
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphoric acid and thermal treatments reveal the peculiar role of surface oxygen anions in lithium and manganese-rich layered oxides†
Jiarong
He‡
a,
Weibo
Hua‡
 *ab,
Aleksandr
Missiul
c,
Georgian
Melinte
de,
Chittaranjan
Das
a,
Akhil
Tayal
f,
Thomas
Bergfeldt
a,
Stefan
Mangold
g,
Xinyang
Liu
a,
Joachim R.
Binder
a,
Michael
Knapp
a,
Helmut
Ehrenberg
a,
Sylvio
Indris
*a,
Björn
Schwarz
a and
Julia
Maibach
*a
*ab,
Aleksandr
Missiul
c,
Georgian
Melinte
de,
Chittaranjan
Das
a,
Akhil
Tayal
f,
Thomas
Bergfeldt
a,
Stefan
Mangold
g,
Xinyang
Liu
a,
Joachim R.
Binder
a,
Michael
Knapp
a,
Helmut
Ehrenberg
a,
Sylvio
Indris
*a,
Björn
Schwarz
a and
Julia
Maibach
*a
aInstitute for Applied Materials (IAM), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: julia.maibach@kit.edu; sylvio.indris@kit.edu
bState Key Laboratory of Electronic Thin Films and Integrated Devices, University of Electronic Science and Technology of China, Chengdu, 610054, China. E-mail: weibo.hua@uestc.edu.cn
cCELLS-ALBA Synchrotron, Cerdanyola del Valles, E-08290 Barcelona, Spain
dInstitute of Nanotechnology, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, D-76344, Eggenstein-Leopoldshafen, Germany
eHelmholtz Institute Ulm (HIU) Electrochemical Energy Storage, Helmholtzstrasse 11, 89081 Ulm, Germany
fDeutsches Elektronen-Synchrotron DESY, Notkestrasse 85, D-22607 Hamburg, Germany
gInstitute for Photon Science and Synchrotron Radiation, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, D-76344 Eggenstein-Leopoldshafen, Germany
First published on 30th September 2020
Abstract
The interplay between cationic and anionic redox activity during electrochemical cycling makes layered Li-rich oxides appealing cathodes for state-of-the-art lithium-ion batteries. However, it remains challenging as the origin of lattice oxygen activity is not yet fully understood. Here we report on the effects of a lithium-deficient layer in the near-surface region of Co-free Li-rich Li[Li0.2Ni0.2Mn0.6]O2 (LLNMO) achieved via a phosphoric acid surface treatment. Our results show that oxidized On− (0 < n < 2) species are formed on the surface of H3PO4-treated LLNMO resulting from Li ion deficiency and lattice distortion. The metastable On− could be easily released from the oxygen surface lattice forming O2via thermal treatment, accompanied by a surface reconstruction and a layered-to-rock-salt/spinel transition. The presented results demonstrate that the surface lattice structure plays a critical role in the electrochemical performance of LLNMO. This information provides new insights into the oxygen redox in LLNMO and opens up an opportunity for Li-rich cathodes to achieve long cycle life toward a broad range of applications in electrical energy storage devices.
Introduction
Alkali-rich transition-metal (TM) oxide materials are of significant interest for advanced battery cathode materials, because they can create excess capacity beyond what is expected from the TM redox alone in stoichiometric cathode materials.1–4 In comparison with the stoichiometric layered oxides (i.e. A[TM]O2, square brackets indicate that the TM is on octahedral sites), some of the TM cations in the TM layer are replaced by alkali ions forming a superstructure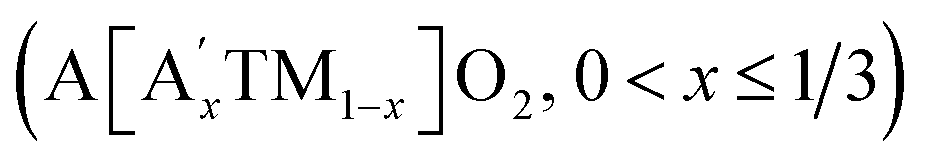 .5–7 It is currently believed that the extra capacity of these cathodes is derived from lattice oxygen redox, which is triggered by the initial charging process at high voltages (e.g. ≥4.5 VLi+/Li).8–10 Among these compounds, Li- and Mn-rich layered oxides (LMLOs) are at the forefront of this area achieving high discharge capacities above 250 mA h g−1.11,12 However, the oxygen redox activity always causes changes in the crystallographic structure, surface inhomogeneity and release of lattice oxygen in LMLOs, thereby resulting in structural instability and severe voltage fading.13,14
.5–7 It is currently believed that the extra capacity of these cathodes is derived from lattice oxygen redox, which is triggered by the initial charging process at high voltages (e.g. ≥4.5 VLi+/Li).8–10 Among these compounds, Li- and Mn-rich layered oxides (LMLOs) are at the forefront of this area achieving high discharge capacities above 250 mA h g−1.11,12 However, the oxygen redox activity always causes changes in the crystallographic structure, surface inhomogeneity and release of lattice oxygen in LMLOs, thereby resulting in structural instability and severe voltage fading.13,14
Recently, extensive research efforts have been devoted to exploring the origin of the structural degradation of LMLOs upon high-voltage cycling. Several hypotheses have been proposed including layered-to-rock-salt/spinel phase transitions,15,16 localization of oxygen electron holes17 or formation of Mn–η1–O2 species,18 TM over-oxidation,19,20 irreversible O2 loss with surface densification,21,22 Li2O removal with “MnO2-like” phase formation,23 generation of peroxo-like O2m− (1 ≤ m ≤ 3) dimers,24,25 O-redox at the interface between the electrolyte and bulk,26etc. Generally, the commonly used carbonate-based electrolytes are not stable up to 5 V versus Li+/Li, which causes interfacial side-reactions at the electrode surface forming a cathode–electrolyte interface (CEI) layer. Thereby, the degradation process of LMLOs is strongly influenced by the structure and reaction chemistry at the cathode surface.27,28 Unfortunately, it is difficult to obtain precise chemical composition and real crystallographic information on the surface of LMLOs during electrochemical cycling.29 Up to now, a clear picture of how the surface lattice oxide ions are oxidized, as lithium ions and electrons are extracted from the layered structure, has been missing.
Recently, chemically driven Li-ion extraction reaction has drawn widespread attention due to its fast reaction rate process. For example, Ramakrishnan et al. achieved an extended interfacial stability of Li-rich oxide cathodes on the surface through a strong H2SO4 acid rinsing, which could suppress the irreversible oxygen evolution and improve the cycling and rate performance.8 Wu et al. tuned the oxygen redox reaction through the inductive effect with proton insertion in Li-rich oxides, thus stabilizing the oxygen activity during charging.30 Paik et al. studied the acid leaching of the layered compound Li2MnO3, showing an H+/Li+ ion exchange with a shearing of the oxygen layers driven by hydrogen bonding, observed by 6Li and 2H MAS NMR measurements in conjunction with X-ray diffraction.31 On the other hand, both Meng's group12 and our group32 found that the high voltage plateau at around 4.5 V of LMLOs in the first charging can be recovered by heating of the cycled electrode, suggesting that the high-voltage functionality of LMLOs is closely tied to their thermal stability. However, how the dilute phosphoric acid and thermal treatments affect the surface structure and chemistry of LMLOs is still unclear.
It is well known that the precipitation process is also one of the fastest reactions in aqueous solution.33 Inspired by the production of a Li3PO4 precipitate,34 we used low-concentration solutions of phosphoric acid to extract lithium ions from Co-free layered Li[Li0.2Ni0.2Mn0.6]O2 oxides (LLNMO) without obvious oxygen loss and H+/Li+ exchange. As determined by high-resolution synchrotron radiation diffraction (SRD), X-ray photoelectron spectroscopy (XPS) and transmission electron microscopy (TEM), the acid treatment leads to a considerably distorted surface lattice of LLNMO after the chemical extraction of lithium ions, which provides the prerequisites to form oxidized On− (0 < n < 2) on the surface (see Scheme 1). To investigate the thermal stability of existing On− in the acid-treated samples, the materials were heated at high temperature. In situ high-temperature SRD (HTSRD) results reveal that the pristine LLNMO is stable during annealing, while a phase transition from a layered structure to a Li-containing rock-salt (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) and/or a Li-containing spinel (Fdm) structure occurs in the H3PO4-treated materials after O2 loss induced by the disproportionation of oxidized On− upon heating, which has never been reported before.
m) and/or a Li-containing spinel (Fdm) structure occurs in the H3PO4-treated materials after O2 loss induced by the disproportionation of oxidized On− upon heating, which has never been reported before.
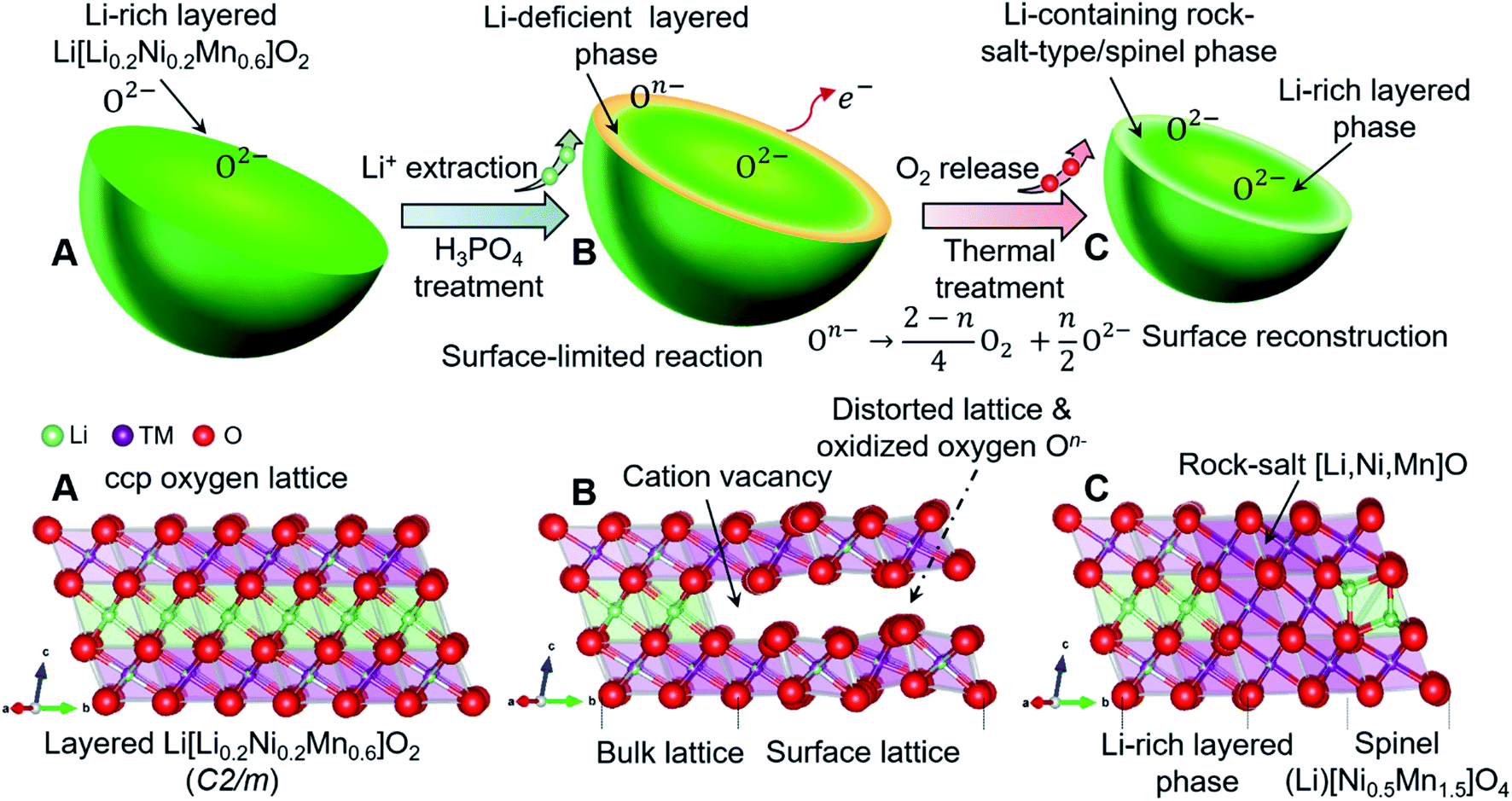 | ||
| Scheme 1 Schematic illustration of chemically and thermally induced structural evolution of layered Li[Li0.2Ni0.2Mn0.6]O2 cathode materials, showing the formation of oxidized On− with surface lattice distortion and lithium deficiency via H3PO4 treatment, and the atomic rearrangement (phase transformation) with recrystallization after thermal treatment. (A) Pristine sample, (B) H3PO4-treated samples, (C) thermally treated sample. | ||
Results and discussion
The pure layered LLNMO (LLNMO-P) was prepared by a hydroxide co-precipitation method followed by a high-temperature lithiation reaction,33,35 as evidenced by the SRD pattern in Fig. 1a. The acid-treated samples were obtained by homogeneously mixing LLNMO-P with different concentrations of H3PO4 solution (4% & 5%, mass ratio). The as-prepared materials were labelled as LLNMO-H4 and LLNMO-H5, respectively. All three samples were subsequently calcined at 900 °C for 2 hours in air to investigate the thermal stability of the oxides, which were marked as LLNMO-TP, LLNMO-TH4 and LLNMO-TH5, respectively. LLNMO-P treated with a high-concentration H3PO4 solution (15%; labelled as LLNMO-H15) and the typical high-voltage spinel LiNi0.5Mn1.5O4 (LNMO) oxide were also prepared for comparison. An impurity phase TMPO4 (TM = Ni, Mn) is found in LLNMO-H15, see ESI Fig. S2g,† suggesting that LLNMO-P would dissolve in high-concentration phosphoric acid. In this work, we focus on the low-concentration H3PO4 treatment. Details of the preparation process are presented in the ESI.†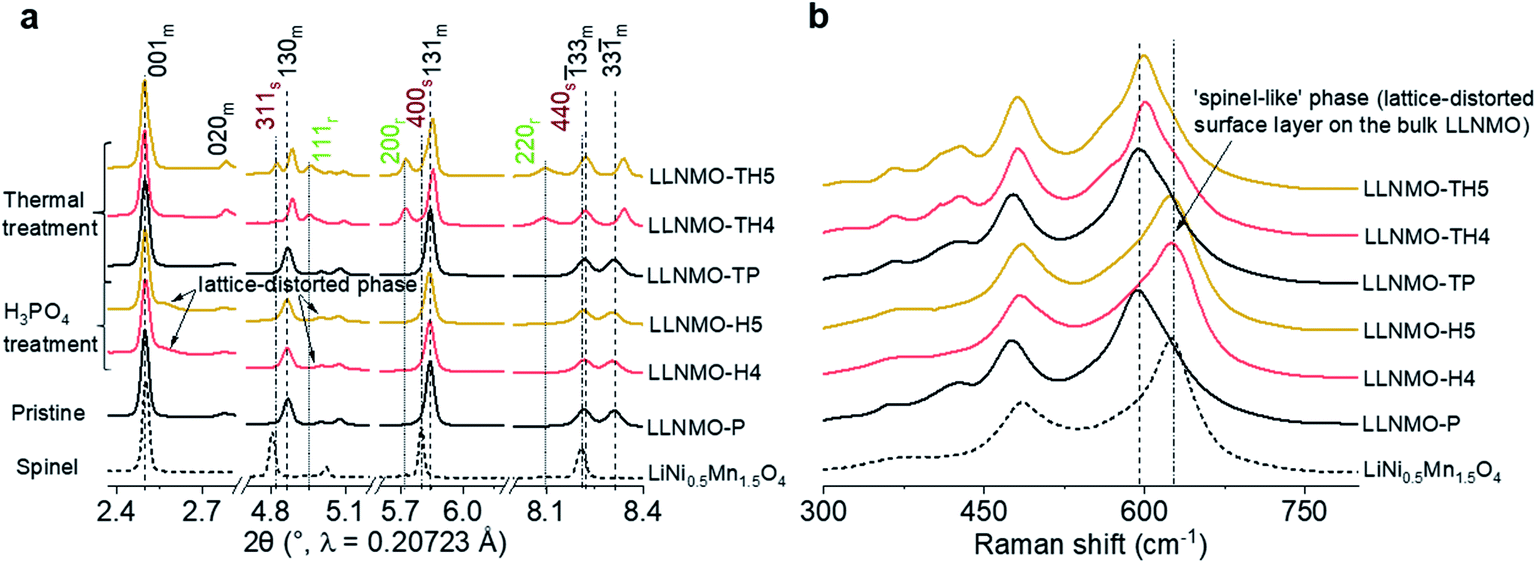 | ||
| Fig. 1 Structural analysis of the prepared samples. (a) SRD patterns and (b) Raman spectra of the samples. In (a), m, r, and s stand for layered monoclinic phase (C2/m), rock-salt-type phase (Fmm), and spinel phase (Fdm). The 020m reflection belonging to the honeycomb superstructure is visible for all products, demonstrating that the acid treatment and the thermal treatment do not affect the bulk monoclinic-layered structure (C2/m). In (b), the grey short dashed line indicates the peak position for the spinel LiNi0.5Mn1.5O4 reference sample. | ||
Compared to the SRD pattern of LLNMO-P, there is almost no shift in the main reflections in the SRD patterns for both LLNMO-H4 and LLNMO-H5. The Ni and Mn oxidation states for the acid-treated materials do not change noticeably, and remain at +2 and +4, respectively, as determined by hard X-ray absorption spectroscopy (XAS, see ESI Fig. S1†). These results demonstrate that the acid treatment does not cause obvious changes in the electronic structure of the bulk oxides. Surprisingly, several additional weak reflections close to the main layered phase appear in the SRD patterns of LLNMO-H4 and LLNMO-H5, e.g. a broad reflection at ∼2.5° on the right side of the 001m reflection. These Bragg reflections are always observed for the LMLOs when they are charged to potentials >4.5 V, proposed to be related to a spinel-like (Fdm) phase on the surface (see the spinel LNMO reference)36. Normally, the 001m reflection of layered LLNMO shifts toward lower scattering angles firstly during charging, and then moves to higher 2-theta angles with further Li-ion extraction, corresponding to an initially increased lattice parameter c caused by increasing electrostatic repulsion and later to a contraction of the c-axis resulting from oxygen oxidation or TM migration.37,38 Therefore, the broad reflection on the right side of the 001m reflection can be ascribed to the over-delithiated surface layer on LLNMO after the H3PO4 treatment. Raman spectra were additionally recorded to further trace the structural changes after the acid treatment (Fig. 1b). The main peak observed for the H3PO4-treated materials, i.e. LLNMO-H4 and LLNMO-H5, shifts from 598 to 630 cm−1 with respect to the LLNMO-P, manifesting the changes in the A1g vibration of the Raman-active mode from a layered to a spinel LNMO phase.39,40 This is regarded as a complementary indicator of spinel-like phase formation on the surface. However, the fundamental question, which naturally arises, is “What is the nature of this often-observed spinel-like (Fdm) phase?”
XPS analyses were carried out to investigate the surface composition and oxidation states of the elements in the specimens (see Fig. 2 and ESI Fig. S3†). In Fig. 2, the peak found at around 52.9 eV can be assigned to the lattice Li+ (Fig. 2a) and the major peaks at 529.6 eV can be ascribed to the lattice oxide ions O2−, while the weak components at higher binding energies (531.9 eV and 533.1 eV) in the O 1s core level spectrum correspond to surface bound species, for example oxygen in carbonates and hydroxides (Fig. 2b). After the acid treatment, the relative intensity of the Li peak at 52.9 eV decreased substantially in the Li 1s spectrum of LLNMO-H4 and LLNMO-H5, indicating that Li ions are extracted from the surface lattice of crystallites. Simultaneously, a peak at ∼530.6 eV appears in the O 1s region which is characteristic of the formation of oxidized On− in the surface lattice of the acid-treated LLNMO samples.25 All samples showed very similar peak shapes and binding energy (BE) positions for Ni 2p3/2, Mn 3p3/2 and Mn 3s (ESI Fig. S3†), indicating that the transition metal oxidation states were the same irrespective of the sample treatment. A multiplet splitting method was adopted to evaluate the oxidation states of Ni and Mn in detail, as demonstrated by Biesinger41 and Azmi et al.42 The fitting results indicate that for all samples the transition metals are present in surface oxidation states of Ni2+ and Mn4+, respectively. The Mn oxidation state is further confirmed in the Mn 3s spectrum as a distinctive satellite appears at 4.5 eV higher binding energy compared to the main Mn 3s peak.
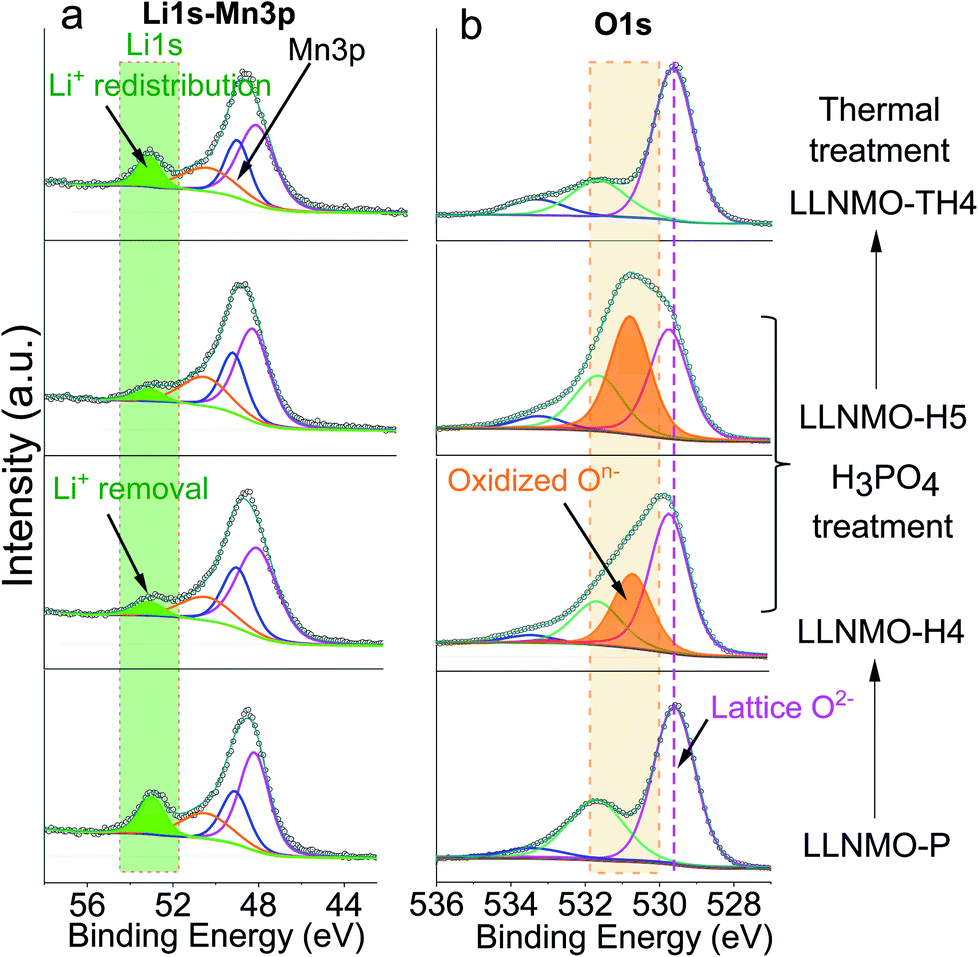 | ||
| Fig. 2 Li 1s (a) and O 1s (b) XP spectra of the pristine and acid treated samples, showing the formation of oxidized On− on the surface of the crystallites with the extraction reaction of lithium ions via the H3PO4 treatment (LLNMO-H4 and LLNMO-H5) and the decomposition of oxidized On− with lithium ion redistribution after thermal treatment (LLNMO-TH4). | ||
To investigate the morphological differences of LLNMO-P and the acid-treated samples, scanning electron microscopy (SEM) was carried out. As shown in Fig. 3a, the LLNMO-P particles are agglomerates consisting of several fine platelet-like crystallites, with a primary crystal size of around 100–500 nm. Fig. 3d displays a high-resolution TEM (HRTEM) image of LLNMO-P along the c-axis. The measured interplanar spacing is approximately 4.75 Å, corresponding to the planar distance between adjacent TM layers of layered LLNMO, i.e. the (001)m plane. Although the crystal morphology and the bulk lattice remain basically unchanged after the H3PO4 treatment (see Fig. 3b and e, and S4†), a severe lattice distortion at the surface is clearly observed for the LLNMO-H5 in the HRTEM image. The distortion results from Li ions being removed from the surface lattice, leaving Li-site cation vacancy defects. The distorted lattice can elongate or shorten the distance between two adjacent oxygen ions (O2−). The former might result from an increased electrostatic repulsive force within the cubic-close packed (ccp) oxygen lattice, while the latter effect might be attributed to the formation of molecular O2 inside the solid18 or localized electron holes on oxygen17 or peroxo oxygen dimers.43
 | ||
| Fig. 3 SEM and HRTEM images of (a and d) LLNMO-P, (b and e) LLNMO-H5 and (c and f) LLNMO-TH5. | ||
ESI Fig. S2† shows the results of the Rietveld refinement against the SRD patterns for the samples presented in Fig. 1. Although in the SRD it is difficult to distinguish between layered and spinel phases on the surface from a nano-scale with the corresponding broadening of reflections,38 HRTEM images show that no TM cations are pronouncedly observed in the Li layer of acid-treated samples (see the HRTEM images above). This suggests that a potential cubic spinel phase (AB2O4, Fdm) as indicated by Raman can actually be excluded since for spinel a quasi-equal distribution of TM is characteristic. Therefore, Rietveld refinements were performed assuming a multiple phase model, i.e. a monoclinic layered Li[Li0.2Ni0.2Mn0.6]O2 (C2/m) and a defective layered Li1−x[TM]O2 (Rm), which seems more appropriate to account for the surface phase than a spinel phase, in LLNMO-H4 and LLNMO-H5. The weight fraction of the Li-defective layered phase is ∼6% for LLNMO-H4, and ∼8% for LLNMO-H5, respectively. Chemical compositions were investigated by inductively coupled plasma optical emission spectroscopy (ICP-OES). ICP-OES results suggest that the atomic ratio of Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mn in the three samples is very close to the theoretical value (1:3, see Table 1). The lithium concentrations in the H3PO4-treated samples decrease significantly, which provides convincing evidence for the chemical extraction of lithium ions from the crystal structure of Li-rich layered oxides during the acid treatment. Therefore, the so-called “spinel-like” phase formed on the surface is most likely a lithium-deficient layered phase with a lattice distortion. Additionally, bubbles are visible during the phosphoric acid treatment. Based on these results, a reduction–oxidation reaction is proposed:
:Mn in the three samples is very close to the theoretical value (1:3, see Table 1). The lithium concentrations in the H3PO4-treated samples decrease significantly, which provides convincing evidence for the chemical extraction of lithium ions from the crystal structure of Li-rich layered oxides during the acid treatment. Therefore, the so-called “spinel-like” phase formed on the surface is most likely a lithium-deficient layered phase with a lattice distortion. Additionally, bubbles are visible during the phosphoric acid treatment. Based on these results, a reduction–oxidation reaction is proposed:
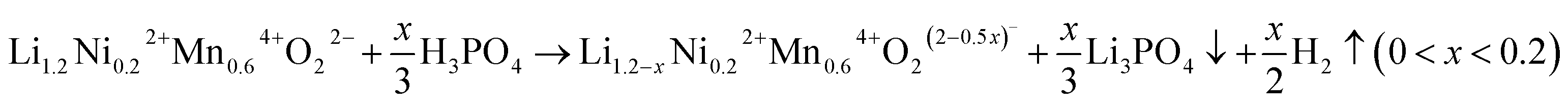 | (1) |
| Samples | Molar ratio | ||||
|---|---|---|---|---|---|
| Li | Ni | Mn | O | P | |
| a Note: determining whether or not oxygen loss occurred through acid treatment could not be deduced from this table because of the larger experimental error in the case of oxygen. | |||||
| LLNMO-P | 1.21 ± 0.03 | 0.20 ± 0.01 | 0.61 ± 0.01 | 2.28 ± 0.18 | — |
| LLNMO-H4 | 1.07 ± 0.03 | 0.20 ± 0.01 | 0.62 ± 0.01 | 2.19 ± 0.18 | 0.01 ± 0.001 |
| LLNMO-H5 | 1.04 ± 0.03 | 0.20 ± 0.01 | 0.63 ± 0.01 | 2.23 ± 0.18 | 0.02 ± 0.002 |
| LLNMO-TP | 1.24 ± 0.03 | 0.20 ± 0.01 | 0.61 ± 0.01 | 2.24 ± 0.18 | — |
| LLNMO-TH4 | 1.07 ± 0.03 | 0.20 ± 0.01 | 0.62 ± 0.01 | 2.17 ± 0.18 | 0.01 ± 0.001 |
| LLNMO-TH5 | 1.04 ± 0.03 | 0.20 ± 0.01 | 0.63 ± 0.01 | 2.17 ± 0.18 | 0.02 ± 0.002 |
In order to gain further insights into the metastable On− on the surface of the Li- and Mn-rich layered oxides, thermal treatment was used and the samples were analysed by SRD and Raman spectroscopy. The reflections in the SRD pattern of LLNMO-TP do not change considerably, as evidenced by comparison with LLNMO-P (see Fig. 1a), revealing a good structural and chemical stability of the prepared Li[Li0.2Ni0.2Mn0.6]O2. In contrast, for the acid-treated samples, in addition to the main monoclinic layered phase (C2/m), a set of reflections, i.e. 111r, 200r and 220r, indexed to the Li-containing rock-salt-type phase (Fmm), is clearly observed in the SRD pattern of the thermally treated LLNMO-TH4. LLNMO-TH5 is found to be a mixture of a Li-rich layered phase (C2/m), a Li-containing rock-salt-type phase (Fmm) and a Li-containing spinel phase (Fdm), and their weight fractions are 73(2), 13(2) and 14(2)% (see ESI Fig. S2 and Table S1†). Thermodynamically, as lithium and oxygen are successively released from the surface lattice, the Li-rich Li[Li0.2Ni0.2Mn0.6]O2 oxide tends to transform into a Li-containing rock-salt-type phase (Fmm) and then into a Li-containing spinel phase (Fdm), which matches precisely with reported results.7 These phase changes again offer compelling evidence for a continuous lithium extraction during the H3PO4 treatment. Interestingly, some reflections in the SRD patterns of LLNMO-TH4 and LLNMO-TH5 (e.g. 001m and ![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 33m) remain at the same diffraction angles (2θ) compared to the SRD patterns of the LLNMO-P, while other reflections such as 130m, 131m and 33m shift toward higher scattering angles. All of this indicates that the average TM–TM inter-slab distance of the monoclinic layered structure (indicative of Li content and c lattice parameter) remains the same after the thermal treatment, but the average TM–TM intra-slab distance of the layered structure becomes smaller (i.e. a and b lattice parameters, see ESI Table S1†). Furthermore, the SRD analysis implies that the cubic rock-salt/spinel phases with a coherent ccp oxygen lattice formed on the surface could compress the a–b plane in the interior of the layered crystal and cause surface reconstruction (see the SEM images in Fig. 3c).
33m) remain at the same diffraction angles (2θ) compared to the SRD patterns of the LLNMO-P, while other reflections such as 130m, 131m and 33m shift toward higher scattering angles. All of this indicates that the average TM–TM inter-slab distance of the monoclinic layered structure (indicative of Li content and c lattice parameter) remains the same after the thermal treatment, but the average TM–TM intra-slab distance of the layered structure becomes smaller (i.e. a and b lattice parameters, see ESI Table S1†). Furthermore, the SRD analysis implies that the cubic rock-salt/spinel phases with a coherent ccp oxygen lattice formed on the surface could compress the a–b plane in the interior of the layered crystal and cause surface reconstruction (see the SEM images in Fig. 3c).
After the thermal treatment, the main peak at ∼630 cm−1 in the Raman spectra observed for the acid treated LLNMO-H4 and LLNMO-H5 moves to 603 cm−1 for LLNMO-TH4 and LLNMO-TH5 (Fig. 1b). It seems that the “spinel-like” phase in LLNMO-H4 and LLNMO-H5 transforms back to the Li-rich layered phase. Indeed, the first interpretation of the Raman spectra after acid treatment indicating a cubic “spinel-like” structure is meagre and not straightforward because no real spinel phase (AB2O4) is formed in the acid-treated samples but rather a lithium-deficient layered phase with a lattice distortion and an O3 structure (ABCABC oxygen stacking sequence, see the discussion above). The results are similar to those reported previously by Yin et al.44 and interpreted by them as the formation of a densified layered phase with the O3 Rm structure. In the XP spectra of LLNMO-TH4 (Fig. 2), the oxygen peak at ∼530.6 eV disappears, signifying the disintegration of oxidized On− on the surface during thermal treatment (i.e.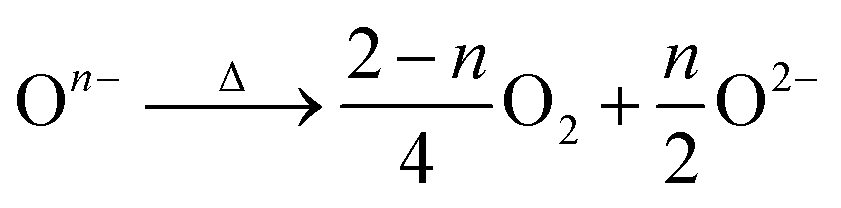 , Δ indicates heating). It is well known that the oxygen anions offer metal coordination sites in the layered structure.11,45 The oxygen release is therefore supposed to be accompanied by surface atomic rearrangement. The intensity of the Li 1s peak in the XP spectra of LLNMO-TH4 increases, compared to that of LLNMO-H4, pointing towards Li ions from the bulk structure migrating into the empty octahedral/tetrahedral sites on the surface during annealing (i.e. lithium redistribution). Moreover, a step and terrace texture is found to form on the surface of LLNMO-TH5, see Fig. 3c, illustrating the surface reconstruction and the recrystallization during the thermal treatment of LLNMO-H5 (see also the SEM-Energy Dispersive X-ray Analysis (EDX) images of selected samples in ESI Fig. S5–S8†). The TEM image of LLNMO-TH5 (Fig. 3f) shows a significant amount of TM migration from the TM layer to the Li layer on the surface that promotes the formation of the cubic spinel/rock-salt structure, which is supposed to accommodate a certain amount of Li ions, as evidenced by the XPS results in Fig. 2.
, Δ indicates heating). It is well known that the oxygen anions offer metal coordination sites in the layered structure.11,45 The oxygen release is therefore supposed to be accompanied by surface atomic rearrangement. The intensity of the Li 1s peak in the XP spectra of LLNMO-TH4 increases, compared to that of LLNMO-H4, pointing towards Li ions from the bulk structure migrating into the empty octahedral/tetrahedral sites on the surface during annealing (i.e. lithium redistribution). Moreover, a step and terrace texture is found to form on the surface of LLNMO-TH5, see Fig. 3c, illustrating the surface reconstruction and the recrystallization during the thermal treatment of LLNMO-H5 (see also the SEM-Energy Dispersive X-ray Analysis (EDX) images of selected samples in ESI Fig. S5–S8†). The TEM image of LLNMO-TH5 (Fig. 3f) shows a significant amount of TM migration from the TM layer to the Li layer on the surface that promotes the formation of the cubic spinel/rock-salt structure, which is supposed to accommodate a certain amount of Li ions, as evidenced by the XPS results in Fig. 2.
In order to trace the chemical and structural evolution of the acid-treated samples with surface defects, thermogravimetric (TG) and in situ high-temperature synchrotron radiation diffraction (HTSRD) measurements were carried out, as shown in Fig. 4 and ESI Fig. S9–S12.† TG curves show that there are two weight loss stages for both LLNMO-H4 and LLNMO-H5 in air up to 900 °C, as compared to an almost constant weight for LLNMO-P during the heating and cooling processes (Fig. 4a). The first weight loss at about 300 °C is attributed to the decomposition of surface oxidized On− into O2; the second weight loss between 700 and 900 °C is most likely due to the release of surface and/or bulk lattice oxygen ions 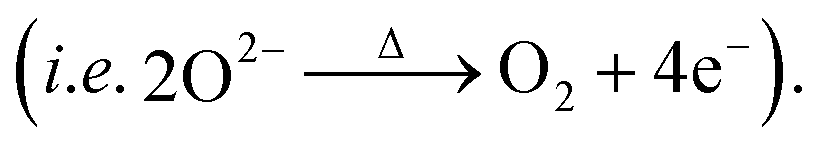 Importantly, a weight gain (i.e. oxygen uptake) occurs for the H3PO4-treated oxides during cooling, leading to an overall weight loss of ∼2% for LLNMO-H4 and ∼3% for LLNMO-H5, respectively, after thermal treatment (heating and cooling). These results match with the theoretical values, e.g.
Importantly, a weight gain (i.e. oxygen uptake) occurs for the H3PO4-treated oxides during cooling, leading to an overall weight loss of ∼2% for LLNMO-H4 and ∼3% for LLNMO-H5, respectively, after thermal treatment (heating and cooling). These results match with the theoretical values, e.g.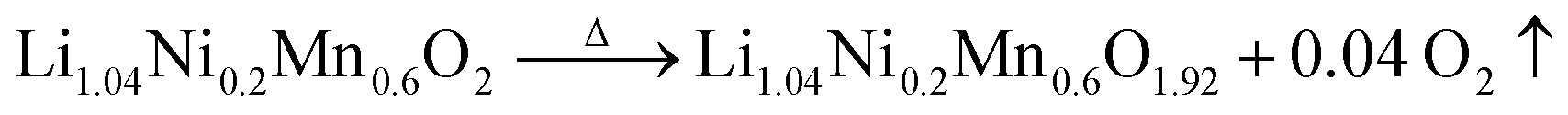 , revealing that lattice oxygen is not released from the crystal structure extensively during H3PO4 treatment. Note that the Ni and Mn valence states on the surface remain almost constant (Ni2+ and Mn4+) after the H3PO4 and thermal treatment (see ESI Fig. S3†), while the amount of lithium ions on the surface is increased in the acid-treated sample after heating (see Fig. 2a). Therefore, the weight gain during cooling reveals that Li ions are gradually redistributed across the entire crystal structure, which causes oxygen incorporation to maintain electroneutrality and provide more coordination sites for the migrated Li ions forming a Li-containing rock-salt/spinel structure on the surface (see the discussion below). Oxygen loss and oxygen uptake at high temperatures >700 °C are consistent with the reversible changes in the lattice parameters of the layered phase during heating and cooling (see ESI Fig. S12†). However, the lattice parameters of LLNMO-H4 and LLNMO-H5 do not return to the initial value after thermal treatment when compared with LLNMO-P, which indicates the destabilization of the bulk material.
, revealing that lattice oxygen is not released from the crystal structure extensively during H3PO4 treatment. Note that the Ni and Mn valence states on the surface remain almost constant (Ni2+ and Mn4+) after the H3PO4 and thermal treatment (see ESI Fig. S3†), while the amount of lithium ions on the surface is increased in the acid-treated sample after heating (see Fig. 2a). Therefore, the weight gain during cooling reveals that Li ions are gradually redistributed across the entire crystal structure, which causes oxygen incorporation to maintain electroneutrality and provide more coordination sites for the migrated Li ions forming a Li-containing rock-salt/spinel structure on the surface (see the discussion below). Oxygen loss and oxygen uptake at high temperatures >700 °C are consistent with the reversible changes in the lattice parameters of the layered phase during heating and cooling (see ESI Fig. S12†). However, the lattice parameters of LLNMO-H4 and LLNMO-H5 do not return to the initial value after thermal treatment when compared with LLNMO-P, which indicates the destabilization of the bulk material.
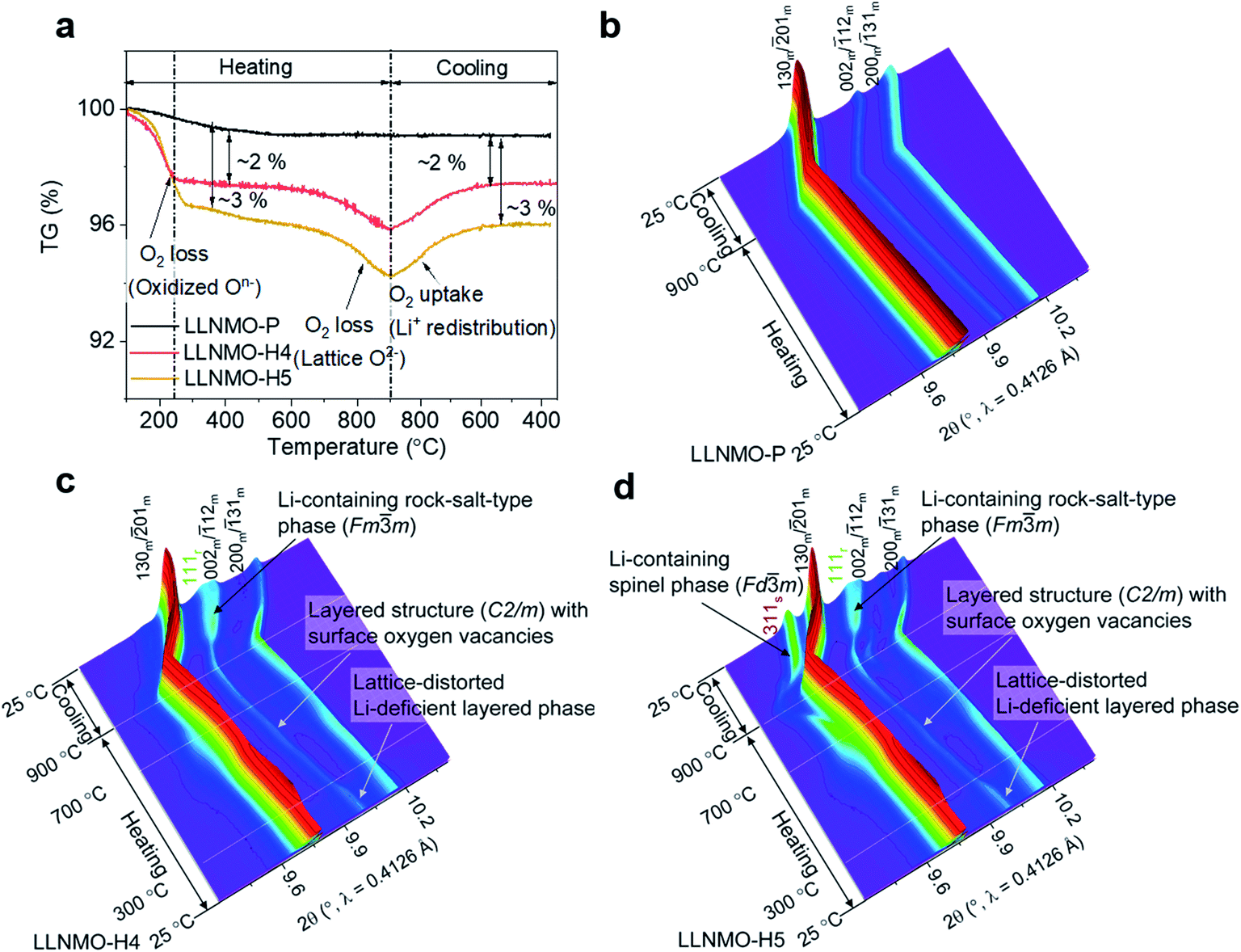 | ||
| Fig. 4 (a) TG curves of the samples, and in situ HTSRD patterns of (b) LLNMO-P, (c) LLNMO-H4 and (d) LLNMO-H5. | ||
In the SRD patterns acquired during continuous heating, all reflections of LLNMO-P move to lower two-theta angles (see Fig. 4b). During cooling, all reflections tend to shift towards their initial position. The difference in lattice parameters a, b, c, and V of LLNMO-P before and after heating is 0.0044(5) Å, 0.0067(5) Å, 0.0039(5) Å, and 0.4772(5) Å3, respectively, again proving the excellent thermal stability of LLNMO-P. Heating of the H3PO4-treated samples up to 300 °C causes the weak broad reflection at around 10°, which corresponds to the Li-deficient layered phase and overlaps with the 002m/12m reflections belonging to the monoclinic layered phase, to vanish, see Fig. 4c and d, in good agreement with the oxygen loss observed in the TG curves. No new reflections in the in situ HTSRD patterns of LLNMO-H4 and LLNMO-H5 appear until the temperature is increased to ∼700 °C, implying that a certain amount of lithium and oxygen vacancies on the surface does not lead to an immediate phase transition in the Li-rich layered oxides (kinetic control). With a further increase of temperature to 900 °C, the 111r reflection of a disordered Li-containing rock-salt structure (Fmm) becomes sharp in both LLNMO-H4 and LLNMO-H5, while a large shift in the 311s reflection corresponding to the Li-containing spinel structure (Fdm) is clearly found in LLNMO-H5. The structural changes are probably related to the defects, more specifically oxygen vacancies, generated on the surface of acid-treated oxides at high temperature (see Fig. 4a). These could effectively contribute to the ionic migration and thus result in the phase transformation from a “Li/O-poor” defective layered structure to a Li-containing cubic rock-salt/spinel structure.45 The increasing intensity of the new reflections during cooling indicates that the oxygen anions are incorporated into the surface lattice to create more coordination sites for TM/Li cations (higher content of Li-containing cubic rock-salt-type/spinel phases, see ESI Table S3†). These results also suggest that the thermal treatment does not affect the lithium content of the oxides noticeably according to the LixNi0.2Mn0.6Oy oxide phase diagram.7 Finally, the formed Li-containing rock-salt/spinel structures are maintained after cooling to room temperature. Therefore, the overall reaction during the thermal treatment can be described as follows:
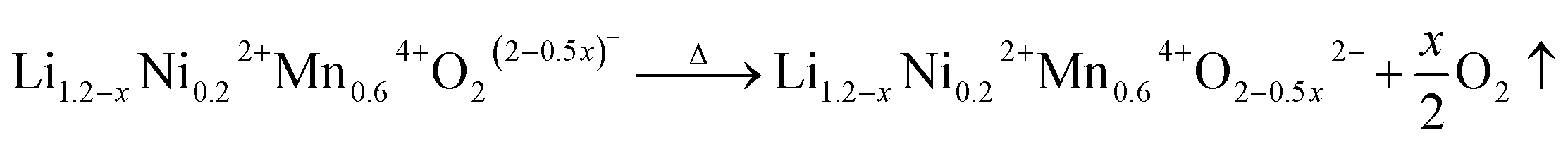 | (2) |
To evaluate the relation between the defect structure and electrochemical properties, electrochemical characterization experiments were performed using CR2032-type coin cells at room temperature. A low current density of 0.01C (1C = 320 mA g−1) and a wide voltage range (2.0–5.0 V) were firstly used to minimize polarization in the charging process and to ensure a fully charged state, as displayed in Fig. 5a and ESI S13.† The initial charge curves of all electrodes show a comparably monotonic region below 4.5 V and a voltage plateau at approximately 4.6 V vs. Li+/Li. Except for the cathodes based on LLNMO-TH4 and LLNMO-TH5, the cathodes exhibit a specific capacity of ∼120 mA h g−1 at voltages <4.5 V, which corresponds to the extraction of 0.4 Li ion from Li[Li0.2Ni0.2Mn0.6]O2 and the oxidation of nickel(II) ions (i.e. 0.2Ni2+ → 0.2Ni4+ + 0.4e−).46 The lower capacity of LLNMO-TH4 and LLNMO-TH5 cathodes in this region could be due to the migration of some Ni2+ cations into the octahedral sites in the rock-salt structure and/or the tetrahedral sites in the spinel structure. The two plateaus at about 4.7 V found in the LLNMO-TH5 cathode (see Fig. 5a and ESI Fig. S13f†) are the characteristics of the spinel LiNi0.5Mn1.5O4 structure. The presence of these plateaus is thus in good agreement with the SRD analysis, which also indicates a spinel structure for LLNMO-TH5. Noticeably, the acid-treated electrodes, LLNMO-H4 and LLNMO-H5, show almost the same capacity as that of LLNMO-P in the low-voltage region (<4.5 V). This provides evidence that the TM cations, at least Ni ions, are not oxidized during the acid treatment. In situ X-ray absorption near-edge structure (XANES) experiments were carried out to investigate the charge compensation mechanism in LLNMO-H5 (see Fig. 5b). There is no significant change in the Mn K-edge spectra of LLNMO-H5 during charging, and all spectra can be assigned to Mn4+. The Ni K absorption edge shifts towards higher energy as the voltage increases to 4.5 V, suggesting electron removal from the Ni2+ ions (Ni2+ → Ni4+ + 2e−). After charging to voltages higher than 4.6 V, no further changes in the XANES spectra are observed, indicating that the nickel valence state does not change considerably. Thus, the extra capacity of LLNMO-H5 (∼180 mA h g−1), originating from the long plateau at 4.6 V, can be ascribed to lattice oxide ion oxidation. The first charge capacity of LLNMO-H4 and LLNMO-H5 with an oxidized oxide surface layer is 324.3 and 293.6 mA h g−1, respectively, which is lower than that of LLNMO-P (398.9 mA h g−1). Except for LLNMO-TH4 and LLNMO-TH5 (self-formation of a Li-containing rock-salt/spinel surface layer), the electrodes exhibit a comparable initial discharge capacity, i.e. 278(3) mA h g−1. Taken together, electrochemically driven oxide oxidation is supposed to participate in the de-lithiation reaction at high voltages (>4.5 V) and start from the surface lattice (a large amount of cation vacancies).
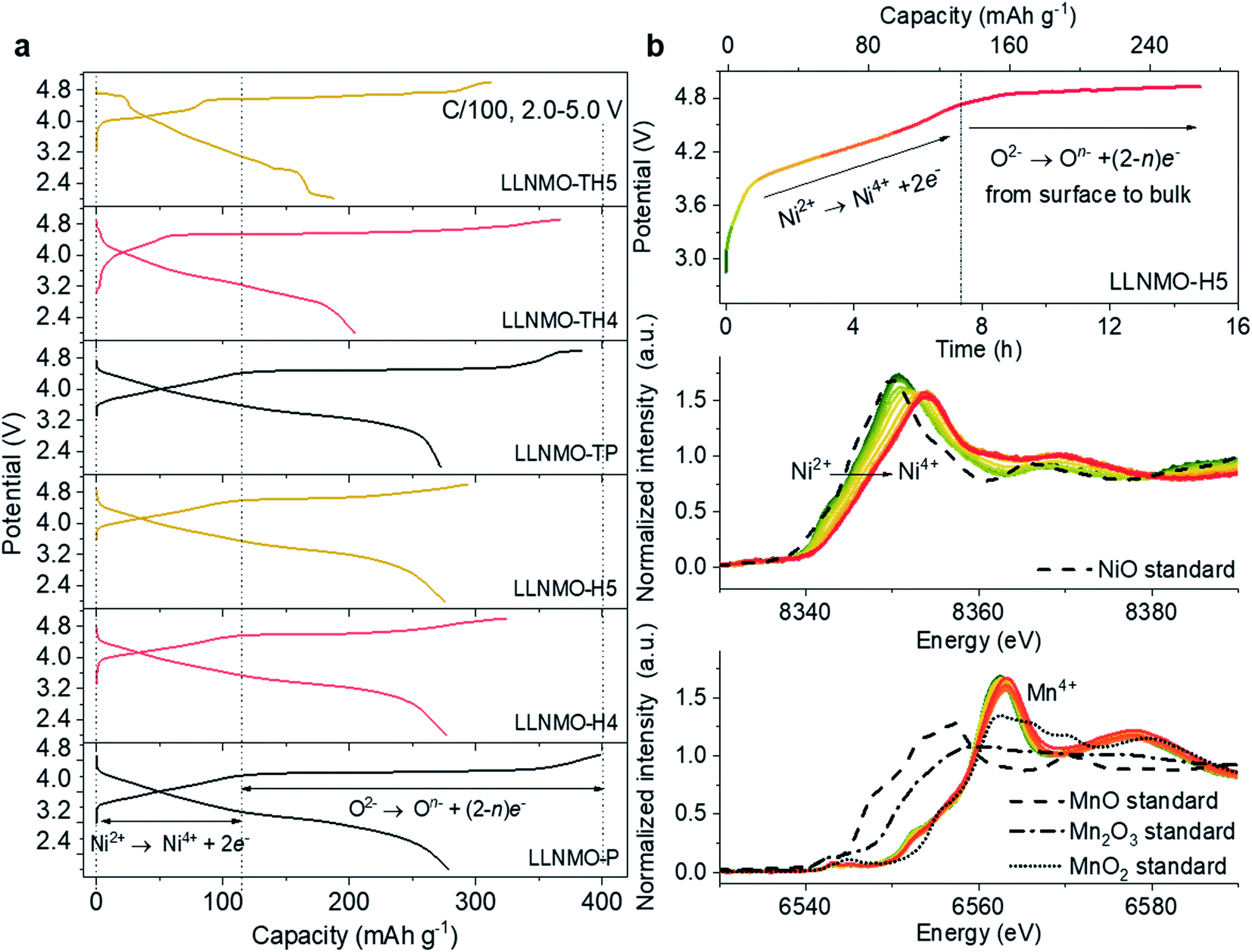 | ||
| Fig. 5 (a) The initial capacity versus voltage profiles of the electrodes at a C-rate of 0.01C; (b) in situ XANES spectra at Ni and Mn K-edges of the LLNMO-H5 electrode during the first charging process. | ||
From a practical application perspective, the fabricated cells were galvanostatically cycled within the narrow voltage range of 2.0–4.8 V at 0.05C. The initial charge–discharge voltage profiles of the electrodes are shown in ESI Fig. S14.† A large overpotential on charging can be observed in the acid-treated electrodes, as supported by an increased charge transfer resistance in the electrochemical impedance spectroscopy (EIS) measurements shown in ESI Fig. S15 and Table S4.† The high polarization is attributed to the oxidized On− surface layer with lattice distortion, which could lead to a high diffusion barrier for Li ions, and thus results in a lower capacity and a shorter plateau at 4.6 V. Previous studies have suggested that the electrochemical performance of the electrodes can be effectively improved by modulating the Li/Ni disorder in layered cathode materials.47–50 After surface reconstruction from a layered to a Li-containing disordered rock-salt structure via thermal treatment, the LLNMO-TH4 electrode exhibits outstanding cycling performance, as shown in Fig. 6a. Even if the coulombic efficiency of LLNMO-TH4 is only around 93% during cycling, a high capacity of 297.2 mA h g−1 can be achieved after 60 cycles, which is much higher than that of LLNMO-P (213.0 mA h g−1). The low coulombic efficiency of LLNMO-TH4 is probably due to the Li-containing rock-salt phase on the surface that acts as a diffusion barrier layer for Li ion transport, which results in a dramatic capacity decrease after 60 cycles (see Fig. 6a). The corresponding derivative capacity–voltage (dQ/dV) plots for the two electrodes at various cycles are shown in Fig. 6b and c. A peak at around 3 V corresponding to the Mn3+/4+ redox activity emerges in both electrodes upon cycling, especially for the LLNMO-TH4 electrode, suggesting that the capacity originating from the bulk lattice oxygen redox activity is reduced to some extent.
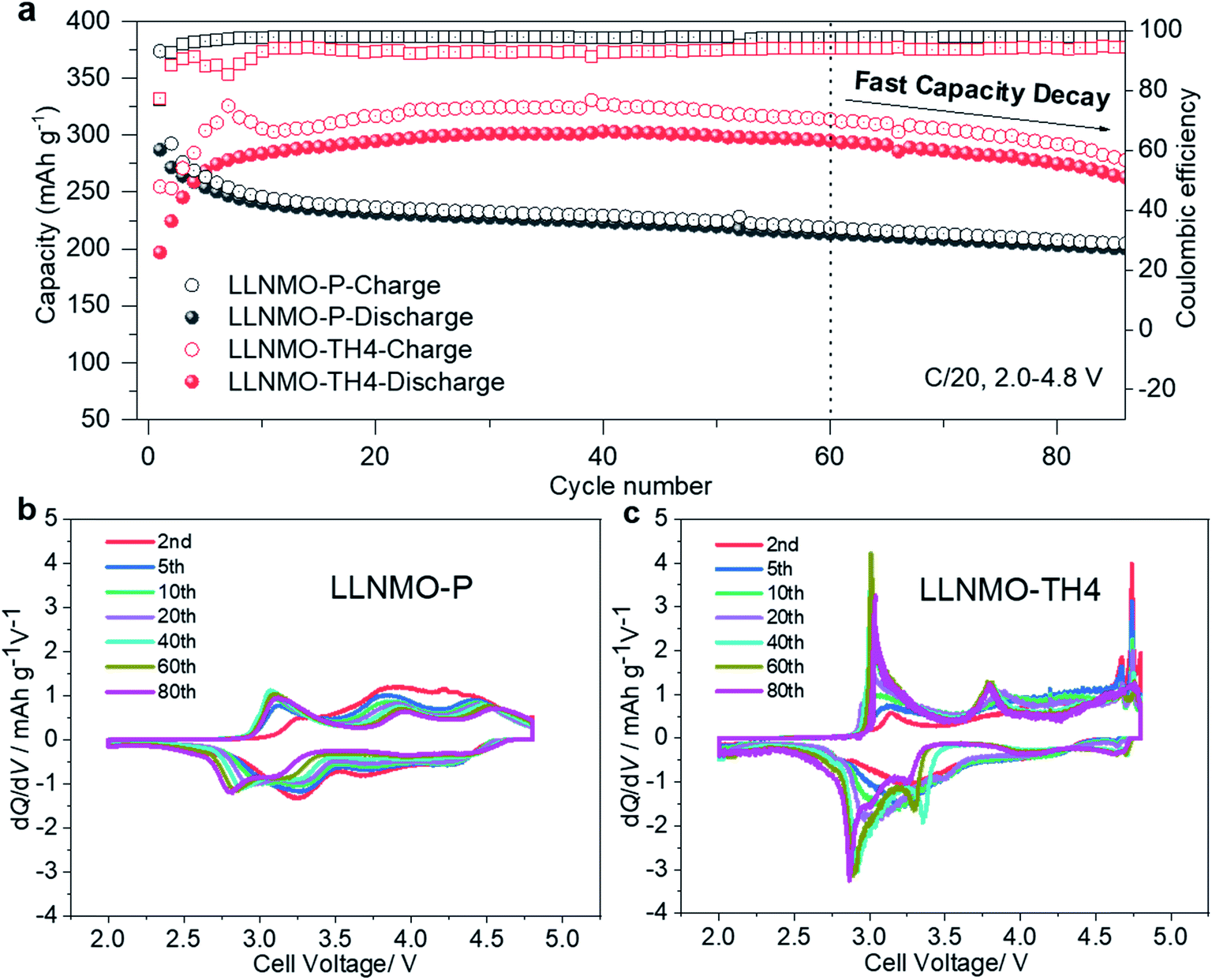 | ||
| Fig. 6 (a) Cycling performance of the LLNMO-P and LLNMO-TH4 electrodes between 2.0 and 4.8 V at 0.05C; dQ/dV plots of (b) LLNMO-P and (c) LLNMO-TH4 samples at different cycles. A pair of sharp peaks at about 4.7 V in the dQ/dV curves of LLNMO-TH4 suggests the presence of a high-voltage spinel phase LiNi0.5Mn1.5O4. | ||
Conclusion
In summary, a simple and reproducible method, i.e. H3PO4 treatment, is proposed for chemical de-intercalation of lithium ions from Li- and Mn-rich layered oxides. Lattice distortion and oxidized On− at the surface of the acid-treated Li-rich oxides are observed. The Li-deficient surface layer, i.e. a defective layered phase with lattice distortion, produces the structural features of a spinel-like phase in the SRD patterns and Raman spectra. Significant structural changes occur on the defective surface during heating and cooling processes. With the release of oxygen, the Li/O-deficient layered structure on the surface of the Li-rich oxides tends to transform into a Li-containing cubic rock-salt/spinel structure, concomitant with atomic rearrangement, surface reconstruction and Li redistribution. These results further demonstrate that the electrochemical properties are closely tied to the surface structure of the Li-rich cathode materials. Surface lattice distortion can block the lithium diffusion channels, while the coherent oxygen lattice with moderate Li/TM cation disorder on the surface after acid and thermal treatment is beneficial for improving the cycling performance of LLNMO cathodes. The knowledge gained from this work on acid and heat surface treatment could help to further improve the cycle life of high-energy Li-rich oxides for Li ion batteries and other electrode materials for post-lithium ion batteries.Author contributions
J. H., W. H., and J. M. oversaw the project; J. H. and W. H. carried out sample preparation and experimental work, and discussed with M. K., J. M., J. R. B., H. E., S. I. and B. S; J. H. W. H., A. M., A. T., S. M., and S. I. performed the synchrotron diffraction measurements; the data were analysed by J. H., W. H., C. D., X. L., J. M., T. B., and S. I.; W. H. wrote the preliminary draft with input from J. H. and J. M.; all authors contributed to interpreting the findings, and reviewing and revising the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
J. H. received financial support from the Sino-German (CSC-DAAD) Postdoc Scholarship Program, 2018 (201800260016). W. H. received financial support from the China Scholarship Council (CSC) and the Helmholtz-OCPC Postdoc-Program. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. We gratefully acknowledge Alexander Schökel and Martin Etter at beamline P02.1 at PETRA-III for the synchrotron-based diffraction experiments, and Wolfgang Caliebe and Vadim Murzin at beamline P64 at PETRA-III for the XAS measurements. Some of these experiments were performed at the MSPD beamline at ALBA Synchrotron with the collaboration of ALBA staff. The XPS, TEM and BTA characterization experiments were performed at the Karlsruhe Nano Micro Facility (KNMF), a Helmholtz research infrastructure operated at KIT. The authors thank Christina Odemer for help with the TG/DSC measurements, Udo Geckle (IAM-ESS, KIT) for help with the SEM experiments, Liuda Mereacre (IAM-ESS, KIT) for help with the Raman measurements and Raheleh Azmi (IAM-ESS, KIT) for the XPS discussion about the Ni/Mn multiplet fitting. This work contributes to the research performed at CELEST (Center for Electrochemical Energy Storage Ulm-Karlsruhe).References
- M. Ben Yahia, J. Vergnet, M. Saubanère and M.-L. Doublet, Nat. Mater., 2019, 18, 496–502 CrossRef CAS.
- G. Van Tendeloo, E. J. Berg, J.-M. Tarascon, A. M. Abakumov, D. Foix, P. Novak, G. Rousse, R. Dominko, D. Gonbeau, M.-L. Doublet, M. Saubanere and E. McCalla, Science, 2015, 350, 1516–1521 CrossRef.
- J. Lee, D. A. Kitchaev, D.-H. Kwon, C.-W. Lee, J. K. Papp, Y.-S. Liu, Z. Lun, R. J. Clément, T. Shi, B. D. McCloskey, J. Guo, M. Balasubramanian and G. Ceder, Nature, 2018, 556, 185–190 CrossRef CAS.
- Y. Liang, C. Zhao, H. Yuan, Y. Chen, W. Zhang, J. Huang, D. Yu, Y. Liu, M. Titirici, Y. Chueh, H. Yu and Q. Zhang, InfoMat, 2019, 1, 6–32 CrossRef CAS.
- X. Rong, J. Liu, E. Hu, Y. Liu, Y. Wang, J. Wu, X. Yu, K. Page, Y. S. Hu, W. Yang, H. Li, X. Q. Yang, L. Chen and X. Huang, Joule, 2018, 2, 125–140 CrossRef CAS.
- Y. Qiao, S. Guo, K. Zhu, P. Liu, X. Li, K. Jiang, C. J. Sun, M. Chen and H. Zhou, Energy Environ. Sci., 2018, 11, 299–305 RSC.
- W. Hua, S. Wang, M. Knapp, S. J. Leake, A. Senyshyn, M. Yavuz, J. R. Binder, C. P. Grey, H. Ehrenberg, S. Indris and B. Schwarz, Nat. Commun., 2019, 10, 5365 CrossRef.
- S. Ramakrishnan, B. Park, J. Wu, W. Yang and B. D. McCloskey, J. Am. Chem. Soc., 2020, 142, 8522–8531 CrossRef CAS.
- Z. W. Lebens-Higgins, H. Chung, M. J. Zuba, J. Rana, Y. Li, N. V. Faenza, N. Pereira, B. D. McCloskey, F. Rodolakis, W. Yang, M. S. Whittingham, G. G. Amatucci, Y. S. Meng, T. L. Lee and L. F. J. Piper, J. Phys. Chem. Lett., 2020, 11, 2106–2112 CrossRef CAS.
- E. Hu, X. Yu, R. Lin, X. Bi, J. Lu, S. Bak, K.-W. Nam, H. L. Xin, C. Jaye, D. A. Fischer, K. Amine and X.-Q. Yang, Nat. Energy, 2018, 3, 690–698 CrossRef CAS.
- W. Hua, M. Chen, B. Schwarz, M. Knapp, M. Bruns, J. Barthel, X. Yang, F. Sigel, R. Azmi, A. Senyshyn, A. Missiul, L. Simonelli, M. Etter, S. Wang, X. Mu, A. Fiedler, J. R. Binder, X. Guo, S. Chou, B. Zhong, S. Indris and H. Ehrenberg, Adv. Energy Mater., 2019, 8, 1803094 CrossRef.
- A. Singer, M. Zhang, S. Hy, D. Cela, C. Fang, T. A. Wynn, B. Qiu, Y. Xia, Z. Liu, A. Ulvestad, N. Hua, J. Wingert, H. Liu, M. Sprung, A. V. Zozulya, E. Maxey, R. Harder, Y. S. Meng and O. G. Shpyrko, Nat. Energy, 2018, 3, 641–647 CrossRef CAS.
- D. Seo, J. Lee, A. Urban, R. Malik and S. Kang, Nat. Chem., 2016, 8, 692–697 CrossRef CAS.
- P. Yan, J. Zheng, Z. K. Tang, A. Devaraj, G. Chen, K. Amine, J. G. Zhang, L. M. Liu and C. Wang, Nat. Nanotechnol., 2019, 14, 602–608 CrossRef CAS.
- A. J. Perez, Q. Jacquet, D. Batuk, A. Iadecola, M. Saubanère, G. Rousse, D. Larcher, H. Vezin, M. L. Doublet and J. M. Tarascon, Nat. Energy, 2017, 2, 954–962 CrossRef CAS.
- J. Zheng, P. Xu, M. Gu, J. Xiao, N. D. Browning, P. Yan, C. Wang and J. G. Zhang, Chem. Mater., 2015, 27, 1381–1390 CrossRef CAS.
- K. Luo, M. R. Roberts, R. Hao, N. Guerrini, D. M. Pickup, Y. S. Liu, K. Edström, J. Guo, A. V. Chadwick, L. C. Duda and P. G. Bruce, Nat. Chem., 2016, 8, 684–691 CrossRef CAS.
- R. A. House, U. Maitra, M. A. Pérez-osorio, J. G. Lozano, L. Jin, J. W. Somerville, L. C. Duda, A. Nag, A. Walters, K. Zhou, M. R. Roberts and P. G. Bruce, Nature, 2019, 577, 502–508 CrossRef.
- M. D. Radin, J. Vinckeviciute, R. Seshadri and A. Van der Ven, Nat. Energy, 2019, 4, 639–646 CrossRef CAS.
- P. Kalyani, S. Chitra, T. Mohan and S. Gopukumar, J. Power Sources, 1999, 80, 103–106 CrossRef CAS.
- A. R. Armstrong, M. Holzapfel, P. Novák, C. S. Johnson, S. H. Kang, M. M. Thackeray and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 8694–8698 CrossRef CAS.
- A. Boulineau, L. Simonin, J. F. Colin, C. Bourbon and S. Patoux, Nano Lett., 2013, 13, 3857–3863 CrossRef CAS.
- S. Hy, F. Felix, J. Rick, W. N. Su and B. J. Hwang, J. Am. Chem. Soc., 2014, 136, 999–1007 CrossRef CAS.
- Y. Wang, Z. Yang, Y. Qian, L. Gu and H. Zhou, Adv. Mater., 2015, 27, 3915–3920 CrossRef CAS.
- M. Sathiya, A. M. Abakumov, D. Foix, G. Rousse, K. Ramesha, M. Saubanère, M. L. Doublet, H. Vezin, C. P. Laisa, A. S. Prakash, D. Gonbeau, G. VanTendeloo and J.-M. Tarascon, Nat. Mater., 2015, 14, 230–238 CrossRef CAS.
- N. Yabuuchi, K. Yoshii, S.-T. Myung, I. Nakai and S. Komaba, J. Am. Chem. Soc., 2011, 133, 4404–4419 CrossRef CAS.
- M. Gauthier, T. J. Carney, A. Grimaud, L. Giordano, N. Pour, H. H. Chang, D. P. Fenning, S. F. Lux, O. Paschos, C. Bauer, F. Maglia, S. Lupart, P. Lamp and Y. Shao-Horn, J. Phys. Chem. Lett., 2015, 6, 4653–4672 CrossRef CAS.
- R. Imhof, J. Electrochem. Soc., 1999, 146, 1702–1706 CrossRef CAS.
- J. Zheng, M. Gu, J. Xiao, P. Zuo, C. Wang and J. G. Zhang, Nano Lett., 2013, 13, 3824–3830 CrossRef CAS.
- J. Wu, X. Zhang, S. Zheng, H. Liu, J. Wu, R. Fu, Y. Li, Y. Xiang, R. Liu, W. Zuo, Z. Cui, Q. Wu, S. Wu, Z. Chen, P. Liu, W. Yang and Y. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 7277–7284 CrossRef CAS.
- Y. Paik, C. P. Grey, C. S. Johnson, J. S. Kim and M. M. Thackeray, Chem. Mater., 2002, 14, 5109–5115 CrossRef CAS.
- F. Sigel, B. Schwarz, K. Kleiner, C. Dräger, L. Esmezjan, M. Yavuz, S. Indris and H. Ehrenberg, Chem. Mater., 2020, 32, 1210–1223 CrossRef CAS.
- W. Hua, Z. Wu, M. Chen, M. Knapp, X. Guo, S. Indris, J. R. Binder, N. N. Bramnik, B. Zhong, H. Guo, S. Chou, Y.-M. Kang and H. Ehrenberg, J. Mater. Chem. A, 2017, 5, 25391–25400 RSC.
- M. H. Lee, T. H. Kim, Y. S. Kim and H. K. Song, J. Phys. Chem. C, 2011, 115, 12255–12259 CrossRef CAS.
- W. B. Hua, X. D. Guo, Z. Zheng, Y. J. Wang, B. H. Zhong, B. Fang, J. Z. Wang, S. L. Chou and H. Liu, J. Power Sources, 2015, 275, 200–206 CrossRef CAS.
- H. Yu, Y. G. So, Y. Ren, T. Wu, G. Guo, R. Xiao, J. Lu, H. Li, Y. Yang, H. Zhou, R. Wang, K. Amine and Y. Ikuhara, J. Am. Chem. Soc., 2018, 140, 15279–15289 CrossRef CAS.
- W. Hua, B. Schwarz, R. Azmi, M. Müller, M. S. Dewi Darma, M. Knapp, A. Senyshyn, M. Heere, A. Missiul, L. Simonelli, J. R. Binder, S. Indris and H. Ehrenberg, Nano Energy, 2020, 78, 105231 CrossRef CAS.
- W. Hua, B. Schwarz, M. Knapp, A. Senyshyn, A. Missiul, X. Mu, S. Wang, J. R. Binder, S. Indris and H. Ehrenberg, J. Electrochem. Soc., 2019, 166, 5025–5032 CrossRef.
- R. Baddour-hadjean, Chem. Rev., 2010, 110, 1278–1319 CrossRef CAS.
- M. H. Lin, J. H. Cheng, H. F. Huang, U. F. Chen, C. M. Huang, H. W. Hsieh, J. M. Lee, J. M. Chen, W. N. Su and B. J. Hwang, J. Power Sources, 2017, 359, 539–548 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- R. Azmi, V. Trouillet, M. Strafela, S. Ulrich, H. Ehrenberg and M. Bruns, Surf. Interface Anal., 2018, 50, 43–51 CrossRef CAS.
- X. Li, Y. Qiao, S. Guo, Z. Xu, H. Zhu, X. Zhang, Y. Yuan, P. He, M. Ishida and H. Zhou, Adv. Mater., 2018, 30, 1705197 CrossRef.
- W. Yin, A. Grimaud, G. Rousse, A. M. Abakumov, A. Senyshyn, L. Zhang, S. Trabesinger, A. Iadecola, D. Foix, D. Giaume and J. M. Tarascon, Nat. Commun., 2020, 11, 1252 CrossRef CAS.
- L. de Biasi, B. Schwarz, T. Brezesinski, P. Hartmann, J. Janek and H. Ehrenberg, Adv. Mater., 2019, 31, 1900985 CrossRef.
- W. Hua, K. Wang, M. Knapp, B. Schwarz, S. Wang, H. Liu, M. Müller, A. Schökel, A. Missyul, D. F. Sanchez, X. Guo, J. R. Binder, J. Xiong, S. Indris and H. Ehrenberg, Chem. Mater., 2020, 32, 4984–4997 CrossRef CAS.
- D. Eum, B. Kim, S. J. Kim, H. Park, J. Wu, S. P. Cho, G. Yoon, M. H. Lee, S. K. Jung, W. Yang, W. M. Seong, K. Ku, O. Tamwattana, S. K. Park, I. Hwang and K. Kang, Nat. Mater., 2020, 19, 419–427 CrossRef CAS.
- Z. Zhu, D. Yu, Y. Yang, C. Su, Y. Huang, Y. Dong, I. Waluyo, B. Wang, A. Hunt, X. Yao, J. Lee, W. Xue and J. Li, Nat. Energy, 2019, 4, 1049–1058 CrossRef CAS.
- R. Chen, S. Ren, M. Knapp, D. Wang, R. Witter, M. Fichtner and H. Hahn, Adv. Energy Mater., 2015, 5, 1–7 Search PubMed.
- J. Lee, A. Urban, X. Li, D. Su, G. Hautier and G. Ceder, Science, 2014, 343, 519–522 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, Rietveld refinement results based on SRD data, crystallographic parameters, SEM-EDX images, XAS spectra and electrochemical performances of the samples. See DOI: 10.1039/d0ta07371g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |