
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProcessing of fast-gelling hydrogel precursors in microfluidics by electrocoalescence of reactive species†
Nicolas
Hauck‡
a,
Talika A.
Neuendorf‡
 a,
Max J.
Männel
a,
Lucas
Vogel
a,
Ping
Liu
b,
Enno
Stündel
a,
Yixin
Zhang
bc and
Julian
Thiele
*a
a,
Max J.
Männel
a,
Lucas
Vogel
a,
Ping
Liu
b,
Enno
Stündel
a,
Yixin
Zhang
bc and
Julian
Thiele
*a
aLeibniz-Institut für Polymerforschung Dresden e.V., Hohe Str. 6, 01069 Dresden, Germany. E-mail: thiele@ipfdd.de
bB CUBE Center for Molecular Bioengineering, Technische Universität Dresden, Tatzberg 41, 01307 Dresden, Germany
cCluster of Excellence Physics of Life, TU Dresden, Dresden, Germany
First published on 19th October 2021
Abstract
Microscopic hydrogels, also referred to as microgels, find broad application in life and materials science. A well-established technique for fabricating uniform microgels is droplet microfluidics. Here, optimal mixing of hydrogel precursor components is crucial to yield homogeneous microgels with respect to their morphology, mechanics, and distribution of functional moieties. However, when processing premixed polymer precursors that are highly reactive, fast or even instantaneous gelation inside fluid reservoirs or the microchannels of the flow cell commonly occurs, leading to an increase of fluid viscosity over time, and thus exacerbating the intrinsic control over fluid flow rates, droplet and microgel uniformity, which are key selling points of microfluidics in material design. To address these challenges, we utilize microflow cells with integrated electrodes, which enable fast addition and mixing of hydrogel precursors on demand by means of emulsion droplet coalescence. Here, two populations of surfactant-stabilized aqueous droplets – the first containing the material basis of the microgel, and the second containing another gel-forming component (e.g., a crosslinker) are formed at two consecutive microchannel junctions and merged via temporary thin-film instability. Our approach provides the ability to process such hydrogel systems that are otherwise challenging to process into uniform droplets and microgels by conventional droplet microfluidics. To demonstrate its versatility, we fabricate microgels with uniform shape and composition using fast hydrogelation via thiol-Michael addition reaction or non-covalent self-assembly. Furthermore, we elucidate the limitations of electrocoalescence of reactive hydrogel precursors by processing sodium alginate, crosslinked by calcium-induced ionic interactions. For this instantaneous type of hydrogelation, electrocoalescence of alginate and calcium ions does not result in the formation of morphologically isotropic microgels. Instead, it enables the creation of anisotropic microgel morphologies with tunable shape, which have previously only been achieved by selective crosslinking of elaborate higher-order emulsions or by aqueous two-phase systems as microgel templates.
Introduction
Hydrogels are three-dimensionally crosslinked networks based on natural or synthetic hydrophilic polymers, which are capable of retaining large amounts of water. Particle-like hydrogels with diameters ranging from 0.1 μm to 100 μm are referred to as microgels according to IUPAC definitions.1 Microgels find broad application in life and materials science, for instance, in drug delivery,2,3 tissue engineering,4,5 and sensing.6,7 Over the last two decades, droplet-assisted microfluidics has evolved into a powerful tool for fabricating microgels with outstanding control over size distribution8 as well as morphology,9,10 mechanics,11,12 swelling properties,13 degradability,14,15 and functionalization.16,17However, processing fast-gelling hydrogel precursor solutions into microgels utilizing droplet microfluidics is still challenging. Hydrogel network formation with very fast reaction kinetics in the range of milliseconds up to a few seconds gelation time includes click chemistry reactions creating covalent bonds, such as the thiol-Michael addition reaction of thiols and maleimides,18 as well as bond formation via supramolecular,19 and ionic interactions.20 A common issue frequently occurring when processing fast-gelling hydrogel precursors in microfluidic cells with well-established microchannel co-flow geometry is the uncontrolled hydrogel formation when two or more reactive hydrogel precursor streams are brought together before being jointly compartmentalized into droplets. Here, the experimenter may observe an undesired transition from a stable dripping regime into uncontrolled jetting of the precursor solutions due to an increase in viscosity caused by their fast gelation in bulk inside a joint fluid reservoir (cf.Fig. 1A and Fig. S1A, ESI†). At one point, the precursor solutions may become so viscous that the force exerted onto the fluid reservoir mechanically by the piston of a syringe or by pressurized air will not be sufficient to move the fluids any further inside the microchannels causing the complete clogging of the microfluidic device (cf.Fig. 1B and Fig. S1B, ESI†). Another issue when processing fast-gelling hydrogel precursors into microemulsions becomes obvious after successful droplet formation, if droplet solidification proceeds faster than droplet mixing. As a result, despite the promise of droplet microfluidics that short mixing times in pico-liter volumes should lead to evenly distributed species inside droplets in sub-milliseconds,21,22 physiochemically heterogeneous microgels may still be obtained from droplet-based templates. To circumvent the issue of solidification of hydrogel precursor solutions inside fluid reservoirs, e.g., syringes, or on-chip of a microfluidic device prior to droplet formation, a liquid flow of two reactive streams needs to be separated until the very moment of droplet formation. This can be achieved by utilizing a three-inflow junction, where two hydrogel precursors, for instance, are separated by a non-reactive central fluid stream, such as water or a buffer solution.18,23 If this fluid stream is sufficiently wide, diffusion across the reactive-non-reactive fluid interface can be neglected, and the reactive hydrogel precursors only meet in the moment of droplet pinch-off at the microchannel junction. While this approach makes continuous and well-controlled microgel fabrication feasible, it is hardly possible to adopt fluid flow to the microchannel design, and this process also requires detailed knowledge of fluid dynamics. The reason for that is the separating fluid stream, which dilutes the hydrogel precursor solutions on-chip. This additional fluid stream potentially hampers reaching the desired material concentration required for gelation as the fluid flow-through is also limited by fluid viscosity. Furthermore, using this approach, the formation of Janus-like microgels is facilitated, if hydrogel network formation in each droplet compartment occurs faster than the mixing of hydrogel precursors across the non-reactive liquid barrier within the droplet resulting from the almost complete absence of convective forces (cf.Fig. 1C and Fig. S1C, ESI†).
 | ||
| Fig. 1 Common challenges in droplet microfluidics-based processing of fast-gelling hydrogel precursors. (A) Fluid jetting instead of geometry-controlled droplet formation at the microchannel cross-junction. The initiation of hydrogelation prior to droplet formation leads to heterogeneous droplets. (B) Clogging of the microfluidic junction due to in-channel hydrogelation. (C) Separation of two reactive aqueous precursor phases by a third flow of an unreactive aqueous spacer phase to prevent gelation prior to droplet formation. In the case of some fast-gelling hydrogel precursors, the resulting microgels may retain a heterogeneous, two-halved arrangement. The scale bars for all panels denote 100 μm. | ||
On this account, we propose a rather different strategy for controlling mixing and solidification of fast-reacting hydrogel precursor solutions in microgel formation via droplet microfluidics. Our approach relies on merging the stream of one hydrogel precursor component with individual, previously formed, surfactant-stabilized droplets containing the second precursor component. The overall microfluidic platform design for this approach is inspired by the work of Abate and coworkers on electrode-driven picoinjection,24 which has led to the application of this technique in droplet RT-PCR,25 high-throughput screening of enzyme libraries,26 and DNA sequence analysis.27 In our approach, the concept of electrocoalescence is utilized for fabricating microgels by merging two hydrogel precursor solutions. While electrocoalescence-induced solidification of fast-gelling precursor solutions is most suitable for forming isotropic microgels via covalent bonds or supramolecular interactions, we also elucidate the limitations of our approach using the example of instantaneous alginate hydrogelation via calcium-induced ionic interactions. Here, electrocoalescence of reactive hydrogel precursor solutions does not lead to the formation of morphologically isotropic microgels. Yet, this approach provides straightforward access to rather complex hydrogel structures like hollow spheres as well as armchair- and hammock-like microgels with tunable inner curvatures that have been previously realized by more complex examples of emulsion templates, e.g., selective polymerization of higher-order emulsions,28 aqueous two-phase systems,29 or precisely controlled de-wetting of emulsion templates.30
Results and discussion
Design of microfluidic device for processing fast-gelling hydrogel systems via electrocoalescence
We realize that the afore-discussed approaches to prevent uncontrolled hydrogelation inside a microfluidic device or a droplet template often merely delay these processes but do not generally rule them out. On this account, we aim for rapid addition and subsequent efficient mixing of hydrogel precursors by completely separating the reactive hydrogel precursors and only bring them together for microgel formation at desired time and location. Through the work of Baret et al.,31 it is well known that droplets can be perceived as individual closed reaction vessels if droplet stability is ensured using a surfactant below the critical micellar concentration (CMC) that significantly reduces the interfacial tension at the oil–water interface.32,33 Our approach utilizes a microfluidic device with integrated electrodes (Fig. 2A) that consists of a flow-focusing junction at which emulsion droplets of a first hydrogel precursor solution (dispersed phase 1, DP1) are formed by a continuous phase (CP) of fluorinated oil to which a surfactant is added for droplet stabilization. From there, the as-formed emulsion droplets pass through a microchannel toward another, T-shaped junction with an integrated pair of a positive and negative electrode opposite to this junction. Here, a second hydrogel precursor solution at defined flow rate (dispersed phase 2, DP2) is added. By adjusting the combined flow rates of DP1 and CP as well as DP2, pairs of droplets initially form, whose surfactant-stabilized interfaces contact each other while they continue to flow downstream through a meander-shaped droplet mixing zone (cf. below). | ||
| Fig. 2 (A) AutoCAD®-made design of microfluidic device for electrocoalescence of droplet pairs containing reactive hydrogel precursor solutions. Arrows indicate fluid flow directions. (B) Droplet formation at a flow-focusing junction from aqueous dispersed phase 1 (DP1). (C) Pairing of droplets of DP1 and DP2, the latter formed at a T-junction. (electrodes are not energized). (D) After electrode activation, DP2 merges with droplets of DP1. The electric field destabilizes the water-oil interfaces inducing coalescence between DP1 and DP2, followed by fast convection-based droplet mixing by passage through a meander-shaped microchannel. The scale bars for all panels denote 100 μm. | ||
To induce merging and thus hydrogelation between a droplet containing DP1 passing by the T-junction where DP2 is ejected, the surfactant layer at the water-oil interface of the existing droplet needs to be destabilized to overcome its interfacial energy. For that, an external electric field is generated through the pair of electrodes opposite of the T-junction to induce thin-film instability and thus electrocoalescence.34
To first characterize the addition and mixing process of hydrogel precursors during electrocoalescence in our flow cell, DP1, which is phosphate-buffered saline (PBS), and DP2, which is PBS colored with the food dye Brilliant Black, are processed. As no potential is applied to the electrodes, and the flow rates of DP1 and DP2 are equal, droplets of DP1 (cf.Fig. 2B) pair exactly with droplets of colored DP2 at the T-junction of the droplet electrocoalescence device (cf.Fig. 2C). When applying a voltage of 240 V to the electrodes, the water-oil interface destabilizes, causing DP2 to be injected into droplets of DP1 instead of forming individual droplets of DP2 (cf.Fig. 2D). When injecting DP2 into droplets of DP1, fluid mixing is enhanced tremendously by convective forces arising from the droplet merging compared to diffusion-based mixing of two fluids brought together at a droplet-forming junction in a co-flow manner.35 For additional enhancement of the mixing speed inside the as-formed droplets, a meander-shaped channel segment is integrated into the microflow cell design. In this segment, droplet mixing is completed by chaotic advection, which can be compared to the process of baker's transformation acting on these moving liquid plugs surrounded by an immiscible oil phase.36 The baker's transformation involves consecutive steps of stretching, folding, and reorientation of the fluid, giving rise to an exponential decrease in the striation length and, therefore, accelerated mixing. For the experiment in Fig. 2, the droplet electrocoalescence cell is operated at flow rates of 400 μL h−1 for CP and 30 μL h−1 for DP1 and DP2, respectively. At these flow rates, the high-speed camera image in Fig. 2D shows that a uniform mixing of the droplet content by electrocoalescence followed by meander-based advection is completed as fast as 89 ms after electrocoalescence of the droplet pair before the droplets pass the third turn of the meander-shaped channel segment.
In the following section, separate processing followed by highly efficient mixing of different aqueous phases via electrocoalescence of droplet pairs is utilized to fabricate microgels with isotropic shape, uniform size, and homogeneous interior based on examples of fast-gelling precursor systems.
Fabrication of isotropic microgels via precursor-phase coalescence based on covalent and supramolecular crosslinking
Microgels with spatial uniformity regarding porosity, mechanics as well as the distribution of functional groups are the basis for reproducible results among large populations of microgels, e.g., as sensor particles, in mechanobiology, or as platforms in cell-free biotechnology.37–39 Poly(ethylene glycol) (PEG)-based precursors are frequently utilized for hydrogel network formation since their terminal functionalities can be crosslinked by means of bio-orthogonal chemistry. Bio-orthogonal reactions can occur inside or in presence of living systems without interfering with native biochemical processes.40 This is essential for hydrogelation in presence of biological machinery, e.g., for cell-free protein synthesis,41 or encapsulation of living cells.42,43 One of these well-established reactions, enabling the formation of a hydrogel network in a bio-orthogonal fashion, is the base-catalyzed thiol-Michael addition click reaction, which is highly efficient and can be performed under mild reaction conditions. However, such reaction, e.g., between thiol-functionalized and maleimide-functionalized PEG, exhibits extremely fast reaction kinetics at physiological pH.44,45 Kharkar et al. reported on a similar multi-arm PEG-based precursor system with terminal thiol and maleimide moieties, whose gel point could not be determined by rheology due to rapid gelation.46 Employing pipetting studies for determining the gel point, Darling et al. reported complete gelation utilizing 4-arm PEGs within 2 s.47Our microfluidic preliminary tests showed that a contact time of just a few milliseconds between the thiol and the maleimide precursor solutions is sufficient to cause hydrogel formation at the interface. Due to this very fast gelation kinetics, this reaction is generally difficult to control in microfluidics to obtain uniform microgels from droplet templates. To overcome the limitations of microgel formation based on thiol-Michael addition in droplets, we employ our droplet electrocoalescence device for processing a starPEG-thiol and a starPEG-maleimide to form uniform microgels via thiol-Michael addition reaction (Fig. 3). For the experiment, starPEG-thiol (2% w/w in PBS) and starPEG-mal (2% w/w in PBS) serve as DP1 and DP2, respectively, while CP is Novec 7500 supplied with 2% (w/w) of a home-made ABA triblock copolymer surfactant. Their flow rates are set to 40 μL h−1, 40 μL h−1, and 800 μL h−1 for DP1, DP2, and CP, respectively. To evaluate the mixing efficiency of the hydrogel precursors inside the merged droplet by fluorescence wide-field and confocal microscopy, Alexa Fluor 488 C5 maleimide is used to pre-functionalize starPEG-thiol. Here, less than 0.02% of the thiol groups are modified with the fluorophore, which has no impact on the gelation process. After reaching a stable droplet-formation regime for DP1 at the first cross-junction, the flow of DP2 is initiated. The electrodes remain turned-off at this point. As droplet flow becomes synchronized between the cross-junction and the T-junction and droplet pairing is observed at the second cross-junction, a voltage of 240 V at 7.5 kHz in AC mode is applied to the electrodes. This value is chosen for all reactive hydrogel precursor solutions in this article to ensure a fast and regular electrocoalescence. Under these conditions, the merging of the droplet pairs proceeds quantitatively, and as shown by the phase-contrast image in Fig. 3B, the as-formed emulsion droplets are uniform in size with diameters of 64.5 ± 1.9 μm. The solidified emulsion content is then transferred into PBS by three consecutive washing steps with perfluorooctanol (PFO) (20% v/v in Novec 7500). Droplets translate into microgels with a similar diameter (65.8 ± 2.0 μm, inset Fig. 3B). The distribution of fluorescent dye incorporated into the hydrogel network is revealed by mid-plane images of the microgels by confocal microscopy (Fig. 3C) and corresponding line scans visualizing the fluorophore distribution within the microgel (Fig. 3D). Uniform dye distribution indicates that mixing the macromolecular hydrogel precursors employing droplet coalescence is faster than hydrogelation by thiol-Michael addition and the method thus suitable to fabricate polymer microgels in a reproducible fashion. This finding renders electrocoalescence suitable to process hydrogel precursors forming a covalently crosslinked network via base-catalyzed thiol-Michael addition reaction.
 | ||
| Fig. 3 Microgel formation from reactive species using the example of thiol-Michael addition. (A) Schematic of hydrogel network formation. (B) Phase-contrast microscopy image of emulsion droplets with gelled content made from starPEG-thiol and starPEG-maleimide. The inset displays a wide-field fluorescence microscopy image of uniform microgels with isotropic shape. (C) Confocal microscopy image of a microgel revealing the homogeneous distribution of Alexa Fluor 488 covalently coupled to starPEG-thiol prior to gelation. (D) Localization of Alexa Fluor 488 inside of the microgel shown in (C) visualized through a line scan. The scale bars for all panels denote 100 μm. | ||
To extend the applicability of our approach beyond the formation of covalently crosslinked microgels, we apply droplet electrocoalescence for microgel formation based on ultra-fast supramolecular crosslinking through non-covalent interactions. We have previously established a non-covalent hydrogel system formed by mixing sulfated polysaccharide and peptide-starPEG conjugate (Fig. 4A).48 The peptide has a minimal motif of (KA)n (K is lysine, A is alanine, and n is the number of repetition). The self-assembly and gelation process is mediated by the binding of (KA)n peptide to the sulfated polymer, which can be used for drug delivery,49 3D cell cultures,50 as well as forming cell-laden hydrogel microparticles.51 The gelation can be remarkably accelerated when lysine is replaced by arginine,48 or a lipid chain is added to (KA)n. The very fast gelation of such systems prevents efficient mixing of the two precursor solutions, and thus hampers broader applicability. As for the covalent hydrogel system discussed above, it was not possible to carry out rheological measurements for determining the gelation point of the non-covalently assembled (KA)n hydrogel system due to the intrinsic speed of gelation. Fig. 4A shows a sketch of the underlying hydrogelation between an octanoic acid-modified (KA)7-starPEG conjugate (KA7-C8-starPEG) and dextran sulfate.
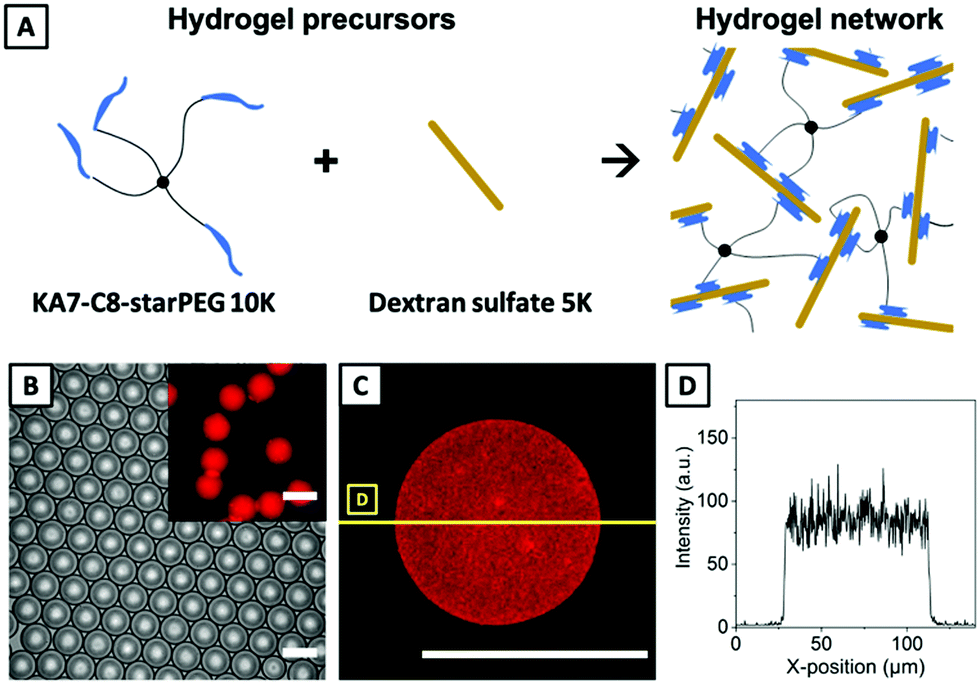 | ||
| Fig. 4 (A) Reaction scheme of hydrogel network formation via supramolecular, non-covalent interaction. (B) Phase-contrast microscopy image of emulsion droplets containing resulting microgels. The inset displays the corresponding fluorescence microscopy image visualizing fluorescent KA7-TAMRA-starPEG incorporated into the hydrogel network. (C) Exemplary confocal microscopy image of a supramolecularly crosslinked microgel revealing a homogeneous distribution of fluorescent TAMRA. (D) Localization of TAMRA inside of the microgel shown in (C) visualized through a line scan. The scale bars for all panels denote 100 μm. | ||
For microfluidic processing, a solution of KA7-C8-starPEG (2.5 mM in DI water) is injected as DP1 into the droplet electrocoalescence device, while DP2 is a solution of 5 mM dextran sulfate in DI water. For evaluating the mixing efficiency between these precursor phases after droplet merging, the KA7-C8-starPEG solution is doped with fluorescent 5-carboxytetramethylrhodamine (TAMRA) labeled KA7-TAMRA-starPEG (1% v/v). Flow rates are set to 80 μL h−1 for DP1, 80 μL h−1 for DP2, and 600 μL h−1 for CP, which is again Novec 7500 supplied with 2% (w/w) of ABA copolymer surfactant. After achieving stable pairing of droplets of DP1 and DP2 at the microchannel T-junction, a potential of 240 V at 7.5 kHz in AC mode is applied to the electrodes. As shown by phase-contrast microscopy (Fig. 4B), emulsion droplets containing as-formed non-covalently crosslinked microgels exhibit a low dispersity with an average diameter of 84.8 ± 1.7 μm. After demulsification by washing with PFO (20% v/v in Novec 7500), the microgels – now in DI water – retain their uniformity with a 1.1-fold increase in diameter and a 1.4-fold increase in volume (95.7 ± 2.3 μm, inset Fig. 4B). We previously reported similar swelling characteristics of polysaccharide-based microgels during droplet-to-microgel transition for hydrogel systems based on hyaluronic acid, chitosan, and alginate.16,39 The cross-section through the mid-plane of a non-covalent microgel (Fig. 4C), imaged by confocal microscopy, as well as the corresponding line scans visualizing the fluorophore distribution within the microgel (Fig. 4D), confirm the uniform distribution of the incorporated KA7-TAMRA-starPEG building block in the hydrogel network, indicating that droplet mixing is faster than the supramolecular assembly process.
The formation of uniform microgels via covalent crosslinking of PEG-based precursors by thiol-Michael addition as well as the supramolecular crosslinking of dextran sulfate and peptide–lipid-starPEG show that separate processing of hydrogel precursors into individual droplet populations followed by their pairing and electric field-induced merging is well-suited for engineering fast-gelling hydrogel systems into microgels with low dispersity and homogeneous distribution of functional moieties. However, hydrogel network formation via ionic interactions between alginate and calcium ions can be considered instantaneous,20 proceeding even faster than the two hydrogel systems used above. Therefore, the mixing of the droplet content upon electrocoalescence will still be slower than gelation of the droplet content. Still, in these cases, the process of droplet electrocoalescence can be utilized to form distinct anisotropic rather than isotropic microgels via crosslinking based on ionic interactions under well-controlled conditions, as discussed next.
Fabrication of anisotropic microgels via ionic interactions
For fabricating anisotropic, alginate-based microgels, a solution of calcium chloride is injected into droplets of sodium alginate by electrocoalescence. Due to instantaneous solidification, uniform, isotropic microgels are only yielded by utilizing chelating agents (cf. below). Instead, to exert control over their anisotropy, we lock the sodium alginate microgels in shape depending on the volume of calcium chloride solution injected into the existing sodium alginate droplet. For that, DP1 is made of sodium alginate (2% w/w), and DP2 is a solution of calcium chloride (6% w/w), both in DI water (Fig. 5, top). In the microfluidic device design, the meander-like segment for droplet mixing by convective forces is removed, as preliminary experiments have not shown to improve the isotropy of the alginate gels. To visualize the microgel shape by confocal microscopy, fluorescein isothiocyanate (FITC)-dextran with a molecular weight of 2 M Da is added to the sodium alginate phase and entrapped in the microgel during hydrogel network formation. The flow rates of DP1 and CP are fixed at 40 μL h−1 and 400 μL h−1, respectively. The flow rate of DP2 is increased in a stepwise fashion starting from 40 μL h−1, over 60 μL h−1 and 80 μL h−1 to 120 μL h−1 to evaluate the impact of the injected volume on the shape and curvature of yielded microgels. A voltage of 240 V at 7.5 kHz in AC mode is applied to the electrodes to induce electrocoalescence. | ||
| Fig. 5 Fabrication of alginate microgels with tailored curvature. Top: AutoCAD®-made design of droplet electrocoalescence device, highlighting the inflow of sodium alginate and calcium chloride. Yellow and green boxes mark the position of microscopy images taken below. Bottom: Electrocoalescence of a solution of calcium chloride in DI water (6% w/w) with droplets formed upstream at a flow-focusing junction containing a solution of sodium alginate in DI water (2% w/w). Instantaneous calcium-induced ionic crosslinking results in hydrogelation directly at the interface of both solutions, whereas the remaining alginate solidifies under geometric control of the droplet template by diffusion. The flow rate of DP1 containing sodium alginate is fixed at 40 μL h−1, DP2 containing calcium chloride is increased continuously from 40 μL h−1 (A) over 60 μL h−1 (B) to 80 μL h−1 (C), and 120 μL h−1 (D). The flow rate of CP is set to 400 μL h−1 in all experiments. The scale bars denote 50 μm. | ||
In the lower part of Fig. 5, time-resolved bright-field microscopy images of the electrocoalescence process of the calcium chloride solution and droplets containing sodium alginate and FITC-dextran are recorded with a high-speed camera. For all flow rates of DP2, a stable interface forms between the sodium alginate droplets and the injected calcium chloride solution, being 2 ms into the coalescence process. We interpret this finding as an instantaneous hydrogel formation at the interface of both solutions due to very fast egg-box-like bridging of the guluronate blocks of the alginate by the divalent calcium ions (cf.Fig. 6), since the observed interface formation is not visible in case of injecting hydrogel precursors that do not gel instantaneously as the hydrogel systems reported in the previous section, for instance. From images taken 5 ms and 12 ms into the coalescence, whereas the latter time point marks the completion of calcium chloride injection, the impact of the injected volume on the droplet and microgel shape can be derived. The comparison of high-speed camera images at 12 ms and corresponding insets of 3D reconstructions from confocal microscopy reveals that the injected volume of calcium chloride determines the final shape of the anisotropic microgel.
 | ||
| Fig. 6 Top: Scheme of Ca2+-based ionic crosslinking of alginate by egg-box-like bridging of guluronate blocks leading to hydrogel network formation. Middle and bottom row: Anisotropic alginate microgels imaged by confocal microscopy. Microgels are formed by electrocoalescence from a solution of calcium chloride in DI water (6% w/w) with droplets containing a solution of sodium alginate in DI water (2% w/w), formed upstream at a flow-focusing junction. Middle: 3D reconstructions as acquired and (bottom) reoriented for simplified comparison of the varying inner curvature between different microgel species. The microgels have been fabricated, as shown in Fig. 5A–D. All scale bars denote 50 μm. | ||
While for all above-discussed hydrogel systems, we can derive the mean diameters and standard deviation of droplets and microgel templates, this is not feasible for emulsion droplets serving as templates for the anisotropic alginate microgels. This is because high salt concentrations lower the solubility of PEG in water, which is the middle block of our triblock copolymer surfactant employed for droplet stabilization.52,53 By lowering its solubility, the effectiveness of the surfactant is reduced, leading to droplet coalescence, and the observed multiple microgels per droplet (ESI,† Fig. S2).
To determine the shape control and inner curvature of the anisotropic alginate microgels by the volume of calcium chloride solution merged with the sodium alginate droplets, we perform confocal microscopy. 3D reconstructions as-acquired (middle) and reoriented for better comparison of the inner curvature (bottom) are shown in Fig. 6. It can be derived that the radius of the inward microgel curvature increases with an increasing flow rate of DP2 and, therefore, increasing injection volume. While for a DP2 flow rate of 40 μL h−1, microgels with small inner cavities are formed, comparable to those which can be fabricated via polymerization of higher-order emulsion templates or aqueous two-phase systems,28,29 concave surfaces with increasing radii are created by a stepwise increase of the flow rate of DP2 up to 120 μL h−1. Here, microgels formed by electrocoalescence with a flow rate of DP2 at 60 μL h−1 exhibit an armchair-like morphology, and microgels formed from DP2 at 120 μL h−1 expose a hammock-like morphology, while at 80 μL h−1, an intermediate architecture of the two aforementioned geometries is obtained. Since the inner curvature of these microgels are not evenly curved due to the fabrication process, no statistically significant quantitative data could be extracted from the 3D reconstructions by using image analysis software, e.g., ImageJ.
To describe and compare the shapes of the anisotropic alginate microgels in more detail, a coordinate system is inserted in Fig. 6 (bottom) to define the directions in space. Here, the diameter of the alginate-based microgels along the x-axis remains at comparable values of approx. 35 μm. This value is not solely determined by the height of the microfluidic channel of 50 μm but also by the spatial location of sodium alginate within the droplet that does not fill out the channel completely due to its curvature. At a process time of 12 ms (cf.Fig. 5), the anisotropic microgels are located mostly in the upper third of the droplet volume along the axis perpendicular to the flow direction. Considering the cylindrical shape of the droplets along the axis of the microfluidic channel and the Marangoni force acting on the droplets due to friction at the channel walls,54 the deviation of the microgel diameter from the total channel height can be explained. While the width of the microgels along the x-axis stays constant, the length of the microgels along the z-axis is prolonged stepwise with increasing injected volume of DP2 from approx. 60 μm at 40 μL h−1, over 62 μm at 60 μL h−1 and 70 μm at 80 μL h−1 to 80 μm at 120 μL h−1, accompanied by increasing radii of the inner curvature and increasing flattening of the overall microgel.
The comparison of the 3D reconstructions in Fig. 6 reveals the variety of microgel populations with different inner curvature that can be generated in one single microfluidic experiment by simply adjusting the flow rate of DP2, and thus the volume that is added to the existing droplet of DP1.
Besides processing sodium alginate into anisotropic microgels with various shapes, our electrocoalescence device can also be employed for fabricating isotropic alginate particles by utilizing EDTA as a chelating agent for calcium ions. Bright-field, fluorescence microscopy, and confocal microscopy images of these as well as the method for fabricating these can be found in the ESI† (ESI† Fig. S3). Alginate-based hydrogels crosslinked via ionic interaction with divalent cations such as Ca2+ find numerous applications in biomedical sciences and engineering due to its favorable properties like biocompatibility, low toxicity, porosity, and degradability.55,56 Especially for mammalian cells, alginate gels can serve as continuous 3D culture systems capable of capturing the complex physiological responses of tissue in vitro.57 However, less is known about how cells interact with cultures that exhibit micron-scale curvature. Therefore, fabricated anisotropic alginate-based microgels with tunable shape and surface curvature could be a suitable experimental platform as it was reported that substrate topography and curvature on the micron-scale affect cell fate as well as cell–cell interactions.58 Microgels fabricated by electrocoalescence of droplet pairs that combine convex as well as concave curvatures in the same microgel particle could enable to evaluate the favored curvature and the accompanied cell fate in one single-cell culturing experiment.
Materials and methods
Materials for microgel synthesis
All chemicals were used without further purification unless stated otherwise. 4-Arm poly(ethylene glycol)-thiol (starPEG-thiol) with a molecular weight of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and 4-arm poly(ethylene glycol)-maleimide (starPEG-maleimide) with 10000 g mol−1 were purchased from Creative PEGWorks (USA) and Jenkem (China). Alexa Fluor 488 C5 maleimide was purchased from Thermo Fisher Scientific (USA). Alginate sodium salt, Brilliant Black, calcium chloride, phosphate-buffered saline (PBS) tablets, dextran sulfate sodium salt with a molecular weight of 5000 g mol−1, and fluorescein isothiocyanate-(FITC-)dextran with an average molecular weight of 2000000 g mol−1 were purchased from Sigma Aldrich/Merck (Germany). Peptide–lipid-starPEGs were synthesized in the Zhang lab (B CUBE, Technische Universität Dresden). Deionized (DI) water was obtained from a Milli-Q water purifier (Direct-Q® 5, Merck Millipore, USA) with a resistance of 18.2 MΩ cm.
000 g mol−1 and 4-arm poly(ethylene glycol)-maleimide (starPEG-maleimide) with 10000 g mol−1 were purchased from Creative PEGWorks (USA) and Jenkem (China). Alexa Fluor 488 C5 maleimide was purchased from Thermo Fisher Scientific (USA). Alginate sodium salt, Brilliant Black, calcium chloride, phosphate-buffered saline (PBS) tablets, dextran sulfate sodium salt with a molecular weight of 5000 g mol−1, and fluorescein isothiocyanate-(FITC-)dextran with an average molecular weight of 2000000 g mol−1 were purchased from Sigma Aldrich/Merck (Germany). Peptide–lipid-starPEGs were synthesized in the Zhang lab (B CUBE, Technische Universität Dresden). Deionized (DI) water was obtained from a Milli-Q water purifier (Direct-Q® 5, Merck Millipore, USA) with a resistance of 18.2 MΩ cm.
Microfluidic device fabrication and setup design
Microflow cells for droplet electrocoalescence were fabricated by standard photo- and soft lithography techniques, followed by subsequent creation of electrodes within these flow cells. A microchannel template, 50 μm in height, was generated by spin-coating the negative photoresist SU-8 2050 (Micro Resist Technology, Germany) onto the polished surface of a 3-inch silicon wafer (Siegert Wafer, Germany). The channel structure was defined by a printed film mask using a mask aligner (MJB3, Suess MicroTec, Germany). After post-exposure bake, the non-hardened photoresist was removed with a developer (mr-Dev 600, Micro Resist Technology, Germany). To confirm the desired microchannel height, the master device was characterized by confocal microscopy (μsurf expert, NanoFocus, Germany). Then, the silicon wafer-based microchannel structure was replicated in poly(dimethylsiloxane) (PDMS) by replica molding. For that, PDMS oligomer and crosslinker (Sylgard 184 silicone elastomer kit, Dow Corning, USA) were mixed in a ratio of 10:1 (w/w), degassed in a planetary centrifugal mixer (ARE-250, Thinky, USA), poured onto the master device and crosslinked in an oven for 2 h at 65 °C. After that, the PDMS replica was cut out of the mold, peeled off from the master, and the inflow and outflow ports were punched with a 1 mm-diameter biopsy punch (KAI Medical, Germany). The side of the PDMS replica exhibiting the microchannel structure was bonded to a microscopy glass slide (76 × 52 mm, Marienfeld, Germany) by exposing both parts to an oxygen plasma (80 W, 15 s, MiniFlecto 10, Plasma Technology, Germany), before gently pressing them together. Electrodes were integrated into the microflow cell by filling selected microchannels with a low-melting-point solder (Indalloy 19, Indium Corporation, USA). To ensure both sufficient wetting of the liquid alloy and uniform filling of the channels, the empty electrode microchannels were pre-treated with a 2% (v/v) solution of (3-mercaptopropyl)trimethoxysilane in acetonitrile (both Sigma Aldrich/Merck, Germany). To fully remove excess solution from the surface treatment, air was blown through the microchannels before placing the device on a hot plate at 85 °C. Then, Indalloy 19 solder (diameter 0.6 mm, 52% In, 32.5% Bi, 16.5% Sn) was injected into the electrode microchannels and allowed to melt. Once the solder filled out a microchannel completely, including the outflow port, pin headers were inserted into the inflow ports to provide an external electrical connection. Electrical contact was verified, measuring the electrical conductivity between the pin header and the alloy at the outflow port of the electrodes by utilizing a digital multimeter. The pin headers were additionally fixed on the flow cell surface by applying Loctite AA 350 (Henkel Adhesives, Germany), and exposing the microfluidic device to UV light for 5 min. Then, 15 min before using the microfluidic device, the surface of all non-electrode microchannels was rendered hydrophobic by injecting a 1% (v/v) solution of (tridecafluoro-1,1,2,2-tetrahydrooctyl)trichlorosilane (Gelest, USA) in Novec 7500 (IoLiTec, Germany) into these microchannels.
To visualize emulsion formation, the flow cell was mounted onto an inverted bright-field microscope (Axio Vert.A1, Carl Zeiss, Germany) equipped with a high-speed digital camera (Miro eX4, Vision Research Inc., USA). Three high-precision syringe pumps (Harvard Apparatus 11 Pico Plus Elite, Harvard Apparatus, USA) were connected to the flow cell via PEG tubing (inner diameter: 0.38 mm, outer diameter: 1.09 mm, Hartenstein, Germany). Two 500 μL-gastight syringes (1750 TLL SYR, Hamilton, USA) were used for the dispersed phases (DPs), DP1 and DP2, and a 3 mL-disposable syringe (BD Luer-Lok tip, Becton Dickinson, USA) was used for the continuous phase (CP). The electrodes were operated by a custom-built power supply with tunable voltage, current, and frequency in AC mode. The entire microfluidic setup was located in a grounded box with a security flap and additionally equipped with two safety switches to comply with general safety regulations for working with high voltage.
General procedure of microgel fabrication
In all microfluidic experiments, a stream of aqueous hydrogel precursor solutions also referred to as DPs, was periodically emulsified by a flow of a second immiscible fluid, regarded as the CP (cf.Fig. 2). While the composition of the DPs varied for different hydrogel systems, the composition of the CP was the same for all experiments, consisting of a home-made triblock copolymer surfactant (Krytox-Jeffamine-Krytox, 2% w/w), synthesized based on a protocol from Holtze et al.,59 dissolved in Novec 7500. For each experiment, both dispersed phases were injected into the flow cell at the same time. Here, droplets of DP1 were formed at a flow-focusing junction, into which the DP2 was added by means of electrocoalescence at a second T-junction downstream of the first junction. The distance between the flow-focusing and the T-junction was 2.8 mm in all experiments. The emulsion resulting from the electrocoalescence of droplet pairs was collected at intervals of 10 min in Eppendorf tubes. Afterward, excess CP was removed with a syringe from the collection vial, and 300 μL of the medium in which the microgels were synthesized was added. These were PBS for covalently crosslinked microgels and DI water for supramolecularly as well as ionically crosslinked microgels. Next, the microgels were transferred into water by breaking the emulsion via the addition of 250 μL Novec 7500 containing 20% (v/v) 1H,1H,2H,2H-perfluorooctanol (PFO, Sigma Aldrich/Merck, USA). This step was repeated twice.Bright-field and wide-field fluorescence microscopy imaging of droplets and microgels was conducted on a DMi8 microscope (Leica, Germany). Confocal microscopy imaging of microgels was performed on an Andor Dragonfly spinning disc confocal microscope (Andor Technology Ltd, Ireland) or on a TCS SP5 (Leica, Germany). For evaluating the initial droplet size and the corresponding size of isotropic microgels produced by thiol-Michael addition or non-covalent interactions, at least 60 droplets and microgels were characterized manually using the software ImageJ.60 From this data, mean diameters and standard deviations were calculated.
Microfluidic fabrication of isotropic covalently assembled microgels
For fabricating isotropic covalently assembled microgels via thiol-Michael addition, a 2% (w/w) solution of starPEG-thiol (MW: 10000 g mol−1) in 1× PBS was used as DP1, and DP2 was a 2% (w/w) solution of starPEG-maleimide (MW: 10000 g mol−1) in 1× PBS. For visualizing the mixing efficiency inside droplets, the starPEG-thiol of DP1 was pre-functionalized with Alexa Fluor 488 C5 maleimide. After forming droplets of DP1 at the flow-focusing junction, DP2 was added into droplets of DP1 right at the point of droplet formation, applying an electrode voltage of 240 V at 7.5 kHz in AC mode. Flow rates were 40 μL h−1 for DP1, 40 μL h−1 for DP2, and 800 μL h−1 for the CP.
Microfluidic fabrication of isotropic supramolecularly assembled microgels
For fabricating isotropic, non-covalently assembled microgels via supramolecular interactions, a solution of 2.5 mM peptide–lipid-starPEG (KA7-C8-starPEG) in DI water was used as DP1, while DP2 was a solution of 5 mM dextran sulfate (MW: 5000 g mol−1) in DI water. For visualizing the mixing efficiency inside droplets, peptide–lipid-starPEG was doped with 1% (v/v) fluorescent dye 5-carboxytetramethylrhodamine labeled KA7-TAMRA-starPEG. After forming droplets of DP1 at the flow-focusing junction, DP2 was added into droplets of DP1, applying an electrode voltage of 240 V at 7.5 kHz in AC mode. Flow rates were 80 μL h−1 for DP1, 80 μL h−1 for DP2, and 600 μL h−1 for the CP.Microfluidic fabrication of anisotropic ionically assembled microgels
For fabricating anisotropic alginate-based microgels via ionic interactions, DP1 was a 2% (w/w) solution of sodium alginate and DP2 a 6% (w/w) solution of calcium chloride, both in DI water. For visualizing the resulting microgel shape by confocal microscopy, 0.2% (w/w) FITC-dextran (MW: 2000000 g mol−1) was added to DP1. After forming droplets of DP1 at the flow-focusing junction, DP2 was added into droplets of DP1, applying an electrode voltage of 240 V at 7.5 kHz in AC mode. Flow rates for forming different populations of alginate-based microgels with varying shape are summarized in Table 1.
| Ratio DP1/DP2 | Flow rate DP1 [μL h−1] | Flow rate DP2 [μL h−1] | Flow rate CP [μL h−1] |
|---|---|---|---|
| 1:1 |
40 | 40 | 400 |
| 1:1.5 |
40 | 60 | 400 |
| 1:2 |
40 | 80 | 400 |
| 1:3 |
40 | 120 | 400 |
Conclusions
Controlled electrocoalescence of droplet pairs containing fast-gelling hydrogel precursors was utilized as a versatile method for processing reactive species into uniform microgels via droplet microfluidics. To highlight that successful droplet coalescence toward uniform microgels largely depends on the speed of gelation rather than on the crosslinking mechanism of hydrogel formation, covalently crosslinked microgels were fabricated via thiol-Michael addition reaction of thiols and maleimides, and supramolecularly crosslinked microgels were fabricated via non-covalent assembly of dextran sulfate and peptide–lipid-starPEG. Both types of hydrogels yielded the desired isotropic microgels with high spatial uniformity of the microgel volume, as indicated by the distribution of fluorescent dye coupled to the hydrogel matrices. By utilizing 3D confocal microscopy scanning through the mid-plane of both types of covalently or non-covalently crosslinked hydrogels, a homogeneous distribution of incorporated dyes throughout the hydrogel volume was confirmed. Homogeneous hydrogels obtained from fast-gelling precursors, particularly those reacting via bio-orthogonal crosslinking based on click-chemistry approaches (e.g., thiol-Michael addition), may further promote their application in cell biology, e.g., in mechanobiology,61,62 where homogeneous hydrogel microenvironments are essential for reproducible force sensing and for comparing results across characterization techniques like real-time deformability cytometry or AFM-based nanoindentation.38Beyond the fabrication of well-defined isotropic microgels, the addition of one reactive hydrogel precursor to a surfactant-stabilized droplet of its corresponding reaction partner, e.g., crosslinking component, was shown to be suitable for producing a special type of anisotropic microgels with a tunable inner and outer curvature that – compared to conventional core–shell or Janus-type microgels – has been challenging to form via standard droplet microfluidics to date. Exemplarily, instantaneous gelation of alginate and calcium ions – exceeding the speed of droplet homogenization prior to droplet gelation – was utilized to lock the microgel shape determined by the injected volume of calcium ion solution to a preexisting droplet template. It was shown that a stepwise increase of the calcium chloride volume injected into the alginate droplet results in an increasing inner curvature of as-formed microgels, such that this curvature can be customized for specific future applications, e.g., in cell culturing experiments to investigate a favorable substrate curvature influencing cell fate.63
Author contributions
N. H., T. A. N., P. L., Y. Z., and J. T. conceived and designed the experiments; N. H., T. A. N., L. V., and P. L performed the experiments and analyzed the data; M. J. M. designed the droplet electrocoalescence device; E. S. built the customized power supply; N. H. and J. T. wrote the paper; all authors reviewed the final version of the submitted manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Federal Ministry of Education and Research (BMBF, Biotechnology2020+: Leibniz Research Cluster, 031A360C), the Volkswagen Foundation (“Experiment!”), and the German Research Foundation (DFG, Research Training Group 1865: Hydrogel-based Microsystems). This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Grant agreement No. 852065). J. T. also acknowledges the Young Investigator Program of TU Dresden. Y. Z. receives funding from the German Federal Ministry of Research and Education (BMBF grants 03Z22E511). N. H. thanks Thomas Heida and Niclas Weigel for providing surfactants, and Martin Schumann for drawing the TOC figure.Notes and references
- J. V. Alemán, A. V. Chadwick, J. He, M. Hess, K. Horie, R. G. Jones, P. Kratochvíl, I. Meisel, I. Mita, G. Moad, S. Penczek and R. F. T. Stepto, Pure Appl. Chem., 2007, 79, 1801–1829 Search PubMed.
- Q. Xu, M. Hashimoto, T. T. Dang, T. Hoare, D. S. Kohane, G. M. Whitesides, R. Langer and D. G. Anderson, Small, 2009, 5, 1575–1581 CrossRef CAS PubMed.
- L. P. B. Guerzoni, J. Bohl, A. Jans, J. C. Rose, J. Koehler, A. J. C. Kuehne and L. De Laporte, Biomater. Sci., 2017, 5, 1549–1557 RSC.
- S. Verbruggen, D. Luining, A. van Essen and M. J. Post, Cytotechnology, 2018, 70, 503–512 CrossRef CAS PubMed.
- L. Zhang, K. Chen, H. Zhang, B. Pang, C.-H. Choi, A. S. Mao, H. Liao, S. Utech, D. J. Mooney, H. Wang and D. A. Weitz, Small, 2018, 14, 1702955 CrossRef PubMed.
- C. Kantak, Q. Zhu, S. Beyer, T. Bansal and D. Trau, Biomicrofluidics, 2012, 6, 022006 CrossRef PubMed.
- N. Visaveliya, S. Lenke and J. M. Köhler, ACS Appl. Mater. Interfaces, 2015, 7, 10742–10754 CrossRef CAS PubMed.
- F. D. Lorenzo and S. Seiffert, Macromol. React. Eng., 2016, 10, 201–205 CrossRef.
- H. C. Shum, A. R. Abate, D. Lee, A. R. Studart, B. Wang, C.-H. Chen, J. Thiele, R. K. Shah, A. Krummel and D. A. Weitz, Macromol. Rapid Commun., 2010, 31, 108–118 CrossRef CAS PubMed.
- S. Xu, Z. Nie, M. Seo, P. Lewis, E. Kumacheva, H. A. Stone, P. Garstecki, D. B. Weibel, I. Gitlin and G. M. Whitesides, Angew. Chem., Int. Ed., 2005, 44, 724–728 CrossRef CAS PubMed.
- M. Chau, M. Abolhasani, H. Thérien-Aubin, Y. Li, Y. Wang, D. Velasco, E. Tumarkin, A. Ramachandran and E. Kumacheva, Biomacromolecules, 2014, 15, 2419–2425 CrossRef CAS PubMed.
- T. Heida, J. W. Neubauer, M. Seuss, N. Hauck, J. Thiele and A. Fery, Macromol. Chem. Phys., 2017, 218, 1600418 CrossRef.
- S. Seiffert, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 435–449 CrossRef CAS.
- Y. Hou, W. Xie, K. Achazi, J. L. Cuellar-Camacho, M. F. Melzig, W. Chen and R. Haag, Acta Biomater., 2018, 77, 28–37 CrossRef CAS PubMed.
- G. A. Foster, D. M. Headen, C. González-García, M. Salmerón-Sánchez, H. Shirwan and A. J. García, Biomaterials, 2017, 113, 170–175 CrossRef CAS PubMed.
- N. Hauck, N. Seixas, S. P. Centeno, R. Schlüßler, G. Cojoc, P. Müller, J. Guck, D. Wöll, L. A. Wessjohann and J. Thiele, Polymers, 2018, 10, 1055 CrossRef PubMed.
- S. Allazetta, T. C. Hausherr and M. P. Lutolf, Biomacromolecules, 2013, 14, 1122–1131 CrossRef CAS PubMed.
- J. W. Neubauer, N. Hauck, M. J. Männel, M. Seuss, A. Fery and J. Thiele, ACS Appl. Mater. Interfaces, 2019, 11, 26307–26313 CrossRef CAS PubMed.
- C. B. Rodell, J. W. MacArthur, S. M. Dorsey, R. J. Wade, L. L. Wang, Y. J. Woo and J. A. Burdick, Adv. Funct. Mater., 2015, 25, 636–644 CrossRef CAS PubMed.
- A. G. Håti, D. C. Bassett, J. M. Ribe, P. Sikorski, D. A. Weitz and B. T. Stokke, Lab Chip, 2016, 16, 3718–3727 RSC.
- H. Song, J. D. Tice and R. F. Ismagilov, Angew. Chem., Int. Ed., 2003, 768–772 CrossRef CAS PubMed.
- L. Jiang, Y. Zeng, H. Zhou, J. Y. Qu and S. Yao, Biomicrofluidics, 2012, 6, 012810 CrossRef PubMed.
- D. Steinhilber, T. Rossow, S. Wedepohl, F. Paulus, S. Seiffert and R. Haag, Angew. Chem., Int. Ed., 2013, 52, 13538–13543 CrossRef CAS PubMed.
- A. R. Abate, T. Hung, P. Mary, J. J. Agresti and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19163–19166 CrossRef CAS PubMed.
- D. J. Eastburn, A. Sciambi and A. R. Abate, PLoS One, 2013, 8, e62961 CrossRef CAS PubMed.
- T. Beneyton, F. Coldren, J.-C. Baret, A. D. Griffiths and V. Taly, Analyst, 2014, 139, 3314–3323 RSC.
- A. R. Abate, T. Hung, R. A. Sperling, P. Mary, A. Rotem, J. J. Agresti, M. A. Weiner and D. A. Weitz, Lab Chip, 2013, 13, 4864 RSC.
- Z. Nie, S. Xu, M. Seo, P. C. Lewis and E. Kumacheva, J. Am. Chem. Soc., 2005, 127, 8058–8063 CrossRef CAS PubMed.
- S. Ma, J. Thiele, X. Liu, Y. Bai, C. Abell and W. T. S. Huck, Small, 2012, 8, 2356–2360 CrossRef CAS PubMed.
- C.-H. Choi, D. A. Weitz and C.-S. Lee, Adv. Mater., 2013, 25, 2536–2541 CrossRef CAS PubMed.
- J.-C. Baret, Lab Chip, 2012, 12, 422–433 RSC.
- M. S. Chowdhury, W. Zheng, S. Kumari, J. Heyman, X. Zhang, P. Dey, D. A. Weitz and R. Haag, Nat. Commun., 2019, 10, 4546 CrossRef PubMed.
- O. Wagner, J. Thiele, M. Weinhart, L. Mazutis, D. A. Weitz, W. T. S. Huck and R. Haag, Lab Chip, 2016, 16, 65–69 RSC.
- C. Priest, S. Herminghaus and R. Seemann, Appl. Phys. Lett., 2006, 89, 134101 CrossRef.
- L. Frenz, A. El Harrak, M. Pauly, S. Bégin-Colin, A. D. Griffiths and J.-C. Baret, Angew. Chem., Int. Ed., 2008, 47, 6817–6820 CrossRef CAS PubMed.
- H. Song, M. R. Bringer, J. D. Tice, C. J. Gerdts and R. F. Ismagilov, Appl. Phys. Lett., 2003, 83, 4664–4666 CrossRef CAS PubMed.
- K. Jiang, P. C. Thomas, S. P. Forry, D. L. DeVoe and S. R. Raghavan, Soft Matter, 2012, 8, 923–926 RSC.
- S. Girardo, N. Träber, K. Wagner, G. Cojoc, C. Herold, R. Goswami, R. Schlüßler, S. Abuhattum, A. Taubenberger, F. Reichel, D. Mokbel, M. Herbig, M. Schürmann, P. Müller, T. Heida, A. Jacobi, E. Ulbricht, J. Thiele, C. Werner and J. Guck, J. Mater. Chem. B, 2018, 6, 6245–6261 RSC.
- T. Heida, T. Köhler, A. Kaufmann, M. J. Männel and J. Thiele, ChemSystemsChem, 2020, 2, e1900058 CAS.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- J. Thiele, Y. Ma, D. Foschepoth, M. M. Hansen, C. Steffen, H. A. Heus and W. T. S. Huck, Lab Chip, 2014, 14, 2651–2656 RSC.
- L. P. B. Guerzoni, J. C. Rose, D. B. Gehlen, A. Jans, T. Haraszti, M. Wessling, A. J. C. Kuehne and L. D. Laporte, Small, 2019, 15, 1900692 CrossRef PubMed.
- Y. Ma, M. P. Neubauer, J. Thiele, A. Fery and W. T. S. Huck, Biomater. Sci., 2014, 2, 1661–1671 RSC.
- L. E. Jansen, L. J. Negrón-Piñeiro, S. Galarza and S. R. Peyton, Acta Biomater., 2018, 70, 120–128 CrossRef CAS PubMed.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- P. M. Kharkar, K. L. Kiick and A. M. Kloxin, Polym. Chem., 2015, 6, 5565–5574 RSC.
- N. J. Darling, Y.-S. Hung, S. Sharma and T. Segura, Biomaterials, 2016, 101, 199–206 CrossRef CAS PubMed.
- R. Wieduwild, M. Tsurkan, K. Chwalek, P. Murawala, M. Nowak, U. Freudenberg, C. Neinhuis, C. Werner and Y. Zhang, J. Am. Chem. Soc., 2013, 135, 2919–2922 CrossRef CAS PubMed.
- R. Wieduwild, W. Lin, A. Boden, K. Kretschmer and Y. Zhang, Biomacromolecules, 2014, 15, 2058–2066 CrossRef CAS PubMed.
- R. Wieduwild, R. Wetzel, D. Husman, S. Bauer, I. El-Sayed, S. Duin, P. Murawala, A. K. Thomas, M. Wobus, M. Bornhäuser and Y. Zhang, Adv. Mater., 2018, 30, 1706100 CrossRef PubMed.
- R. Wieduwild, S. Krishnan, K. Chwalek, A. Boden, M. Nowak, D. Drechsel, C. Werner and Y. Zhang, Angew. Chem., 2015, 127, 4034–4038 CrossRef.
- P. Alexandridis and J. F. Holzwarth, Langmuir, 1997, 13, 6074–6082 CrossRef CAS.
- G. Etienne, M. Kessler and E. Amstad, Macromol. Chem. Phys., 2017, 218, 1600365 CrossRef.
- A. Karbalaei, R. Kumar and H. J. Cho, Micromachines, 2016, 7, 13 CrossRef PubMed.
- K. Y. Lee and D. J. Mooney, Prog. Polym. Sci., 2012, 37, 106–126 CrossRef CAS PubMed.
- N. Huebsch, C. J. Kearney, X. Zhao, J. Kim, C. A. Cezar, Z. Suo and D. J. Mooney, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9762–9767 CrossRef CAS PubMed.
- T. Andersen, P. Auk-Emblem and M. Dornish, Microarrays, 2015, 4, 133–161 CrossRef CAS PubMed.
- G. Altay, S. Tosi, M. García-Díaz and E. Martínez, Front. Bioeng. Biotechnol. DOI:10.3389/fbioe.2020.00294.
- C. Holtze, A. C. Rowat, J. J. Agresti, J. B. Hutchison, F. E. Angilè, C. H. J. Schmitz, S. Köster, H. Duan, K. J. Humphry, R. A. Scanga, J. S. Johnson, D. Pisignano and D. A. Weitz, Lab Chip, 2008, 8, 1632–1639 RSC.
- C. T. Rueden, J. Schindelin, M. C. Hiner, B. E. DeZonia, A. E. Walter, E. T. Arena and K. W. Eliceiri, BMC Bioinf., 2017, 18, 529 CrossRef PubMed.
- K. Wagner, S. Girardo, R. Goswami, G. Rosso, E. Ulbricht, P. Müller, D. Soteriou, N. Träber and J. Guck, Soft Matter, 2019, 15, 9776–9787 RSC.
- S. Kühn, J. Sievers, A. Stoppa, N. Träber, R. Zimmermann, P. B. Welzel and C. Werner, Adv. Funct. Mater., 2020, 30, 1908857 CrossRef.
- D. Baptista, L. Teixeira, C. van Blitterswijk, S. Giselbrecht and R. Truckenmüller, Trends Biotechnol., 2019, 37, 838–854 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sm01176f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |