
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConcurrent self-regulated autonomous synthesis and functionalization of pH-responsive giant vesicles by a chemical pH oscillator†
E.
Poros-Tarcali
 *a and
J.
Perez-Mercader
*ab
*a and
J.
Perez-Mercader
*ab
aDepartment of Earth and Planetary Science and Origins of Life Initiative, Harvard University, 20 Oxford Street, Cambridge, Massachusetts 02138, USA. E-mail: jperezmercader@fas.harvard.edu; eporostarcali@fas.harvard.edu
bSanta Fe Institute, Santa Fe, New Mexico 87501, USA
First published on 5th March 2021
Abstract
The semibatch BrO3−–SO32− pH oscillator serves as the radical source for the in situ polymerization of the pH-responsive 2-(diisopropylamino)-ethyl methacrylate monomer on poly(ethylene-glycol)-macroCTA chain and generates an amphiphilic block copolymer. These building blocks concurrently self-assemble to micelles and then transforms to vesicles as the chain length of the hydrophobic block growths. Large amplitude oscillations in the concentration of H+ by the semibatch BrO3−–SO32− are provoked when the conditions in the system are favorable. The oscillations control the protonation state of the tertiary amine group in the core segment of the block copolymer. Rhythmic assembly-disassembly of the polymer structures is observed. All processes, from the time- regulated autonomous formation of the building blocks, their self-assembly and the rhythmic disassembly-reassembly are governed by the same simple chemical system, in the same reaction vessel, without complicated multi step procedures and are fueled and kept out of equilibrium by the uniform inflow of SO32−.
1. Introduction
Inspired by how natural living systems operate, during the past decade self-assembly has been demonstrated in a variety of far from thermodynamic equilibrium chemical systems. Although the number of examples is still limited, surprisingly versatile transient structures have been designed.1–4 These structures form in an energetically uphill process using an energy supply which generates transient structures while the energy is dissipated. Because of the time-dependence, this class of phenomena has the potential to enable temporal control of the self-assembly and self-assembly related functions. The energy source can be light,5–8 ultrasound9 or the consumption of a chemical fuel.10,11 Furthermore, the experimental environmental conditions under which this takes place remind one of general scenarios for the origin of life.12Living systems are spatially finite and thermodynamically open. This allows them to maintain a free-energy gradient with the environment where they exist while avoiding the “Arithmetic Demon”13,14 and concentration problems for their chemical operation. In natural life this is enabled via the presence of a phospholipid membrane which can be regulated, and provides the necessary open system character to guarantee, as needed, the execution of the system's life cycle. For many applications, such as in the synthesis of autonomous functional materials, in artificial, synthetic or proto-life work it is interesting to substitute the complex natural cell membrane by a membrane made by less complex artificial and synthetic amphiphilic block copolymers (ABC). Due to their robustness and tunable properties,15–17 ABCs are excellent molecules to study molecular self-organization, dynamical self-assembly or their combination. Indeed, the opportunities offered by ABCs open a vast window into the understanding and applications of dissipative phenomena with completely autonomous self-regulation and functionality of complex chemical systems in a dynamic environment.
The relatively novel technique of polymerization-induced self-assembly (PISA) makes ABCs even more attractive. PISA, an intrinsically out of equilibrium process, enables the autonomous synthesis of ABCs objects from the nano- to the micron scales with different morphologies (micelles, worms, vesicles, etc.), compositions and features in a one-step, uncomplicated and efficient process in several solvents, including water.18–24 The generation of amphiphiles in PISA is based on the extension of an originally soluble polymer block by the polymerization of another monomer to a nonsoluble copolymer block, which results in the accompanying self-assembly of polymeric structures. Recently, besides the commonly used initiators (e.g. light, heat), oscillatory chemical reactions have been shown to be a powerful source of radicals for the PISA process.25–30 Among the about 200 known oscillating chemical reactions,31 the Belousov–Zhabotinsky redox oscillator was the first with which micelles and vesicles made of various ABCs have been generated using PISA in batch and continuously stirred-tank reactors (CSTR).25–27,29,30 Then PISA initiated by a chemical pH oscillator (represents a different family of oscillatory chemical reactions), specifically by the semibatch BrO3−–SO32− chemical oscillator has been demonstrated by us.28
Chemical oscillators provide remarkable examples of transient self-assembly regulating chemical fuels. They necessarily operate far from thermodynamic equilibrium and bring with their redox or pH oscillations an internal time regulation mechanism that can be coupled to a reaction network. The concentration oscillations in these systems induce periodic transitions in the state of self-assembly.
Self-oscillating gels,32 micelles33 and vesicles34 driven by redox oscillations in the Belousov–Zhabotinsky reaction have been developed. In addition, pH oscillators have been reported to control the pH-responsive aggregation of coated gold nanoparticles,35 the micelle-to-vesicle oscillation of oleic acid-based surfactants,36 the self-assembly of ABCs37 and supra-amphiphiles.38 However, all the above examples require multi-step, multi-pot procedures. The building blocks are synthesized beforehand in a separate reaction, in contrast with biological systems which rely on precise, temporally and spatially synchronized reaction networks.4,39,40
In this paper we report on a one-pot completely non-biochemical chemical reaction network, which autonomously generates its own building blocks and kinetically controls its transient stimulus-responsive self-assembly into an out of equilibrium system with its own internal time-regulatory mechanism.
The reaction network is a combination of a pH-oscillator driven and regulated (A) synthesis of a pH-responsive amphiphilic diblock copolymer, (B) their self-assembly into cooperative polymeric structures in PISA, and (C) which is followed by the transient periodic pH-responsive disassembly and reassembly of the structures.
The core BrO3−–SO32− pH oscillator41,42 generates large amplitude (3–4 pH units) pH oscillations in the pH range of ∼3–7. The oscillations in pH arise from the two-way oxidation of SO32− by BrO3− (Table S1, ESI†).41 The complete oxidation to SO42− generates H+ in an autocatalytic way and lowers the pH (R3, R4). The delayed consumption of the autocatalyst, H+, by the partial oxidation of SO32− to S2O62− (R5) and the protonation of constantly inflowed SO32− introduce a negative feedback process. Since SO32− is consumed in each oscillatory period, its continuous supply is necessary to maintain the system and start a new period; otherwise after an autocatalytic pH drop the system reaches thermodynamic equilibrium and stays in the low pH state region. The consumption of SO32− is the reason why the BrO3−–SO32− pH oscillator, just as any other chemical pH oscillator, does not work in a batch reactor and only in a flow reactor. The BrO3−–SO32− pH oscillator is known to generate free radicals and able to initiate polymerization.28,43
For functionalization purposes the ABC generated by PISA in our system must respond to the pH oscillator and switch between amphiphilic and doubly hydrophilic. This requires a pH-responsive polymer block whose pKa is within the working pH range of the pH oscillator. The pKa of the tertiary amine group of the core segment of the poly(ethylene-glycol)-block-poly(2-(diisopropylamino)-ethyl methacrylate) (PEG-b-PDPA) is ∼6.37,44 and decreases to 5.7 at high salt concentration.45 The degree of polymerization (DP, the number of monomeric units in a polymer) of the PDPA block has no effect on its pKa.16 This value therefore is an ideal match with the working pH range of the BrO3−–SO32− pH oscillator. At pH values above the pKa, the PDPA block has no charge and is highly hydrophobic. When the pH is lower than the pKa, the PDPA block becomes protonated and turns hydrophilic. This reversible protonation induces transient changes in the amphiphilicity of the block copolymer which result in the self-assembly and disassembly of the polymer structures being modulated by the core pH oscillator.
This is the first time that this pH-responsive PEG-b-PDPA amphiphilic copolymer has been coupled to any pH oscillator, or that the BrO3−–SO32− pH oscillator has been coupled to a process for the (autonomous) PISA synthesis and pH responsive assembly of amphiphilic copolymers. The novelty of this system, reported here for the first time, is the highly complex reaction network in which both autonomous functions of the pH oscillator, the generation of radicals and high amplitude pH oscillations, are incorporated into one system in a one pot reaction.
2. Materials and methods
2.1. Materials
Poly(ethylene glycol)methyl ether (4-cyano-4-pentanoate dodecyl trithiocarbonate) (PEG-mCTA, Sigma-Aldrich), 2-(diisopropylamino)ethyl methacrylate (Sigma-Aldrich), sodium bromate (NaBrO3, Sigma-Aldrich), sodium sulfite (Na2SO3, anhydrous min. 98%, Sigma-Aldrich), sulfuric acid (H2SO4, 10 Normal, Ricca Chemical Company), methanol-d4 (Cambridge Isotope Laboratories, Inc) were used without further purification. The stock solutions were made with ultra-pure water (Hardy Diagnostics). Sodium sulfite (Na2SO3) solution was freshly prepared and used on the same day.2.2. Experimental setup
The experiments were conducted in a semibatch reactor, where 15 mL of a solution containing the NaBrO3 were thermostated to 45 °C (IKA HB100) and mixed with a magnetic stirrer at 600 rpm stir rate (2mag MIXdrive 1eco). After starting to inflow the solution of Na2SO3 and H2SO4 by an iPump i150 peristaltic pump on a constant flow rate (0.08 ml min−1), the PEG- mCTA and the neutralized PDA (with the addition of H2SO4) were added immediately. The pH change of the aqueous solution was monitored by a Sper Scientific Benchtop pH/mV Meter equipped with a pH Electrode with BNC + PIN connector (Hanna Instruments HI1330B), pH values were collected once per second. The intensity of the scattered light was measured with an Ocean Optics USB2000 + VIS-NIR spectrophotometer. The light source was a green laser (532 nm) positioned perpendicular to the detector. The light absorbance was measured by the same Ocean Optics USB2000 + VIS-NIR spectrophotometer, but the light source (Thorlabs QTH10) was aligned with detector. The 1H-NMR spectra of polymers after the removal of water and monomer by freeze drying were recorded on a 500 MHz Varian Unity/Inova 500B spectrometer in methanol-d4 solvent. Dynamic light scattering (DLS) analysis was conducted on a Malvern Zetasizer Nano ZS. Morphology of the self-assembled structures was observed under a transmission electron microscope (Hitachi 7800 TEM) after making it negatively stained with 2% phosphotungstic acid.3. Results and discussion
3.1. Autonomously driven coupling of subsystems
The main features of our chemical reaction network are presented on Fig. 1. The reaction network system is driven autonomously by the semibatch BrO3−–SO32− pH oscillator, which generates the pH-responsive PEG-b-PDPA building blocks, furthermore regulates and controls the self-organization and the rhythmic dissipative self-assembly of the functional polymer vesicles. In the semibatch reactor the oxidant BrO3− is mixed with the mCTA and the DPA, while the reductant, SO32− is continuously fed at a constant but slow flow rate. There is no outflow from the system, the volume is constantly increasing. In our experiments this reactor setup has some advantages compared with a batch-like or continuous stirred-tank reactor (CSTR) configuration. It allows us to maintain the continuous supply of SO32− at a precisely controlled feed rate while preventing the outwash of the products. | ||
| Fig. 1 Schematic illustration of the pH oscillator driven polymerization induced self-assembly and pH-responsive disassembly and reassembly of PEG-b-PDPA. (A) pH vs. time; Phase I induction period; Phase II oscillatory state. (B) Illustration of the time-regulated steps of the system (C) polymerization of the DPA monomer on PEG-CTA RAFT agent. (D) Protonation and deprotonation of the DPA. (E) Calculated species distribution of H-DPA+ and DPA as a function of the pH. | ||
As was to be expected, the introduction of a system coupled to the pH oscillator, competing for resources, and using the oscillator's chemical output required a readjustment of the working conditions in the core oscillators. This coupling affected greatly the measured pH oscillations (Fig. S1A, ESI†). In our system, one experiment can be divided into two phases. Phase I and II operate in different states, each of which is controlled by different dominating processes. In Phase I, an induction period with high pH is generated instead of oscillations. The oscillations are inhibited by the contribution of the high rate radical consuming polymerization reactions and the buffering effect of the DPA monomers. The HSO3* and OH* radicals are generated by the redox reaction between the BrO3− and the continuously inflowed SO32−. These radicals initiate the polymerization between the PEG-mCTA and DPA (Fig. 1C). Their consumption during the polymerization inhibits the oscillations in Phase I. The ongoing RAFT polymerization results in the growth and extension of the hydrophobic PDPA chain on the originally soluble PEG block attached to the RAFT agent (Fig. 1B and C). After a critical chain length and the critical micelle concentration in the solution are reached, the amphiphiles self-assemble into micelles, and subsequently their morphology transitions to that of vesicles. The transformation to vesicles from micelles is driven by the increase in the DP of the hydrophobic group with a constant hydrophilic block length which, as is well known, leads to a decrease in the effective volume of the stabilizer block and an increase in the packing parameter.46
Between Phases I and II, at the end of Phase I, the oscillatory subsystem switches from its high pH quasi-steady state induction period to an oscillatory regime. This is essentially due to the decrease in the rate of polymerization. The pH drop per se is however the result of the complete oxidation of HSO3−/H2SO3 by BrO3− (Table S1, ESI,† R3 and R4). But now the delayed partial oxidation of H2SO3 (Table S1, ESI,† R5) and the inflow of the HSO3− slowly increase the pH. The pH of the system starts oscillating due to the periodic alteration of the autocatalytic H+ producing and consuming reactions. The changes in the pH govern the protonation and deprotonation of the tertiary amine groups in the PDPA chain (Fig. 1D). When the pH drops, the amphiphilic character of the block copolymer is switched off and becomes doubly hydrophilic (Fig. 1B) due to the protonation of the PDPA chain. This results in the deactivation of the building blocks and disassembly of the previously formed vesicles. However, now the slow delayed negative feedback and the constant inflow of SO32− increases the pH and with it a higher and higher fraction of the total length of the monomers in the PDPA chain becomes unprotonated. This activates the building blocks by controlling their hydrophobicity and switches on their reorganization. The combined effect of all this activity results in that the periodic change in pH, due to the pH oscillator, induces multiple cycles of assembly and disassembly of the polymer structures which are now under its control. Fig. 1E shows the calculated distribution of the H-DPA+ and DPA as a function of the pH within the working pH range of our pH oscillator.47
In our experimental conditions, after 2 oscillatory periods, the system slips out of the oscillatory regime and enters a high pH steady state. Experiments were carried out to understand the chemical background of the transitions. The monomer conversion (%) was determined by 1H-NMR at the end of Phases I and II. The conversion (%) of the sample collected before the first pH drop was calculated to be 69.90% by the area ratio of A(–N(CH(CH3)2)/A(PEG) (chemical shift at ∼3 ppm). The conversion at the end of the second phase was found to be 80.45% (Fig. S3, ESI†). The time interval for Phase I and Phase II is about the same (∼4000 s), but the changes in conversion in these phases are significantly different, which indicates that the transition from the induction period to the oscillatory region is caused by the decreasing rate of polymerization as the concentration of the unreacted monomer is decreased by the end of Phase I. These values also indicate that the polymerization reaction continues to occur on a slower rate during the oscillatory state in Phase II. The reason for the second transition from oscillatory state to steady-state at the end of Phase II, is the consumption of the BrO3−. This is also supported by an experiment in which the addition of extra BrO3− revived the oscillations, as seen in Fig. S2 (ESI†).
We have also checked that the number of oscillatory periods, their amplitude and period are controllable by changing the experimental conditions. For example, increasing the concentration of the BrO3− decreases the period length and increases the total number of oscillatory periods (Fig. S1B, ESI†).
3.2. Changes generated in optical transmission
The polymerization induced self-assembly and the pH change induced disassembly/reassembly results in a significant change in the optical transmission properties of the solution. UV-Vis spectroscopy is a useful tool to follow and study these processes (Fig. 2).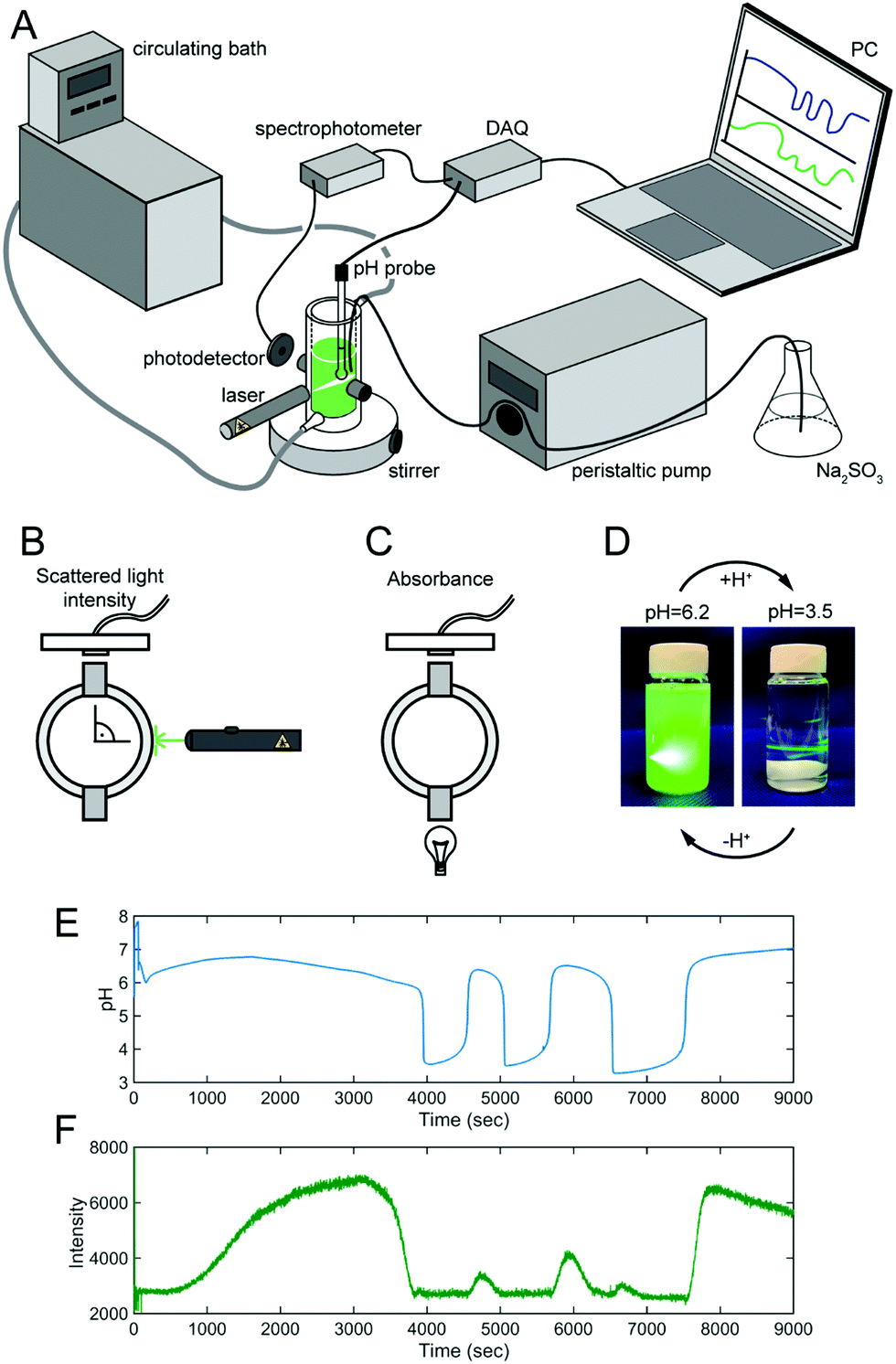 | ||
| Fig. 2 Optical transmission measurments in the system. (A) Illustration of the experimental setup by which the pH and the light transmission can be measured at the same time. (B) The position of the detector and the light source in scattered light intensity measurements. (C) The position of the detector and the light source in light absorbance measurements. (D) The appearance of the reaction mixture at pH = 6.2 and at pH = 3.5 illuminated with the laser light source (λ = 532 nm). (E) Measured pH oscillations when the solution of Na2SO3 (c = 2.0 M) and H2SO4 (c = 0.0675 M) was inflowed at a rate of 0.08 ml min−1 to 15.0 cm3 solution of 0.22 M NaBrO3 containing 6 μmol PEG45-CTA and 360 μmol DPA at T = 40 °C. (F) The intensity of the scattered light in time. | ||
Fig. 2A shows our setup, where changes in the pH and the optical transmission in our system are measured simultaneously. Fig. 2E shows the pH change recorded in the system for DPt = 60 (target degree of polymerization, given by the ratio of mCTA and monomer concentrations). The photodetector of the spectrophotometer was positioned at 90° angle with respect to the laser light source for the measurements of scattered light intensity (Fig. 2B). The changes in the scattered light intensity (caused by the change in solution turbidity as the polymerization progresses), are shown in Fig. 2F. The concomitantly induced self-assembly causes an increase in the turbidity of the reaction mixture. The decrease in pH is accompanied by a decrease in the light intensity. Once the pH drops and the polymeric structures disassemble, the solution becomes clear due to the solubility of the doubly hydrophilic copolymer. Fig. 2D shows the significant difference in the turbidity of the solution at the higher and lower pH. The deprotonation of the PDPA block caused by the increase in the solution pH brings about an increase in the turbidity of the solution caused by the recurrent formation of the polymeric self-assembled structures. The synchronicity between the maximum in pH and scattered light intensity indicates the slower (bulk) intermolecular reorganization of the structures (Fig. 2E and F). This is also supported by our observations concerning the lack of a visible increase in turbidity when the BrO3− concentration of the solution is higher, and the oscillatory period is faster (Fig. S1B, ESI†). We also infer that the appearance of a double peak in the light intensity in Fig. 2F during the second oscillatory period (between 5800 and 7000 s) is, most probably, a sign of a morphology transition. The decrease in scattered light intensity in the high pH state after 8000 s is caused by the aggregation of the polymersomes. The further continuation of SO32− inflow results in a visible precipitation. Fig. S4 (ESI†) shows the changes in light absorbance at two wavelengths (470 and 650 nm) measured in the same experimental configuration but the light source aligned with the photodetector as illustrated in Fig. 2C. Similarly, as in measurements of scattered light intensity, a synchronized increase in the absorbance of the reaction mixture was observed during Phase I with the proceeding polymerization. In Fig. S4 (ESI†) a double peak appears which implies that morphology changes in Phase I take place. Indeed, concurrently with the pH drop the absorbance decreases as the solution becomes clear. Then the absorbance increases as the solution turns turbid again controlled by the increase in pH.
Fig. S5 (ESI†) shows the change in the absorbance of a different system, which consists on the polymerization induced self-assembly of the poly(ethylene glycol)-block-poly(butyl acrylate)-block-poly(acrylic acid) (PEG-b-PBA-b-PAA) copolymer driven by the semibatch BrO3−–SO32−–Fe(CN)64− pH oscillator, as a comparison. In this system, there is no induction period but the pH oscillations start immediately as the SO32− is inflowed to the reaction mixture of BrO3−, Fe(CN)64−, PEG-mCTA and the monomers (AA, BA). Stepwise increments in the absorbance at three different wavelengths (480 nm, 600 nm and 700 nm) are detected, which indicates the presence of a periodic polymerization driven by the pH oscillator. Although the acrylic acid (DPt = 10) makes the hydrophobic block of the copolymer pH-responsive, the third non-responsive PBA block (DPt = 65) prevents the pH-responsive disassembly, since the copolymer remains amphiphilic at any pH. This is supported by the measured time dependence of the absorbance as well, which never returns to the initial value but shows an increase.
3.3. Changes in the average size of the self-assembled functional polymer structures
The change in the average size of the polymeric objects as a function of elapsed time was determined by measuring the particle size distribution (in relative %) using the Dynamic Light Scattering (DLS), which corroborates the formation and the pH-responsive self-assembly and disassembly of these cooperative and self-assembled structures (Fig. 3B).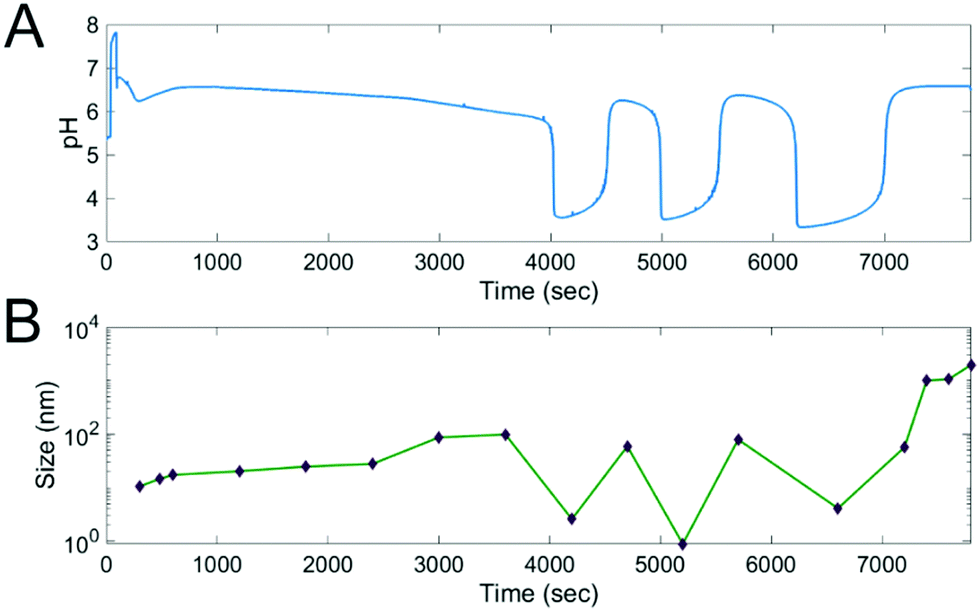 | ||
| Fig. 3 Synchronized changes in the pH and in the average size of the polymer structures. (A) pH in time when the solution of Na2SO3 (c = 2.0 M) and H2SO4 (c = 0.0675 M) was inflowed at a rate of 0.08 ml min−1 to 15.0 cm3 solution of 0.22 M NaBrO3 containing 6 μmol PEG45-CTA and 360 μmol DPA at T = 40 °C. (B) Average sizes of the polymer structures in time determined by DLS in samples collected within 10 min intervals and at characteristic pH values. | ||
In Phase I a slow increment in size was measured (33–121 nm) caused by the increase in the DP. In Phase II the self-assembled object sizes are oscillating and are synchronized with, and by, the periodic pH changes. The pH drop is accompanied by a large decrease in size, indicating the disassembly of the above structures. In contrast, the pH increase is accompanied by an increase in size. At low pH values, the measured average size was 1–9 nm, which is in the size range of the unimers. At high pH values in the oscillatory state, the measured average sizes were 58 nm and 68 nm an fold. The system is highly sensitive to the pH, and after returning to the high pH steady state a large growth can be measured at slightly higher pH values. This high sensitivity to pH can arguably be attributed to changes in the (%) fraction of the protonated and unprotonated DPA at higher pH, which greatly affects the ratio of the hydrophobic and hydrophilic blocks in the synthesized amphiphile (Fig. 1E).
3.4. Morphology of the self-assembled functional polymer structures
The panels B–G in Fig. 4 show the morphology of the polymeric structures characterized by TEM in samples collected at different, selected time points. | ||
| Fig. 4 Size and morphology of the pH oscillator-generated PEG-b-PDPA polymer structures. (A) DLS curves of samples collected at different times (the relating pH in time curve is shown on Fig. 3A). (B–G) TEM images about the formed polymer structures (B = 600 s; C = 2400 s; D = 3600 s; E = 4100 s; F = 4800 s; G = 8700 s). | ||
The measured sizes of the observed structures are in good agreement with the sizes measured by DLS (Fig. 4A). In Phase I, besides the transition from micelles (Fig. 4B), a size growth of the objects can be seen (Fig. 4C and D). (Note the different scale bars.) At low pH, no self-assembled structures can be observed (Fig. 4E). At high pH, reassembly can be seen. The decrease in the number of objects (Fig. 4F) is also a harbinger of incomplete reversibility, along with the determined increase in the polydispersity index (PDI) of samples collected after the disassembly (at 4100 s, 4800 s and 8700 s) (Fig. 4A). Although, we point out that these images only provide us with statistical information on the number of objects, as it is not possible to observe the whole sample under TEM. In samples collected at 8700 s (Fig. 4G) a 900% increase in the diameter of vesicles, were seen. Size and morphology are determined by multiple control parameters. In addition to the DP of the hydrophobic block, the pH of the environment, the continuous changes in the concentration of the solution by the reaction, the inflow and the dilution, all have an impact on the size and the duration of the reorganization.
The role of the pH oscillator in this system goes beyond what we have discussed up to now. In addition to the pH-responsivity of the building blocks, the generated vesicles are functionalized by the PISA process. During their self-assembly they entrap the active reaction mixture. Although the self-assembled vesicles encapsulate the reactants, after this, their contents are cut off from the continuous SO32− supply. This results in different reaction kinetics in the inner and outer media, and a gradient on the two sides of the vesicle layer. The appearance of double peaks in Fig. 2F (between 5800 and 7000 s) and S4 (between 3000 s and 4000 s) indicates a morphology transition driven by the autocatalytic reaction in the inner medium of the active vesicle microreactors.
3.5. Dynamical co-evolution of the free energy landscapes
The BrO3−–SO32− pH oscillator fueled (and controlled) RAFT polymerization of the hydrophobic PDPA on the initially hydrophilic PEG block results in the formation of the amphiphilic building blocks (Fig. 5A). The block copolymer unimers reside higher in the free energy landscape than the water-soluble PEG-mCTA. Their self-assembly into micelles and, later into vesicles is driven by a gain in free energy. | ||
| Fig. 5 Gibbs free energy landscapes. (A) During the polymerization induced self-assembly of the PEG-b-PDPA copolymer driven by the BrO3−–SO32− semibatch pH oscillator; and (B) during the pH-responsive disassembly/reassembly of PEG-b-PDPA regulated by the BrO3−–SO32− semibatch pH oscillator. (B) is an adaptation of Fig. 1C in ref. 1. | ||
In Phase II the H+ production by the autocatalytic processes causes the protonation of the hydrophobic PDPA block of the building blocks and deactivates their action as collective encapsulators due to the induced electrostatic repulsion and change from solvophobic to solvophilic. Energetically these non-assembling doubly hydrophilic building blocks lay on a global minimum of the thermodynamic landscape (Fig. 5B).
The delayed, H+ consuming, negative feedback processes deprotonate the tertiary amine groups of DPA and reactivate the higher energy state building blocks, and therefore induces repetitive self-assembly. The alternation in time of the H+ consuming and producing reactions between the same reactants act as a rhythmic actuator on the protonation state of the pH-responsive polymer segment, repetitively activating and deactivating the building blocks and regulating the periodic assembly-disassembly episodes during the time evolution under the control of the pH oscillator. The inflow of SO32− serves as “fuel” for the reaction network and keeps the system far from equilibrium and oscillating. Discontinuation of the SO32− inflow results in a pH drop at any time, the system reaches its equilibrium state after the consumption of SO32−, the building blocks becomes deactivated and the structures disassemble.
4. Conclusions
Inspired by natural living systems, we have presented in this paper an autonomous and out of equilibrium chemical system designed to couple self-assembly and self-regulation at microscopic length scales. In it, the reaction-network builds its own building blocks and kinetically controls the transient self-assembly of the blocks in a time-synchronized one-pot reaction. The core system, a semibatch BrO3−–SO32− pH oscillator, acts as a radical-source for the RAFT polymerization of a PEG-b-PDPA amphiphilic block copolymer, whose molecules eventually self-assembled dissipatively as micelles and vesicles. This generates a gradient of free energy between the vesicles’ internal and external environments, and an increase in order as the self-assembly takes place. Besides running the PISA process, the pH oscillator has a second role in the system because of its capacity to generate high amplitude pH oscillations as it consumes chemical fuel. The PDPA-block of the copolymer is pH-responsive so that the self-assembled structures into which the amphiphiles self-organize can be functionalized and regulated by changes in pH. Due to this functionalization, the self-assembled structures are also highly responsive to periodic pH changes. Therefore, the pH governs pH-dependent episodes of assembly and disassembly.The core pH oscillator makes the system operate far from thermodynamic equilibrium and is the source of rhythmicity in this system where every process is fueled by the continuous SO32− inflow. The system operates autonomously and there is no need for intermittence of inflow or repetitive addition of chemical fuel: once started, chemistry alone does all the job.
Multiple design control parameters exist in our system. The length of the hydrophobic block, the constant change in the pH of the environment, the continuous changes in the concentrations, and the timescale at which the above happen all have effects on the size, type and function of the structures.
In a semibatch reactor the consumption of the reactants stops the oscillations after two cycles. While we were able to obtain more oscillatory periods at different concentrations, the period times were too fast to observe reorganization of the resulting supramolecular structures in those experiments. Experiments were also performed in a CSTR configuration, in which the oscillations are maintained, and the degree of polymerization is determined by the residence time. Although the semibatch reactor has limitations in terms of the number of periods the reaction undergoes, it allowed us to reach a higher degree of polymerization and consequently the formation of vesicles. All the experience we have gained about the behavior and the challenges of characterization of this system in a semibatch reactor can help in the future to extend the system to work in a CSTR reactor configuration and project it into idealizations of naturally occurring scenarios.
In the future our methodology can be seen as a novel approach to the synthesis of time controlled and programmed smart materials. Besides the functionalization of the layer by the inclusion of a pH-responsive segment into the copolymer, these vesicles have an additional remarkable feature. Their generation in PISA provides a powerful way to create, by encapsulating the reactants, a compartmentalized system containing an active reaction.
Our approach can be extended to different pH-responsive polymers (e.g. ABA, BAB, ABC triblock copolymers) to generate more complex architectures, with new properties and new behaviors, such as rhythmic size oscillations or morphological transitions without the use of biochemistry.
Recently, oscillatory chemistry also has been shown to be capable of doing “native chemical computation”, i.e., advanced computations carried out (at the Turing machine level) by chemistry unaided by external constructs.48,49 Thus, pH oscillations offer the possibility of expanding our system with the important new feature of information-processing, another of the basic properties of living systems.
This opens the door to extend this class of self-encapsulated functional systems to research areas such as the origin of life. For example in geochemical settings, where the changes in pH are induced by complex geochemical processes, some of the properties of the system presented here could be transferred to be under the control of the external geophysical pH oscillator, which would in turn also regulate the behavior of the PISA generated functional vesicles or of pH sensitive factors integrated within the amphiphilic block copolymers.
Funding
This work is supported by Repsol S. A. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. This work was performed in part at the Center for Nanoscale Systems (CNS), a member of the National Nanotechnology Coordinated Infrastructure Network (NNCI), which is supported by the National Science Foundation under NSF award no. 1541959. CNS is part of Harvard University.Author contributions
E. P.-T. and J. P.-M. designed the project, analyzed the data, and wrote the manuscript. E. P.-T. performed the experiments, processed the experimental data, and designed the figures. J. P.-M. coordinated the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the funders for their support for this work. We thank Dr Jinshan Guo for help in 1H-NMR measurements, Dr Chenyu Lin for his help in building the photometric setup and Dr Gong Cheng the useful discussions. J. P.-M. thanks Drs Anders N. Andersen, Bishnu Bastakoti and Jan K. Szymanski for discussions on PISA and its diagnosis.References
- S. A. P. van Rossum, M. Tena-Solsona, J. H. van Esch, R. Eelkema and J. Boekhoven, Chem. Soc. Rev., 2017, 46, 5519–5535 RSC.
- G. J. M. Koper, J. Boekhoven, W. E. Hendriksen, J. H. van Esch, R. Eelkema, I. Pagonabarraga, J. M. Rubi and D. Bedeaux, Int. J. Thermophys., 2013, 34, 1229–1238 CrossRef CAS.
- B. A. Grzybowski, K. Fitzner, J. Paczesny and S. Granick, Chem. Soc. Rev., 2017, 46, 5647–5678 RSC.
- B. Rieß, R. K. Grötsch and J. Boekhoven, Chem, 2020, 6, 552–578 Search PubMed.
- R. Klajn, P. Wesson, K. Bishop and B. A. Grzybowski, Angew. Chem., Int. Ed., 2009, 48, 7035–7039 CrossRef CAS PubMed.
- D. Samanta and R. Klajn, Adv. Opt. Mater., 2016, 4, 1373–1377 CrossRef CAS.
- P. K. Kundu, S. Das, J. Ahrens and R. Klajn, Nanoscale, 2016, 8, 19280–19286 RSC.
- L. L. Yang, Y. H. Bai, X. X. Tan, Z. Q. Wang and X. Zhang, ACS Macro Lett., 2015, 4, 611–615 CrossRef CAS.
- C. G. Pappas, T. Mutasa, P. W. J. M. Frederix, S. Fleming, S. Bai, S. Debnath, S. Kelly, A. Gachagan and R. V. Ulijn, Mater. Horiz., 2015, 2, 198–202 RSC.
- G. Wang and S. Liu, ChemSystemsChem, 2020, 2, e1900046 CAS.
- L. Heinen and A. Walther, Sci. Adv., 2019, 5, eaaw0590, DOI:10.1126/sciadv.aaw0590.
- S. Dimitar, J. Grotzinger and J. Sutherland, Sci. Adv., 2020, 6, eaax3419, DOI:10.1126/sciadv.aax3419.
- R. E. Ireland, Organic Synthesis, Prentice-Hall, Englewood Cliffs, 1969, p. 29 Search PubMed.
- J. Xicart and F. Serratosa, Organic Chemistry in Action: The Design of Organic Synthesis, Elsevier Science, 1996 Search PubMed.
- D. E. Discher and A. Eisenberg, Science, 2002, 9, 967–973 CrossRef PubMed.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- Y. Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- R. T. Pearson, N. J. Warren, A. L. Lewis, S. P. Armes and G. Battaglia, Macromolecules, 2013, 46, 1400–1407 CrossRef CAS.
- J. Yeow and C. Boyer, Adv. Sci., 2017, 4, 1700137 CrossRef PubMed.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS PubMed.
- G. Cheng and J. Perez-Mercader, Macromol. Rapid Commun., 2019, 40, 1800513 CrossRef PubMed.
- S. Pearce and J. Pérez-Mercader, Polym. Chem., 2021, 12, 29–49 RSC.
- J. K. Szymanski and J. Perez-Mercader, Polym. Chem., 2016, 7, 7211–7215 RSC.
- A. N. Albertsen, J. K. Szymański and J. Pérez-Mercader, Sci. Rep., 2017, 7, 41534 CrossRef CAS PubMed.
- B. P. Bastakoti and J. Perez-Mercader, Angew. Chem., Int. Ed., 2017, 56, 12086–12091 CrossRef CAS PubMed.
- B. P. Bastakoti and J. Perez-Mercader, Adv. Mater., 2017, 29, 1704368 CrossRef PubMed.
- B. P. Bastakoti, S. Guragain and J. Perez-Mercader, Chem. – Eur. J., 2018, 24, 10621–10624 CrossRef CAS PubMed.
- J. Guo, E. Poros-Tarcali and J. Perez-Mercader, Chem. Commun., 2019, 55, 9383–9386 RSC.
- L. Hou, M. Dueñas-Díez, R. Srivastava and J. Perez-Mercader, Commun. Chem., 2019, 2, 139 CrossRef CAS.
- G. Cheng and J. Perez-Mercader, Chem, 2020, 6, 1160–1171 CAS.
- M. Orbán, K. Kurin-Csörgei and I. R. Epstein, Acc. Chem. Res., 2015, 48, 593–601 CrossRef PubMed.
- R. Yoshida, T. Sakai, S. Ito and T. Yamaguchi, J. Am. Chem. Soc., 2002, 124, 8095–8098 CrossRef CAS PubMed.
- T. Ueki, M. Shibayama and R. Yoshida, Chem. Commun., 2013, 49, 6947–6949 RSC.
- R. Tamate, T. Ueki, M. Shibayama and R. Yoshida, Angew. Chem., Int. Ed., 2014, 53, 11248–11252 CrossRef CAS PubMed.
- H. Nabika, T. Oikawa, K. Iwasaki, K. Murakoshi and K. Unoura, J. Phys. Chem. C, 2012, 116, 6153–6158 CrossRef CAS.
- I. Lagzi, D. Wang, B. Kowalczyk and B. A. Grzybowski, Langmuir, 2010, 26, 13770–13772 CrossRef CAS PubMed.
- G. Wang, Y. Liu, Y. Liu, N. Xia, W. Zhou, Q. Gao and S. Liu, Colloids Surf., A, 2017, 529, 808–814 CrossRef CAS.
- G. T. Wang, B. Tang, Y. Liu, Q. Y. Gao, Z. Q. Wang and X. Zhang, Chem. Sci., 2016, 7, 1151–1155 RSC.
- T. Ganti, The Principles of Life, Oxford University Press, 2003 Search PubMed.
- D. Deamer, On the origin of systems. Systems biology, synthetic biology and the origin of life, EMBO Rep., 2009, 10, S1–S4 CrossRef CAS PubMed.
- T. G. Szántó and G. Rábai, J. Phys. Chem. A, 2005, 109, 5398–5402 CrossRef PubMed.
- E. Poros, K. Kurin-Csörgei, I. Szalai, G. Rábai and M. Orbán, Chaos, 2015, 25, 064602 CrossRef PubMed.
- A. R. Mukherjee, P. Ghosh, S. C. Chadha and S. R. Palit, Makromol. Chem., 1966, 97, 202–208 CrossRef CAS.
- W. Wu, W. G. Wang, S. Li, J. T. Wang, Q. J. Zhang, X. H. Li, X. L. Luo and J. S. Li, J. Polym. Res., 2014, 21, 494 CrossRef.
- C. Giacomelli, L. Le Men, R. Borsali, J. Lai-Kee-Him, A. Brisson, S. P. Armes and A. L. Lewis, Biomacromolecules, 2006, 7, 817–828 CrossRef CAS PubMed.
- J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press, Elsevier, 2011 Search PubMed.
- I. Puigdomenech, HYDRA and MEDUSA Programs, Royal Institute of Technology, Stockholm, 2006, http://www.kemi.kth.se/medusa/.
- M. Dueñas-Diez and J. Pérez-Mercader, iScience, 2019, 19, 514–526 CrossRef PubMed.
- M. Dueñas-Díez and J. Pérez-Mercader, Sci. Rep., 2020, 10, 6814 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sm00150g |
| This journal is © The Royal Society of Chemistry 2021 |