleton. In comparison, ZnS quantum dots are undetectable in a control sample of C@S (Fig. S1 in the ESI†). The particle size of ZnS is around 20 nm, and the selected area electron diffraction (SAED) pattern further reveals the (111), (101), (001), and (110) crystalline planes of ZnS (Fig. S2e†).25 To calculate the particle size of ZnS in ZnS/C@S, we randomly took several images (Fig. S2†) of ZnS/C@S, which suggests that the ZnS particles are mainly over 15 nm (86%) (inset Fig. 1b). EDS mapping of ZnS/C@S is also shown in Fig. S3.†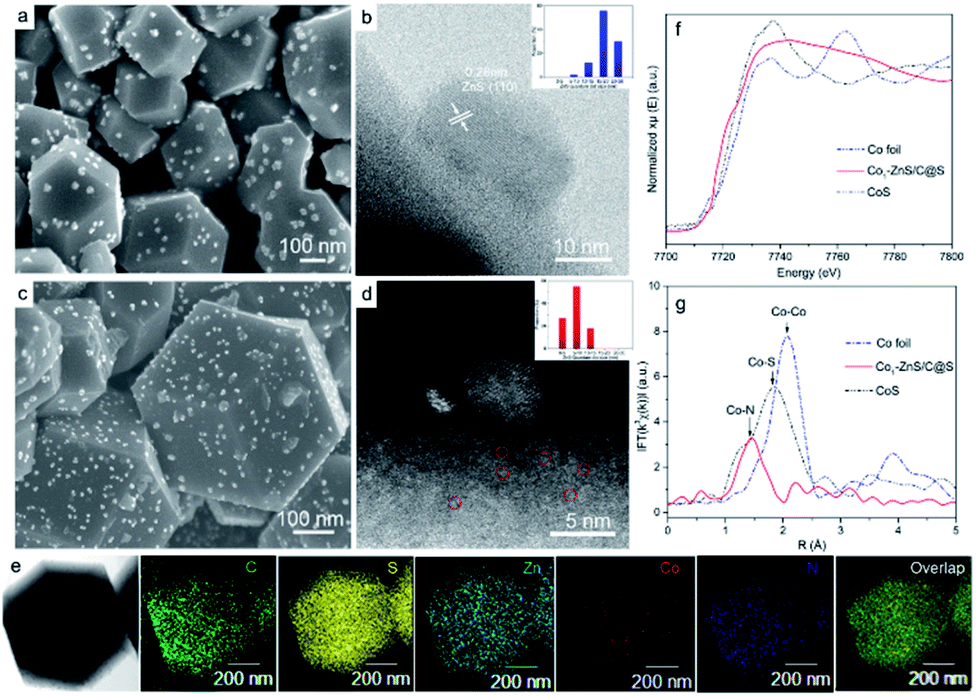 | ||
| Fig. 1 (a) Representative SEM image and (b) STEM image of ZnS/C@S. (c) SEM image and (d) STEM image of Co1–ZnS/C@S. (e) EDS mapping of Co1–ZnS/C@S. (f) Co K-edge XANES spectra and (g) EXAFS spectra of Co foil, Co1–ZnS/C@S and CoS. The insets of (b) and (d) show the corresponding particle size distributions. | ||
Fig. 1c and d show images of Co1–ZnS/C@S. With the doping of atomic Co, the particle size of the ZnS quantum dots shrinks according to our counting (Fig. S4†). The size of the ZnS quantum dots is mainly below 10 nm (83%). The Co1 doping originates from Co2+, which takes the place of part of the Zn2+ and coordinates with N (Fig. S5†). During the sulfuration process, neighboring Zn atoms gather and react with S, resulting in ZnS quantum dots, but the isolated Co2+ is confined by nitrogen bond and becomes atomic Co. According to the energy-dispersive X-ray spectroscopy (EDS) mapping (Fig. 1e), the five elements are finely dispersed in a nanoparticle skeleton. The elemental calculations confirm a large proportion of S, which accounts for 73.9% of the total mass, while atomic Co only accounts for 1.8% but evenly dispersed in a nanoparticle skeleton (Fig. S6†). A large number of bright dots (∼3 Å) are randomly distributed in the carbon matrix, which also exist in the sample of Co1/C@S (Fig. S7 and S8†), suggesting the existence of Co1 atom. The single Co atoms are connected with the N-doped carbon matrix to maintain the stable energy state that exists in the nanoparticles.26
Thermogravimetric analysis (TGA) further confirms a large amount of S contained in Co1/C@S, Co1–ZnS/C@S and ZnS/C@S with 60, 65, and 62 wt%, respectively (Fig. S9†). Once sulfur has been removed by immersing the samples in CS2, the Brunauer–Emmett–Teller (BET) analysis further confirms the specific surface areas of Co1/C, Co1–ZnS/C and ZnS/C with 756, 1012 and 928 m2 g−1 respectively (Fig. S10†). The high surface area makes sure considerable porous space for storing sulfur, which is fundamental for the cathode host in RT Na–S batteries. According to X-ray powder diffraction (XRD), sulfur existing in the three samples is crystalline S (PDF: 04-012-1107) (Fig. S11†).27 Besides, X-ray photoelectron spectroscopy (XPS) in the Zn 2p region (Fig. S12†) shows that Co1–ZnS/C@S has two peaks located at 1044.5 and 1021.4 eV, which are assigned to Zn 2p1/2 and Zn 2p3/2 of ZnS, respectively.28 In the S 2p region (Fig. S13†), three peaks at 167.9, 165.2, and 163.3 eV are assigned to sulfide, and S 2p1/2, and S 2p3/2 respectively for Co1/C@S.29 After doping with ZnS, two new peaks situated at 169.8 and 164.2 eV correspond to S2− 2p1/2 and S2− 2p3/2, respectively. Raman and Fourier-transform infrared (FT-IR) spectroscopy also confirm the presence of ZnS in the Co1–ZnS/C@S (Fig. S14 and S15†).30 In the case of atomic Co, the X-ray adsorption near-edge structure (XANES) spectra (Fig. 1f) show that the position of Co1–ZnS/C@S is located between those of Co foil and CoS, indicating that the valence state of atomic Co is situated between that of Co0 and Co2+.31 The extended X-ray adsorption fine structure (EXAFS) spectrum exhibits Co–N coordination with a peak at 1.42 Å for Co1–ZnS/C@S, while there are Co–Co and Co–S bonds at 2.15 Å and 1.84 Å for Co foil and CoS, respectively (Fig. 1g).32 The assembled coin cells were tested in the voltage window of 0.8–2.8 V, and the mass of sulfur was calculated according to the TGA result. The Co1–ZnS/C@S electrode evidently shows the highest coulombic efficiency among the four samples during the first 150 cycles. Consequently, the Co1–ZnS/C@S delivers better capacity retention and cycling stability, achieving a remarkable capacity of 640 mA h g−1 over 500 cycles at a current density of 0.1 A g−1 (Fig. 2a).
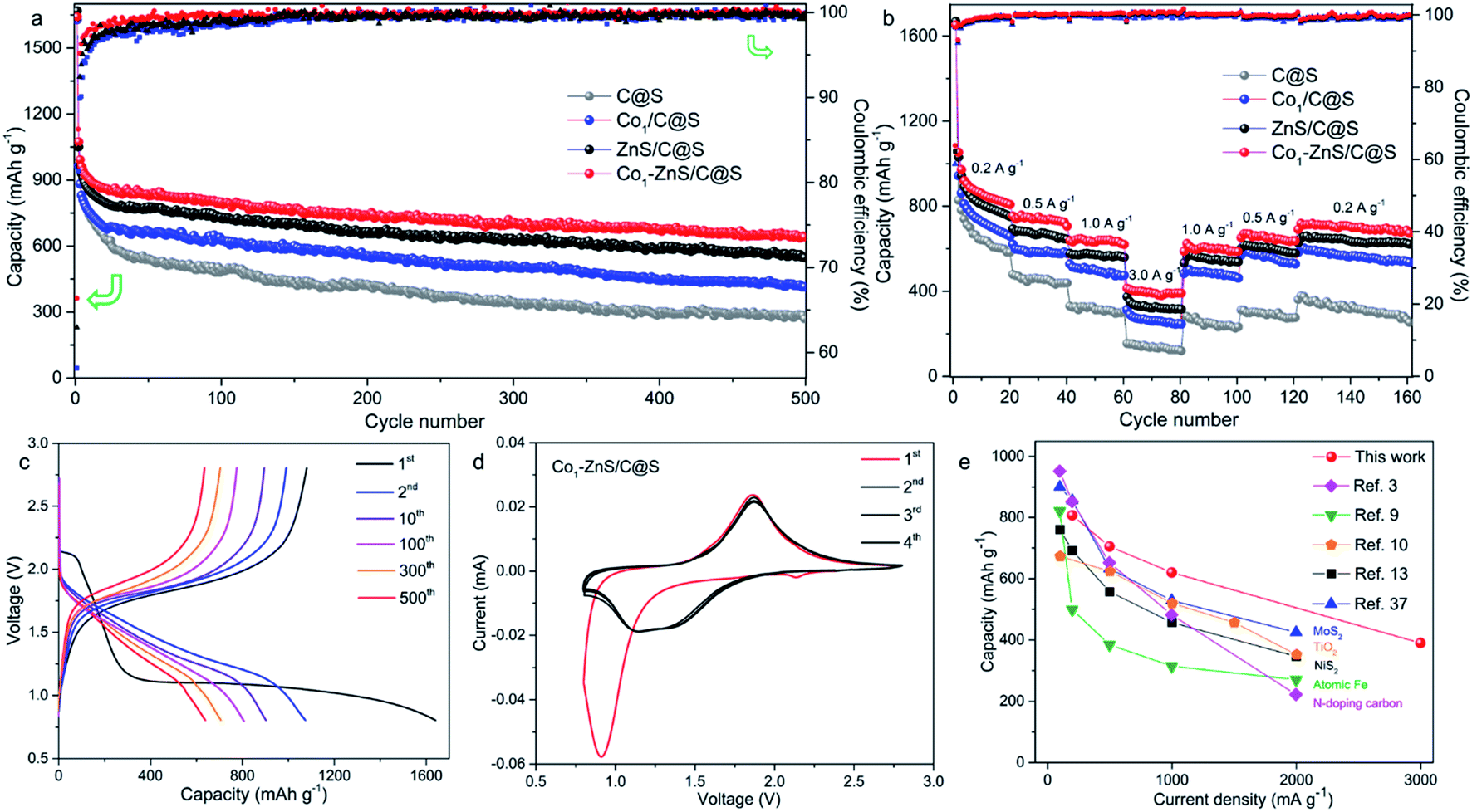 | ||
| Fig. 2 (a) Cycling performance at 0.1 A g−1 and (b) rate performance of C@S, Co1/C@S, ZnS/C@S, and Co1–ZnS/C@S. (c) Discharge/charge curves of Co1–ZnS/C@S at 0.1 A g−1. (d) Cyclic voltammetry curves of Co1–ZnS/C@S at 0.1 mV s−1. (e) Comparisons of the rate capacities of previously reported room-temperature sodium-sulfur batteries with our work. | ||
The excellent cycling performance of the Co1–ZnS/C@S electrode is reliably attributed to the strong chemical bonding between NaPSs and ZnS quantum dots. Meanwhile, the doping with Co atoms can decrease the particle size of ZnS quantum dots and effectively improve the conductivity, according to the electrochemical impedance spectroscopy (EIS) (Fig. S17†). Moreover, the Co1–ZnS/C@S electrode also presents excellent rate performance, delivering a reversible capacity of 807, 705, 620, and 390 mA h g−1 at current densities of 0.2, 0.5, 1.0, and 3.0 A g−1, respectively (Fig. 2b). Upon reverting to 0.2 A g−1, it shows a fully restored capacity of 680 mA h g−1 after 160 cycles. When we investigate the cycling process, the charge/discharge plateau (Fig. 2c) can be clearly distinguished for Co1–ZnS/C@S electrode at 0.1 A g−1. In the first cycle, discharge plateaus above 2.0 V are attributed to the reduction from sulfur to long-chain polysulfides. As the voltage drops below 1.5 V, the plateau corresponds to the formation of short-chain NaPSs. Interestingly, the plateau above 2.0 V disappears in the following cycles and a well-defined plateau around 1.5 V becomes clear and is highly repeatable. This suggests that the transition between long-chain and short-chain NaPSs is highly reversible, in which most of the reversible capacity is generated at a scan rate of 0.1 mV s−1, as shown in Fig. 2d. There are two prominent peaks centered at 2.2 and 0.9 V during the first cathodic scan. The peak around 2.2 V corresponds to the transition from crystalline sulfur to long-chain NaPSs, while the sharp peak at 0.9 V corresponds to the formation of short-chain NaPSs during further sodiation. In the following three cathodic scans, two major repeatable reduction peaks appear at 1.4 and 1.1 V, which correspond to the conversion from long-chain to short-chain NaPSs and the formation of Na2S, respectively. The highly repeatable scans without current attenuation indicate a reversible reaction mechanism with high capacity retention in this system. A comparison of the rate capability of Co1–ZnS/C@S with the state-of-the-art electrode reported in the literature is presented in Fig. 2e; such an exceedingly high rate capability of RT-Na/S batteries has promising applications. Energy density is another important parameter to evaluate the performance of cathode materials. In the voltage range from 0.8 to 2.8 V, the Co1–ZnS/C@S cathode whose energy density can reach 541 W h kg−1 after 500 cycles shows promising energy density retention of 60.1% after activation (Fig. S18†). Some concerns may focus on the role of ZnS-QDs, which may generate the capacity instead of S. However, ZnS-QDs account for 8% mass of the Co1–ZnS/C@S cathode and hardly generate sufficient capacity (Fig. S6†). To explain this point, we prepare Co1–ZnS/C by evaporating S out of Co1–ZnS/C@S via annealing the sample at 275 °C under N2 flow. The resulting Co1–ZnS/C electrode shows only 25 mA h g−1 reversible capacity (Fig. S19†) in the voltage range from 0.8 to 2.8 V, indicating the ZnS-QD function as a catalyst within the Co1–ZnS/C@S cathode for accelerating the reaction of soluble NaPSs. To gain chemical insight into the highly reversible reactions of Co1–ZnS/C@S, in situ synchrotron XRD (λ = 0.688 Å) was carried out on the Powder Diffraction Beamline (Australian Synchrotron) to investigate the charge/discharge products and intermediate phases (Fig. 3a).33
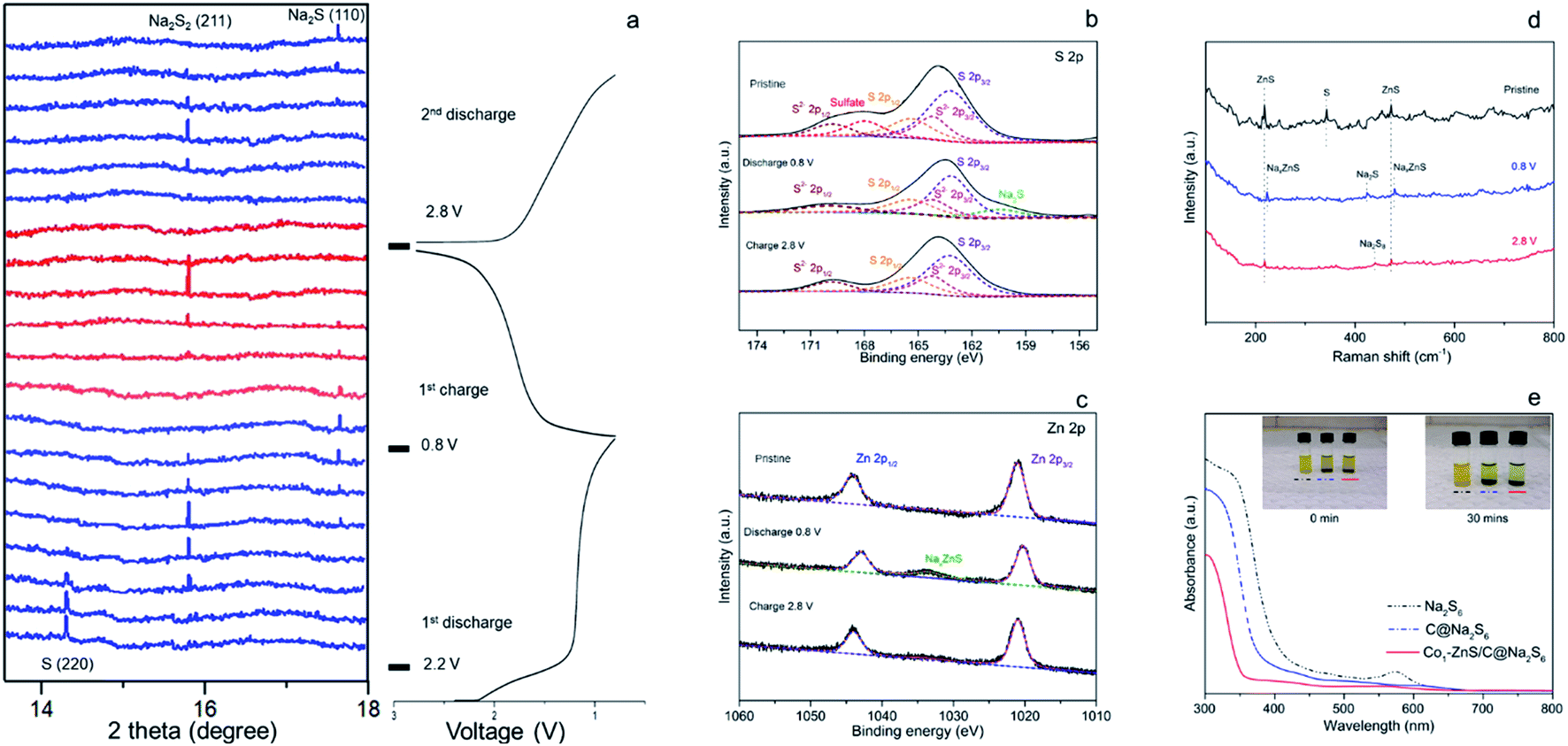 | ||
| Fig. 3 (a) In situ synchrotron X-ray diffraction patterns of Co1–ZnS/C@S with the initial galvanostatic charge/discharge curves at 0.5 A g−1. XPS spectra of Co1–ZnS/C@S in the (b) S 2p and (c) Zn 2p regions when half coin-cells were charged/discharged to different voltages. (d) Raman spectra of Co1–ZnS/C@S when the half coin-cells were charged/discharged to different voltages. (e) Photographs (inset) and ultraviolet-visible spectra of the Na2S6 solution before and after exposure to C and Co1–ZnS/C. | ||
There is a strong peak in the pristine cell located at 14.5°, which can be indexed to the (013) plane of sulfur (PDF: 04-012-1107).34 The transition from S to long-chain NaPSs starts from 2.2 V.35 During this voltage range, no indication of long-chain NaPSs can be indexed to the XRD pattern indicative of a fast transition from long-chain NaPSs to short-chain NaPSs. As the voltage drops down to 1.2 V, a new peak at 15.2 V appears with the sacrifice of sulfur. This new peak, indexed to the (100) plane of Na2S2 (PDF: 01-081-1771), becomes strong and reaches its maximum at the voltage of 1.0 V.13,36 However, the peak of Na2S2 gradually fades as the signal of Na2S (PDF: 01-070-7161) emerges with further discharging to 0.8 V, which signifies the final discharged product in the Co1–ZnS/C@S cathode.33,37
Compared with previous reports, no signal referring to long-chain polysulfides and Na2S4 can be traced according to in situ XRD, indicative of a fast transition of these mid-products.16,23 The fast transition of soluble NaPSs can effectively prevent the shuttle effect; therefore, the Co1–ZnS/C@S cathode achieves ultra-high capacity in the first discharge. Such a high efficiency of long-chain NaPS transition is benefited from the catalysis of ZnS-QDs, which will be further studied by the means of DFT calculation. Combined with previous CV analysis, we confirm that sulfur mainly undergoes three intermediate phases in the Co1–ZnS/C@S cathode from long-chain NaPSs to Na2S2 and finally Na2S in the initial discharge.38
During the charging process, the peak of Na2S2 appears again, suggesting a good reversibility in this system. As the voltage is charged back to 2.8 V, peaks of non-conductive Na2S2 and Na2S are hardly seen, indicative of the outstanding conductivity in the Co1–ZnS/C@S electrode. The shuttle effect of soluble NaPSs and the irreversibility of poor conductive Na2S are the main reasons for low reversible capacity in RT Na–S batteries.39 However, owing to the co-catalysis of ZnS-QDs and the Co atom, the shuttle effect is significantly prevented by ZnS-QDs while the irreversibility is improved by the conductive Co atom. Therefore, the Co1–ZnS/C@S electrode shows good reversibility in chemical changes, which is essential to maintain outstanding cyclability in RT Na–S batteries.
To further prove the reversible reaction, ex situ XPS was further applied to study the changes in the binding energy in the S 2p (Fig. 3b) and Zn 2p (Fig. 3c) regions. Pristine Co1–ZnS/C@S shows five peaks in the S 2p region located at 169.8, 168.0, 165.4, 164.2 and 163.2 eV, which are assigned to S2− 2p1/2, soluble sulfate, S 2p1/2, S2− 2p3/2 and S 2p3/2, respectively.40 As the voltage of the Co1–ZnS/C@S electrode dropped to 0.8 V, Na+ insertion leads to the formation of Na2S, which is reflected in a new peak at 160.4 eV, while soluble sulfate dissolves into the electrolyte.22 This result is coordinated with in situ XRD. As voltage is charged back to 2.8 V, the intensity of Na2S falls off, indicating that nonconductive Na2S has been reversibly transformed into long-chain NaPSs with an increasing intensity of S 2p1/2 and S 2p3/2.41 Previous reports on RT Na–S batteries show that Na2S is difficult to reverse to long-chain NaPSs because of poor conductivity.42 Thanks to good dispersion of the Co atom on the Co1–ZnS/C@S particle (Fig. 1e), the conductivity of Co1–ZnS/C@S is significantly improved (Fig. S17†); therefore, nonconductive Na2S can be reversibly transformed into long-chain NaPSs. Regarding the Zn 2p region, two peaks at 1044.1 and 1021.9 eV correspond to Zn 2p1/2 and Zn 2p3/2, respectively. When the coin cell is discharged to 0.8 V, a new peak at 1033.6 eV arises while the intensities of Zn 2p1/2 and Zn 2p3/2 are a little bit decreased. Since this peak appeared with the insertion of Na+, it should be assigned to NaxZnS, which explains where the reversible capacity of Co1–ZnS/C comes from (Fig. S19†).43 As voltage charges back to 2.8 V, the signal of NaxZnS is disappeared, which suggests a reversible reaction for Na-ion inserting/extracting in the ZnS lattice.
Although previous chemical tests have witnessed a reversibly chemical reaction in the Co1–ZnS/C@S electrode, the final product after charging back to 2.8 V is still obscured. To find out the specific composition of long-chain NaPSs in 2.8 V, ex situ Raman spectra were applied. Fig. 3d shows three peaks corresponding to ZnS and S of Co1–ZnS/C@S in the pristine stage. As the voltage drops down to 0.8 V, the peaks of ZnS slightly shift upward, which may correspond to Na-ion insertion. As the voltage charged back to 2.8 V, they shift backward to the pristine stage, which is consistent with the sodium insertion and extraction processes.44 Meanwhile, the chemical changes from S to Na2S and Na2S8 during discharge/charge processes are proved in the Raman test. Thanks to Co atom, non-conductive Na2S can be fully transformed to Na2S8 while ZnS-QDs offer strong adsorption toward soluble Na2S8, preventing the shuttle effect. Furthermore, the strong adsorption of the ZnS-QDs toward NaPSs is evidenced by the ultraviolet-visible (UV-vis) spectra (Fig. 3e).
The Na2S6 solution exposed to Co1–ZnS/C powder for 30 min exhibits this more clearly than that after exposure to blank C, suggesting the effective adsorption capability of ZnS quantum dots towards NaPSs. Based on the above-mentioned studies, the conclusion can be reached that the benefit of ZnS-QDs contributes to adsorb soluble NaPSs and suppresses the shuttle effect. The strong adsorption is further visualized in the STEM images (Fig. 4a). After 500 cycles at 0.1 A g−1, the Co1–ZnS/C@S electrode is discharged to 0.8 V. The resulting particle is dispersed in the electrolyte solvent and studied by the STEM test. The Co1–ZnS/C@S particle retains a stable microstructure, and there are numerous crystals dispersed on the surface, which have been further observed by EDS mapping (Fig. S20†). According to the crystal plane spacing, the planes of ZnS(002), NaxZnS(002), and Na2S(110) are indexed.45 To further illustrate the strong confining effect in the Co1–ZnS/C@S electrode, we disassembled the different battery cells after 500 cycles in an argon-filled glove box and took a close look at the separators (Fig. S21†). The separator in C@S electrode shows obvious yellow colour indicative of a strong shuttle effect of polysulfides. The Co1/C@S separator only shows slight yellow color around the center area. For ZnS/C@S and Co1–ZnS/C@S, the separator is almost colourless, indicating the strong polysulfide entrapment of ZnS/C and Co1–ZnS/C hosts.
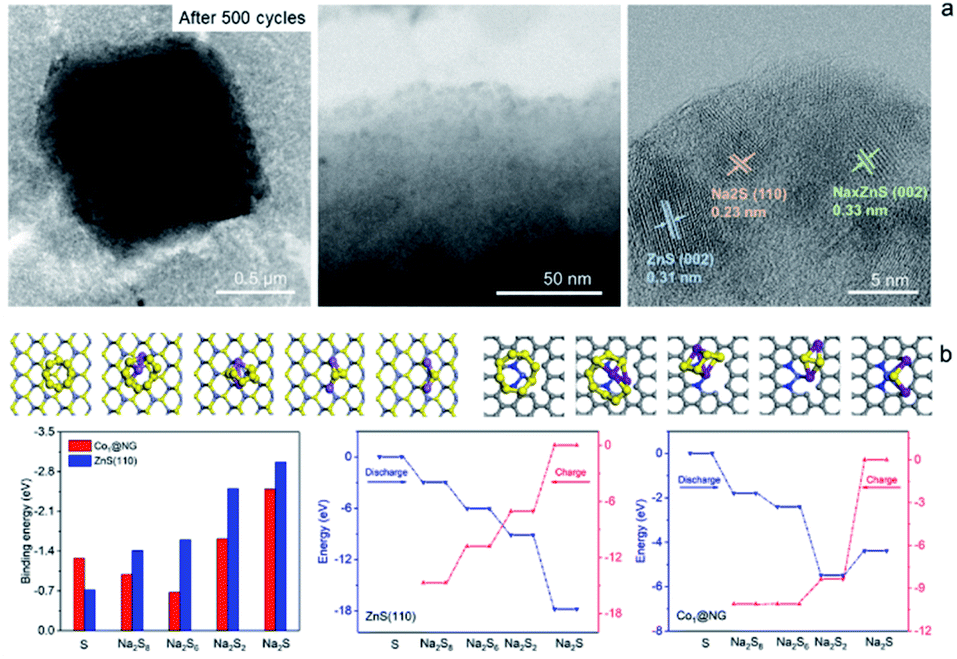 | ||
| Fig. 4 (a) STEM images of Co1–ZnS/C@S after 500 cycles. (b) Top panels: atomic structures of polysulfides adsorbed on ZnS (left) and Co1@NG (right); bottom panels: Comparison of binding energies of polysulfides on ZnS(110) and Co1@NG. Comparison of energy diagrams of charging/discharging processes on ZnS(110) and Co1@NG. | ||
In order to gain atomistic insight into the mechanism behind the ZnS-QDs and polysulfides, DFT calculations were performed to explore their composite structure and determine the role of ZnS-QDs in the adsorption and transformation of polysulfides. As the ZnS-QDs synthesized in our experiment have diameters of a few nanometers, we consider slab models of ZnS crystals in the zinc blende structure. To explore the catalytic activities of ZnS-QDs towards polysulfide conversion, we consider the (110) surface of ZnS, which are the most stable facets. The structures of various Na2Sx (x = 1, 2, 6, 8) adsorbed on these model structures are simulated, and their binding energies are calculated as:
| Eb = E(catalyst+Na2Sx) − ENa2Sx − Ecatalyst |
 | ||
| Fig. 5 In situ TEM images of Co1–ZnS/C@S during (a) sodiation and (b) desodiation processes. (c) In situ SAED patterns of Co1–ZnS/C@S at various states. | ||
In addition, a new diffraction spot with a recognizable reflection of the (002) planes of NaxZnS is observed after 189 s, when Na ions are introduced into Co1–ZnS/C@S. Traces of NaxZnS are found to coexist with ZnS in the sodiation process, demonstrating that Na+ partially intercalates into ZnS. While Na is extracted out of the nanoparticle (Fig. 5b), the diffraction spot of NaxZnS also disappears, along with a reduction in volume from 986 nm to 908 nm.46 This intercalation-type ZnS quantum dots therefore have fast kinetics, since NaxZnS has high polaron mobility.15 Meanwhile, Na2S disappears after sodium extraction, indicating the benefit of conductive Co1 atom catalyzing the transition of non-conductive Na2S. Consequently, the affinity for NaPSs of the novel host can be attributed to the intercalation-type ZnS quantum dots, conductive Co atoms, and stable carbon nanoparticles, thereby suppressing the shuttling effect and resulting in stable cyclability.
Conclusion
In summary, the as-prepared Co1–ZnS/C@S features high surface area (1012 m2 g−1), a small particle size but well dispersion of ZnS quantum dot (∼10 nm), and conductive atomic Co doping. In situ synchrotron XRD, in situ TEM, and X-ray adsorption spectroscopy (XAS) results confirm that the hierarchical microparticles coupled with the effective ZnS quantum dots and conductive single-atom Co1, which simultaneously endows the Co1–ZnS/C@S cathode with outstanding reversible capacity and long lifespan. Significantly, the soluble NaPSs can be strongly adsorbed and predominantly catalyzed on the polar surfaces of the ZnS quantum dots, while the conductive Co1 atom catalyzes the transition of non-conductive Na2S during charging. Therefore, Co1–ZnS/C@S undergoes a fast sodiation process to form Na2S2 intermediate with low diffusion barriers and to subsequently transform it into the final discharge product, Na2S, preventing the active material from dissolving in the carbonate electrolyte. Consequently, the S cathode achieves much enhanced Na-storage properties, in terms of high accessible capacity (640 mA h g−1 after 500 cycles), long cycling lifespan, and excellent energy density (541 W h kg−1). By introducing atomic Co doping and downsizing the ZnS-QD catalyst in the host, this work will open up a new avenue to optimize S cathodes for superior RT Na–S batteries.Experimental
Chemicals
Analytical grade zinc nitrate hexahydrate (Zn(NO3)2·6H2O), cobalt nitrate hexahydrate (Co(NO3)2·6H2O), and 2-methylimidazole were obtained from Sigma-Aldrich.Characterization
The morphologies of the samples were investigated by SEM (JEOL 7500) and STEM (JEOL ARM-200F, 200 keV). The XRD patterns were collected by powder XRD (GBC MMA diffractometer) with Cu Kα radiation at a scan rate of 1.5° min−1. XPS measurements were carried out using Al Kα radiation and a fixed analyzer transmission mode: the pass energy was 60 eV for the survey spectra and 20 eV for the specific elements. The electrochemical tests were conducted by assembling coin-type half-cells in an argon-filled glove box. The slurry was prepared by fully mixing 80 wt% active materials, 10 wt% carbon black, and 10 wt% carboxymethyl cellulose (CMC) in an appropriate amount of water using a planetary mixer (KK-250S). Then, the obtained slurry was pasted on an Al foil using a doctor blade with a thickness of 150 μm, which was followed by drying at 60 °C in a vacuum oven overnight. The working electrode was prepared by punching the electrode film into discs of 0.97 cm diameter with ∼1 mg sulfur in each electrode. The sodium foil was employed as both reference and counter electrodes. The electrodes were separated using a glass fiber separator. The electrolyte included 1.0 M NaClO4 in 95 wt% diethyl carbonate (DEC)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethylene carbonate (EC) in a volume ratio of 1:1 and 5 wt% fluoroethylene carbonate (FEC) additive (DEC & EC + 5 wt% FEC). About 50 μl electrolyte is dropped onto each coin cell. The electrochemical performance was tested using a LAND Battery Tester with a voltage window of 0.8–2.8 V. All the capacities of cells have been normalized based on the weight of sulfur. CV was performed using a Biologic VMP-3 electrochemical workstation. The Na2S6 solution was prepared by adding sulfur and Na2S powder into the DEC solvent under stirring for 2 h.
:ethylene carbonate (EC) in a volume ratio of 1:1 and 5 wt% fluoroethylene carbonate (FEC) additive (DEC & EC + 5 wt% FEC). About 50 μl electrolyte is dropped onto each coin cell. The electrochemical performance was tested using a LAND Battery Tester with a voltage window of 0.8–2.8 V. All the capacities of cells have been normalized based on the weight of sulfur. CV was performed using a Biologic VMP-3 electrochemical workstation. The Na2S6 solution was prepared by adding sulfur and Na2S powder into the DEC solvent under stirring for 2 h.
In the in situ synchrotron XRD measurements, the cells were similar to the above-mentioned coin cells for electrochemical performance testing. To enhance the diffraction peak intensity, a thicker layer of cathode material was loaded on the Cu foil, with a loading up to 5 mg cm−2. To guarantee that the X-ray beams could penetrate the whole cell and that the electrochemical reactions could be monitored, three holes of 4 mm diameter were punched in the negative and positive caps as well as the spacer. Then, the Kapton film (only showing low-intensity responses in XRD patterns) was used to cover the holes in the negative and positive caps, and AB glue was used for complete sealing. The charged/discharged process was conducted with a battery test system (Neware) that was connected to the cell.
Preparation of C@S
In a typical procedure, Zn(NO3)2·6H2O (2.38 g) and 1.31 g 2-methylimidazole were dissolved in 200 ml methanol and stirred for 5 min. After aging for 12 h, the as-obtained precipitates were centrifuged and washed several times with ethanol and dried in vacuum at 70 °C overnight. The as-obtained white powder was annealed at 1000 °C for 5 h at a heating rate of 2 °C min−1 in a N2 atmosphere. After cooling down, the black powder was mixed with sulfur in a weight ratio of 1:2 and sealed in a glass tube. The tube was heated at 155 °C for 12 h and 275 °C for 1 h resulting in C@S.
Preparation of Co1/C@S
In a typical procedure, Zn(NO3)2·6H2O (0.87 g), Co(NO3)2·6H2O (0.89 g) and 1.2 g 2-methylimidazole were dissolved in 70 ml methanol, stirred for 5 min, transferred into a 100 ml Teflon-lined stainless-steel autoclave and then heated at 120 °C for 4 h. The as-obtained precipitates were centrifuged and washed several times with ethanol and dried in vacuum at 70 °C overnight. The as-obtained powder was annealed at 1000 °C for 5 h at a heating rate of 2 °C min−1 in a N2 atmosphere. After cooling down, the black powder was mixed with sulfur in a weight ratio of 1:3 and sealed in a glass tube. The tube was heated at 155 °C for 12 h and 275 °C for 1 h resulting in Co1/C@S.
Preparation of Co1–ZnS/C@S
In a typical procedure, Zn(NO3)2·6H2O (0.87 g), Co(NO3)2·6H2O (0.89 g) and 1.2 g 2-methylimidazole were dissolved in 70 ml methanol, stirred for 5 min, transferred into a 100 ml Teflon-lined stainless-steel autoclave and then heated at 120 °C for 4 h. The as-obtained precipitates were centrifuged and washed several times with ethanol and dried in vacuum at 70 °C overnight. The as-obtained powder was annealed at 800 °C for 2 h at a heating rate of 2 °C min−1 in an Ar atmosphere. After cooling down, the black powder was mixed with sulfur in a weight ratio of 1:3 and sealed in a glass tube. The tube was heated at 155 °C for 12 h and 275 °C for 1 h resulting in Co1–ZnS/C@S.
Preparation of ZnS/C@S
In a typical procedure, Zn(NO3)2·6H2O (2.38 g) and 1.31 g 2-methylimidazole were dissolved in 200 ml methanol and stirred for 5 min. After aging for 12 h, the as-obtained precipitates were centrifuged and washed several times with ethanol and dried in vacuum at 70 °C overnight. The as-obtained white powder was annealed at 800 °C for 2 h at a heating rate of 2 °C min−1 in an Ar atmosphere. After cooling down, the black powder was mixed with sulfur in a weight ratio of 1:3 and sealed in a glass tube. The tube was heated at 155 °C for 12 h and 275 °C for 1 h, resulting in ZnS/C@S.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Australian Research Council (ARC) (DE170100928) and the Australian Renewable Energy Agency (ARENA) Project (G00849) and ARC DP170101467. The authors acknowledge the use of the facilities at the UOW Electron Microscopy Center (LE0882813 and LE0237478) and Dr Tania Silver for critical reading of the manuscript.Notes and references
- Y. X. Wang, B. Zhang, W. Lai, Y. Xu, S. L. Chou, H. K. Liu and S. X. Dou, Adv. Energy Mater., 2017, 7, 1602829 CrossRef.
- Y. X. Wang, W. H. Lai, Y. X. Wang, S. L. Chou, X. Ai, H. Yang and Y. Cao, Angew. Chem., Int. Ed., 2019, 58, 18324–18337 CrossRef CAS PubMed.
- W. Xue, Z. Shi, L. Suo, C. Wang, Z. Wang, H. Wang, K. P. So, A. Maurano, D. Yu and Y. Chen, Nat. Energy, 2019, 4, 374–382 CrossRef CAS.
- C. Niu, H. Lee, S. Chen, Q. Li, J. Du, W. Xu, J.-G. Zhang, M. S. Whittingham, J. Xiao and J. Liu, Nat. Energy, 2019, 4, 551–559 CrossRef CAS.
- H. Wang, B. D. Adams, H. Pan, L. Zhang, K. S. Han, L. Estevez, D. Lu, H. Jia, J. Feng and J. Guo, Adv. Energy Mater., 2018, 8, 1800590 CrossRef.
- Y.-M. Chen, W. Liang, S. Li, F. Zou, S. M. Bhaway, Z. Qiang, M. Gao, B. D. Vogt and Y. Zhu, J. Mater. Chem. A, 2016, 4, 12471–12478 RSC.
- Q. Lu, X. Wang, J. Cao, C. Chen, K. Chen, Z. Zhao, Z. Niu and J. Chen, Energy Storage Materials, 2017, 8, 77–84 CrossRef.
- Z. Qiang, Y.-M. Chen, Y. Xia, W. Liang, Y. Zhu and B. D. Vogt, Nano Energy, 2017, 32, 59–66 CrossRef CAS.
- S. Wei, S. Xu, A. Agrawral, S. Choudhury, Y. Lu, Z. Tu, L. Ma and L. A. Archer, Nat. Commun., 2016, 7, 11722 CrossRef CAS PubMed.
- X. Yu and A. Manthiram, ChemElectroChem, 2014, 1, 1275–1280 CrossRef CAS.
- X. Yu and A. Manthiram, Chem. Mater., 2016, 28, 896–905 CrossRef.
- B. W. Zhang, T. Sheng, Y. X. Wang, S. Chou, K. Davey, S. X. Dou and S. Z. Qiao, Angew. Chem., 2019, 131, 1498–1502 CrossRef.
- D. Ma, Y. Li, J. Yang, H. Mi, S. Luo, L. Deng, C. Yan, M. Rauf, P. Zhang and X. Sun, Adv. Funct. Mater., 2018, 28, 1705537 CrossRef.
- Z. Hu, Q. Liu, S. L. Chou and S. X. Dou, Adv. Mater., 2017, 29, 1700606 CrossRef PubMed.
- Z. Yan, Y. Liang, J. Xiao, W. Lai, W. Wang, Q. Xia, Y. Wang, Q. Gu, H. Lu and S. L. Chou, Adv. Mater., 2020, 1906700 CrossRef CAS PubMed.
- Z. Yan, J. Xiao, W. Lai, L. Wang, F. Gebert, Y. Wang, Q. Gu, H. Liu, S.-L. Chou and H. Liu, Nat. Commun., 2019, 10, 1–8 CrossRef PubMed.
- H. Liu, Y. Zou, L. Tao, Z. Ma, D. Liu, P. Zhou, H. Liu and S. Wang, Small, 2017, 13, 1700758 CrossRef PubMed.
- L. M. Bloi, J. Pampel, S. Dörfler, H. Althues and S. Kaskel, Adv. Energy Mater., 2020, 10, 1903245 CrossRef CAS.
- H. Liu, L. Tao, Y. Zhang, C. Xie, P. Zhou, H. Liu, R. Chen and S. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 36849–36856 CrossRef CAS PubMed.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- Y. Lin, P. Liu, E. Velasco, G. Yao, Z. Tian, L. Zhang and L. Chen, Adv. Mater., 2019, 31, 1808193 CrossRef PubMed.
- L. Zhang, D. Liu, Z. Muhammad, F. Wan, W. Xie, Y. Wang, L. Song, Z. Niu and J. Chen, Adv. Mater., 2019, 31, 1903955 CrossRef CAS PubMed.
- B.-W. Zhang, T. Sheng, Y.-D. Liu, Y.-X. Wang, L. Zhang, W.-H. Lai, L. Wang, J. Yang, Q.-F. Gu and S.-L. Chou, Nat. Commun., 2018, 9, 4082 CrossRef PubMed.
- W. Luo, Y. Wang, S. Chou, Y. Xu, W. Li, B. Kong, S. X. Dou, H. K. Liu and J. Yang, Nano Energy, 2016, 27, 255–264 CrossRef CAS.
- D. Su, K. Kretschmer and G. Wang, Adv. Energy Mater., 2016, 6, 1501785 CrossRef.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong and Z. Deng, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- H. Liu, K. Hu, D. Yan, R. Chen, Y. Zou, H. Liu and S. Wang, Adv. Mater., 2018, 30, 1800295 CrossRef PubMed.
- X. Li, X. Li, B. Zhu, J. Wang, H. Lan and X. Chen, RSC Adv., 2017, 7, 30956–30962 RSC.
- H. Liu, W.-H. Lai, H.-L. Yang, Y.-F. Zhu, Y.-J. Lei, L. Zhao, J. Peng, Y.-X. Wang, S.-L. Chou and H.-K. Liu, Chem. Eng. J., 2020, 127348 Search PubMed.
- Z. Chen, Y. Ha, H. Jia, X. Yan, M. Chen, M. Liu and R. Wu, Adv. Energy Mater., 2019, 9, 1803918 CrossRef.
- H. Hu, B. Y. Guan and X. W. D. Lou, Chem, 2016, 1, 102–113 CAS.
- Y. Hua, H. Jiang, H. Jiang, H. Zhang and C. Li, Electrochim. Acta, 2018, 278, 219–225 CrossRef CAS.
- A. Kumar, A. Ghosh, A. Roy, M. R. Panda, M. Forsyth, D. R. MacFarlane and S. Mitra, Energy Storage Materials, 2019, 20, 196–202 CrossRef.
- T. H. Hwang, D. S. Jung, J.-S. Kim, B. G. Kim and J. W. Choi, Nano Lett., 2013, 13, 4532–4538 CrossRef CAS PubMed.
- S. Wei, L. Ma, K. E. Hendrickson, Z. Tu and L. A. Archer, J. Am. Chem. Soc., 2015, 137, 12143–12152 CrossRef CAS PubMed.
- S. Wenzel, H. Metelmann, C. Raiß, A. K. Dürr, J. Janek and P. Adelhelm, J. Power Sources, 2013, 243, 758–765 CrossRef CAS.
- J. Xu, W. Zhang, H. Fan, F. Cheng, D. Su and G. Wang, Nano Energy, 2018, 51, 73–82 CrossRef CAS.
- C.-W. Park, J.-H. Ahn, H.-S. Ryu, K.-W. Kim and H.-J. Ahn, Electrochem. Solid-State Lett., 2006, 9, A123–A125 CrossRef CAS.
- S. Zheng, P. Han, Z. Han, P. Li, H. Zhang and J. Yang, Adv. Energy Mater., 2014, 4, 1400226 CrossRef.
- Q. Guo and Z. Zheng, Adv. Funct. Mater., 2019, 1907931 Search PubMed.
- T. Yang, B. Guo, W. Du, M. K. Aslam, M. Tao, W. Zhong, Y. Chen, S. J. Bao, X. Zhang and M. Xu, Adv. Sci., 2019, 6, 1901557 CrossRef CAS.
- H. Liu, W. Pei, W.-H. Lai, Z. Yan, H. Yang, Y. Lei, Y.-X. Wang, Q. Gu, S. Zhou and S. Chou, ACS Nano, 2020, 14, 7259–7268 CrossRef CAS.
- Y. Zhang, P. Wang, Y. Yin, X. Zhang, L. Fan, N. Zhang and K. Sun, Chem. Eng. J., 2019, 356, 1042–1051 CrossRef CAS.
- S. Dong, C. Li, X. Ge, Z. Li, X. Miao and L. Yin, ACS Nano, 2017, 11, 6474–6482 CrossRef CAS PubMed.
- S. Dong, C. Li, Z. Li, L. Zhang and L. Yin, Small, 2018, 14, 1704517 CrossRef PubMed.
- X. Tian, J. Wen, S. Wang, J. Hu, J. Li and H. Peng, Mater. Res. Bull., 2016, 77, 279–283 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta08748c |
| This journal is © The Royal Society of Chemistry 2021 |