
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAu3+-Induced gel network formation of proteins†
Laura M. I.
Schijven
 ab,
Vittorio
Saggiomo
b,
Aldrik H.
Velders
b,
Johannes H.
Bitter
a and
Constantinos V.
Nikiforidis
*a
ab,
Vittorio
Saggiomo
b,
Aldrik H.
Velders
b,
Johannes H.
Bitter
a and
Constantinos V.
Nikiforidis
*a
aBiobased Chemistry and Technology, Wageningen University and Research, Bornse Weilanden 9, 6708 WG Wageningen, The Netherlands. E-mail: costas.nikiforidis@wur.nl
bBioNanoTechnology, Wageningen University and Research, Bornse Weilanden 9, 6708 WG Wageningen, The Netherlands
First published on 11th October 2021
Abstract
The formation of protein gel networks in aqueous systems is a result of protein intermolecular interactions after an energy input, like heating. In this research, we report that a redox reaction between Au3+ ions and proteins can also lead to the formation of a protein gel network. Amino acids, like cysteine and tyrosine, get oxidized and form covalent bonds with neighboring protein molecules, while Au3+ ions get reduced to Au+ and Au0, nucleate and form gold nanoparticles. The protein gel network formation occurs within 2 h at room temperature and can be tuned by varying Au3+/protein ratio and accelerated by increasing the incubation temperature. The proposed Au3+-induced gel network formation was applied to different proteins, like egg yolk high-density lipoprotein, bovine serum albumin and whey protein. This research opens new insights for the investigation of the metal–protein interactions and may aid in the design of novel hybrid-soft nanocomposite materials.
1. Introduction
Protein–protein interactions in aqueous systems can lead to the formation of a three-dimensional network, leading to soft gel structures. The most common technique to induce protein–protein interactions is by heating the system over the denaturation point of the proteins, resulting in their (partial) unfolding.1,2 Unfolded proteins expose residual functional groups of amino acids, such as polar, non-polar and thiol groups, and hydrogen bond donor–acceptor pairs, which leads to an increase of attractive forces between amino acids of neighboring proteins, bond formation and subsequently to a three-dimensional gel network.3,4Interactions between amino acids can also be induced with the use of metal ions. Metal ions, such as Cu2+ and Fe2+/3+ can oxidize thiol- or amino-containing side groups of amino acids (e.g. cysteine, methionine, histidine)5 leading to di-amino acid covalent bonding.6,7
In this research, we aimed to take advantage of the interactions of metal ions with amino acids, and use it to create covalent bonds between proteins in aqueous solutions. In this way, we can develop a new technique to create protein gel networks. For that, we have used Au3+ ions, which have strong oxidizing properties.8 Additionally, it has been reported that several available amino acid groups (e.g. tryptophan, tyrosine, aspartic acid, or phenylalanine)9,10 can act as reduction sites of Au3+ and synthesize gold nanoparticles (AuNPs).11,12 AuNPs are more stable forms than Au3+ ions13 and their formation limits the protein oxidation rate. On top of that, AuNPs have unique chemical and physical properties, e.g. localized surface plasmon resonance (LSPR), and are attractive contrast agents for medical applications since they can be visualized with different techniques.14–16 Most of the studies have focused on proteins in solution as reducing and stabilizing agents for AuNPs synthesis.17–19 The use of Au3+ ions for covalently cross-linking protein and protein gel network formation, however, has not been reported before.
The interactions between Au3+ ions and proteins for the formation of protein gel networks were studied, using egg yolk high-density lipoprotein (HDL), a rather inactive lipoprotein. First, different Au3+/HDL ratios were tested for the formation of a protein gel network. The formed gel networks were characterized by confocal laser scanning microscopy (CLSM), rheology and UV-vis. The reduced Au3+ formed AuNPs and their formation was followed in time by UV-vis. Additionally, the temperature influence on the gel network and AuNPs formation were studied. The interactions between the amino acids and Au3+ ions, which could be involved in the gel network formation were studied by Fourier-Transform Infrared spectroscopy (FTIR), gel electrophoresis and fluorescence spectroscopy. Finally, the Au3+-induced gel network formation was also successfully applied to two other proteins, namely bovine serum albumin (BSA) and whey protein isolate (WPI).
In this work, we investigated the formation of protein gel networks through the addition of Au3+ ions. Our research reveals that Au3+ ions rapidly coordinate to the proteins and upon reduction of the Au3+ ions, AuNPs are formed and the proteins are covalently cross-linked through oxidation. This research can open new paths towards the design of controlled protein gel networks for multiple applications in materials science. Even inactive proteins like HDL can be used, while the ability to incorporate AuNPs in protein gel networks provides dual functionality to the design, since it can be further used for advanced biosensing and catalysis.
2. Experimental
2.1 Materials and methods
Fresh hen eggs were purchased from a local organic farm De Hoge Born, Wageningen, the Netherlands. Sodium chloride (NaCl, ≥99.5%), 1 M NaOH solution and potassium cyanide (KCN, ≥97%) were purchased from VWR international B.V. Gold(III) chloride hydrate (HAuCl4·xH2O, 99.5%), Fast green FCF fluorescent dye (≥85%) and 76 mm dialysis tubing cellulose membrane (MWCO = 14 kDa) were purchased from Sigma Aldrich. LDS sample buffer, a 10–250 kDa pre-stained PageRuler protein ladder, pre-casted NuPAGE 4–12% Bis–Tris gels, 20× MOPS SDS running buffer and Coomassie R-250 staining solution were purchased from ThermoFisher Scientific. Glacial acetic acid (100%) was purchased from Biosolve B.V. Methanol (99.98%) was purchased from LPS. All chemicals were used without further purification and deionized water was used throughout the experiments.The crude protein content was determined by Dumas (Thermo Quest NA 2100 Nitrogen and Protein Analyser) using a protein-to-nitrogen conversion factor of 6.25. For CLSM, the samples were stained with 10 μL of 1.2 mM Fast Green FCF, imaged using a Leica SP8-SMD CLSM with at λex = 633 nm, using a 63× objective, and analyzed with FIJI Is Just ImageJ software.20 Rheological measurements of the Au–HDL and heat-set HDL gel were done on an Anton-Paar® 302 rheometer, using rotational parallel-plate devices, using a shear-stress amplitude of 0.1% and frequency of 1 Hz. For the heat-set gel, the HDL solution was heated to T = 90 °C, kept constant for 1 h and cooled down to T = 20 °C (heat and cool rate = 5 °C min−1). For TEM, the sample was cast on a carbon-coated hexagonal 400 mesh copper grid. The grid was then transferred to a JEOL JEM1400+ microscope and the image was analyzed with FIJI software. UV-vis measurements were done on a Hitachi U-2010 UV-visible spectrophotometer using 1 mm glass cuvettes. Fluorescence spectroscopy measurements were done on an Agilent Cary Eclipse Fluorescence Spectrophotometer, using 10 mm quartz cuvettes. Excitation spectra were measured from λ = 250–390 nm with λem = 410 nm, emission spectra were measured from λ = 350–500 nm with λex = 325 nm, slits were set at 5 nm. For KCN treatments, 10 μL (1.8 μmol, 10 molar equivalents of Au3+) of 178 mM KCN solution (11.6 mg mL−1) was added to 3 mL of 0.01% (w/v) HDL solution and Au–HDL mixture and were measured after 1 h of exposure to air. FTIR spectra were measured using a Tensor 27 Fourier transform spectrophotometer over a spectral range 4000–500 cm−1 with a resolution of 4 cm−1 at ambient conditions. The samples were freeze-dried prior to the measurement and prepared in KBr pellets. Denaturing SDS-PAGE gel electrophoresis was done according to the manufacturer's protocol. For the sample preparation, 10 × 3.0% (w/v) Au–HDL gels were prepared in separate microcentrifuge tubes. After each time period (0–240 minutes), 1 mL LDS sample buffer was added so that the total concentration of protein in each sample was 1 mg mL−1. The samples were kept in the freezer till loading.
2.2 Extraction of HDL
High-density lipoprotein (HDL) was extracted from egg yolk according to the method developed by Castellani,21 with slight modifications. Hen eggs were cracked and the egg yolks were separated manually from the albumen. The egg yolks were carefully rolled on a paper tissue to remove the chalazae and adhering albumen. The yolk membranes were then punctured, using a glass pipette, and their contents were collected and pooled in a beaker cooled in iced water. The liquid yolks were diluted with three volumes of 0.17 M NaCl (1% (w/v)) and homogenized by stirring for 1 h. The yolk was fractionated into plasma and granules by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 45 minutes at 4 °C, using a Thermoscientific Sorvall Legend XFR centrifuge. The pellet (granules) was washed with 0.17 M NaCl and centrifuged once more. The granules were suspended in 1.71 M NaCl (10% (w/v)) and the solution's pH was adjusted to 7.25, using 1 M NaOH. The mixtures were collected in dialysis tubings and were dialyzed against deionized water with 3 changes over 24 h. The HDLs were precipitated. The content of the dialysis tubings were collected and centrifuged. The pellets, rich in HDL, were collected and freeze-dried, using a Salmenkipp alpha 2–4 plus freeze-dryer at a temperature of −76 °C and pressure of 0.0090 mbar for 72 h.
000 × g for 45 minutes at 4 °C, using a Thermoscientific Sorvall Legend XFR centrifuge. The pellet (granules) was washed with 0.17 M NaCl and centrifuged once more. The granules were suspended in 1.71 M NaCl (10% (w/v)) and the solution's pH was adjusted to 7.25, using 1 M NaOH. The mixtures were collected in dialysis tubings and were dialyzed against deionized water with 3 changes over 24 h. The HDLs were precipitated. The content of the dialysis tubings were collected and centrifuged. The pellets, rich in HDL, were collected and freeze-dried, using a Salmenkipp alpha 2–4 plus freeze-dryer at a temperature of −76 °C and pressure of 0.0090 mbar for 72 h.
2.3 Preparation of Au–HDL gel networks
In a typical experiment, HDL was prepared as a homogeneous 6% (w/v) solution by dissolving 60 mg of HDL (0.14 μmol, mHDL = 422480 kDa)22 in 1 mL of 1.71 M NaCl in a 20 mL glass vial.
Au3+ solutions, of 10, 100, 150, 200 and 250 molar equivalents to HDL, were prepared by using a 0.1 M HAuCl4 stock solution. For this, 14, 71, 142, 213, 284 and 355 μL of Au stock solution was added to 500 μL of 1.71 M NaCl. The pH of the Au solutions was adjusted to 7, using a 1 M NaOH solution. The total, final volume of the Au3+ solutions was kept constant at 1 mL. The Au3+ solutions were rapidly injected into the HDL solutions to have a final HDL concentration of 3.0% (w/v).
3. Results and discussion
3.1 Au–HDL interactions and gel network formation
HDL is a globular-like protein with a proximate protein composition of 83.3 wt%.23Au3+ ions were added as a solution at pH 7 to a saline, aqueous dispersion of HDL (with a final concentration of 3.0% (w/v) at different molar ratios). Between 10 and 100 molar equivalents of Au3+ to HDL, it was observed that HDL aggregated and precipitated. At 150 molar equivalents of Au3+, the viscosity of the HDL solution increased instantly, while after storage of 24 h, a homogeneous, soft-solid material was formed. For 250 molar equivalents of Au3+, a soft-solid gel was formed 2 h after addition into the solution. These findings indicate extensive interactions of Au3+ with HDL, even at room temperature, which subsequently lead to the formation of an HDL network. Without the presence of Au3+, HDL was rather inactive and did not form a network.
3.2 Properties of Au–HDL gel networks
The transition of the HDL dispersions from a liquid to a soft gel due to the addition of 250 molar equivalents of Au3+ was qualitatively assessed by a vial tilting method. The initial HDL dispersion appeared translucent, yellow-colored, and flowed when the vial was tilted. When the Au3+ ions were added, the dispersion became cloudy, but the sample was still flowing. After 2 h, the sample appeared yellow, opaque and did not flow anymore (Fig. 1A). The cloudiness, that was observed directly after the addition of Au3+, indicates that the protein–protein interactions and the formation of aggregates started instantly. However, an extensive gel network was only formed after 2 h.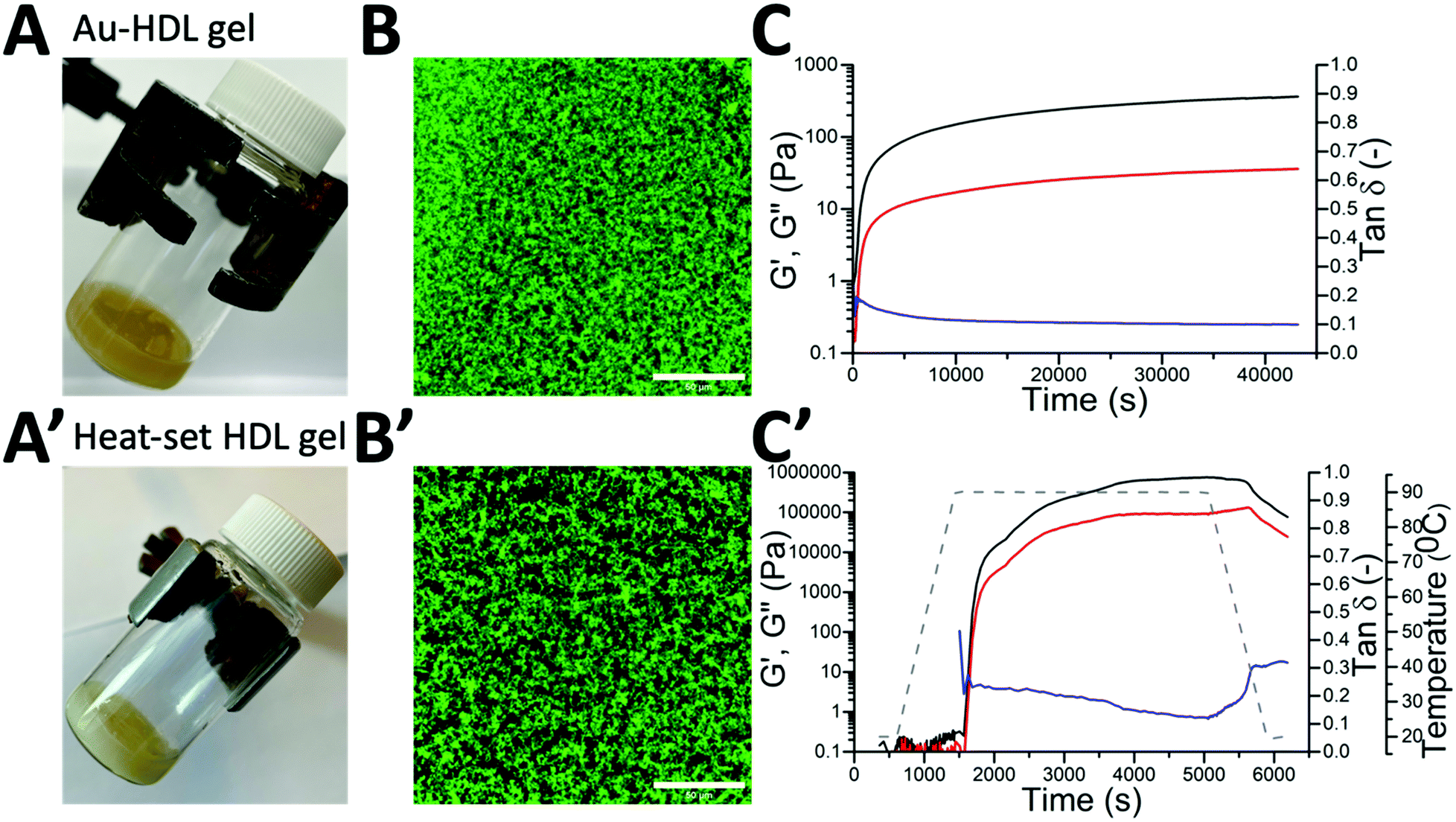 | ||
| Fig. 1 Pictures of tilted vials containing (A) Au–HDL gel network (3.0% (w/v), 250 equivalents of Au3+) after 2 h of preparation and (A′) heat-set HDL gel (3.0% (w/v) after heating at T = 90 °C for 1 h). CLSM image of (B) Au–HDL gel network after 1 day of preparation and (B′) heat-set HDL gel. The gels were stained with Fast Green FCF, scale bars are 50 μm. Time-dependent function of storage modulus (G′, black line), loss modulus (G′′, red line), and loss tangent (tanδ, blue line) of the (C) Au–HDL gel network formation and (C′) heat-set HDL gel during heating at T = 90 °C for 1 h (grey dashed line). | ||
The microstructure of the formed Au–HDL gel network was imaged by CLSM (Fig. 1B). In the image, a high contrast difference between the Fast Green FCF stained protein (green colored) and background (black pores) was observed. The addition of Au3+ of HDL resulted in the formation of a uniformly packed, dense protein network.
Besides the visual observation, the network formation of the Au–HDL mixture was quantitatively investigated by applying a controlled shear stress test (Fig. 1C). To simulate the behavior of the Au–HDL mixture at rest, a very low shear was applied. During the first seconds of the measurement, the graph of the loss modulus (G′′, viscous-like behavior) was above the storage modulus (G′, solid-like behavior) graph, indicating that the mixture had a viscous response. Within seconds, a cross-over was observed, where G′ was significantly higher than G′′. The cross-over indicates stronger interactions in the system, which induced the formation of a soft gel structure. When 2 h passed after the addition of Au3+ ions, the formed soft-solid structure was reaching an equilibrium since the G′ and G′′ didn’t significantly change after that point. This finding corresponds to the visual observation that during tilting the dispersion was not flowing. At the point of 2 h, the loss factor (tanδ = G′′/G′) was found at 0.1, which directly shows that the material exhibited more solid-like than liquid-like properties.
The Au3+-induced gelation method was compared to urea- and heat-induced gelation methods. It was found that a rather concentrated HDL solution was required (>10% (w/v)) for urea-induced gelation of HDL. Urea destabilizes hydrophobic interactions and hydrogen bonds in proteins and forms disulfide bonds by cysteine oxidation.24 For the Au3+-induced gelation, a concentration of 3.0% (w/v) was sufficient to form a gel network, which suggests that stronger protein–protein interactions were involved than in urea-induced gelation.
When the heat-induced gelation was applied to HDL, a white, opaque gel was formed (Fig. 1A′). The microstructure of the heat-set gel appeared irregular and contained more pores (Fig. 1B′). This suggests that there were fewer inter-protein connections in the heat-set HDL gel. The heat-set HDL gel was also applied to a shear stress test. During heating, the G′ > G′′ cross-over was found at T = 75 °C, where HDL starts to unfold.3 After 1 h of heating, the gel reached an equilibrium (Fig. 1C′). Due to its rigid, globular structure, HDL is less sensitive to heat and the protein unfoldment requires a lot of energy.3 While cooling down the gel, the G′ and G′′ decreased and the loss factor increased, indicating that the gel became more liquid-like. For the Au3+-induced gelation, this phenomenon was not observed because the protein–protein interactions remained stable after the formation of the protein gel network. This confirms that the heat-set HDL gel is constituted of weak protein–protein interactions compared to the Au–HDL gel.
3.3 AuNPs formation
24 h after the preparation of the Au–HDL network, the soft material gradually changed color from dark yellow to red (Fig. 2A). It was hypothesized that the red color of the Au–HDL network originates from the characteristic LSPR absorption of formed AuNPs, which can be found in the absorption region of λ = 500–600 nm.25 The gradual color change of the Au–HDL soft material was followed by UV-vis absorbance at λ = 300–800 nm for 7 days (Fig. 2B). Two days after preparation of the Au–HDL mixture, a small band was observed in the region of λ = 500–600 nm, confirming the formation of AuNPs. The intensity of this LSPR peak further increased in time, which corresponds to the formation and growth of AuNPs.26 To confirm the presence of AuNPs, TEM was used by taking images of a mixture of Au–HDL at low concentrations (0.1% (w/v)) to observe the AuNPs. In the image, AuNPs of different sizes were observed (Fig. S1, ESI†). These findings indicate that Au3+ ions not only induce the formation of a protein network, but the HDL also reduces the Au3+ ions and synthesizes AuNPs. Compared to proteins in solution, the synthesis of AuNPs requires a longer reaction time, after HDL gel network formation. This could be due to the rigid protein structure of HDL in the gel network, causing steric hindrance, which slows down the reaction with Au3+ ions. Next to the reducing properties, HDL stabilizes the AuNPs through steric stabilization effects, adsorption to the surface and by coordinating Au3+ to numerous functional amino acid groups (e.g. amine and carboxylate groups). Therefore, it was suggested that the AuNPs are embedded in the protein gel network and could not be recovered. | ||
| Fig. 2 (A) Pictures of vials containing Au–HDL gel networks after 1, 2 and 7 days of preparation. The soft material turned from dark yellow (1 day) to dark red (2 and 7 days). (B) Solid UV-vis absorbance spectra of Au–HDL gel network followed in time from 0–7 days. The characteristic LSPR peak of AuNPs increased in time, as indicated by the arrow. | ||
3.4 Influence of temperature on gel network and AuNPs formation
The Au3+-induced gel network and subsequent AuNPs formation were observed and investigated at room temperature. However, it was expected that heating could accelerate the interactions of HDL with Au3+ ions. Accordingly, the Au–HDL mixtures were incubated at different temperatures (T = 4, 20, 40, and 60 °C) and monitored for 5 h using UV-vis at λ = 300–800 nm (Fig. 3). For samples incubated at T = 4 and 20 °C, there was no red color visually observed within 5 h and the LSPR peaks were absent (Fig. 3A, B and E). Moderately heating the Au–HDL mixture at T = 40 °C resulted in the appearance of a slight red color of the mixture and the LSPR peak after 4 h (Fig. 3C and E). Heating at T = 60 °C resulted in the appearance of a red color of the mixture and the LSPR in the spectra after 1 h (Fig. 3D and E). Increasing the temperature resulted in notably higher reaction rates, which resulted in a shorter reaction time of AuNPs formation.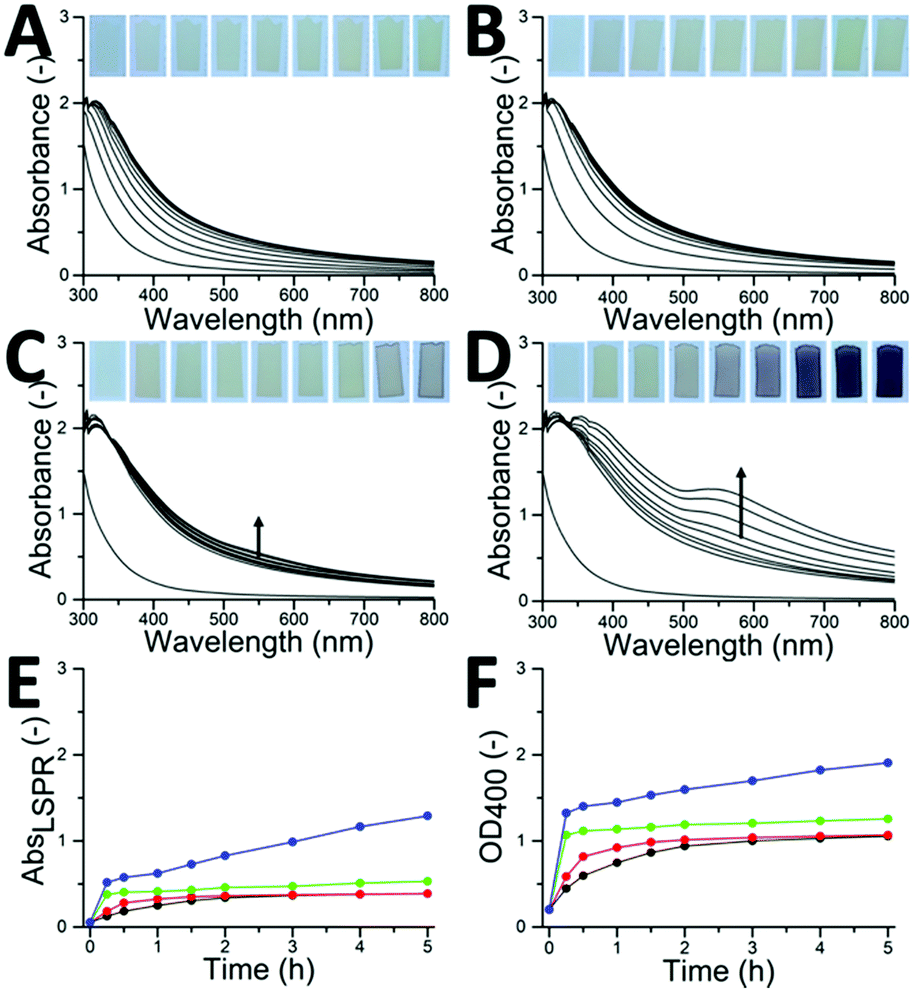 | ||
| Fig. 3 (A–D) Solid UV-vis absorbance and (E) derived LSPR absorbance (at λ = 555 nm) and (F) OD400 spectra of Au–HDL gel networks incubated at T = (A) 4 °C (black dotted line), (B) 20 °C (red dotted line), (C) 40 °C (green dotted line) and (D) 60 °C (blue dotted line) followed for 5 h. (insets) Optical photographs showing gradual color change of the Au–HDL mixtures at different time intervals. | ||
Regardless of the incubation temperature, the UV-vis absorption bands increased in time. This increase is caused by scattering of the light, which is known as optical density (OD). When proteins form aggregates, the light is more scattered compared to dispersed, single proteins (ODaggregates > ODproteins). When a dense gel network is formed, the light scatters even more (ODgel > ODaggregates).27 It was hypothesized that the gel network formation can be followed by an increase in OD, which corresponds to the unfolding, formation of aggregates and finally gel networks. Accordingly, the network formation process was characterized by plotting the OD against time. The OD was taken at λ = 400 nm, where there was no inherent absorption of the HDL and AuNPs (Fig. 3F). For the Au–HDL mixture incubated at T = 20 °C, which is known to form a soft gel, the OD400 gradually increased during the first 2 h and stabilized around 1.0. This result is complementary to the results obtained with the vial tilting method and shear stress test, indicating that the increase in OD400 is related to the gel network formation. When the Au–HDL mixture was incubated at T = 4 °C, the OD400 also increases gradually up to 1.0. Moderately heating the Au–HDL mixture at T = 40 °C resulted in an increase of the OD400 to 1.0 within 15 minutes, which then stabilized. Incubation of the Au–HDL mixture at T = 60 °C resulted in increase of OD400 to 1.3, which then further increased. Those results indicate that Au3+ ions induce protein–protein interactions not only at room temperature but also at T = 4 °C and when incubated at higher temperatures. Comparing the OD400 values for spectra of the Au–HDL mixtures incubated at T = 4–40 °C, the OD400 values were stable for 5 h. Based on that result, we hypothesize that the network formation is completed at OD400 = 1.0 and it is expected that OD400 values for the Au–HDL mixtures incubated at T = 4 and 20 °C would still increase after 5 h because of the AuNPs formation and growth. When incubating the Au–HDL mixture at T = 60 °C, the OD400 further increased to 2.0. This increase was assigned to the contribution of LSPR absorbance due to the AuNPs formation. As a control, HDL solutions (in the absence of Au3+) were incubated at T = 4–60 °C and the OD400 values were measured for 5 h (Fig. S2A, ESI†). No observable differences between spectra were found within 5 h, indicating that HDL doesn’t unfold while moderately heating and the increase in OD400 is only attributed to Au3+ induced gel network formation.
When heating the HDL solution at T = 80 °C, the OD400 started to increase after 30 min and further increased in time till OD400 = 1.8 (Fig. S2A, ESI†). In the rheology experiment of the heat-set HDL gel, it was observed that HDL starts to unfold above T = 75 °C.3 For this reason, the influence of heat-induced gelation on the Au3+-induced gelation was investigated by incubating the Au–HDL mixture at T = 80 °C. A red color of the sample was already visually observed after 15 min together with the appearance of the LSPR peak (Fig. S2B and C, ESI†). The OD400 value for the Au–HDL mixture heated at T = 80 °C was significantly higher than for the Au–HDL mixtures incubated at T = 4–60 °C (Fig. S2D, ESI†). This could be assigned to the combination of the unfolding of HDL by heating, the Au3+-induced network formation and the AuNPs formation.
3.5 Nature of Au–HDL interactions
To investigate which amino acids of HDL interact with Au3+ ions and contribute to the protein–protein interactions, FTIR spectra of HDL and the Au–HDL gel network were measured (Fig. S3, ESI†). The pattern of group vibrations of HDL corresponds to a typical FTIR spectrum of lipoproteins.28 The FTIR spectra of HDL and the Au–HDL network were very similar, however, the absorbance of the band at region 2260–2010 cm−1, corresponding to SH stretching vibrations, was decreased after the addition of Au3+ ions. The decrease of free SH groups could indicate that cysteine is oxidized by Au3+ and forms disulfide bonds29,30 or forms Au–S bonds.To further investigate whether other bonds than disulfide bonds are responsible for the protein–protein interactions and subsequent network formation, Au–HDL fractions were qualitatively analyzed using an SDS electropherogram (Fig. S4, ESI†). Aliquots of the Au–HDL mixture were taken at different time frames and were dispersed into β-mercaptoethanol solutions. The β-mercaptoethanol reduces the disulfide bonds and allows for investigating their role in the protein–protein network. SDS-PAGE separation of native HDL revealed five major bands, ranging in molecular weight from 28 to 110 kDa (lane 2).23 After the addition of Au3+ ions to the HDL, the pattern in the gel electropherogram remained unchanged (lane 3). Further extending the reaction time to 10 minutes (lane 4), larger protein bands were observed (>185 kDa), while the smaller bands (28–75 kDa) decreased in intensity. The β-mercaptoethanol breaks down the disulfide bridges, however, there were still large protein fragments present in lane 4–12. This indicates that there are other types of bonds present in the protein network, probably of covalent nature. It was expected that those covalent bonds are derived from other oxidized amino acid groups, such as tyrosine.
Tyrosine exhibits strong reducing properties9 and in order to evaluate the effect of Au3+ on tyrosine of HDL, the physical–chemical properties of HDL and Au–HDL were investigated. Oxidation of tyrosine results in the formation of tyrosine–tyrosine cross-linking (dityrosine) (Fig. 4A).31 Dityrosine has characteristic fluorescent properties with excitation at λex = 325 nm and emission at λem = 410 nm.31 When the fluorescent spectra of the HDL and Au–HDL mixtures were measured, no dityrosine fluorescence was detected (Fig. S5, ESI†). This could be caused by the spectral overlap of the absorbance of Au3+ with the excitation band of the dityrosine, known as the inner filter effect (Fig. S6A, ESI†).31 It has to be noted that the absorption at λ = 280 nm decreased in a period of 0–5 h, due to Au3+ complexation and reduction. Extraction of Au3+ is required to avoid possible interferences or quenching of the dityrosine fluorescence. Therefore, KCN was added to Au–HDL to form an Au(CN) complex.32 As a control, KCN was added to HDL. It was observed that the absorbance and excitation spectra of HDL were unaffected by the KCN treatment, indicating that the KCN does not damage the tertiary HDL structure (Fig. S6, ESI†). After Au3+ extraction of the Au–HDL mixture, the absorbance at λ = 280 nm was decreased because of the formation of the Au(CN)2− complex, which exhibits no absorbance (Fig. S6A′, ESI†). When measuring the fluorescent properties of the Au–HDL mixtures after KCN treatment, an excitation peak at λ = 328 nm and emission peak at λ = 400 nm were observed (Fig. 4B and C). Those peaks were not observed for native HDL before and after addition of KCN (Fig. S5 and S6, ESI†). The intensity of those peaks further increased in time and stabilized after 1.5 h. Those results confirm that the addition of Au3+ to HDL results in the formation of dityrosine, which can lead to the formation of extensive, stable inter- and intra-protein cross-linkages.6
 | ||
| Fig. 4 (A) Scheme of dityrosine cross-linking between neighboring proteins. Fluorescence (B) excitation (λem = 410 nm) and (C) emission (λex = 325 nm) spectra of 0.01% (w/v) HDL (dashed line) and Au–HDL mixture (solid line) in 1.71 M NaCl after KCN treatment at different time intervals (0–5 h, every 30 minutes). The arrow indicates the increase in intensity in time. | ||
However, to further verify the binding of Au3+ to other amino acids in the Au–HDL system, more research needs to be done, e.g. combination studies of X-ray crystallography, isotope labeling NMR and mass spectrometry.33–36
3.6 Interactions of Au3+ ions with other proteins
To investigate whether Au3+ ions could induce network formation of other globular proteins, the same method as for HDL was applied to BSA and WPI, (a mixture of α- and β-lactalbumin, β-lactoglobulin, BSA and immunoglobulin).37 After the addition of Au3+ ions to BSA and WPI, a red color and LSPR peaks were observed after 2 days (Fig. S7A and B, ESI†). This was also observed for HDL, which suggested that the AuNPs formation is not affected by the protein source and potentially the same amino acids are involved in Au3+ reduction. However, after the addition of Au3+ ions to BSA, the mixture remained liquid-like for 2 days and formed a soft-solid material only after 7 days, which was also observed in the OD400 graph (Fig. S7C, ESI†). The Au–WPI mixture started to form a soft material after 1 day. Based on those results, we found that Au3+ ions could induce network formation for different protein sources, but the reaction times and potentially the mechanism differed. More research needs to be done for studying the Au3+-induced gelation for different protein systems.4. Conclusions
In summary, we have demonstrated that a redox reaction can take place between Au3+ and proteins, inducing protein–protein interactions and a subsequent gel protein network. The Au3+-induced protein network formation does not require additional heating and even occurs at T = 4 °C. The network formation in time was revealed by vial tilting, rheology and OD measurements. The results show that Au3+-induced gel network formation was faster as opposed to the heat-induced gelation of proteins. The interactions between Au3+ ions and amino acids, responsible for the Au–HDL network formation, were further characterized by FTIR, SDS-PAGE, UV-vis absorbance, and fluorescence spectroscopy, showing that there were cross-links between cysteine and tyrosine. For 2 days, there was no Au3+ reduction, and only then it starts forming AuNPs. The network formation can be further tuned by parameters, such as Au3+/protein ratio, protein source and temperature. The Au3+-induced protein–protein interactions provide new insights into protein network formation and may contribute to the design and construction of novel hybrid-soft nanocomposite materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research presented in this article was financially supported by the Graduate School VLAG. The authors would like to thank Simha Sridharan and Eleni Ntone for their help with laboratory experiments and discussions and Wageningen Electron Microscopy Centre for their support with the TEM measurements.Notes and references
- T. Nicolai, Adv. Colloid Interface Sci., 2019, 270, 147–164 CrossRef CAS PubMed.
- D. Durand, J. C. Gimel and T. Nicolai, Phys. A, 2002, 304(1–2), 253–265 CrossRef CAS.
- V. Kiosseoglou and A. Paraskevopoulou, Food Hydrocolloids, 2005, 19(3), 527–532 CrossRef CAS.
- F. Cordobés, P. Partal and A. Guerrero, Rheol. Acta, 2004, 43(2), 184–195 CrossRef.
- E. Shacter, Drug Metab. Rev., 2000, 32(3–4), 307–326 CrossRef CAS PubMed.
- W. Zhang, S. Xiao and D. U. Ahn, Crit. Rev. Food Sci. Nutr., 2013, 53(11), 1191–1201 CrossRef CAS PubMed.
- A. Bradshaw, M. Salt, A. Bell, M. Zeitler, N. Litra and A. M. Smith, J. Exp. Biol., 2011, 214(10), 1699–1706 CrossRef CAS PubMed.
- C. Gabbiani, A. Casini and L. Messori, Gold Bull., 2007, 40(1), 73–81 CrossRef CAS.
- Y. N. Tan, J. Y. Lee and D. I. Wang, J. Am. Chem. Soc., 2010, 132(16), 5677–5686 CrossRef CAS PubMed.
- J. J. Warren, J. R. Winkler and H. B. Gray, FEBS Lett., 2012, 586(5), 596–602 CrossRef CAS PubMed.
- D. M. Chevrier, A. Chatt and P. Zhang, J. Nanophotonics, 2012, 6(1), 064504 CrossRef.
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131(3), 888–889 CrossRef CAS PubMed.
- P. Zhao, N. Li and D. Astruc, Coord. Chem. Rev., 2013, 257(3–4), 638–665 CrossRef CAS.
- R. De La Rica and A. H. Velders, Small, 2011, 7(1), 66–69 CrossRef CAS.
- R. de laRica, R. M. Fratila, A. Szarpak, J. Huskens and A. H. Velders, Angew. Chem., Int. Ed., 2011, 50(25), 5704–5707 CrossRef CAS.
- J. Hainfeld, D. Slatkin, T. Focella and H. Smilowitz, Br. J. Radiol., 2006, 79(939), 248–253 CrossRef CAS.
- M. Annadhasan, T. Muthukumarasamyvel, V. Sankar Babu and N. Rajendiran, ACS Sustainable Chem. Eng., 2014, 2(4), 887–896 CrossRef CAS.
- H. Kawasaki, K. Hamaguchi, I. Osaka and R. Arakawa, Adv. Funct. Mater., 2011, 21(18), 3508–3515 CrossRef CAS.
- J. Xie, J. Y. Lee and D. I. Wang, J. Phys. Chem. C, 2007, 111(28), 10226–10232 CrossRef CAS.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld and B. Schmid, Nat. Methods, 2012, 9(7), 676–682 CrossRef CAS PubMed.
- O. Castellani, C. Guérin-Dubiard, E. David-Briand and M. Anton, Food Chem., 2004, 85(4), 569–577 CrossRef CAS.
- D. H. Ohlendorf, R. F. Wrenn and L. J. Banaszak, Nature, 1978, 272(5648), 28–32 CrossRef CAS.
- M. Anton, High-density lipoproteins (HDL) or lipovitellin fraction, in Bioactive egg compounds, Springer, 2007, pp. 13–16 Search PubMed.
- A. Totosaus, J. G. Montejano, J. A. Salazar and I. Guerrero, Int. J. Food Sci. Technol., 2002, 37(6), 589–601 CrossRef CAS.
- H. Wei, Z. Wang, J. Zhang, S. House, Y.-G. Gao, L. Yang, H. Robinson, L. H. Tan, H. Xing and C. Hou, Nat. Nanotechnol., 2011, 6(2), 93 CrossRef CAS PubMed.
- J. B. ten Hove, L. M. Schijven, J. Wang and A. H. Velders, Chem. Commun., 2018, 54(95), 13355–13358 RSC.
- S. Mleko, Eur. Food Res. Technol., 1999, 209(6), 389–392 CrossRef CAS.
- D. Krilov, M. Balarin, M. Kosović, O. Gamulin and J. Brnjas-Kraljević, Spectrochim. Acta, Part A, 2009, 73(4), 701–706 CrossRef PubMed.
- B. Đ. Glišić, U. Rychlewska and M. I. Djuran, Dalton Trans., 2012, 41(23), 6887–6901 RSC.
- H. Le, L. Ting, C. Jun and W. Weng, Int. J. Food Prop., 2018, 21(1), 277–288 CrossRef CAS.
- D. Malencik and S. Anderson, Amino Acids, 2003, 25(3–4), 233–247 CrossRef CAS PubMed.
- X.-B. Wang, Y.-L. Wang, J. Yang, X.-P. Xing, J. Li and L.-S. Wang, J. Am. Chem. Soc., 2009, 131(45), 16368–16370 CrossRef CAS PubMed.
- D. Loreto, G. Ferraro and A. Merlino, Int. J. Biol. Macromol., 2020, 163, 970–976 CrossRef CAS PubMed.
- G. Marcon, L. Messori, P. Orioli, M. A. Cinellu and G. Minghetti, Eur. J. Biochem., 2003, 270(23), 4655–4661 CrossRef CAS PubMed.
- L. Messori and A. Merlino, Coord. Chem. Rev., 2016, 315, 67–89 CrossRef CAS.
- A. Pratesi, D. Cirri, D. Fregona, G. Ferraro, A. Giorgio, A. Merlino and L. Messori, Inorg. Chem., 2019, 58(16), 10616–10619 CrossRef CAS PubMed.
- C. I. Butré, P. A. Wierenga and H. Gruppen, Process Biochem., 2014, 49(11), 1903–1912 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sm01031j |
| This journal is © The Royal Society of Chemistry 2021 |