
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiscale simulations of polyzwitterions in aqueous bulk solutions and brush array configurations†
Aristotelis P.
Sgouros
 a,
Stefan
Knippenberg
b,
Maxime
Guillaume
b and
Doros N.
Theodorou
*a
a,
Stefan
Knippenberg
b,
Maxime
Guillaume
b and
Doros N.
Theodorou
*a
aSchool of Chemical Engineering, National Technical University of Athens, 9 Heroon Polytechniou Street, Zografou Campus, GR-15780 Athens, Greece. E-mail: doros@central.ntua.gr; Fax: +30 210 772 3112; Tel: +30 210 772 3157
bSolid State Battery Applicability Laboratory, Solvay SA, 310 Rue de Ransbeek, B-1120 Brussels, Belgium. E-mail: maxime.guillaume@solvay.com
First published on 2nd November 2021
Abstract
Zwitterionic polymers are very promising candidates for antifouling materials that exhibit high chemical stability as compared to polyethylene glycol-based systems. A number of simulation and experimental studies have emerged over recent years for the investigation of sulfobetaine-based zwitterionic polymers. Investigating the structural and thermodynamic properties of such polymers requires access to broad time and length regimes, thus necessitating the development of multiscale simulation strategies. The present article advocates a mesoscopic dissipative particle dynamics (DPD) model capable of addressing a wide range of time and length scales. The mesoscopic force field was developed hand-in-hand with atomistic simulations based on the OPLS force field through a bottom-up parameterization procedure that matches the atomistically calculated strand-length, strand-angle and pair distribution functions. The DPD model is validated against atomistic simulations conducted in this work, and against relevant atomistic simulation studies, theoretical predictions and experimental correlations from the literature. Properties examined include the conformations of SPE polymers in dilute bulk aqueous solution, the density profile and thickness of brush arrays as functions of the grafting density and chain length. In addition, we compute the potential of mean force of an approaching hydrophilic or hydrophobic foulant via umbrella sampling as a function of its position relative to the poly-zwitterion-covered surface. The aforementioned observables lead to important insights regarding the conformational tendencies of grafted polyzwitterions and their antifouling properties.
1. Introduction
Antifouling, i.e., preventing the accumulation of biomolecules, microorganisms, plants, algae, or even small animals on surfaces, is extremely important in applications ranging from maintaining the separation efficiency of polymeric membranes to excluding pathogens from medical devices.1–4 Antifouling is typically accomplished by coating the surface of a structure or product with a layer that can eliminate or repel foulants (FLs). Generally, there are two classes of antifouling layers: biocidal5,6 or non-biocidal.7–9 Among non-biocidal layers—which are considered to be less toxic7—one can find materials that are antifouling because of surface structuration (such as super-slippery sharklet technology),10 or superhydrophobicity,11,12 or high surface hydrophilicity.13Among hydrophilic non-biocidal coatings, zwitterionic polymers14–22 are considered to be one of the two major classes of antifouling materials that are accessible and relatively easy to implement, the other being hydrophilic polymers based on poly(ethylene glycol) (PEG).23,24 Brushes of zwitterionic polymers terminally grafted to a surface have been found to be effective antifouling agents, exhibiting higher chemical stability than PEG-based layers.23 A recent review paper highlights the development of biocompatible and bioactive materials for antifouling surfaces as an important and timely topic; it points out that significant efforts have been made for the development of these materials, but “much of the work retains an empirical flavor due to the complexity of experiments and the lack of robust theoretical models”.23 These systems involve macromolecules of a complex chemical constitution, interfaces, electrolyte solutions, and chemical reactions. Each of these aspects is considered to be a major challenge for molecular simulation.
Promising polyzwitterionic candidates with antifouling properties are carboxybetaine methacrylate (CBMA), 2-methacryloyloxyethyl phosphorylcholine (MPC), and (sulfobetaine methacrylate) (SBMA).23 The latter, more correctly named 3-[N-(2-methacryloyloxy)ethyl-N,N-dimethyl] ammonio propane-1-sulfonate, is also called SPE. We will use this acronym for both the monomer and its polymer in this work.
During the last decade there have been several atomistic16,19–22 and a few mesoscopic25 simulations of the antifouling action of zwitterionic membranes. In a series of papers, Shao et al. examine the influence of charged groups16 and ions20 on polyzwitterions, as well as the effect of the latter on proteins.22 In a recent paper by Xiang et al.,19 brushes of terminally grafted sulfobetaine polymers were considered at different surface densities. The structure of the brushes, including ion pairs between sulfobetaine zwitterions, was explored. Interactions between two such brushes were computed in vacuo, in water, and in a salt solution. Furthermore, the mean force experienced by a foulant (FL) as a function of its distance from the surface was computed by steered molecular dynamics (SMD).19 The FL in the work of Xiang et al.19 was represented as an alginate gel.
The relevant time and length scales governing the thermodynamics and dynamics of long-chain polyzwitterions in solution, their adsorption or chemical grafting onto solid surfaces, and their antifouling action exceed by far the range of microseconds and tens of nanometers that can be addressed by atomistic simulations on readily available computational platforms;26,27 judicious coarse-graining is thus necessary.
A wide variety of mesoscopic techniques have been developed for addressing the properties of soft matter systems for time scales up to seconds and length scales up to micrometers. A promising technique of this kind for simulating anti-biofouling polyzwitterionic layers is Dissipative Particle Dynamics (DPD), in which beads interact with each other through soft conservative and pairwise dissipative and random forces.28–33 A definite advantage of DPD is that it correctly represents hydrodynamic interactions. The DPD method has been applied to a variety of problems involving polymers and copolymers,32,34,35 nanocomposites,34 water–oil interfaces,36,37 brush arrays,38,39 liquid/solid40 and liquid/vacuum41 interfaces, bilayers/membranes,31,42–46 and liquid flow in bulk47 and microchannels.48
The objective of the present study is to develop a multiscale molecular modeling and simulation methodology that can facilitate the design of polyzwitterionic brush arrays capable of repelling bacteria and other bio-fouling agents from solid surfaces in aqueous environments. The modeling approach is able to predict how the length and concentration of the polymer brushes affect the structure of the layers formed on the surface, and the repulsive potential of mean force experienced by biofoulants due to these layers. Given the chemical complexity of the systems of interest and the broad range of length and time scales governing their physicochemical behavior, a multiscale modeling and simulation methodology is adopted, employing two interconnected representations: (i) a detailed atomistic representation based on a suitable force field, simulated with molecular dynamics (MD); (ii) a mesoscopic representation cast in terms of coarse-grained “beads,” simulated with DPD.
Parameterizing a DPD simulation so as to represent a specific chemical system is not a trivial undertaking.30,35–37,42–46,48–50 A common practice is to tune the repulsive parameters of a ramp potential aij for each interaction pair ij in a way that reproduces specific Flory–Huggins parameters χij, using the correlation proposed by Groot et al.30 to link χij with the repulsion parameters, aij, of the DPD potential. Weiß et al.51 propose a strategy which initially maps out the surface charge density distribution by quantum chemical methods around each moiety to be coarse-grained into a bead, then invokes the COSMO-RS framework to calculate the residual Gibbs energy of mixing of binary systems of beads, then fits a χ factor to the latter, and finally determines the DPD parameter a so as to match this χ factor. An alternative bottom-up approach to restore the nonbonded interactions is to employ a tabulated pair potential that can be parameterized from the pair-distributions of an MD simulation with iterative Boltzmann inversion52,53 or with reverse Monte Carlo.54,55
Given that the estimation of the χ parameters for charged SPEs is not straightforward due to the presence of electric charges, we opted to incorporate a hybrid bottom-up parameterization procedure in which: (i) we retained the usual ramp potential of DPD but, (ii) we optimized the aij parameters in a way that best matches the pair-distribution functions between MD and DPD. Optimization of the repulsive coefficients was performed via brute force and machine-learning-based parameterization procedures (Bayesian optimization) which minimized the standard deviation between the pair-distributions from MD and DPD. The effective bonded interactions, on the other hand, were parameterized based on a Monte Carlo optimization scheme which best matches the strand-length and strand-angle distributions between MD and DPD.
Predicted properties include the conformations of long SPE chains in aqueous bulk solutions, the density profiles of brush arrays and the brush dimensions and grafted layer structure by means of the root mean square brush height 〈hg2〉0.5 and the span 〈h99%〉,53,56,57 and the potential of mean force (PMF) between a solid substrate covered by the brush array and an approaching mock foulant (suspended particle) as a function of the foulant's position relative to the surface. PMF is a direct measure of the layer's effectiveness as an antifouling agent. A repulsive PMF is desirable; the longer the range of repulsion and the steeper the potential of mean force, the better the antifouling properties. The repulsive/attractive tendencies between the foulants and the antifouling brushes are governed by an interplay of brush–solution, brush–foulant and foulant–solution interactions. To discern and validate these tendencies we performed a sensitivity analysis of the PMF in terms of varying the strength of the repulsive foulant–water and foulant–SPE interactions; hence, tuning in that way the hydrophobicity/hydrophilicity of these substances. Extensive validation efforts were performed between the MD and DPD levels of modeling and between simulations19 and available experimental evidence14,15 from the literature.
To the authors’ knowledge, this is the first mesoscale study of SPE polyzwitterions that addresses the following aspects in the field of dissipative particle dynamics and antifouling:
• It proposes a hybrid IBI/MC/machine learning approach for the bottom-up parameterization of the DPD model, which does not require knowing the Flory's χ-parameters.
• It provides indirect indications for the transferability of the free energy potential to different concentrations.
• It displays predictions concerning poly-zwitterions of high molar mass, whose simulation exceeds by far the capabilities of atomistic molecular dynamics.
• It assesses the scaling laws of the brush measures over a broad range of molar masses and grafting densities and provides comparisons with theoretical predictions.
• It demonstrates the applicability of umbrella sampling techniques for assessing the antifouling properties of polyzwitterionic layers.
The article is structured as follows: Section 2 presents an introduction to DPD; Section 3 introduces the atomistic and mesoscopic models and discusses the bottom-up parameterization procedure that bridges the atomistic levels of description with the mesoscopic ones. Section 4 includes in-depth details regarding the MD and DPD simulations. Section 5 presents the main findings of this work for bulk solutions and brush arrays. Finally, Section 6 concludes the manuscript.
2. Dissipative particle dynamics
DPD was initially introduced by Hoogerbrugge and Koelman28 and reformulated by Español and Warren.29 In DPD, the dissipative and random forces are related through the fluctuation–dissipation theorem, leading to conservation of the momentum, which is a prerequisite for a correct description of hydrodynamics.58 In this model a set of point particles move and interact with each other with three types of pairwise forces: | (1) |
| fDij = −γijwD(eij·vij)eij | (2) |
| fRij = σijwRζijeij | (3) |
The standard description of nonbonded soft conservative interactions in DPD involves a repulsive ramp force that has a finite value at zero distance and decays linearly with increasing distance to zero at a characteristic cutoff distance, rc.
 | (4) |
3. Model systems
3.1 Atomistic model
The chemical formula of SPE is illustrated schematically in Fig. 1. The sulfobetaine group contained in SPE is zwitterionic, consisting of a positively charged quaternary ammonium and a negatively charged sulfonate tethered by three methylene units. During the polymerization the double bond opens, connecting the SPEs across the CH2 into a backbone. | ||
| Fig. 1 Chemical structure of an SPE monomer. During the polymerization the double bond at the backbone opens, forming a new bond with the next SPE monomer. | ||
DREIDING is considered to be relatively inexpensive computationally and generic; thus, it is readily applicable over a vast range of polymer types. Nonetheless, the fact that it is generic makes its accuracy for describing uncommon molecular architectures questionable. DREIDING does not offer a specific recipe for assigning charges to individual monomers; thus the charges were assigned using the generic Gasteiger62 scheme.
The OPLS force field was initially developed by Jorgensen's group60,61 and has been parameterized for describing accurately very similar polymers; in particular, the same sulfobetaine groups, but shorter by one methylene segment,21 and the same SPE group with a different backbone attached to Au nanoparticles.61 Thus, the potential employed herein was derived by mixing the original OPLS parameters with the force fields in ref. 21 and 61 (see Section S1, ESI†).63–65 The OPLS force field was applied via an in-house code that was developed for the purposes of this project.
The primary atomistic simulations based on which we parameterized our mesoscopic model were conducted with the OPLS force field. A few representative cases were simulated with DREIDING as well for comparison purposes.
3.2 Mesoscopic model
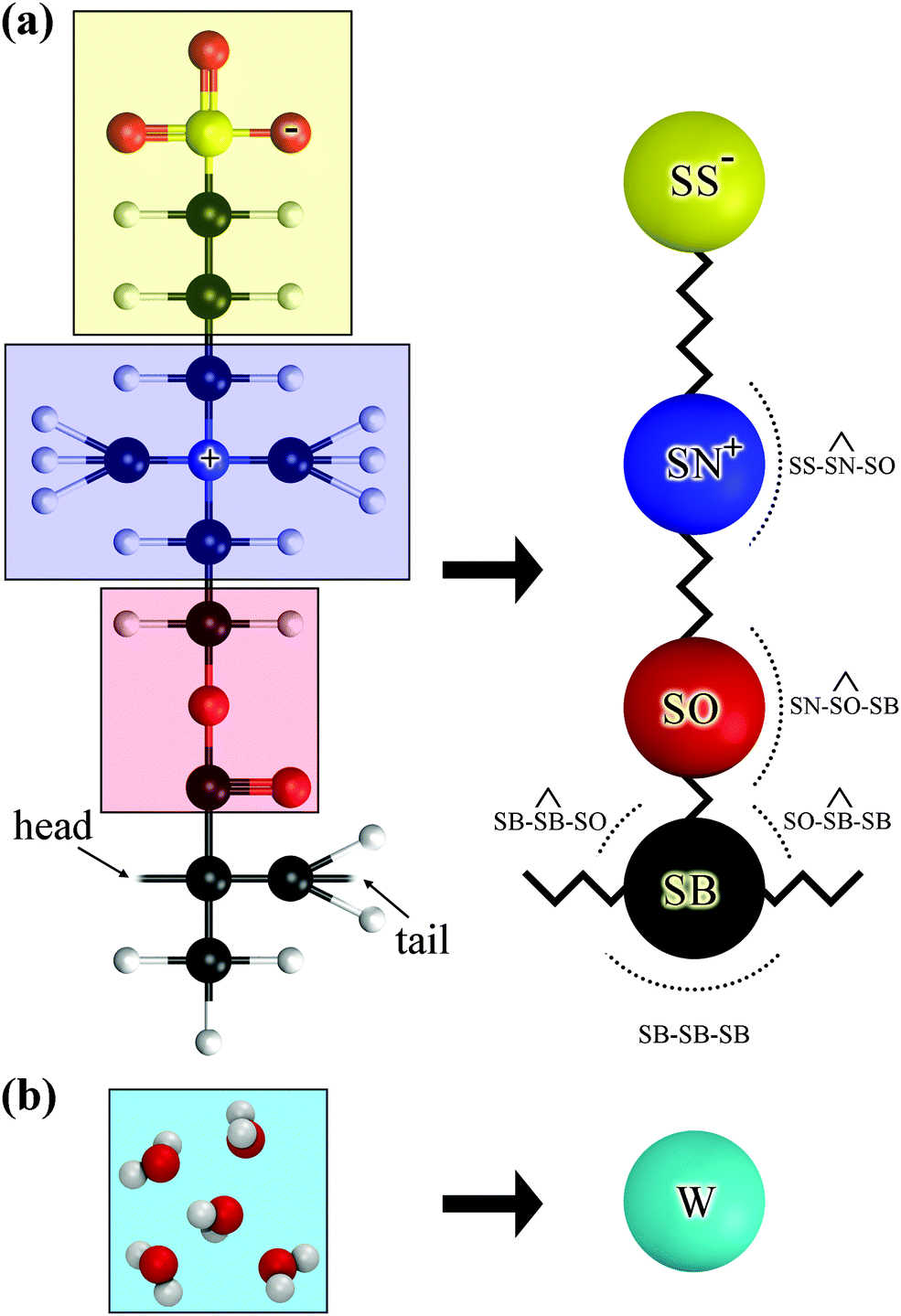 | ||
| Fig. 2 Atomistic (left) and mesoscopic (right) representation an (a) SPE and (b) water molecules. | ||
 ,
, 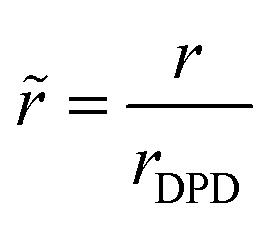 ,
,  ,
,  ,
,  . The reference bead mass, mDPD = Nm·mH2O, is set equal to the mass of the water bead describing Nm water molecules (mH2O).
. The reference bead mass, mDPD = Nm·mH2O, is set equal to the mass of the water bead describing Nm water molecules (mH2O).  is the cutoff distance of the nonbonded interactions with VH2O being the volume per water molecule as calculated from the density of bulk liquid water under the conditions of interest.31 Thus, we find the physical size of the interaction radius. τDPD is a reference time and is usually set equal to
is the cutoff distance of the nonbonded interactions with VH2O being the volume per water molecule as calculated from the density of bulk liquid water under the conditions of interest.31 Thus, we find the physical size of the interaction radius. τDPD is a reference time and is usually set equal to  . εDPD is usually set to kBT. Note that the aforementioned quantities are functions of the dimensionless DPD number density, which is set to
. εDPD is usually set to kBT. Note that the aforementioned quantities are functions of the dimensionless DPD number density, which is set to ![[small rho, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e4.gif) DPD = ρDPDrc3 = 3, as in most DPD studies.30,31,37,70 It should be noted that DPD is a free parameter of the model that can be set to any value equal to or larger than 3, which is the lowest value for which the excess pressure retains its proportionality to DPD2.30 Setting DPD > 3, however, has been deemed impractical because the number of interactions scales as ∼DPD2.30
DPD = ρDPDrc3 = 3, as in most DPD studies.30,31,37,70 It should be noted that DPD is a free parameter of the model that can be set to any value equal to or larger than 3, which is the lowest value for which the excess pressure retains its proportionality to DPD2.30 Setting DPD > 3, however, has been deemed impractical because the number of interactions scales as ∼DPD2.30
The commonly used dimensionless friction factor, ![[small gamma, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0dd.gif) ij = 4.5 (in
ij = 4.5 (in  units) leads to a very good temperature control; however, it predicts overly enhanced diffusivity for water.31,45 The effect is demonstrated in Table 1 which depicts the self-diffusion coefficient of experimental71 and simulated water via MD and DPD. The diffusion coefficient of the DPD water beads, multiplied by Nm = 5, is much higher than the experimental one.
units) leads to a very good temperature control; however, it predicts overly enhanced diffusivity for water.31,45 The effect is demonstrated in Table 1 which depicts the self-diffusion coefficient of experimental71 and simulated water via MD and DPD. The diffusion coefficient of the DPD water beads, multiplied by Nm = 5, is much higher than the experimental one.
| System | Method | D (nm2 ns−1) | D renorm (nm2 ns−1) |
|---|---|---|---|
| H2O | EXP71 | 2.430 | — |
| H2O | MD-SPCE | 2.595 | — |
| W (H2O)5 | DPD | 10.47 | 0.486 |
| SPE1 | MD-OPLS | 0.261 | — |
| SPE1 | DPD | 4.531 | 0.210 |
An approach to overcome this problem and reproduce the diffusivity of water exactly is to renormalize the reference time accordingly:31,45
 | (5) |
 and
and  are the simulated and the experimental self-diffusion coefficients of a water bead and a water molecule, respectively.71 For Nm = 5, τDPD = 4.6 ps, whereas
are the simulated and the experimental self-diffusion coefficients of a water bead and a water molecule, respectively.71 For Nm = 5, τDPD = 4.6 ps, whereas  was estimated at ∼10.47 × 10−9 m2 s−1 from our simulations (1/6 of the slope of mean square displacement with respect to time). Using these values in eqn (5), the reference time, τrenormDPD, increases by ∼21.5 times (see Table 2). Interestingly, rescaling the time by eqn (5) yields DPD self-diffusivities for SPE which are very close to those obtained by atomistic MD (see the two last rows of Table 1). This reinforces the idea that the rescaling of eqn (5) is physically meaningful.
was estimated at ∼10.47 × 10−9 m2 s−1 from our simulations (1/6 of the slope of mean square displacement with respect to time). Using these values in eqn (5), the reference time, τrenormDPD, increases by ∼21.5 times (see Table 2). Interestingly, rescaling the time by eqn (5) yields DPD self-diffusivities for SPE which are very close to those obtained by atomistic MD (see the two last rows of Table 1). This reinforces the idea that the rescaling of eqn (5) is physically meaningful.
| Quantity | Real units | DPD units |
|---|---|---|
| N m | 5 water molecules | 5 |
| m DPD | 0.09 kg mol−1 | 1 |
| τ renormDPD | 99.245 × 10−12 s | 1 |
| r DPD = rc | 7.66 × 10−10 m | 1 |
| ε DPD = kBT | 4.14 × 10−21 J | 1 |
| |qDPD| | 0.84e = 1.346 × 10−19 C | 7.16 |
| ρ DPD | 997 kg m−3 | 3 |
| γ ij | 1.46 × 10−13 kg s−1 | 4.5 |
| σ ij | 3.48 × 10−17 (J kg s−1)0.5 | 3 |
| a WW | 7.34 × 10−13 N | 135.868 |
Regarding the nonbonded interactions among water beads, a common practice is to fit the coefficient aij in eqn (4) to a value that in the case of water reproduces the experimental compressibility of the liquid:
 | (6) |
 being the dimensionless experimental inverse compressibility of water, where κT is the real isothermal compressibility and ρn the number density of molecules.30 According to Groot and Warren,30 setting α = (PDPD − ρDPDkBT)/(aWWρDPD2) to 0.101rc4 provides a good approximation for DPD > 2, with PDPD being the pressure predicted by the DPD simulation. In this work we recalculated it to α = 0.0919rc4, which provides a more accurate estimation of the ratio for DPD = 3.
being the dimensionless experimental inverse compressibility of water, where κT is the real isothermal compressibility and ρn the number density of molecules.30 According to Groot and Warren,30 setting α = (PDPD − ρDPDkBT)/(aWWρDPD2) to 0.101rc4 provides a good approximation for DPD > 2, with PDPD being the pressure predicted by the DPD simulation. In this work we recalculated it to α = 0.0919rc4, which provides a more accurate estimation of the ratio for DPD = 3.
| Ustrand = kstrand(r − l0)2 | (7) |
| Uangle = kangle(θ − θ0)2 | (8) |
Regarding the electrostatics, because of the softness of short-range nonbonded interactions, point charges cannot be used in DPD, for they would bring about collapse of oppositely charged beads upon each other and negatively infinite electrostatic energy. Instead, it is common practice to smear the charge of a bead according to a prescribed density function within a sphere of the prescribed radius. The electrostatic field resulting from the smeared distributions is then summed using the particle–particle particle-mesh (PPPM) or the three-dimensional Ewald summation method (EW3DC).72 In our model, the DPD beads experience electrostatic interactions which are described by the following potential68 which was introduced by the authors in LAMMPS for the purposes of this project:
 | (9) |
 with λ being the second moment of the charge distribution.68 Note that in the limit λ → 0, the potential recovers its original Coulomb expression, while according to the DPD literature,45,73,74 the optimal value for λ is 0.25rc.
with λ being the second moment of the charge distribution.68 Note that in the limit λ → 0, the potential recovers its original Coulomb expression, while according to the DPD literature,45,73,74 the optimal value for λ is 0.25rc.
In our implementation the DPD nonbonded interactions are zero between 1st neighboring (directly bonded) beads in a molecule, 50% between 2nd bonded neighbors and 100% everywhere else. Regarding the Coulomb interactions, these are zero between 1st–3rd bonded neighbors.
Based on the parameterization procedure that is reported in Appendix A we obtained the coefficients for the mesoscopic force field shown in Tables 3–5. Finally, for the case of brush arrays, the nonbonded coefficients of the atoms comprising the solid surface were set to ãAA = 200 (in DPD units), whilst the interactions with beads of type j were derived by applying the geometric mixing rule; 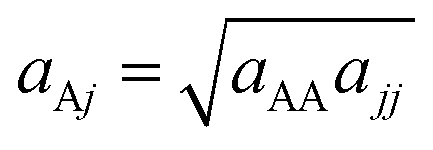 .
.
| Bond type | k strand | l 0 |
|---|---|---|
| SB–SB | 993.893 | 0.390 |
| SB–SO | 6536.214 | 0.375 |
| SN–SO | 707.171 | 0.526 |
| SN–SS | 1810.595 | 0.663 |
| Angle type | k angle | θ 0 (°) |
|---|---|---|
| SB–SB–SB | 2.744 | 115.252 |
| SB–SB–SO | 6.434 | 82.982 |
| SB–SO–SN | 2.585 | 130.594 |
| SO–SN–SS | 3.094 | 134.925 |
| SB | SO | SN | SS | W | Charges | Mass | |
|---|---|---|---|---|---|---|---|
| SB | 63.69 | 61.59 | 62.16 | 103.44 | 98.26 | 0.00 | 0.456 |
| SO | 61.07 | 39.87 | 89.33 | 103.33 | 0.00 | 0.645 | |
| SN | 61.46 | 77.35 | 54.45 | 7.16 | 0.801 | ||
| SS | 129.03 | 135.24 | −7.16 | 1.201 | |||
| W | 135.86 | 0.00 | 1.000 |
It is worth mentioning here that our implementation involves variable bead-size,75 charged DPD beads, and variable aii coefficients; thus, it is not straightforward to interpret the physical meaning of the repulsive coefficients listed in Table 5. In detail, the derived aij contains information about several molecular-level aspects such as:
• Bead size
• Enthalpic interactions
• Entropic interactions
• Charges and hydrogen-bonding effects
• Geometric constraints imposed by the branched architecture that generate correlations
Even though these aspects are highly convoluted, one could nonetheless attempt to discern some basic trends. Overall, the coefficients increase with bead mass (rightmost column of Table 5) since the beads occupy more space and thus aij must become more repulsive. Interestingly, even though mSN > mSB and mSO, their repulsive coefficients are similar. This is attributed to SN being positively charged; the SN–SN repulsive coefficient remains relatively low in order to compensate for electrostatic repulsion between positively charged SN–SN pairs. The same reasoning can be applied to SS beads as well: even though mSS > mW, we see that aSS < aWW in order to compensate for the repulsive interaction between negatively charged SS beads. Interactions between the beads at the end of the zwitterion (SN and SO) with its backbone (SB) are somewhat more repulsive (e.g., compare SN–SO with SN–SB, and SS–SO with SS–SB, whilst keeping in mind that mSO > mSB). This effect could be attributed to the molecular architecture of the polyzwitterions and to excluded volume effects arising due to the presence of the intermediate SO beads.
Finally, the pair-wise friction factors of the SPE beads were set to ij = 4.5. Interestingly, even though we based the time-mapping solely on the dynamics of water, the renormalized diffusion coefficients of the coarse-grained SPEs are in reasonable agreement with the diffusion coefficients of the atomistic ones; e.g., compare the renormalized DPD diffusion coefficient with the atomistic diffusion coefficient in Table 1. The agreement could be potentially improved by optimizing the ij with the target to match the diffusion coefficient of the atomistic and the mesoscopic SPEs.
4. Simulation details
4.1 Atomistic molecular dynamics
The initial configurations were generated with the Amorphous Builder program,76 as implemented in the MAPS77 package by Scienomics. During the generation phase (chain growth and insertion of water molecules), the system temperature was set to T = 450 K, the density to ρ = 0.8 g mol−1, and the interactions were described by the DREIDING force field59 with Gasteiger charges.62 For simulations with the OPLS force field the bonded and nonbonded OPLS coefficients were applied using an in-house code.The atomistic simulations consisted of three main stages:
1. The initial configurations were subjected to sequential energy minimizations, in which the bond and angle stiffness was gradually increased, until these hard degrees of freedom practically reached their equilibrium values. In particular, the initial coefficients for the harmonic, ![[scr V, script letter V]](https://www.rsc.org/images/entities/char_e149.gif) bond = kbond(l − l0)2, SPCE bonds for water were set to kbond = 100 kcal mol−1 Å−1 and l0 = 1 Å, and the initial coefficients for the harmonic SPCE angles, angle = kangle(θ − θ0)2, were set to kangle = 100 kcal mol−1 rad−1 and θ0 = 109.47 Å. Furthermore, over subsequent energy minimizations, the bond and angle stiffness was increased by powers of 2 until these geometric characteristics practically reached their equilibrium lengths corresponding to infinite stiffness.
bond = kbond(l − l0)2, SPCE bonds for water were set to kbond = 100 kcal mol−1 Å−1 and l0 = 1 Å, and the initial coefficients for the harmonic SPCE angles, angle = kangle(θ − θ0)2, were set to kangle = 100 kcal mol−1 rad−1 and θ0 = 109.47 Å. Furthermore, over subsequent energy minimizations, the bond and angle stiffness was increased by powers of 2 until these geometric characteristics practically reached their equilibrium lengths corresponding to infinite stiffness.
2. The energy-minimized atomistic configurations were then simulated for moderate periods of time until they reached their equilibrium properties. In detail, the equilibration stages were the following: (i) first, the samples were simulated for 10 ps using the efficient velocity rescaling thermostat; (ii) then, the samples were simulated in the NVT ensemble using the Nosé–Hoover thermostat for another 10 ps; (iii) finally, the samples were simulated in the NPT ensemble using the Nosé–Hoover thermostat and barostat for up to 160 ps, until they reached their equilibrium density, which is about equal to the density of water, 1 g cm−3.78,79
3. The equilibrated samples were simulated for extended periods of time (up to 0.1 μs).
Note that, in stages (2) and (3), the equations of motion were integrated using the velocity-Verlet algorithm80 with a time step set to 1 fs. In addition, the SHAKE algorithm81 was enabled which applies bond and angle constraints on the bonds and angles of the SPCE water molecules. Throughout the MD simulations we investigated SPEs with 1 up to 64 repeating units at mass ratios ∼10 wt%. Unless otherwise stated, it is assumed that the atomistic simulations are conducted with the OPLS force field, based on which we parameterized our mesoscopic model.
4.2 Mesoscopic simulations
1. The initial mesoscopic configurations for bulk solutions were generated using an in-house builder, PyMeso, which was developed for the purposes of the current project. The code is written in Python3.8 and can generate chains of arbitrary connectivity and chemical constitution based on user input. There is no restriction regarding the number of chain types, polydispersity and the periodic boundary conditions applied at the wall boundaries. For more details about PyMeso see Section S2 (ESI†).82
2. Subsequently, the configurations were energy minimized and then equilibrated for up to ![[small tau, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e5.gif) = 104 (∼1 μs).
= 104 (∼1 μs).
3. Finally, the trajectories were simulated for extended time periods up to = 2 × 105 (∼20 μs), for the larger systems considered here.
The equations of motion were integrated using the velocity-Verlet algorithm80 in conjunction with the multiple time step Reference System Propagator Algorithm (RESPA),80,83 with one hierarchical level having a time step equal to 0.005τDPD for the bonded interactions and another hierarchical level having a time step equal to 0.015τDPD for the nonbonded interactions. The cutoff of the long range interactions was set to rc = 1 (in DPD units). The summation of the electrostatics was carried out using the Particle–Particle Particle-Mesh (PP-PM) solver84 (“pppm/cg” style in LAMMPS which is identical to the “pppm” style except that it incorporates an optimization for systems where most particles are uncharged) with a G-Ewald parameter for the dispersion equal to 1.8, and a maximum relative error of 10−4. The simulations were performed for SPEs with 1 up to 512 repeating units at 10 wt%.
 | ||
| Fig. 3 Coarse-grained representation of a brush array with area/chain = 1.44 nm2. The SPE brushes comprise SB (black), SO (red), SN (blue) and SS (yellow) beads, while white beads represent the wall surface. The beads of the wall surface and the grafted beads (SB beads marked with “×”) remain immobile throughout the simulation. zS and zg denote the position of the wall surface and the grafted point-wall surface distance, respectively. The violet beads comprise a model foulant particle at zFL. Water beads are omitted for clarity. | ||
1. The initial mesoscopic configurations for the brush arrays were generated with PyMeso. The solid substrate was modeled by a slice of an 11 × 11 BCC lattice with a lattice constant of 0.8542rDPD (ρ ∼ 3.2 in DPD units), as shown in Fig. 3. The SPE brushes emanate from grafting points which are distributed periodically across a lattice spanning the lateral directions of the solid surface, with a grafted bead-grafted bead closest distance,  where area/chain = σg−1, and σg is the grafting density.
where area/chain = σg−1, and σg is the grafting density.
2. The brush-arrays were then equilibrated in conjunction with a Newton–Raphson scheme that varied the normal dimensions of the simulation box until the normal component of the system pressure matched the pressure of bulk DPD water [PN ∼ 121.3εDPD/rDPD3 for Nm = 5]. The tolerance value for pressure differences was set to 0.1 and the optimization lasted for up to 7 cycles.
3. The initial snapshots for the umbrella sampling were extracted by Steered Molecular Dynamics (SMD) simulations. In SMD the z-component of the center of mass of the foulant (FL), zFL, was tethered to the surface with a harmonic spring starting at zg with stiffness kspring and equilibrium length lspring, without applying any constraint along the lateral dimensions; a similar treatment is employed in ref. 19. By adjusting the equilibrium length of the spring one can tune the distance of the FL from the solid surface. The simulations were performed in three stages: (i) the system was initially equilibrated for 500τDPD with lspring = zinit and kspring rising gradually from 0 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (in DPD units). (ii) The system was equilibrated for an additional 100τDPD with the same lspring and kspring. (iii) The length of the spring decreased gradually from zinit to zfinal at a rate Δz/τequil, with Δz = (zinit − zfinal)/nbins being the bin width for the umbrella sampling simulations, nbins being the number of bins, and τequil = 100τDPD; the FL was thereby gradually moved towards zg and snapshots of the systems were stored with a frequency τequil−1.
000 (in DPD units). (ii) The system was equilibrated for an additional 100τDPD with the same lspring and kspring. (iii) The length of the spring decreased gradually from zinit to zfinal at a rate Δz/τequil, with Δz = (zinit − zfinal)/nbins being the bin width for the umbrella sampling simulations, nbins being the number of bins, and τequil = 100τDPD; the FL was thereby gradually moved towards zg and snapshots of the systems were stored with a frequency τequil−1.
4. Starting from the jth snapshot from the SMD simulations an umbrella sampling simulation was performed for 400τDPD, lspring = (zfinal − zinit)/j, and kspring = 500 whilst storing values of (zFL − zg) along the generated trajectory.
5. Finally, trajectories started at each snapshot were processed by the implementation of the Weighed Analysis Histogram Method (WHAM) by Grossfield.85
Each umbrella sampling experiment was repeated two times with different initial configurations in order to assess the deviation. The latter is very small, probably due to the sufficient equilibration time and relatively large system sizes. It should be noted that, according to our benchmarks, the PMF arising from pulling the FL away from the antifouling layer to infinity (in the reverse direction) is practically identical.
During the simulations both the beads comprising the solid surface and the grafted beads were kept frozen. This treatment, however, can generate artifacts in the calculation of system temperature and pressure; thus, these properties were recomputed based on the atomic velocities and virial of the mobile atoms, respectively. For the calculation of pressure the effective volume of the system was scaled by  ; an alternative treatment would be to consider the effective volume of the mobile atoms as the sum of their Voronoi volumes. The systems were periodic along the lateral and aperiodic along the normal directions, whereas reflective (specular) boundary conditions were implemented along the normal direction. The incorporation of fixed wall particles in conjunction with reflective boundaries has been shown to be an effective approach for preventing DPD beads from crossing the top and bottom faces of the simulation box and for enforcing the no slip boundary condition.33,86,87
; an alternative treatment would be to consider the effective volume of the mobile atoms as the sum of their Voronoi volumes. The systems were periodic along the lateral and aperiodic along the normal directions, whereas reflective (specular) boundary conditions were implemented along the normal direction. The incorporation of fixed wall particles in conjunction with reflective boundaries has been shown to be an effective approach for preventing DPD beads from crossing the top and bottom faces of the simulation box and for enforcing the no slip boundary condition.33,86,87
During these simulations the equations of motion were integrated using the velocity-Verlet algorithm80 with a time step of 0.005τDPD. The estimation of the electrostatic interactions in these aperiodic systems was performed with the algorithm by Yeh and Berkowitz88 that inserts an empty volume between the images of the system along the aperiodic directions and as a result removes the dipole interactions among the slabs. The parameters of the brush arrays examined in the present work are displayed in Table 6.
| N SPE | Area/chain (nm2) | a latt (nm) | σ g (nm−2) | L T (nm) |
|---|---|---|---|---|
| 5, 10, 15, 20 | 36.9 | 6.075 | 0.027 | 6.075 |
| 5, 10, 15, 20 | 16.4 | 4.05 | 0.061 | 8.1 |
| 5, 10, 15, 20 | 7.29 | 2.7 | 0.137 | 8.1 |
| 5, 10, 15, 20 | 3.24 | 1.8 | 0.309 | 7.2 |
| 5, 10, 15, 20 | 1.44 | 1.2 | 0.694 | 7.2 |
| 5, 10, 15, 20 | 0.64 | 0.8 | 1.562 | 7.2 |
5. Results
5.1 Bulk solutions
Fig. 4 depicts the mean square radius of gyration, 〈Rg2〉, of free SPE in dilute aqueous solution from DPD, MD and from the experimental correlations derived by Mary et al.14,15 The latter are based on measurements in the presence of NaCl and they refer to the conditions of temperature, SPE and electrolyte concentration under which the upper critical solution temperature of each system exhibits a maximum with respect to the electrolyte concentration. In addition, representative configurations from the atomistic (OPLS) and mesoscopic (DPD) models are displayed in Fig. 5. Overall, atomistic chains as described by the DREIDING and the OPLS force fields exhibit very similar behavior. The size of the atomistic chains—as quantified by 〈Rg2〉—increases sub-linearly with respect to their length in the short chain regime, while for longer chains it follows a scaling law very close to ∼N1, characteristic of random coils. As far as the mesoscopic chains are concerned, the estimated 〈Rg2〉 values are in very good quantitative agreement with the atomistic ones (compare green triangles with red squares and circles), while for larger chain lengths the scaling exponent becomes slightly higher. The accessible chain length regime from DPD features significant overlap with the experimental correlations by Mary et al.14,15 Moreover, the predicted square radii of gyration are in quantitative agreement with the experimental correlation which is built-in in the model proposed by Mary et al.: Rg = aMbα with a = 0.69 nm, b = 0.48 and Mα being the weight-average (Mw) or number-average (Mn) molar mass.14,15 (all SPE polymers studied experimentally in ref. 15 were quite polydisperse, with polydispersity indices ranging from 2 to 3.) Small Angle X-ray Scattering (SAXS) measurements, on the other hand, are for NSPE = 1611 and are a better match with the predictions based on the Mn averaged degree of polymerization. | ||
| Fig. 4 Mean square radius of gyration from MD with OPLS (○) and DREIDING (□), DPD calculations (▲) and experimental data from ref. 15 (×, + and ★). The experimental data were derived from the correlation Rg = aMbα with a = 0.69 nm and b = 0.48, with Mα being either the weight- (Mw, ×) or number- (Mn, +) average molar mass. | ||
 | ||
| Fig. 5 Atomistic (MD with OPLS) and mesoscopic (DPD) representation of representative SPE polymers investigated in this work. | ||
5.2 Brush arrays
Throughout our DPD simulations we investigated polyzwitterionic brush arrays exposed to water similar to those examined by Xiang et al.,19 over a broad range of molecular parameters such as the area/grafted molecule (i.e., the inverse of the grafting density, σg) and the polymerization degree of the grafted SPE chains (NSPE).(1) The MD profiles include the additional contribution of the saturated benzene rings with the azide group (C20H19O3N4) representing the root of the brush and the polyamide membrane surface.19
(2) At low grafting densities the polyamide membrane surface employed in the atomistic MD simulations becomes very sparse and as a result the sulfobetaine groups can penetrate it (e.g., see Fig. 6c in ref. 19).
(3) Another difference between these systems is that the atomistic SPEs simulated by Xiang et al.19 are smaller by one methylene unit,  in order to facilitate comparisons with previous simulation works by Shao et al.16,20–22
in order to facilitate comparisons with previous simulation works by Shao et al.16,20–22
These differences have to be accounted for in order to perform meaningful comparisons between these simulations. Therefore, to facilitate comparisons among these models the density profiles in Fig. 6 are visualized against the effective segment-wall distance:
| h = z − zS | (10) |
 | (11) |
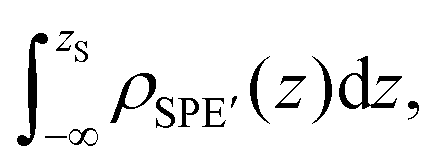 corresponds to the mass of the polyamide membrane per unit area. Note that comparisons between
corresponds to the mass of the polyamide membrane per unit area. Note that comparisons between  from MD and DPD are not especially meaningful, since the polyamide membrane is not exactly the same as the solid substrate used in our DPD simulations (see the lattice in Fig. 3).
from MD and DPD are not especially meaningful, since the polyamide membrane is not exactly the same as the solid substrate used in our DPD simulations (see the lattice in Fig. 3).
 | ||
| Fig. 6 Density profiles of SPE (red) and water (blue) from DPD (solid lines) and atomistic MD19 (dots) for area/chain = (a) 3.24, (b) 1.44 and (c) 0.64 nm2. The thick vertical line illustrates the effective position of the wall, zS, whereas the dashed one depicts the grafting point in DPD. The width of the bins of the profile was set to 0.5rDPD ∼ 0.383 nm. | ||
According to Fig. 6, the density profiles from DPD are in reasonable agreement with the MD19 density profiles considered by Xiang et al.19 The DPD profiles appear to be slightly more expanded as compared to the MD ones; the difference can be attributed to the SPEs from the MD study19 being shorter by one CH2 segment. In addition, DPD profiles feature a narrow low density region near the surface, since in that case the SPE chains are grafted to a planar solid surface instead of the mobile saturated benzene rings in Xiang et al.19 As has been demonstrated by numerous works in the past,38,89–91 the density profiles tend to form steep peaks near rigid planar surfaces. These peaks arise due to excluded volume effects which lead to the formation of layers.70,92–94 The effect becomes even more intense in DPD with increasing coarse-graining because the interactions among beads become more repulsive94 in order to maintain the compressibility at a constant density; e.g., see eqn (6). Nonetheless, setting the width of the profile bin to half the radius of a DPD bead leads to overall smooth profiles, as shown in Fig. 6. In the case corresponding to the highest grafting density examined here, the SPE chains experience intense confinement and as a result some of their beads penetrate the wall surface at the lowest boundary of the simulation box (e.g., see the density states below the solid vertical line in Fig. 6c). The efficacy of DPD to sample the configurations of these densely grafted brushes was quantified in terms of the autocorrelation function of the tangential (with respect to the surface) components of the end-to-end vector of the SPE chains. These results are shown in the Section S3 (ESI†) and demonstrate that the dynamics becomes more sluggish with increasing grafting density, albeit the autocorrelation drops below 10% at times longer than 1000τDPD. Another difference between the DPD and MD19 profiles is the tendency of the water molecules to penetrate the SPE brushes, especially in the dense brush regime. In MD19 the water molecules can penetrate the SPE brushes over all the grafting densities considered there. In DPD, however, the water beads cannot penetrate the dense brush in Fig. 6c because the water beads are much larger (5 times) than individual water molecules. The profiles of the water beads are similar to those from MD for the sparser brushes (Fig. 6a and b).
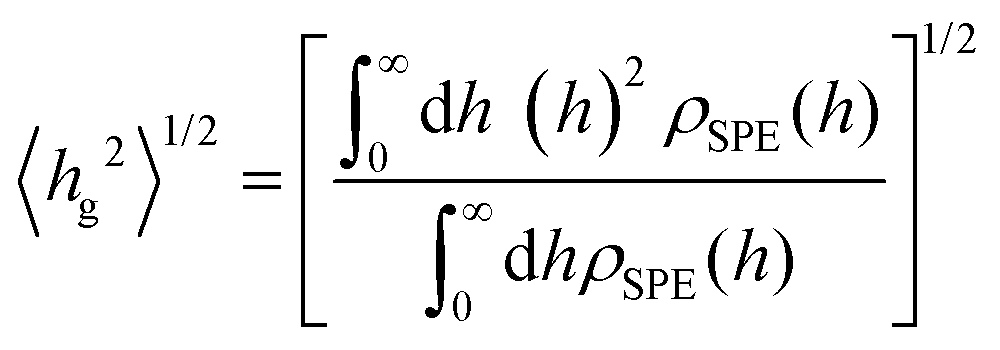 | (12) |
 | ||
| Fig. 7 (a) 〈hg2〉1/2versus NSPEσg1/2 from MD19 (filled symbols) and DPD (hollow symbols). (b) 〈hg2〉1/2, (c) 〈hg,99%〉, and (d) 〈hg,99%〉/〈hg2〉1/2versus NSPEσg1/5. The dashed line in (d) illustrates the prediction from Alexander's model57,95,96 for incompressible brushes. The markers denote grafting densities, σg = 0.027 (●), 0.061 (▲), 0.137 (★), 0.309 (■), 0.694 (+), and 1.562 (×) nm−2, whereas colors denote different chain lengths, NSPE = 5 (red), 10 (blue), 15 (green) and 20 (violet). The error bars depict the standard deviation derived from three DPD simulations for each case starting with different initial configurations. | ||
Overall, the brush thickness is a close match between MD19 and DPD; e.g., compare the red data points for NSPE = 5. The brush thickness scales linearly with NSPE, as is indicated by markers with the same shapes and different colors in Fig. 7a; this linear scaling has been shown to hold regardless of whether the solvent is good, theta or poor.99 The Θ-scaling with surface grafting density 〈hg2〉1/2/NSPE ∼ σg1/2, however, does not seem to collapse our data fully satisfactorily.
Based on our simulations, the brush thickness is practically insensitive to σg at low grafting densities, a feature which indicates that the sparsely grafted chains interact weakly with each other (mushroom regime).100 At larger grafting densities the chains experience significant confinement and thus the brush thickness increases with σg. Nevertheless, this increase appears to be weaker than the prediction for flexible polymer brushes in Θ solvents97–99 and as a result considerable scattering is seen in Fig. 7a.
Plotting the brush thickness against NSPE1σng with a scaling exponent n < 1/2 leads to significantly lower scatter. This is clearly demonstrated in Fig. 7b, which depicts 〈hg2〉1/2 plotted against N1σg1/5. The master plot of all data for the brush height in this figure is very satisfactory. Even though long SPE/water solutions behave as Θ solutions under the temperature of interest, the shorter SPE chains considered in Fig. 7 deviate from the expected Θ-solution scaling for brushes of long, randomly coiled chains. This can be attributed to the finite extensibility of the short chains comprising our brushes. Finite extensibility plays a significant role here because of the considerable conformational stiffness exhibited by our SPE chains in water. The inflexibility of our chains is attributable to the large branches on the SPE monomers (SO–SN–SS side groups), which lead to thick and twisted chain contours, and thus to persistent polymer backbones. This effect is especially evident when considering relatively short chains; e.g., please inspect the atomistic and mesoscopic configurations for NSPE < 40 in Fig. 5.
Finite extensibility leads to a weaker scaling with σg. To highlight this, we have performed a simple scaling analysis for a brush of finitely extensible chains in a Θ solvent following ref. 101, which we present in Section S4 (ESI†). A similar effect has been reported for good solvents as well, where, in situations with enhanced chain stretching and orientation, the brush thickness scales more weakly with σg, and can even become independent of σg in extreme cases.99,101 It should be noted, however, that in the limit of very high chain molar masses, for which the chains assume random-coil-like configurations, the dependence of 〈hg2〉1/2 on grafting density could potentially become stronger, approaching the Θ-chain 〈hg2〉1/2 ∼ NSPE1σg1/2 scaling.
Another measure for quantifying the shape of a grafted polymer brush is the brush span hg,99%, which can be defined for planar geometries as the segment-wall distance that encloses 99% of grafted SPE mass:
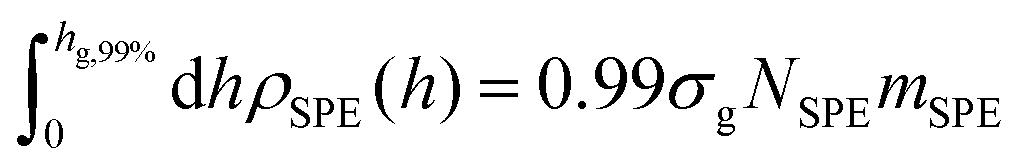 | (13) |
 which is the prediction from Alexander's model for incompressible brushes.57,95,96
which is the prediction from Alexander's model for incompressible brushes.57,95,96
The DPD foulant is modeled as an icosahedron formed by DPD beads connected with harmonic springs. Unless otherwise stated, the spring constant equals kspring,FL = 500εDPD/rDPD2 and the equilibrium length equals, l0,FL = 0.77 nm (1rDPD). The repulsive/attractive nature of the resulting mean force depends on an interplay among the strengths of the enthalpic and entropic FL–W, FL–AFL and the W–AFL interactions. In order to explore these tendencies, the repulsive FL–W and FL–AFL interactions were varied systematically, based on the following scaled geometric mixing rules:
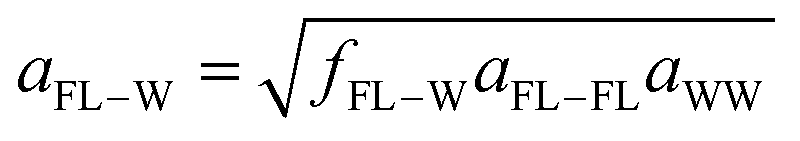 | (14) |
 | (15) |
 | (16) |
Fig. 8 depicts the MF as obtained from the umbrella sampling DPD simulations on (a and d) sparsely, (b and e) moderately and (c and f) densely grafted brushes, as a function of the separation distance between the FL's center of mass and the wall.
 | ||
| Fig. 8 Mean force (MF) from DPD as a function of the foulant's center of mass-wall surface distance, for area/chain set to (a and d) 3.24 nm2, (b and e) 1.44 nm2 and (c and f) 0.64 nm2. Positive/negative MF indicates attraction/repulsion. Different lines/colors indicate a different combination of (aFL–FL/aWW, fFL–W, fFL–AFL) as indicated in the legends. In all cases, the equilibrium length of the FL–FL bonds was set to 0.77 nm (rDPD). The horizontal lines are guides to the eye. The error bars depict the deviation from the mean based on two DPD simulations starting with different initial configurations. | ||
Panels a–c in Fig. 8 depict situations where aFL–FL is set to 0.5aWW (circles), aWW (triangles), and 2aWW (squares), and fFL–W = fFL–AFL = 1. It appears that, regardless of the magnitude of aFL–FL, the MF remains strictly repulsive. This is to be expected, since the relative strengths of the FL–W and FL–AFL interactions are invariant to aFL–FL, therefore the overall attractive/repulsive tendencies of the MF are not affected. In practice, since fFL–W/fFL–AFL = const, increasing aFL–FL has the effect of varying the effective bead size of the FL, thus shifting the MF towards slightly larger distances.
The right-hand panels of Fig. 8(d–f) present the effect of varying the relative strength of the FL–W and FL–AFL interactions, whilst fixing aFL–FL to aWW. Decreasing fFL–W/fFL–AFL (making the foulant more hydrophilic) has the effect of shifting the MF towards larger separation distances, indicating stronger effective FL–AFL repulsion. Increasing fFL–W/fFL–AFL, on the other hand (making the foulant more hydrophobic) leads to the emergence of a more complicated picture which could be explained as follows:
(1) At the edge of the AFL brush the MF becomes positive; thus indicating that there is a tendency for the FL to stick on it.
(2) Below a threshold separation distance (which becomes larger with increasing brush density) the MF begins to decrease (becomes more repulsive) due to the manifestation of excluded volume effects; i.e., the foulant is displaced to a region that is already occupied by the brush segments. The effect is more pronounced when considering the densest brush in Fig. 8f, where the MF becomes negative (strictly repulsive) below a distance of 3 nm.
(3) At distances of ∼1 nm where the entire foulant has been submerged into the sparse brush, the MF forms a minimum and begins to rise again (becomes more attractive). This is attributed to the density of the AFL brush having reached approximately its maximum value (where its effect on the MF becomes minor), whereas the density of the (repulsive) water phase declines; hence, favoring the occupation of these low water density-regions.
It is noteworthy that the MF remains strictly positive (attractive) across the full range when considering the sparser brush studied here (Fig. 8d).
The AFL–W interactions play a significant role for the resulting MF, as well. However, we opted not to vary them during our tests since our model has been parameterized for a specific chemical constitution of the antifoulant (AFL). Nonetheless, it is expected that, as the AFL–W interactions become less repulsive there is a tendency for the FL and the AFL to repel each other, since the latter will prefer to interact with the solvent molecules instead of with the FL. More repulsive AFL–W interactions are expected to promote the opposite behavior; i.e., the FL tending to stick to the AFL layer.
Fig. 9 provides qualitative comparisons between the MF from our DPD simulations and from the steered atomistic MD simulations by Xiang et al.19 In MD, the FL is modeled by an alginate gel via the OPLS force field.19 In DPD, the equilibrium length of the harmonic springs was set to either l0,FL = 0.38 nm (0.5rDPD) or 0.77 nm (rDPD) in order to explore the effect of varying the size of the foulant. For these comparisons, the FL–AFL and FL–W interactions were estimated via the original geometric mixing rule (fFL–W = fFL–AFL = 1). The strength of the repulsive interactions among the beads that comprise the foulant were set to two times those of water (aFL–FL = 2aWW), albeit as long as fFL–W and fFL–AFL are fixed the overall qualitative behavior is practically the same for lower and higher aFL–FL values (e.g., see the demonstrations in Fig. 8a–c).
 | ||
| Fig. 9 Mean force (MF) from atomistic MD19 (circles) and DPD with l0,FL = 0.38 nm (blue triangles) and 0.77 nm (green squares) as a function of the foulant's center of mass-wall surface distance, for area/chain = (a) 3.24 nm2, (b) 1.44 nm2 and (c) 0.64 nm2. Positive/negative MF indicates attraction/repulsion. The horizontal lines are guides to the eye. The error bars depict the deviation from the mean based on two DPD simulations starting with different initial configurations. | ||
The MF is qualitatively similar between DPD and MD; the MF assumes negative (repulsive) values as the foulant approaches the SPE brushes, while it decreases more steeply with increasing grafted density, indicating an enhancement of the antifouling properties of the SPE brush. Increasing the size of the FL by increasing l0,FL makes the MF slightly more repulsive, albeit the effect is minor. The difference between DPD and MD is to be expected, since the DPD foulant has not been parameterized from an alginate gel, and thus there might be differences in the size and geometry of the foulants considered in each case. For reference, the radius of gyration of the DPD foulant is about  with
with  being the golden ratio. It is interesting that, given the simplicity of the biofoulants employed in DPD, the corresponding MFs are in reasonable qualitative agreement with those from MD. The estimation of the PMF arising from the interaction among grafted polyzwitterions and more complex biofoulants (in terms of their chemistry and structure) as a function of their concentration, and in the presence of ionic solutions, is a subject for future research.
being the golden ratio. It is interesting that, given the simplicity of the biofoulants employed in DPD, the corresponding MFs are in reasonable qualitative agreement with those from MD. The estimation of the PMF arising from the interaction among grafted polyzwitterions and more complex biofoulants (in terms of their chemistry and structure) as a function of their concentration, and in the presence of ionic solutions, is a subject for future research.
6. Concluding remarks
Sulfobetaine-based polyzwitterions constitute promising candidate materials for the design of surfaces with antifouling properties. Several atomistic studies have been conducted in recent years addressing the structure and thermodynamics of these polymers over atomistic length scales.16,19–22 Nonetheless, the relevant time and length scales of long polyzwitterions are far greater than those that can be reached by conventional atomistic simulations. Hence, there is a need for the development of mesoscopic simulation approaches involving aggressive coarse-graining.The present work develops a multiscale simulation strategy with two levels of description, entailing an atomistic and a mesoscopic regime. The atomistic regime is investigated with molecular dynamics simulations (MD) using the OPLS force field, whereas the mesoscopic regime is explored by dissipative particle dynamics (DPD) simulations. These two different levels of description are capable of providing access to a very broad spectrum of time- and length-scales, which constitutes an absolute necessity for describing the structural features and thermodynamics of SPE poly-zwitterions. The mesoscopic model is parameterized hand-in-hand with atomistic molecular dynamics simulations across the overlapping regime of chain lengths. More specifically, the coefficients of the mesoscopic force field are optimized to match the atomistically computed strand-length, strand-angle and pair distribution functions via Monte Carlo and machine-learning based approaches.
Overall, the developed potential exhibits good transferability among different concentrations as indicated indirectly, by the good match with molecular dynamics, theory and experiments for both sparse and dense systems. In detail, the structural features of the mesoscopic SPE chains in dilute aqueous solutions reproduce those from the underlying OPLS atomistic model and agree with the experimental findings of Mary et al.14,15 For sufficiently long SPE chains, the scaling exponent between the mean squared radius of gyration and chain length in dilute aqueous solution is close to 1. The brush array configurations studied herein are investigated in terms of the corresponding brush thickness measures, the local density profiles, and the mean force they exert on an approaching foulant. Results from our DPD simulations are compared with a relevant molecular dynamics study by Xiang et al.19 In detail, the root mean square brush thickness and the span of the density profiles as quantified by hg,99% increase approximately linearly with the product of the degree of polymerization and the grafting density to the power of 1/5, a feature that can be explained by scaling considerations for brushes of finitely extensible chains exposed to theta solvents. For very high grafting densities the structure of the brush approaches that of an Alexander-type incompressible brush, also obtained from self-consistent field theory treatment of compressible dense brushes53,57 in planar geometries.
The repulsive potential of mean force derived from umbrella sampling simulations of a foulant upon approaching the polyzwitterionic brush is in good qualitative agreement with the atomistic findings by Xiang et al.,19 whereas the discrepancies between atomistic and mesoscopic simulations can be traced to differences between the geometric aspects and the overall parameterization of the model systems.
The repulsive/attractive tendencies of the foulant are governed by an interplay among the foulant–solvent, foulant–antifoulant and the solvent–antifoulant interactions (both enthalpic and entropic). A general design rule for effective nonbiocidal antifoulants would be to enhance antifoulant–solution and the foulant–solution interactions with respect to the antifoulant–foulant ones. For biocidal antifoulants, on the other hand, opposite design rules would be applicable, since sticking the foulant to the antifouling brush would facilitate the biocidal process.
Conflicts of interest
There are no conflicts of interest to declare.Appendix A
The mesoscopic force field described in Section 3.2.3 was parameterized through a bottom-up mapping procedure aimed at matching the bond and bond-angle distributions and the pair correlation functions between DPD and atomistic trajectories. For the purpose of this matching, the polymer configurations in each atomistic trajectory were geometrically mapped onto coarse-grained configurations. This coarse-graining of atomistic trajectories was performed by lumping the SPE segments into coarse-grained beads (see Fig. 2a) based on the center of mass of the corresponding segments using a code developed in-house: | (17) |
1. Derivation of the distributions concerning bonded interactions
2. Boltzmann inversion of the bonded distributions
3. Calibration of the coefficients for the bonded interactions
4. Calibration of the coefficients for the nonbonded interactions
5. Recalibration of the coefficients for the bonded interactions
Details about each step of the mapping procedure are presented in Appendices A1–A5, respectively.
A1. Derivation of the distributions concerning the bonded interactions
Our initial attempt was to extract the strand length (ρstrand) and strand-angle (ρangle) distributions from a coarse-grained trajectory of a single SPE8 chain immersed in 850 water molecules for extended simulation times (∼0.1 μs). In doing so, we observed that the strand-angle distributions exhibited nonergodic behavior within the time span of the atomistic molecular dynamics; thus, the distributions would not converge within these simulation times. In other words, configurations sampled by the atomistic MD within the accessible simulation time appeared to be trapped in the vicinity of local energy minima, retaining strong memory of their initial characteristics (e.g., angle distributions along consecutive SB–SB–SB triplets differed substantially). Example nonergodic strand-angle distributions are illustrated in Section S5 (ESI†). To circumvent this issue we incorporated a temperature annealing scheme at constant volume and number of particles which proceeds as follows: (i) equilibration of 120 independently generated atomistic samples at T = 900 K with different initial velocities for 2 ns; (ii) temperature annealing from T = 900 K down to 300 K over 2 ns. (iii) Additional relaxation at T = 300 K for 0.5 ns. (iv) Sampling at T = 300 K for 2 ns. Subsequently, the strand length and strand-angle distributions were calculated based on the 120 temperature-annealed atomistic trajectories by placing beads at the centers of mass of atomistically represented groups, configuration by configuration, according to eqn (17). The resulting distributions for strands and strand-angles differed very little across the polymer backbone. The converged ρstrand and ρangle from MD are illustrated in Fig. 10 (red circles). It should be noted that the corresponding box size for this concentration is ∼4 times the radius of gyration of an SPE8 chain, and as a result interactions between different images of the same chain segments are minimal.90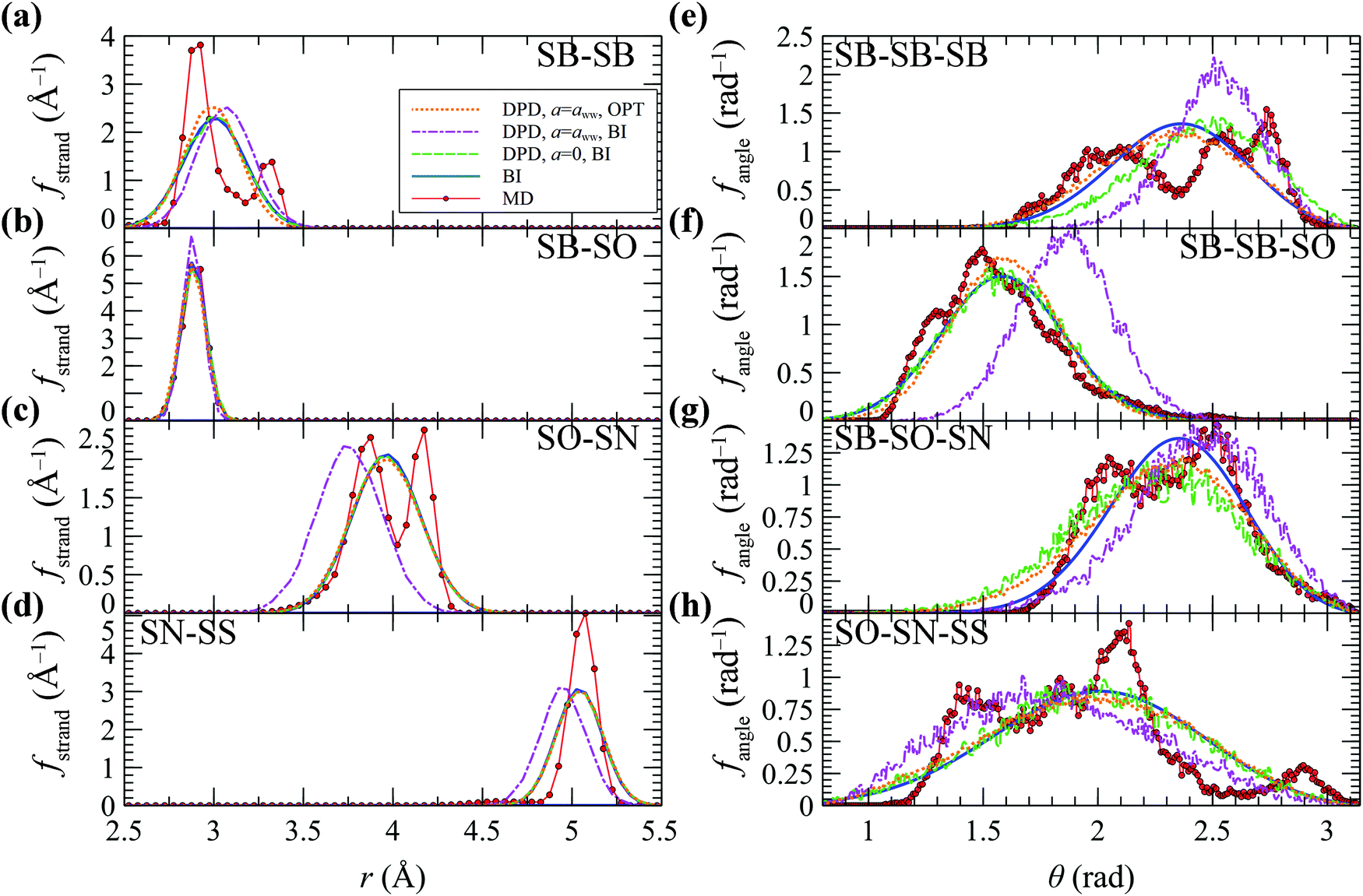 | ||
| Fig. 10 Strand length (a–d) and strand-angle (e–h) distributions from MD with OPLS (red circles); BI (blue lines); DPD, aij = 0, coefficients from BI (green dashes); DPD, aij = aww, coefficients from BI (purple, dot-dashes); DPD, optimized aij coefficients after the final recalibration (orange, dots). | ||
A2. Boltzmann inversion of the distributions
The free energy of the atomistically calculated strand and strand-angle distributions up to an additive constant is given by the following equation:| U(r) = −kBTlnf(r) | (18) |
 for strands, and
for strands, and 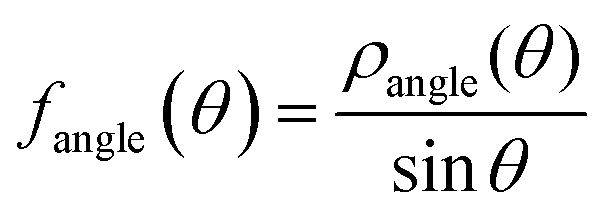 for angles, respectively. Assuming that f follows a Gaussian distribution,
for angles, respectively. Assuming that f follows a Gaussian distribution,  then the kind-α strands and the strand-angles can be described by harmonic free energy functions of the form: Uα(x) = kα(x − μα)2, with x being the strand length or the angle. Therefore, the stiffness of the strand/strand-angle potential equals kα = kBT/(2σα2), and the reference length/angle equals μα. The blue lines in Fig. 10 depict the theoretical predictions based on the kα and μα coefficients from the Boltzmann inversion (BI). It is worth mentioning that some of the distributions derived from atomistic MD are seen to display complex features. Notable is the bimodality in SB–SB and SO–SN, originating in torsion angles along the backbone and along the side chain adopting trans versus gauche states. These details are not kept in the coarse-grained model.
then the kind-α strands and the strand-angles can be described by harmonic free energy functions of the form: Uα(x) = kα(x − μα)2, with x being the strand length or the angle. Therefore, the stiffness of the strand/strand-angle potential equals kα = kBT/(2σα2), and the reference length/angle equals μα. The blue lines in Fig. 10 depict the theoretical predictions based on the kα and μα coefficients from the Boltzmann inversion (BI). It is worth mentioning that some of the distributions derived from atomistic MD are seen to display complex features. Notable is the bimodality in SB–SB and SO–SN, originating in torsion angles along the backbone and along the side chain adopting trans versus gauche states. These details are not kept in the coarse-grained model.
A3. Calibration of the coefficients for the bonded interactions
As seen in Fig. 10, the strand distributions from DPD with the set of kα and μα for each strand extracted from BI and in the absence of the nonbonded interactions (aij = 0) are in good match with the Boltzmann inverted distributions from MD (compare blue lines with the green dashed lines). The strand-angle distributions for SB–SB–SB and SB–SO–SN triplets exhibit a slightly worse match and this is attributed to cross-correlations among different angle types. The introduction of the nonbonded interactions perturbs the distributions significantly; e.g., compare green dashes (aij = 0) with the purple dot-dashed lines (aij = aww) in Fig. 10. To circumvent this issue we developed an optimization scheme that finds the optimal strand and strand-angle coefficients that best reproduce the BI distributions from MD. The scheme optimizes all the coefficients for the target strands and strand-angles at the same time in the presence of the nonbonded interactions, while the whole procedure takes 100–200 iterations. The orange dotted lines in Fig. 10 depict the distributions after the final recalibration (see Appendix A5), which are in excellent match with the fitted (BI) distribution. In other words, the DPD distributions have been optimized with the target to replicate the mean and the standard deviation of their corresponding MD distributions. Introducing more complex functional expressions would allow one to replicate these distributions more closely, but this would introduce additional complications to the model and potentially require even smaller time steps due to the enhanced roughness of the potential hypersurface. Details about the optimization scheme for the bonded interactions are illustrated in S6 (ESI†). An alternative approach for tackling these cross-correlations among different intramolecular components would be to invoke the MRG-GG (molecular renormalization group coarse-graining) scheme devised by Sevalyev et al.102A4. Calibration of the coefficients for the nonbonded interactions
As we discussed in the Introduction section, the χij for the SPE polymers under investigation are inaccessible, and thus we opted for a more direct parameterization approach based on matching the partial pair distribution functions (gij) between MD and DPD; the former is shown in Fig. 11 (dashes) and was derived from coarse-graining atomistic trajectories of 4 × SPE monomers in 800 water molecules (concentration ∼7.8 wt%) with a duration of 0.3 μs. The objective function is illustrated in S7 (ESI†). For SPE the objective function to be optimized has as input npair = ntypes(ntypes + 1)/2 − 1 = 14 free parameters (the “−1” has to do with keeping aWW fixed), while its output (cost) is proportional to the mean squared error between gij from MD and DPD averaged over all the npair interactions,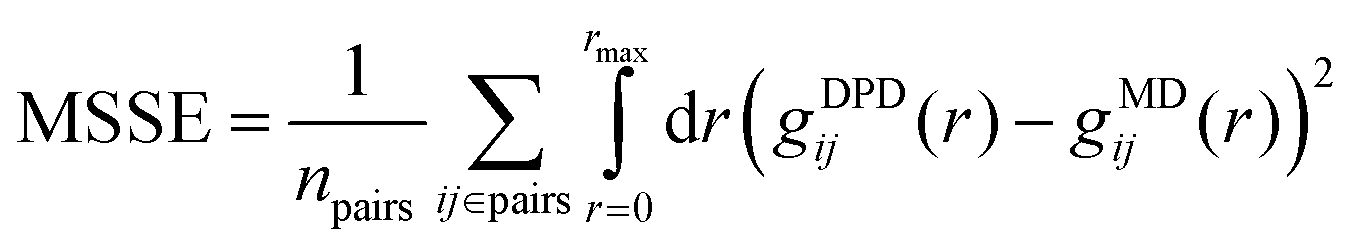 . To minimize the objective function, initially we performed ∼5000 bounded evaluations for 10 < aij < 180, and then we performed Bayesian optimization for ∼3000 steps (having fed the optimized output of the first ∼5000 iterations) to guide us closer to the global minimum using the implementation by Nogueira.103 It is worth mentioning that the efficiency of the bounded optimization was comparable to the Bayesian one, since the former was performed in parallel using 128 cpu cores (1 core per simulation). Details about Bayesian optimization can be found in the S8 (ESI†).103–106
. To minimize the objective function, initially we performed ∼5000 bounded evaluations for 10 < aij < 180, and then we performed Bayesian optimization for ∼3000 steps (having fed the optimized output of the first ∼5000 iterations) to guide us closer to the global minimum using the implementation by Nogueira.103 It is worth mentioning that the efficiency of the bounded optimization was comparable to the Bayesian one, since the former was performed in parallel using 128 cpu cores (1 core per simulation). Details about Bayesian optimization can be found in the S8 (ESI†).103–106
 | ||
| Fig. 11 Pair radial distribution functions from MD with OPLS (dashes) and DPD (solid lines). | ||
With few exceptions, the optimized DPD coefficients allow us to capture very reasonably the overall shape and probability amplitude of the RDFs from MD. The largest discrepancy arises in the SN–SS term of the RDF, wherein the corresponding beads assume opposite charges. The agreement between the atomistic and DPD pair distribution functions could be potentially improved by continuing the Bayesian optimization for much longer times, given that the 14-D objective function is sensitive (spans ∼5 orders of magnitude as shown in Fig. S5, ESI†), somewhat noisy, and that Bayesian optimization lacks appropriate convergence criteria. In addition, one could attempt to introduce additional degrees of freedom, such as varying the absolute values of the charges or the charge smearing radius λ. Here we opted to retain the original bottom-up charges as in most DPD studies, in order to avoid introducing additional complexity to the model. In this way, the interactions with charged foulants are straightforward to describe and do not necessitate additional parameterization. Nonetheless, the aforementioned discrepancies do not appear to affect the performance of the model much, as indicated by the good match of the long range conformational characteristics between MD and DPD presented below in Fig. 4, and the similar density profiles discussed in Section 5.2.1.
Another potential approach for describing the nonbonded interactions would be to incorporate a more complicated expression for the description of the repulsive interactions, or even to invoke tabulated potentials.52–55
A5. Recalibration of the coefficients for the bonded interactions
The variation of the nonbonded coefficients during the optimization introduced minor perturbations to the strand and strand/angle distributions. To refine these parameters and restore the unperturbed distributions, we performed one last recalibration to the coefficients for the bonded interactions in the presence of the optimized aij using the algorithm in S6 (ESI†). This recalibration step had a very minor effect on the partial radial distribution functions; note that the orange dots in Fig. 10 and the red lines in Fig. 11 where calculated using the final form of the potential, after the recalibration process.Acknowledgements
The authors are grateful for the fruitful discussion with Dr Jean-Yves Delannoy. The contribution of Dr Denis Bendejacq in discussing ref. 14 and 15 with us and pointing our attention to experimental correlations for the SPE radii of gyrations in solution has been valuable. A. P. Sgouros thanks SOLVAY, Brussels, Belgium, for financial support.References
- F. Siedenbiedel and J. C. Tiller, Polymers, 2012, 4, 46–71 CrossRef CAS.
- O. P. Abioye, C. A. Loto and O. S. I. Fayomi, J. Bio- Tribo-Corros., 2019, 5, 22 CrossRef.
- A. U. Khan, M. Khan, N. Malik, M. H. Cho and M. M. Khan, Bioprocess Biosyst. Eng., 2019, 42, 1–15 CrossRef CAS PubMed.
- Z. K. Zander and M. L. Becker, ACS Macro Lett., 2018, 7, 16–25 CrossRef CAS.
- K. Thomas, in Woodhead Publishing Series in Metals and Surface Engineering, ed. C. Hellio and D. Yebra, Woodhead Publishing, 2009, pp. 522–553 Search PubMed.
- S. Brooks and M. Waldock, in Woodhead Publishing Series in Metals and Surface Engineering, ed. C. Hellio and D. Yebra, Woodhead Publishing, 2009, pp. 492–521 Search PubMed.
- A. G. Nurioglu, A. C. C. Esteves and G. de With, J. Mater. Chem. B, 2015, 3, 6547–6570 RSC.
- M. Lejars, A. Margaillan and C. Bressy, Chem. Rev., 2012, 112, 4347–4390 CrossRef CAS PubMed.
- S. Nir and M. Reches, Curr. Opin. Biotechnol, 2016, 39, 48–55 CrossRef CAS PubMed.
- G. D. Bixler and B. Bhushan, Philos. Trans. Ser. A, Math. Phys. Eng. Sci., 2012, 370, 2381–2417 CAS.
- A. Marmur, Biofouling, 2006, 22, 107–115 CrossRef CAS PubMed.
- G. B. Hwang, K. Page, A. Patir, S. P. Nair, E. Allan and I. P. Parkin, ACS Nano, 2018, 12, 6050–6058 CrossRef CAS PubMed.
- G. Zhao and W. N. Chen, RSC Adv., 2017, 7, 37990–38000 RSC.
- P. Mary, D. D. Bendejacq, M. P. Labeau and P. Dupuis, J. Phys. Chem. B, 2007, 111, 7767–7777 CrossRef CAS PubMed.
- P. Mary and D. D. Bendejacq, J. Phys. Chem. B, 2008, 112, 2299–2310 CrossRef CAS PubMed.
- Q. Shao and S. Jiang, J. Phys. Chem. B, 2014, 118, 7630–7637 CrossRef CAS PubMed.
- H. Y. Yu, Y. Kang, Y. Liu and B. Mi, J. Membr. Sci., 2014, 449, 50–57 CrossRef CAS.
- M. Ajmal, S. Demirci, M. Siddiq, N. Aktas and N. Sahiner, Colloids Surf., A, 2015, 486, 29–37 CrossRef CAS.
- Y. Xiang, R. Xu and Y. Leng, Langmuir, 2018, 34, 2247–2257 CrossRef PubMed.
- Q. Shao, Y. He and S. Jiang, J. Phys. Chem. B, 2011, 115, 8358–8363 CrossRef CAS PubMed.
- Q. Shao, Y. He, A. D. White and S. Jiang, J. Phys. Chem. B, 2010, 114, 16625–16631 CrossRef CAS PubMed.
- Q. Shao, Y. He, A. D. White and S. Jiang, J. Chem. Phys., 2012, 136, 225101 CrossRef PubMed.
- S. Chen, L. Li, C. Zhao and J. Zheng, Polymer, 2010, 51, 5283–5293 CrossRef CAS.
- S. Lowe, N. M. O’Brien-Simpson and L. A. Connal, Polym. Chem., 2015, 6, 198–212 RSC.
- J. Huo, Z. Chen and J. Zhou, Langmuir, 2019, 35, 1973–1983 CrossRef CAS PubMed.
- H. A. L. Filipe, M. J. Moreno and L. M. S. Loura, Molecules, 2020, 25, 3424 CrossRef CAS PubMed.
- S. Osella and S. Knippenberg, Biochim. Biophys. Acta, Biomembr., 2021, 1863, 183494 CrossRef CAS PubMed.
- P. J. Hoogerbruuge and J. M. V. A. Koelman, EPL, 1992, 19, 155 CrossRef.
- P. Español and P. B. Warren, J. Chem. Phys., 2017, 146, 150901 CrossRef PubMed.
- R. D. Groot and P. B. Warren, J. Chem. Phys., 1997, 107, 4423–4435 CrossRef CAS.
- R. D. Groot and K. L. Rabone, Biophys. J., 2001, 81, 725–736 CrossRef CAS PubMed.
- R. D. Groot, J. Chem. Phys., 2003, 118, 11265–11277 CrossRef CAS.
- M. B. Liu, G. R. Liu, L. W. Zhou and J. Z. Chang, Arch. Comput. Methods Eng., 2015, 22, 529–556 CrossRef.
- A. Karatrantos, N. Clarke, R. J. Composto and K. I. Winey, Soft Matter, 2013, 9, 3877–3884 RSC.
- N. Moreno, S. Nunes and V. M. Calo, Procedia Comput. Sci., 2014, 29, 728–739 CrossRef.
- F. Alvarez, E. A. Flores, L. V. Castro, J. G. Hernández, A. López and F. Vázquez, Energy Fuels, 2011, 25, 562–567 CrossRef CAS.
- Y. Ruiz-Morales and O. C. Mullins, Energy Fuels, 2015, 29, 1597–1609 CrossRef CAS.
- F. Alarcón, E. Pérez and A. Gama Goicochea, Soft Matter, 2013, 9, 3777–3788 RSC.
- C. Ibergay, P. Malfreyt and D. J. Tildesley, J. Phys. Chem. B, 2010, 114, 7274–7285 CrossRef CAS PubMed.
- T. H. Lin, W. P. Shih, C. S. Chen and Y. T. Chiu, 2006 1st IEEE Int. Conf. Nano/Micro Eng. Mol. Syst., 2006, pp. 571–574.
- M. Liu, P. Meakin and H. Huang, Phys. Fluids, 2006, 18, 017101 CrossRef.
- J. C. Shillcock and R. Lipowsky, J. Chem. Phys., 2002, 117, 5048–5061 CrossRef CAS.
- S. Yamamoto and S. A. Hyodo, Polym. J., 2003, 35, 519–527 CrossRef CAS.
- E. O. Johansson, T. Yamada, B. Sundén and J. Yuan, Int. J. Hydrogen Energy, 2015, 40, 1800–1808 CrossRef CAS.
- M. T. Lee, A. Vishnyakov and A. V. Neimark, J. Chem. Phys., 2016, 144, 014902 CrossRef PubMed.
- M. T. Lee, A. Vishnyakov and A. V. Neimark, J. Phys. Chem. B, 2013, 117, 10304–10310 CrossRef CAS PubMed.
- M. H. Nafar Sefiddashti, M. Boudaghi-Khajehnobar, B. J. Edwards and B. Khomami, Sci. Rep., 2020, 10, 1–13 CrossRef PubMed.
- A. Kumar, Y. Asako, E. Abu-Nada, M. Krafczyk and M. Faghri, Microfluid. Nanofluid., 2009, 7, 467–477 CrossRef.
- A. P. Sgouros, G. G. Vogiatzis, G. Megariotis, C. Tzoumanekas and D. N. Theodorou, Macromolecules, 2019, 52, 7503–7523 CrossRef CAS.
- R. M. Füchslin, H. Fellermann, A. Eriksson and H. J. Ziock, J. Chem. Phys., 2009, 130, 214102 CrossRef PubMed.
- H. Weiß, P. Deglmann, P. J. in’t Veld, M. Cetinkaya and E. Schreiner, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 65–86 CrossRef PubMed.
- A. K. Soper, Chem. Phys., 1996, 202, 295–306 CrossRef CAS.
- A. P. Sgouros, C. J. Revelas, A. T. Lakkas and D. N. Theodorou, Polymers, 2021, 13, 1197 CrossRef CAS PubMed.
- A. P. Lyubartsev and A. Laaksonen, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1995, 52, 3730–3737 CrossRef CAS PubMed.
- A. Lyubartsev, A. Mirzoev, L. J. Chen and A. Laaksonen, Faraday Discuss., 2010, 144, 43–56 RSC.
- G. G. Vogiatzis and D. N. Theodorou, Macromolecules, 2013, 46, 4670–4683 CrossRef CAS.
- A. T. Lakkas, A. P. Sgouros, C. J. Revelas and D. N. Theodorou, Soft Matter, 2021, 17, 4077–4097 RSC.
- P. Español and P. Warren, EPL, 1995, 30, 191 CrossRef.
- S. L. Mayo, B. D. Olafson and W. A. Goddard, J. Phys. Chem., 1990, 94, 8897–8909 CrossRef CAS.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- T. Yamanaka, A. De Nicola, G. Munaò, T. A. Soares and G. Milano, Chem. Phys. Lett., 2019, 731, 136576 CrossRef CAS.
- J. Gasteiger and M. Marsili, Tetrahedron Lett., 1978, 19, 3181–3184 CrossRef.
- J.-P. Ryckaert and A. Bellemans, Faraday Discuss. Chem. Soc., 1978, 66, 95–106 RSC.
- M. J. Abraham, E. Lindahl, B. Hess and D. van der Spoel, GROMACS 2021.1 Manual, 2021 DOI:10.5281/zenodo.5053220.
- C. Klein, A. Z. Summers, M. W. Thompson, J. B. Gilmer, C. McCabe, P. T. Cummings, J. Sallai and C. R. Iacovella, Comput. Mater. Sci., 2019, 167, 215–227 CrossRef CAS.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, J. Phys. Chem., 1987, 91, 6269–6271 CrossRef CAS.
- C. Li, X. Fu, W. Zhong and J. Liu, ACS Omega, 2019, 4, 10216–10224 CrossRef CAS PubMed.
- R. Vaiwala, S. Jadhav and R. Thaokar, J. Chem. Phys., 2017, 146, 124904 CrossRef PubMed.
- K. Shi, C. Lian, Z. Bai, S. Zhao and H. Liu, Chem. Eng. Sci., 2015, 122, 185–196 CrossRef CAS.
- M. González-Melchor, E. Mayoral, M. E. Velázquez and J. Alejandre, J. Chem. Phys., 2006, 125, 224107 CrossRef PubMed.
- J. R. Partington, R. F. Hudson and K. W. Bagnall, Nature, 1952, 169, 583–584 CrossRef CAS.
- C. Ibergay, P. Malfreyt and D. J. Tildesley, J. Chem. Theory Comput., 2009, 5, 3245–3259 CrossRef CAS PubMed.
- P. B. Warren and A. Vlasov, J. Chem. Phys., 2014, 140, 084904 CrossRef PubMed.
- Z. Posel, Z. Limpouchová, K. Šindelka, M. Lísal and K. Procházka, Macromolecules, 2014, 47, 2503–2514 CrossRef CAS.
- T. P. Liyana-Arachchi, S. N. Jamadagni, D. Eike, P. H. Koenig and J. Ilja Siepmann, J. Chem. Phys., 2015, 142, 044902 CrossRef PubMed.
- D. N. Theodorou and U. W. Suter, Macromolecules, 1985, 18, 1467–1478 CrossRef CAS.
- MAPS®, Scienomics, Paris, 2019.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- W. G. Hoover, A. J. C. Ladd and B. Moran, Phys. Rev. Lett., 1982, 48, 1818–1820 CrossRef CAS.
- M. Tuckerman, B. J. Berne and G. J. Martyna, J. Chem. Phys., 1992, 97, 1990–2001 CrossRef CAS.
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- R. Mukundan, Proceedings of the 7th Asian Technology Conference in Mathematics 2002, 2002, pp. 97–106.
- G. J. Martyna, M. E. Tuckerman, D. J. Tobias and M. Klein, Mol. Phys., 1996, 87, 1117–1157 CrossRef CAS.
- R. W. Hockney and J. W. Eastwood, Computer Simulation Using Particles, CRC Press, Bristol, 1988 Search PubMed.
- A. Grossfield, WHAM: the weighted histogram analysis method, v 2.0.10, http://membrane.urmc.rochester.edu/wordpress/?page_id=126.
- X. Fan, N. Phan-Thien, S. Chen, X. Wu and T. Yong Ng, Phys. Fluids, 2006, 18, 63102 CrossRef.
- X. Fan, N. Phan-Thien, N. T. Yong, X. Wu and D. Xu, Phys. Fluids, 2002, 15, 11–21 CrossRef.
- I.-C. Yeh and M. L. Berkowitz, J. Chem. Phys., 1999, 111, 3155–3162 CrossRef CAS.
- K. C. Daoulas, V. A. Harmandaris and V. G. Mavrantzas, Macromolecules, 2005, 38, 5796–5809 CrossRef.
- A. P. Sgouros, G. G. Vogiatzis, G. Kritikos, A. Boziki, A. Nikolakopoulou, D. Liveris and D. N. Theodorou, Macromolecules, 2017, 50, 8827–8844 CrossRef CAS.
- G. Kritikos, N. Vergadou and I. G. Economou, J. Phys. Chem. C, 2016, 120, 1013–1024 CrossRef CAS.
- Particles in Flows, ed. T. Bodnár, P. G. Galdi and Š. Nečasová, Birkhäuser, Cham, 1st edn, 2017 Search PubMed.
- G. E. Karniadakis, A. Beskok and N. Aluru, Systematic coarse-graining of molecular models by the Newton inversion method, Springer, New York, 2nd edn, 2005 Search PubMed.
- I. V. Pivkin and G. E. Karniadakis, Phys. Rev. Lett., 2006, 96, 206001 CrossRef PubMed.
- S. Alexander, J. Phys., 1977, 38, 983–987 CrossRef CAS.
- P. G. de Gennes, Macromolecules, 1980, 13, 1069–1075 CrossRef CAS.
- P. Lai and K. Binder, J. Chem. Phys., 1992, 97, 586–595 CrossRef CAS.
- G. S. Grest and M. Murat, Macromolecules, 1993, 26, 3108–3117 CrossRef CAS.
- B. Zhao and W. J. Brittain, Prog. Polym. Sci., 2000, 25, 677–710 CrossRef CAS.
- M. Aubouy, G. H. Fredrickson, P. Pincus and E. Raphael, Macromolecules, 1995, 28, 2979–2981 CrossRef CAS.
- D. V. Kuznetsov and Z. Y. Chen, J. Chem. Phys., 1998, 109, 7017–7027 CrossRef CAS.
- A. Savelyev and G. A. Papoian, Biophys. J., 2009, 96, 4044–4052 CrossRef CAS PubMed.
- F. Nogueira, Bayesian Optimization: Open source constrained global optimization tool for Python, 2014, https://github.com/fmfn/BayesianOptimization.
- P. Frazier, 2018, arXiv:1807.02811.
- C. E. Rasmussen and K. I. Williams, Gaussian Processes for Machine Learning, the MIT Press, Massachusetts Institute of Technology, 2006 Search PubMed.
- L. P. Kaelbling, M. L. Littman and A. W. Moore, J. Artif. Int. Res., 1996, 4, 237–285 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: (S1) OPLS coefficients; (S2) details about the mesoscopic polymer builder PyMeso; (S3) autocorrelation function of the tangential components of the end-to-end vector; (S4) brush-thickness scaling arguments for finitely extensible chains in a theta solvent; (S5) nonergodic angle distributions; (S6) calibration of the bonded coefficients; (S7) optimization of the nonbonded coefficients; (S8) details about the Bayesian optimization. See DOI: 10.1039/d1sm01255j |
| This journal is © The Royal Society of Chemistry 2021 |