
Fluorene-based enamines as low-cost and dopant-free hole transporting materials for high performance and stable perovskite solar cells†
Sarune
Daskeviciute‡
a,
Cristina
Momblona‡
 b,
Kasparas
Rakstys
a,
Albertus Adrian
Sutanto
b,
Maryte
Daskeviciene
a,
Vygintas
Jankauskas
c,
Alytis
Gruodis
c,
Giedre
Bubniene
a,
Vytautas
Getautis
*a and
Mohammad Khaja
Nazeeruddin
*b
b,
Kasparas
Rakstys
a,
Albertus Adrian
Sutanto
b,
Maryte
Daskeviciene
a,
Vygintas
Jankauskas
c,
Alytis
Gruodis
c,
Giedre
Bubniene
a,
Vytautas
Getautis
*a and
Mohammad Khaja
Nazeeruddin
*b
aDepartment of Organic Chemistry, Kaunas University of Technology, Radvilenu pl. 19, Kaunas 50254, Lithuania. E-mail: vytautas.getautis@ktu.lt
bGroup for Molecular Engineering of Functional Material, Institute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne, CH-1951 Sion, Switzerland,. E-mail: mdkhaja.nazeeruddin@epfl.ch
cInstitute of Chemical Physics, Vilnius University, Sauletekio al. 3, Vilnius 10257, Lithuania
First published on 23rd November 2020
Abstract
The power conversion efficiency of perovskite solar cells is approaching the Shockley–Queisser limit, and therefore this technology is next to the commercialization stage. Inexpensive and stable hole transporting materials are highly desirable for the successful scale-up. Most high performing devices generally employ expensive hole conductors that are synthesized via cross-coupling reactions which require expensive catalysts, inert reaction conditions and time-consuming sophisticated product purification. In a quest to employ cost-effective chemistry to combine the building blocks, we explore enamine-based small molecules that can be synthesized in a simple condensation reaction from commercially available materials leading to an estimated material cost of a few euros per gram. The synthesized fluorene-based enamines exhibit a very high hole mobility up to 3.3 × 10−4 cm2 V−1 s−1 and enable the fabrication of perovskite solar cells with a maximum power conversion efficiency of 19.3% in a doped configuration and 17.1% without doping. In addition, both PSC systems demonstrate superior long-term stability compared to spiro-OMeTAD. This work shows that hole transporting materials prepared via a simple condensation protocol have the potential to compete in terms of performance with materials obtained via expensive cross-coupling methods at a fraction of their cost and deliver exceptional stability of the final device. This work provides a design strategy for the further development of novel, low-cost semiconductors.
Introduction
In their first decade, hybrid organic–inorganic halide perovskite-based solar cells (PSCs) continually improved. They are now approaching the Shockley–Queisser power conversion efficiency (PCE) limit with the current best certified PCE of 25.5%.1 The improvement in PCE can be attributed to the outstanding optoelectronic properties such as high absorption coefficient, long carrier diffusion length, small exciton binding energy, and high charge carrier mobility of the perovskite material and rapid advances in the device engineering.2–6 Owing to the combination of highly abundant precursor materials and simple preparation methods it is believed that the commercialization of PSC technology during the second decade is inevitable.7–9While the very high efficiency obtained using perovskites is a significant achievement, issues related to the high price of the device components such as hole transporting materials (HTMs) and long-term stability against moisture, heat, and light are still a concern for the commercial application of the technology; therefore, breaking these bottlenecks is a must for the realization of cost-effective and stable devices.10–14
Until now, most of the highly efficient PSCs are based on either the small organic molecule 2,2′,7,7′-tetrakis(N,N-di-p-methoxyphenylamine)-9,9′-spirobifluorene (spiro-OMeTAD) or conjugated macromolecule poly[bis(4-phenyl)(2,4,6-trimethylphenyl)amine] (PTAA) HTMs, both of which are very expensive.14–17 For example, spiro-OMeTAD is synthesized in a multi-step reaction scheme that requires a low temperature (−78 °C) and sensitive (n-butyllithium) and aggressive (Br2) reagents, resulting in a relatively high material cost and consequently leading to a significant contribution to the total device cost.18–20 Additionally, the tedious synthesis hampers large scale production and thereby could impede the commercial success of PSCs.
Therefore, huge interest of many research groups has been directed towards new HTM candidates to find an ideal HTM, which would be easily scalable for reasonable cost, including spiroxanthene-,21–23 fluorene-,24,25 carbazole-,26–31 silane-,32 bifluorene-,33 pyrene-,34,35 and bifluorenylidene-based36 examples. However, most of the conjugated HTMs are generally designed by linking together building blocks of the conjugated central core with costly diphenylamine- or triphenylamine-containing methoxy-substituted side groups using C–N or C–C cross-coupling chemistry, respectively. These coupling reactions generally require stringent reaction conditions resulting in several disadvantages, such as inert reaction conditions, expensive transition metal catalysts and extensive purification procedures. The commonly used purification methods involve sublimation or repeated column chromatography due to the inherent formation of side products that are usual for this type of reaction and a tiny amount of metal catalyst residues that may remain in the hole transporting layer. The metal catalyst residues act as traps that deteriorate the charge-transporting properties of the synthesized HTMs and negatively affect the performance of the resulting devices as well as greatly reducing the material yield and therefore further increasing the final product costs. Also the high processing cost in turn results in a significant cost contribution of HTMs, making them industrially less interesting.
To this extent, significant effort is now being put towards finding simplified synthetic protocols to reduce the cost of HTM synthesis without sacrificing the efficiency. Recently, several research groups have focussed on tuning the structure by decreasing the number of synthetic steps, thus reducing the synthetic complexity, cost of materials and the environmental impact.37,38 This includes reports based on azomethine,39 hydrazone,40 and amide41 by Petrus et al. and our previously explored aniline42 and carbazole43,44 enamines prepared by a facile condensation reaction. In this sense, condensation chemistry is an excellent perspective moving away from palladium-catalysed reactions since water is the only side-product and expensive catalysts are not required. Moreover, simple product workup and purification drastically reduce the cost of the final product.
In this work, we further explore the potential of enamine family HTMs employing fluorene as the central scaffold. Five different substituents containing fluorene-based HTMs were successfully synthesised employing facile synthesis using commercially available and cheap reagents. Their optical, thermal, electrophysical, and photovoltaic properties were thoroughly investigated by combining experimental and simulation methods. Moreover, the impact of the differently substituted central fluorene core on different properties of the synthesized molecules, in comparison to our earlier reports, has been systematically investigated. All these enamine-based HTMs have been successfully applied in PSCs with and without additives, showing a photovoltaic performance of up to 19.3% and 17.1%, respectively, with excellent long-term stability in both cases. With this we demonstrate that both simple chemistry and product purification result in estimated material costs of a few euros per gram without sacrificing the efficiency and in contrast enhancing the stability.
Results and discussion
Fluorene enamines were synthesized using straightforward chemistry with excellent yields and high purity. As shown in Fig. 1, V1275 only required one-pot reaction condensing inexpensive commercially available reagents 2,7-diaminofluorene and 2,2-bis(4-methoxyphenyl)acetaldehyde in the presence of camphor sulfonic acid. The reaction is performed under ambient conditions and water is the only by-product separated from the reaction mixture using a Dean–Stark trap, accelerating the formation of the final product and significantly reducing the reaction duration. Moreover, we note that column chromatography or vacuum sublimation processes are avoided for the purification, and the simplicity of the condensation chemistry reduces batch-to-batch variations. | ||
| Fig. 1 Straightforward reaction scheme of fluorene enamine HTMs and their molecular structures. | ||
V1275 was further reacted with different alkylating agents to yield methyl-, propyl-, hexyl-, and benzyl-substituted fluorene enamines as final HTMs V1237, V1235, V1236, and V1227, respectively. The chemical structures of the synthesized products were verified by NMR spectroscopy and elemental analysis. Detailed synthetic procedures and analysis are reported in the ESI.† In order to assess the price of the synthesized materials, we performed a cost-analysis on a lab-scale synthesis (Table S1†).45 The estimated cost of V1275 is ∼10€ per g and that of the alkylated product V1235 is around 22€ per g, which are a fraction of the cost of spiro-OMeTAD (∼92€ per g)46 and less than that of our previously developed double-armed carbazole enamines mainly due to the less expensive 2,7-diaminofluorene starting reagent.44
Quantum chemical calculations were performed with Gaussian 09 software to establish the most probable molecular geometry and absorption spectrum.47 The density functional theory (DFT) method B3LYP/6-31G was used for geometry optimization. V1275, V1237, and V1227 have been chosen as model compounds for computations, and as the different lengths of the aliphatic substituents should not affect the electronic properties they were ignored. Due to fragmental motions, a large number of different conformers could be formed. Only two typical and the most probable conformers of each compound (a and b, after ground state geometry optimization) are presented in Fig. S1–S3.† Conformers could be formed due to the following condition: enamine subfragments are formed as quite well expressed π-conjugated fragments >C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–N*–CHC< oriented quasi linearly, and this bridge between two O–Ph subfragments must be treated as the important factor for fragment displacement in space. Two chains of O–Ph subfragments of the left and right fragments could be oriented in the shape of the upper-side roof according to the fluorene core (see V1237 and V1275, a and b, bottom part), but other O–Ph subfragments are oriented chaotically (see V1227, a and b, top part). Due to the presence of benzyl substituents in V1227, it is necessary to conclude that phenyl fragments are not included in the core π-conjugated system (connection through two single –C–C– bonds with high lability). The possibility to orient the phenyl on the fluorene core could be realized in many ways. This factor of indeterminacy of the phenyl position creates the condition of non-ordered distribution of enamine subfragments in the V1227 structure. For all three derivatives, b conformers are more ordered than a conformers. The semiempirical TD method (for singlets only) was used for the simulation of the electronic absorption spectrum. Table S2† shows the excitation parameters for the three lowest excited states S1, S2, and S3: transition energy and oscillator strength. For all compounds (including both conformers), S0 → S1 transitions are allowed and partially allowed (oscillator strength in the interval 0.57–0.71) and the transition energy is approximately 2.89–2.92 eV. Table S3† presents the scheme of the population of excited electronic states and the corresponding set of MO.
CH–N*–CHC< oriented quasi linearly, and this bridge between two O–Ph subfragments must be treated as the important factor for fragment displacement in space. Two chains of O–Ph subfragments of the left and right fragments could be oriented in the shape of the upper-side roof according to the fluorene core (see V1237 and V1275, a and b, bottom part), but other O–Ph subfragments are oriented chaotically (see V1227, a and b, top part). Due to the presence of benzyl substituents in V1227, it is necessary to conclude that phenyl fragments are not included in the core π-conjugated system (connection through two single –C–C– bonds with high lability). The possibility to orient the phenyl on the fluorene core could be realized in many ways. This factor of indeterminacy of the phenyl position creates the condition of non-ordered distribution of enamine subfragments in the V1227 structure. For all three derivatives, b conformers are more ordered than a conformers. The semiempirical TD method (for singlets only) was used for the simulation of the electronic absorption spectrum. Table S2† shows the excitation parameters for the three lowest excited states S1, S2, and S3: transition energy and oscillator strength. For all compounds (including both conformers), S0 → S1 transitions are allowed and partially allowed (oscillator strength in the interval 0.57–0.71) and the transition energy is approximately 2.89–2.92 eV. Table S3† presents the scheme of the population of excited electronic states and the corresponding set of MO.
Distributions of electron density for the HOMO−1 and HOMO as well as the LUMO and LUMO+1 for V1275, V1237, and V1227 structures are presented in Fig. S4–S6.† Any pure CT charge redistribution behaviour was established for both a and b conformers, and only charge redistribution between the first and second enamine subfragments of the left fragment takes place. It is necessary to point out that the enamine subfragments could play the role of charge donors and charge acceptors, depending on the fragment orientation to the central core fluorene unit (a conformer, HOMO → LUMO transition). Also, charge redistributions between the enamine fragment and fluorene core take place (b conformer, HOMO → LUMO transition and a conformer, HOMO → LUMO+1 transition). In general, charge redistribution between the left and right enamine substituents and the central core fluorene unit is typical for both conformers.
The thermal behaviour of HTMs was evaluated by thermogravimetric analysis (TGA) (Fig. 2a) and differential scanning calorimetry (DSC) (Fig. S7†) techniques. TGA analysis has shown that V1275 has the highest thermal stability among the series with a decomposition temperature (Tdec) of 403 °C at 5% weight loss. The introduction of the aliphatic substituents to the central fluorene position has led to the deteriorated thermal resistance. However, we note that the instant weight loss at around 400 °C is observed for all new materials suggesting that they may undergo sublimation rather than decomposition, enabling them to be vacuum-deposited. The thermal transitions of V-series molecules were determined by DSC. Interestingly, it was found that V1275, V1236, and V1227 exist both in the crystalline and the amorphous state, while V1237 and V1235 tend to crystallize. Only the glass transition temperature (Tg) was investigated for all compounds during the second heating scan, while V1237 has the most stabilized amorphous state with the glass transition detected at 153 °C.
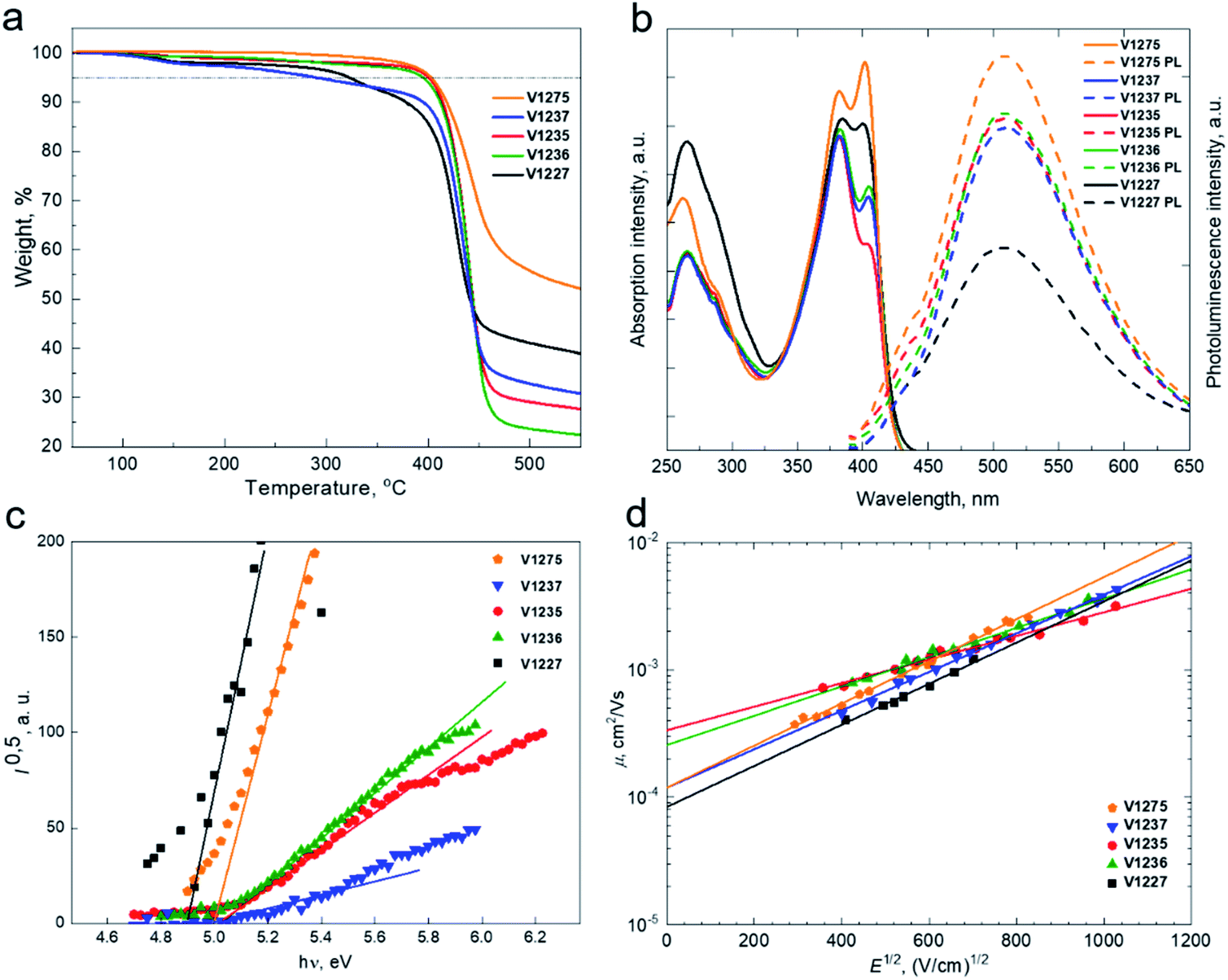 | ||
| Fig. 2 (a) Thermogravimetric analysis (TGA) data (heating rate of 10 °C min−1, N2 atmosphere); (b) UV-Vis absorption (solid line) and photoluminescence (dashed line) spectra of V-series HTMs in THF solution (10−4 M); (c) photoemission in air spectra of the charge transporting layers V1275, V1237, V1235, V1236, and V1227; (d) electric field dependencies of the hole-drift mobility in the synthesized HTMs. | ||
The UV-visible absorption and photoluminescence (PL) spectra of the synthesized HTMs in THF solutions and solid films are depicted in Fig. 2b and S8,† respectively. All HTMs have two major absorption peaks at approximately 260 nm and 400 nm. The less intense absorption peak at the shorter wavelength corresponds to localised π–π* transitions. The longer wavelength arises from more intensive delocalisation from the conjugated scaffold and is assigned to n–π* transitions. Change of the different aliphatic fragments has not influenced the conjugation, and therefore, spectra of all molecules are almost identical; however, there is a significant difference in the absorption intensity ratio of V1227 peaks arising from benzyl moieties. The PL spectra of all compounds are similar to the peak at 510 nm, showing that significantly large Stokes shifts of approximately 100 nm are observed for all molecules, and therefore, significant changes in the geometry of the molecules are desired upon excitation. The optical gaps (Eg) of HTMs were calculated from the intersection of absorption and PL spectra of thin films and were found to be identical for all the materials at around 2.8 eV.
To understand the energy level alignment of the HTMs in PSCs, we next measured solid-state ionization potential (IP) using the electron photoemission in air of the thin films (PESA) with the experimental data shown in Fig. 2c. V1275, V1237, V1235, V1236, and V1227 were found to have IP values of 5.01, 5.0, 5.03, 5.03 and 4.9 eV, respectively, which ideally align with the valence band (VB) energy of the triple cation-based perovskite (∼5.70 eV), and therefore, efficient hole transfer from the perovskite to the cathode should be ensured.48 Spiro-OMeTAD has been measured as well and was found to have a very similar ionization potential of 5.00 eV (Fig. S9†). Additionally, to reveal the effect of the p-dopant as the electron acceptor we have evaluated the ionization potentials of doped layers  since they are known to control the HOMO energy level by removing electrons from the HOMO to generate holes of an intrinsic HTM, enhancing the device efficiency.14,49–51 HTMs were doped in the same manner as in the device fabrication part detailed in the ESI.† As expected, upon doping, ionization potentials were stabilized by around 0.3 eV further reducing the overpotential with the VB of the perovskite thus expectedly increasing the VOC in doped HTL-containing devices including spiro-OMeTAD (Fig. S10 and S11†). Based on the solid-state optical gap and IP values, we calculated the electron affinities (Eea) of the enamine materials to be 2.22, 2.21, 2.22, 2.24, and 2.11 eV for V1275, V1237, V1235, V1236, and V1227, respectively. Importantly, the electron affinities of the compounds are smaller than the conduction band energy of the perovskite (−4.10 eV), and therefore, they should effectively block the electron transfer from the perovskite to the anode.48 We next measured the charge mobility of the V-series using the xerographic time of flight (XTOF) technique. Dependences of hole drift mobility on electric field strength are depicted in Fig. 2d. The zero-field hole drift mobility (μ0) for V1275, V1237, V1235, and V1236 was determined to be at 10−4 cm2 V−1 s−1, while the propyl-substituted V1235 was found to have the highest hole mobility of 3.3 × 10−4 cm2 V−1 s−1 among the series. We note that this is also the highest hole mobility compared with that of our previous aniline and carbazole enamine reports and it outperforms that of spiro-OMeTAD (μ0 = 1.3 × 10−4 cm2 V−1 s−1).36,44 Switching to the aromatic benzyl substitution has negatively influenced the hole drift mobility as V1227 showed the lowest result of 8 × 10−5 cm2 V−1 s−1 due to the larger energetic disorder. The thermal, optical, and photoelectrical properties of the novel HTMs are listed in Table 1.
since they are known to control the HOMO energy level by removing electrons from the HOMO to generate holes of an intrinsic HTM, enhancing the device efficiency.14,49–51 HTMs were doped in the same manner as in the device fabrication part detailed in the ESI.† As expected, upon doping, ionization potentials were stabilized by around 0.3 eV further reducing the overpotential with the VB of the perovskite thus expectedly increasing the VOC in doped HTL-containing devices including spiro-OMeTAD (Fig. S10 and S11†). Based on the solid-state optical gap and IP values, we calculated the electron affinities (Eea) of the enamine materials to be 2.22, 2.21, 2.22, 2.24, and 2.11 eV for V1275, V1237, V1235, V1236, and V1227, respectively. Importantly, the electron affinities of the compounds are smaller than the conduction band energy of the perovskite (−4.10 eV), and therefore, they should effectively block the electron transfer from the perovskite to the anode.48 We next measured the charge mobility of the V-series using the xerographic time of flight (XTOF) technique. Dependences of hole drift mobility on electric field strength are depicted in Fig. 2d. The zero-field hole drift mobility (μ0) for V1275, V1237, V1235, and V1236 was determined to be at 10−4 cm2 V−1 s−1, while the propyl-substituted V1235 was found to have the highest hole mobility of 3.3 × 10−4 cm2 V−1 s−1 among the series. We note that this is also the highest hole mobility compared with that of our previous aniline and carbazole enamine reports and it outperforms that of spiro-OMeTAD (μ0 = 1.3 × 10−4 cm2 V−1 s−1).36,44 Switching to the aromatic benzyl substitution has negatively influenced the hole drift mobility as V1227 showed the lowest result of 8 × 10−5 cm2 V−1 s−1 due to the larger energetic disorder. The thermal, optical, and photoelectrical properties of the novel HTMs are listed in Table 1.
| ID | T m (°C) | T c (°C) | T g (°C) | T dec (°C) | λ abs (nm) | λ em (nm) | I P (eV) |
|
E g (eV) | E ea (eV) | μ 0 (cm2 V−1 s−1) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Melting (Tm), crystallization (Tc), glass transition (Tg) and decomposition (Tdec) temperatures observed from DSC and TGA, respectively (10 °C min−1, N2 atmosphere). b Absorption and emission (excitation = λabs max) spectra were measured in THF solution (10−4 M). c Ionization energies of the films measured using PESA without doping. d Ionization energies of the films measured using PESA with doping. e E g estimated from the intersection of absorption and emission spectra of solid films. f E ea = IP − Eg. g Mobility value at zero field strength. | |||||||||||
| V1275 | 255 | — | 150 | 403 | 262, 381, 401 | 508 | 5.01 | 5.39 | 2.79 | 2.22 | 1.2 × 10−4 |
| V1237 | 247, 267, 272 | 198 | 153 | 285 | 265, 382, 404 | 510 | 5.0 | 5.32 | 2.79 | 2.21 | 1.2 × 10−4 |
| V1235 | 273 | 159 | 120 | 399 | 266, 382, 404 | 509 | 5.03 | 5.39 | 2.81 | 2.22 | 3.3 × 10−4 |
| V1236 | 173, 195 | — | 90 | 393 | 265, 383, 404 | 508 | 5.03 | 5.25 | 2.79 | 2.24 | 2.6 × 10−4 |
| V1227 | 330 | — | 116 | 321 | 265, 384, 400 | 507 | 4.9 | 5.34 | 2.79 | 2.11 | 8 × 10−5 |

The new V-series HTMs were implemented in n–i–p solar cells with the following layout: fluorine-doped tin oxide (FTO)/compact TiO2 (c-TiO2)/mesoporous TiO2 (m-TiO2)/SnO2/perovskite/HTM/Au, where the HTMs were doped with tert-butylpyridine (tBP), tris(bis(trifluoromethylsulfonyl)imide) (LiTFSI) and tris(2-(1H-pyrazol-1-yl)-4-tert-butylpyridine)cobalt(III) (FK209). The detailed fabrication procedure can be found in the ESI.†
Cross-sectional scanning electron microscopy (SEM) imaging was performed on the complete devices fabricated with the V-series and spiro-OMeTAD (Fig. 3a and S12†). The analysis of the images shows that the HTM layers are compact and uniform on top of the perovskite layer with a thickness of ∼150 nm for V1275, V1237, V1235 and V1236, ∼120 nm for V1227, and ∼260 nm for spiro-OMeTAD. The surface morphology was evaluated by scanning electron microscopy (Fig. S13†), with the doped-HTM layers deposited on top of FTO-glass under the same deposition conditions as in the device fabrication. All the images of doped-HTM thin films show homogeneous and complete surface coverage without the presence of material aggregation.52 The energy levels of the complete devices with the studied HTMs as well as spiro-OMeTAD can be found in Fig. 3b.48,53
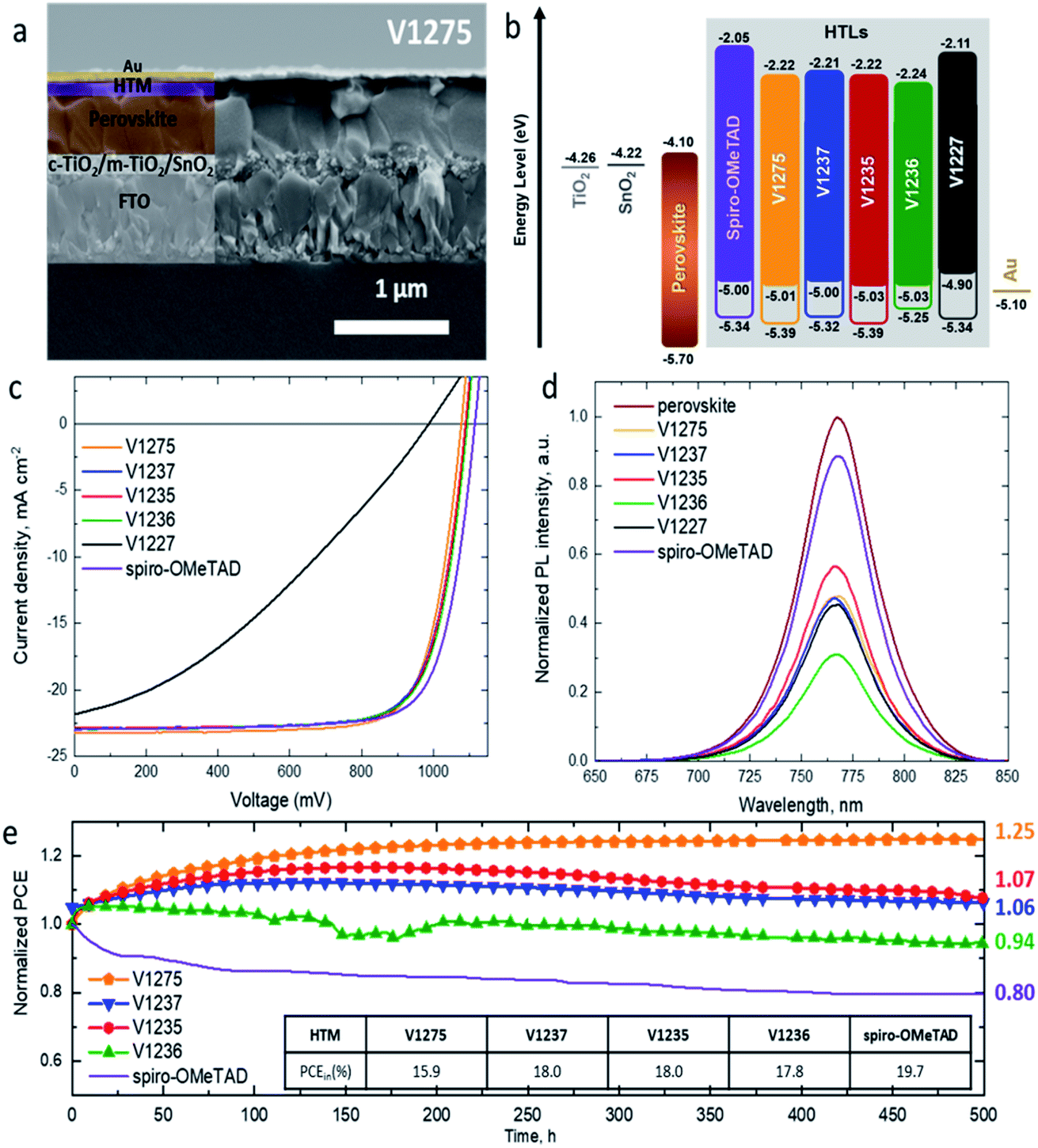 | ||
| Fig. 3 (a) Cross-sectional SEM image of the photovoltaic device fabricated with the doped V1275 HTM; (b) schematic energy level diagram of the device representing the HOMO/LUMO and the stabilized HOMO of doped layers; (c) J–V curves recorded for the champion devices for each doped HTM; (d) normalized steady-state photoluminescence spectra of perovskite and perovskite/HTM layers deposited on glass (λexc = 625 nm); (e) stability test of unencapsulated devices under continuous 1 sun illumination with the initial PCE values shown in the insert. | ||
The initial J–V curves and the corresponding photovoltaic parameters are presented in Fig. S14 and Table S4.† The initial PCEs are 15.9% for V1275 (non-alkylated HTM), 18.0% for V1237 and V1235 (methyl- and propyl-based, respectively) and 17.8% for V1236 hexyl-based devices. This initial performance of the devices might indicate that the alkyl functionalization of the HTM is required for an appropriate perovskite/HTM interface for enhanced charge extraction through the device. Once the maximum efficiency was reached, the corresponding photovoltaic parameters were extracted and are presented in Table 2. The devices containing V1275, alkylated-HTMs V1237, V1235, and V1236, and spiro-OMeTAD present comparable photovoltaic behaviour with the maximum PCE around 19%. The comparison of the devices at the best performing efficiency is confirmed by statistical data (Fig. S15†). The performance is greater than that of the benzyl-substituted V1227-based device with 12.5% efficiency. Note that this trend can be related to the hole mobility values obtained for the HTMs, ranging from 1.2 × 10−4 cm2 V−1 s−1 to 3.3 × 10−4 cm2 V−1 s−1 for V1275 and V1235, and the value for spiro-OMeTAD (1.3 × 10−4 cm2 V−1 s−1) is similar, but lower for the bulky analogue V1227 with a hole mobility value of 8 × 10−5 cm2 V−1 s−1.44
| HTM | V OC (mV) | J SC (mA cm−2) | FF | PCE (%) | R S (Ω) | HTM thickness (nm) | |
|---|---|---|---|---|---|---|---|
| Doped-HTM | V1275 | 1077 | 23.24 (23.11) | 0.77 | 19.3 | 5.9 | ∼150 |
| V1237 | 1090 | 22.97 (22.74) | 0.76 | 19.2 | 6.2 | ∼150 | |
| V1235 | 1089 | 22.86 (22.85) | 0.77 | 19.2 | 5.1 | ∼150 | |
| V1236 | 1094 | 22.95 (22.94) | 0.76 | 19.1 | 5.2 | ∼150 | |
| V1227 | 1024 | 22.32 (21.82) | 0.55 | 12.6 | 26.0 | ∼120 | |
| Spiro-OMeTAD | 1115 | 22.97 (21.86) | 0.77 | 19.7 | 5.7 | ∼260 | |
| Dopant-free HTM | V1275 | 1033 | 22.95 (22.81) | 0.72 | 17.1 | 22.2 | ∼70 |
| V1237 | 1038 | 22.98 (22.93) | 0.71 | 16.9 | 22.9 | ∼70 | |
| V1235 | 1029 | 23.09 (22.81) | 0.70 | 16.6 | 24.5 | ∼70 | |
| V1236 | 1022 | 23.02 (22.88) | 0.69 | 16.2 | 25.2 | ∼70 | |
| Spiro-OMeTAD58 | 972 | 22.83 | 0.47 | 10.4 |
The highest efficiency reached for each HTM (Table 2) shows that the V1275 and alkyl-based devices have almost identical short-circuit current density values (JSC) in the range from 23.24 mA cm−2 for V1275 to 22.86 mA cm−2 for V1235, respectively. This suggests that the substitution with an alkyl chain and the increasing alkyl chain length from methyl to hexyl do not strongly influence the charge collection properties of the perovskite layer. However, the benzyl-based V1227 results in devices having a slightly lower JSC value of 22.32 mA cm−2. Such behaviour is also confirmed by the external quantum efficiency (EQE) spectra (Fig. S16a†). The corresponding integrated current densities from the respective EQEs (Fig. S16†) are presented in Table 2 in brackets, and the values are in good agreement with the measured values from the J–V characteristics (within 5% error). Similar fill-factor (FF) values are obtained for the devices employing V1275, V1237, V1235 and V1236, with values ranging between 0.76 and 0.77. However, the FF in the device with V1227 is reduced to 0.55, this can be explained by the presence of benzyl groups in V1227, which causes a less ordered packing of the molecules in the film and in the interface with the perovskite, and this also leads to a greater dispersion of solar cell parameters (Fig. S15†).54 The main parameter leading to the slightly lower performance of the V-series in comparison to spiro-OMeTAD is the open-circuit voltage (VOC). The cells incorporating V1275, V1237, V1235 and V1236 exhibit lower values of VOC (1077, 1090, 1089 and 1094 mV, respectively) than the reference device containing spiro-OMeTAD (1115 mV).
In order to evaluate the photogenerated hole extraction efficiency of the new HTMs, we performed thin-film steady-state PL measurements. Perovskite layers were deposited on top of glass and the PL spectra of the films were recorded under 625 nm excitation wavelength. Afterwards, the HTMs were deposited on top of the pristine perovskite layers under the same conditions as in the device fabrication. The quenching effect was analysed in comparison with the corresponding pristine perovskite layer and it is presented in Fig. 3d, and the percentage of PL quenching for each HTM is shown in Table S5.† In spite of the perovskite/HTM energy level mismatch, the decrease of PL intensity suggests a good hole-extraction capability and confirms the efficient extraction of holes across the interface from the VB of the perovskite into the HOMO of enamine-based HTMs, which is attributed to good perovskite/HTM contact.55
The device stability was evaluated for the solar cells containing V1275 and the alkyl-substituted HTMs (V1237, V1235 and V1236) due to their higher efficiency than that of the benzyl-substituted HTM V1227. As a reference, the long-term stability of the spiro-OMeTAD-based device was also evaluated. All the devices were unencapsulated and kept under constant 1 sun illumination in a N2 atmosphere for 500 hours. The results of the long-term stability tests of the devices are presented in Fig. 3e. For comparison, the study was carried out with all the devices kept under the same light source. The device containing the non-substituted V1275 HTM shows a continuous increase of PCE achieving 125% of its original efficiency after 500 h under operation, being not only the device having the most thermally stable HTM of the series but also the most stable device under constant light illumination among them. The introduction of the methyl and propyl groups (V1237 and V1235) reduces this increase, obtaining 107 and 106% of its original efficiency after 500 h. However, the PCE of the device containing the longest insulating hexyl chain V1236 of the series is only reduced to 94% of its initial efficiency after 500 h under illumination. Interestingly, although the length of the alkyl chains only slightly influences the maximum device performance (see Table 2), it displays an influence on the time to reach this value and the long-term device stability. The introduction of an alkyl group and with a longer alkyl chain results in a shorter time to reach the highest efficiency, but the long-term light stability is deteriorated. Such behaviour may suggest that the stabilization/degradation of the layer is enhanced with a higher/lower degree of packing due to the longer insulating chains. In addition, these changes might also be influenced by different relaxation processes in the thin film56 and/or photo/oxygen-doping during operation.57
The outstanding light stability of the doped V-series HTMs is increased when compared with the most widely used HTM in PSCs, spiro-OMeTAD, whose efficiency is reduced to 80% from its initial performance after 500 h under constant illumination. With this result we can confirm that the use of dopants does not negatively affect the long-term stability of PSCs, and this fact contradicts previously reported studies.14
In order to evaluate if the dopants chemically interact with the HTMs and detrimentally affect the long-term stability of the devices, the most promising HTMs from the series, V1275, V1237, V1235 and V1236, were studied without the use of additives.59–61 The initial J–V curves are presented in Fig. S17† and the corresponding photovoltaic parameters are listed in the insert of Fig. 4b and Table S6.† Once the maximum efficiency is reached, all of them showed efficiencies exceeding 16% (Fig. 4a and Table 2) with improved device performance compared with the reported 10.39% PCE for dopant-free spiro-OMeTAD-based devices previously reported by our group (Table 2).58 The corresponding EQEs and the integrated short-circuit current density are presented in Fig. S16b† and the values in Table 2. The statistical data (Fig. S18†) compared at the maximum efficiency show that the gradual increase of the length of the alkyl substituents results in a slight deterioration of the device performance. Devices fabricated with the non-alkylated HTM V1275 are the most efficient of the series, whereas the efficiency is gradually reduced by the introduction of the methyl, propyl, and hexyl groups, V1237, V1235 and V1236, respectively, mainly due to lower VOC and FF values. This difference can be attributed to different packing of the molecules compared to the equivalent doped devices.
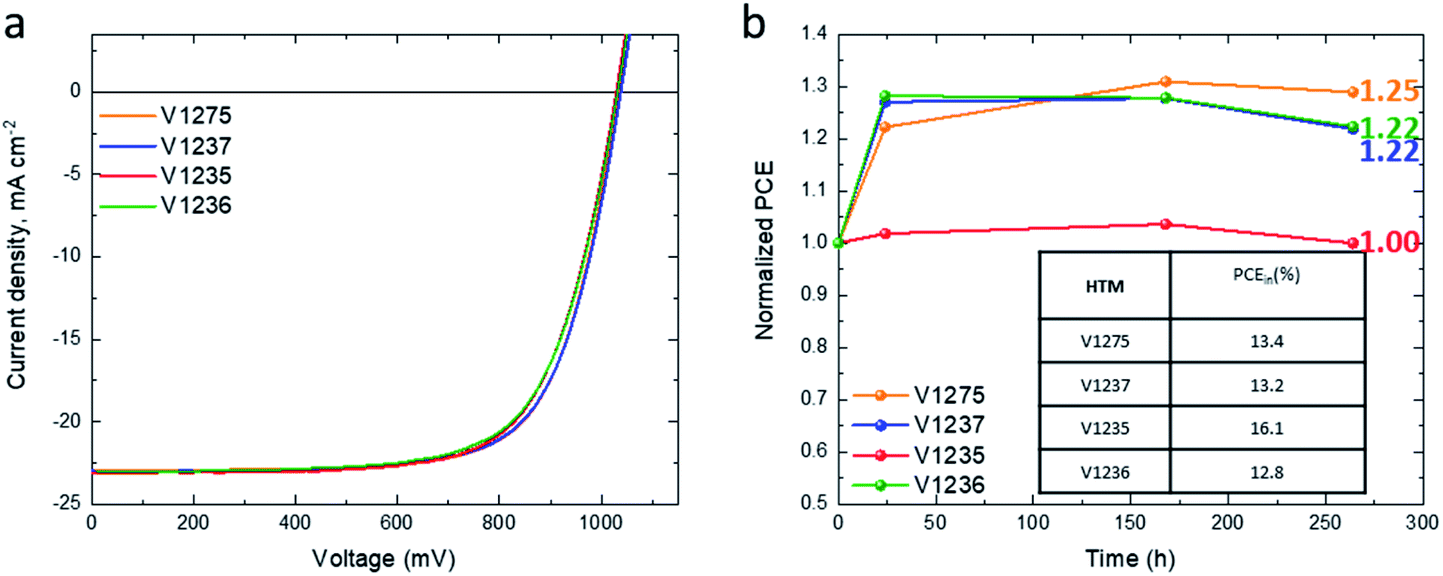 | ||
| Fig. 4 (a) J–V curves recorded for the champion devices for each dopant-free HTM; (b) shelf-lifetime of unencapsulated devices fabricated with each dopant-free HTM (stored in the dark and dry environment RH < 10%) with the initial PCE values shown in the insert. | ||
While the JSC is similar to that of the equivalent doped devices, the VOC and FF values are significantly lower. The series resistance (RS) of the devices was calculated from the corresponding J–V curves. The RS is reduced with the incorporation of dopants in the HTM layer due to the increase of the hole mobility in the layer.62,63 The lowest series resistance values of 5.9, 6.2, 5.1 and 5.2 Ω are obtained for devices made with doped-V1275, V1237, V1235 and V1236 HTMs and are comparable to the value registered for the spiro-OMeTAD device (5.7 Ω). The series resistance in the doped V1227-based device is similar to the values obtained in the dopant-free series, denoting a charge carrier transport issue in the device. The presence of the bulky benzyl group can induce a less ordered packing of the molecules in the film, also confirmed by DFT calculations, hence resulting in a possible poor charge transfer between the perovskite and the doped V1227 layer. This interface issue might reduce the transport of charge carriers through the perovskite/HTM interface ultimately limiting the overall solar cell performance.56
On the other hand, devices containing the dopant-free HTMs suffer from 4–5 times higher resistance than the doped counterparts. The dopant-free HTM layer presents higher resistance values even when we tried to reduce the series resistance by lowering the HTM solution concentration from 20 mM for the doped HTM to 15 mM for the dopant-free HTM to generate a thinner HTM layer. These higher values are reflected in the lower device performance for dopant-free HTMs. The use of the longer alkyl chain for the devices containing dopant-free HTMs increases the series resistance of the device. This suggests less ordered packing of the molecules in the film reducing the carrier transport through the perovskite/HTM interface, and therefore the HTM layer itself. This trend is not observed for the doped HTM counterparts.56 The higher resistance of the dopant-free HTM layer reduces the hole extraction and increases the interfacial recombination at the perovskite/HTM contact reflected in the drop of FF and VOC in all the devices.64
The HTM thicknesses of the dopant-free devices were extracted from their respective cross-sectional SEM images (Fig. S19†), obtaining films of ∼70 nm. To shed light on the surface morphology, scanning electron microscopy was performed (Fig. S20†). As mentioned above, the dopant-free HTM layers were deposited on top of FTO-glass under the same deposition conditions as in the device fabrication. A homogeneous surface morphology was observed for the dopant-free HTM layers, but due to the thinner layer thickness than in the doped counterparts, the layer underneath can be intuited. The stability of the dopant-free devices was tested by storing non-encapsulated devices in the dark under dry air (RH < 10%) and periodically testing under a relative humidity of 45% (Fig. 4b). The PCE of all HTMs shows no degradation after 250 h from their fabrication. After 250 h, the device efficiency increases to reach 125%, 122%, 100%, and 122% from the initial device efficiency for the V1275, V1237, V1235 and V1236 HTMs, respectively. This result demonstrates that V-series HTMs can also efficiently work as dopant-free HTMs in PSCs with excellent stability.
Conclusions
We report the synthesis and a systematic study of the fluorene-based hole transporting enamines. Novel HTMs are easily attainable by a straightforward synthetic scheme ensuring cost-effective scale-up; in particular, V1275 only required a one-pot reaction condensing inexpensive commercially available reagents leading to the extremely low synthetic cost of approximately 10€ per g. The impact of different substitution in the central fluorene was revealed through the optical, electrochemical, photophysical, and photovoltaic measurements. PSCs using the V-series compounds were fabricated in doped and dopant-free configurations. The synthesized materials exhibit a very high hole mobility up to 3.3 × 10−4 cm2 V−1 s−1 leading to a light-to-energy power conversion efficiency exceeding 19% with the doped non-functionalized V1275, which is on par with that of the widely researched spiro-OMeTAD, with a remarkable improvement in the long-term stability. While the efficiency of the spiro-based device dropped down to 80% of the original PCE after 500 h without encapsulation and constant light illumination, the doped V-based devices achieved up to 125% of the original PCE in the same ageing time. The devices fabricated with dopant-free HTMs showed high efficiency, exceeding 17%, and also excellent shelf-lifetime stability. The results presented here show that HTMs prepared via a simple condensation protocol can compete in performance with materials obtained via expensive cross-coupling methods at a fraction of their cost and may be very attractive low-cost and stable semiconductors solving one of the concerns for the near future commercialization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie grant agreement no. 754462. We acknowledge the Swiss National Science Foundation (SNSF) funding through the Synergia Grant EPISODE (Grant No. CRSII5_171000). The authors (M. Daskeviciene and G. Bubniene) acknowledge the funding from the Research Council of Lithuania (grant no. MIP-19-14). Computations were performed on resources at the High Performance Computing Center “HPC Sauletekis” (Faculty of Physics, Vilnius University). Prof. R. Buonsanti is acknowledged for the use of the Fluorolog system and Dr E. Kamarauskas is acknowledged for ionisation potential measurements. KR acknowledges the funding received from the MJJ Foundation.Notes and references
- NREL, Photovoltaic Research: Best Research Cell Efficiency Chart, https://www.nrel.gov/pv/cell-efficiency.html, accessed 28 April 2020 Search PubMed.
- J. Burschka, N. Pellet, S. J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Grätzel, Nature, 2013, 499, 316–319 CrossRef CAS.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. Il Seok, Nature, 2015, 517, 476–480 CrossRef CAS.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef CAS.
- C. Wehrenfennig, G. E. Eperon, M. B. Johnston, H. J. Snaith and L. M. Herz, Adv. Mater., 2014, 26, 1584–1589 CrossRef CAS.
- M. Saliba, T. Matsui, J. Y. Seo, K. Domanski, J. P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- M. A. Green, A. Ho-Baillie and H. J. Snaith, Nat. Photonics, 2014, 8, 506–514 CrossRef CAS.
- B. Chen, Z. J. Yu, S. Manzoor, S. Wang, W. Weigand, Z. Yu, G. Yang, Z. Ni, X. Dai, Z. C. Holman and J. Huang, Joule, 2020, 4, 850–864 CrossRef CAS.
- W. S. Yang, B. W. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim, J. H. Noh and S. Il Seok, Science, 2017, 356, 1376–1379 CrossRef CAS.
- W. W. Liu, T. H. Wu, M. C. Liu, W. J. Niu and Y. L. Chueh, Adv. Mater. Interfaces, 2019, 6, 1801758 CrossRef.
- D. H. Kim, J. B. Whitaker, Z. Li, M. F. A. M. van Hest and K. Zhu, Joule, 2018, 2, 1437–1451 CrossRef CAS.
- L. Qiu, L. K. Ono and Y. Qi, Mater. Today Energy, 2018, 7, 169–189 CrossRef.
- Y. Rong, Y. Hu, A. Mei, H. Tan, M. I. Saidaminov, S. Il Seok, M. D. McGehee, E. H. Sargent and H. Han, Science, 2018, 361, 6408 CrossRef.
- K. Rakstys, C. Igci and M. K. Nazeeruddin, Chem. Sci., 2019, 10, 6748–6769 RSC.
- U. Bach, D. Lupo, P. Comte, J. E. Moser, F. Weissörtel, J. Salbeck, H. Spreitzer and M. Grätzel, Nature, 1998, 395, 583–585 CrossRef CAS.
- W. S. Yang, J. H. Noh, N. J. Jeon, Y. C. Kim, S. Ryu, J. Seo and S. Il Seok, Science, 2015, 348, 1234–1237 CrossRef CAS.
- G. W. Kim, H. Choi, M. Kim, J. Lee, S. Y. Son and T. Park, Adv. Energy Mater., 2020, 10, 1903403 CrossRef CAS.
- T. P. I. Saragi, T. Spehr, A. Siebert, T. Fuhrmann-Lieker and J. Salbeck, Chem. Rev., 2007, 107, 1011–1065 CrossRef CAS.
- J. Salbeck, F. Weissörtel and J. Bauer, Macromol. Symp., 1998, 125, 121–132 CrossRef CAS.
- J. Salbeck, N. Yu, J. Bauer, F. Weissörtel and H. Bestgen, Synth. Met., 1997, 91, 209–215 CrossRef CAS.
- B. Xu, D. Bi, Y. Hua, P. Liu, M. Cheng, M. Grätzel, L. Kloo, A. Hagfeldt and L. Sun, Energy Environ. Sci., 2016, 9, 873–877 RSC.
- D. Bi, B. Xu, P. Gao, L. Sun, M. Grätzel and A. Hagfeldt, Nano Energy, 2016, 23, 138–144 CrossRef CAS.
- B. Xu, J. Zhang, Y. Hua, P. Liu, L. Wang, C. Ruan, Y. Li, G. Boschloo, E. M. J. Johansson, L. Kloo, A. Hagfeldt, A. K. Y. Jen and L. Sun, Chem, 2017, 2, 676–687 CAS.
- T. Malinauskas, M. Saliba, T. Matsui, M. Daskeviciene, S. Urnikaite, P. Gratia, R. Send, H. Wonneberger, I. Bruder, M. Graetzel, V. Getautis and M. K. Nazeeruddin, Energy Environ. Sci., 2016, 9, 1681–1686 RSC.
- K. Rakstys, S. Paek, G. Grancini, P. Gao, V. Jankauskas, A. M. Asiri and M. K. Nazeeruddin, ChemSusChem, 2017, 10, 3825–3832 CrossRef CAS.
- A. Magomedov, S. Paek, P. Gratia, E. Kasparavicius, M. Daskeviciene, E. Kamarauskas, A. Gruodis, V. Jankauskas, K. Kantminiene, K. T. Cho, K. Rakstys, T. Malinauskas, V. Getautis and M. K. Nazeeruddin, Adv. Funct. Mater., 2018, 28, 1704351 CrossRef.
- J. A. Christians, P. Schulz, J. S. Tinkham, T. H. Schloemer, S. P. Harvey, B. J. Tremolet De Villers, A. Sellinger, J. J. Berry and J. M. Luther, Nat. Energy, 2018, 3, 68–74 CrossRef CAS.
- X. Li, M. Cai, Z. Zhou, K. Yun, F. Xie, Z. Lan, J. Hua and L. Han, J. Mater. Chem. A, 2017, 5, 10480–10485 RSC.
- L. Guan, X. Yin, D. Zhao, C. Wang, Q. An, J. Yu, N. Shrestha, C. R. Grice, R. A. Awni, Y. Yu, Z. Song, J. Zhou, W. Meng, F. Zhang, R. J. Ellingson, J. Wang, W. Tang and Y. Yan, J. Mater. Chem. A, 2017, 5, 23319–23327 RSC.
- F. Wu, Y. Shan, J. Qiao, C. Zhong, R. Wang, Q. Song and L. Zhu, ChemSusChem, 2017, 10, 3833–3838 CrossRef CAS.
- C. Lu, I. T. Choi, J. Kim and H. K. Kim, J. Mater. Chem. A, 2017, 5, 20263–20276 RSC.
- R. Xue, M. Zhang, G. Xu, J. Zhang, W. Chen, H. Chen, M. Yang, C. Cui, Y. Y. Li and Y. Y. Li, J. Mater. Chem. A, 2018, 6, 404–413 RSC.
- J. Zhang, Y. Hua, B. Xu, L. Yang, P. Liu, M. B. Johansson, N. Vlachopoulos, L. Kloo, G. Boschloo, E. M. J. Johansson, L. Sun and A. Hagfeldt, Adv. Energy Mater., 2016, 6, 1601062 CrossRef.
- D. Li, J. Y. Shao, Y. Y. Li, Y. Y. Li, L. Y. Deng, Y. W. Zhong and Q. Meng, Chem. Commun., 2018, 54, 1651–1654 RSC.
- B. Bin Cui, C. Zhu, S. Yang, Y. Han, N. Yang, L. Zhang, Y. Wang, Y. Jia, L. Zhao and Q. Chen, ACS Omega, 2018, 3, 10791–10797 CrossRef.
- K. Rakstys, M. Saliba, P. Gao, P. Gratia, E. Kamarauskas, S. Paek, V. Jankauskas and M. K. Nazeeruddin, Angew. Chem., Int. Ed., 2016, 55, 7464–7468 CrossRef CAS.
- F. Zhang, Z. Wang, H. Zhu, N. Pellet, J. Luo, C. Yi, X. Liu, H. Liu, S. Wang, X. Li, Y. Xiao, S. M. Zakeeruddin, D. Bi and M. Grätzel, Nano Energy, 2017, 41, 469–475 CrossRef CAS.
- F. Zhang, S. Wang, H. Zhu, X. Liu, H. Liu, X. Li, Y. Xiao and S. M. Zakeeruddin, ACS Energy Lett., 2018, 3, 1145–1152 CrossRef CAS.
- M. L. Petrus, A. Music, A. C. Closs, J. C. Bijleveld, M. T. Sirtl, Y. Hu, T. J. Dingemans, T. Bein and P. Docampo, J. Mater. Chem. A, 2017, 5, 25200–25210 RSC.
- M. L. Petrus, M. T. Sirtl, A. C. Closs, T. Bein and P. Docampo, Mol. Syst. Des. Eng., 2018, 3, 734–740 RSC.
- M. L. Petrus, K. Schutt, M. T. Sirtl, E. M. Hutter, A. C. Closs, J. M. Ball, J. C. Bijleveld, A. Petrozza, T. Bein, T. J. Dingemans, T. J. Savenije, H. Snaith and P. Docampo, Adv. Energy Mater., 2018, 8, 1–11 Search PubMed.
- D. Vaitukaityte, Z. Wang, T. Malinauskas, A. Magomedov, G. Bubniene, V. Jankauskas, V. Getautis and H. J. Snaith, Adv. Mater., 2018, 30, 1–7 CrossRef.
- M. Daskeviciene, S. Paek, Z. Wang, T. Malinauskas, G. Jokubauskaite, K. Rakstys, K. T. Cho, A. Magomedov, V. Jankauskas, S. Ahmad, H. J. Snaith, V. Getautis and M. K. Nazeeruddin, Nano Energy, 2017, 32, 551–557 CrossRef CAS.
- M. Daskeviciene, S. Paek, A. Magomedov, K. T. Cho, M. Saliba, A. Kizeleviciute, T. Malinauskas, A. Gruodis, V. Jankauskas, E. Kamarauskas, M. K. Nazeeruddin and V. Getautis, J. Mater. Chem. C, 2019, 7, 2717–2724 RSC.
- T. P. Osedach, T. L. Andrew and V. Bulović, Energy Environ. Sci., 2013, 6, 711–718 RSC.
- M. L. Petrus, T. Bein, T. J. Dingemans and P. Docampo, J. Mater. Chem. A, 2015, 3, 12159–12162 RSC.
- A. C. Recommendations, Gaussian 09 Citation, Gaussian.com, https://gaussian.com/g09citation/, accessed 15 May 2020 Search PubMed.
- J. Urieta-Mora, I. García-Benito, I. Zimmermann, J. Aragó, J. Calbo, G. Grancini, A. Molina-Ontoria, E. Ortí, N. Martín and M. K. Nazeeruddin, J. Mater. Chem. C, 2019, 7, 6656–6663 RSC.
- K. Walzer, B. Männig, M. Pfeiffer and K. Leo, Chem. Rev., 2007, 107, 1233–1271 CrossRef CAS.
- B. Lüssem, M. Riede and K. Leo, Phys. Status Solidi A, 2013, 210, 9–43 CrossRef.
- I. Salzmann, G. Heimel, M. Oehzelt, S. Winkler and N. Koch, Acc. Chem. Res., 2016, 49, 370–378 CrossRef CAS.
- J. Y. Feng, K. W. Lai, Y. S. Shiue, A. Singh, C. H. P. Kumar, C. T. Li, W. T. Wu, J. T. Lin, C. W. Chu, C. C. Chang and C. C. Su, J. Mater. Chem. A, 2019, 7, 14209–14221 RSC.
- Y. Lee, S. Paek, K. T. Cho, E. Oveisi, P. Gao, S. Lee, J. S. Park, Y. Zhang, R. Humphry-Baker, A. M. Asiri and M. K. Nazeeruddin, J. Mater. Chem. A, 2017, 5, 12729–12734 RSC.
- K. Rakstys, A. Abate, M. I. Dar, P. Gao, V. Jankauskas, G. Jacopin, E. Kamarauskas, S. Kazim, S. Ahmad, M. Grätzel and M. K. Nazeeruddin, J. Am. Chem. Soc., 2015, 137, 16172–16178 CrossRef CAS.
- G. Xing, N. Mathews, S. Sun, S. S. Lim, Y. M. Lam, M. Grätzel, S. Mhaisalkar and T. C. Sum, Science, 2013, 342, 344–347 CrossRef CAS.
- N. Drigo, C. Roldan-Carmona, M. Franckevičius, K. H. Lin, R. Gegevičius, H. Kim, P. A. Schouwink, A. A. Sutanto, S. Olthof, M. Sohail, K. Meerholz, V. Gulbinas, C. Corminboeuf, S. Paek and M. K. Nazeeruddin, J. Am. Chem. Soc., 2020, 142, 1792–1800 CrossRef CAS.
- W. Zhou, Z. Wen and P. Gao, Adv. Energy Mater., 2018, 8, 1702512 CrossRef.
- T. Braukyla, R. Xia, T. Malinauskas, M. Daskeviciene, A. Magomedov, E. Kamarauskas, V. Jankauskas, Z. Fei, C. Roldán-Carmona, C. Momblona, M. K. Nazeeruddin, P. J. Dyson and V. Getautis, Sol. RRL, 2019, 3, 1900224 CrossRef.
- A. Magomedov, E. Kasparavičius, K. Rakstys, S. Paek, N. Gasilova, K. Genevičius, G. Juška, T. Malinauskas, M. K. Nazeeruddin and V. Getautis, J. Mater. Chem. C, 2018, 6, 8874–8878 RSC.
- E. Kasparavicius, A. Magomedov, T. Malinauskas and V. Getautis, Chem.–Eur. J., 2018, 24, 9910–9918 CrossRef CAS.
- A. K. Jena, M. Ikegami and T. Miyasaka, ACS Energy Lett., 2017, 2, 1760–1761 CrossRef CAS.
- D. Shi, X. Qin, Y. Li, Y. He, C. Zhong, J. Pan, H. Dong, W. Xu, T. Li, W. Hu, J. L. Brédas and O. M. Bakr, Sci. Adv., 2016, 2, e1501491 CrossRef.
- M. Namatame, M. Yabusaki, T. Watanabe, Y. Ogomi, S. Hayase and K. Marumoto, Appl. Phys. Lett., 2017, 110, 123904 CrossRef.
- J. Zhang, Q. Daniel, T. Zhang, X. Wen, B. Xu, L. Sun, U. Bach and Y. B. Cheng, ACS Nano, 2018, 12(10), 10452–10462 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta08452b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |