
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Keratins determine network stress responsiveness in reconstituted actin–keratin filament systems†
Iman
Elbalasy‡
 *ab,
Paul
Mollenkopf‡
ac,
Cary
Tutmarc
ac,
Harald
Herrmann
de and
Jörg
Schnauß
*acf
*ab,
Paul
Mollenkopf‡
ac,
Cary
Tutmarc
ac,
Harald
Herrmann
de and
Jörg
Schnauß
*acf
aPeter-Debye Institute for Soft Matter Physics, Leipzig University, 04103 Leipzig, Germany
bFaculty of Science, Cairo University, 12613, Giza, Egypt
cFraunhofer Institute for Cell Therapy and Immunology, 04103 Leipzig, Germany
dMolecular Genetics, German Cancer Research Centre, 69120 Heidelberg, Germany
eDepartment of Neuropathology, University Hospital Erlangen, 91054, Erlangen, Germany
fUnconventional Computing Lab, Department of Computer Science and Creative Technologies, UWE, Bristol BS16 1QY, UK
First published on 5th March 2021
Abstract
The cytoskeleton is a major determinant of cell mechanics, and alterations in the central mechanical aspects of cells are observed during many pathological situations. Therefore, it is essential to investigate the interplay between the main filament systems of the cytoskeleton in the form of composite networks. Here, we investigate the role of keratin intermediate filaments (IFs) in network strength by studying in vitro reconstituted actin and keratin 8/18 composite filament networks via bulk shear rheology. We co-polymerized these structural proteins in varying ratios and recorded how their relative content affects the overall mechanical response of the various composites. For relatively small deformations, we found that all composites exhibited an intermediate linear viscoelastic behaviour compared to that of the pure networks. In stark contrast, when larger deformations were imposed the composites displayed increasing strain stiffening behaviour with increasing keratin content. The extent of strain stiffening is much more pronounced than in corresponding experiments performed with vimentin IF as a composite network partner for actin. Our results provide new insights into the mechanical interplay between actin and keratin filaments in which keratin provides reinforcement to actin. This interplay may contribute to the overall integrity of cells. Hence, the high keratin 8/18 content of mechanically stressed simple epithelial cell layers, as found in the lung and the intestine, provides an explanation for their exceptional stability.
Introduction
The complex mechanical behaviour of eukaryotic cells is largely determined by the three principal filament systems of the cytoskeleton, i.e. actin filaments (F-actin), microtubules, and intermediate filaments (IFs) as well as their regulated interplay.1 In addition to establishing a cell's specific functional and tissue-related shape, these components and their distinct structures are also crucial for numerous general cellular processes such as stability within tissues as well as cell motility, division, and signal transduction.2 Alterations of these key constituents are the cause of numerous human diseases. It is somehow perplexing that these very same cytoskeletal components can lead to very static, stable cell conformations, which allow, for instance, the formation of stable tissue layers such as the epithelium, or highly dynamic cells during wound healing and cancer metastasis.3 Switching between these physically seemingly contradictory cell states highly depends on the ratios of the individual cytoskeletal components. This becomes especially apparent during embryogenesis when cells need to constantly switch between stable epithelial states and motile mesenchymal states to form complex tissue structures.4 During these switching events, which are the so-called epithelial to mesenchymal transition (EMT) along with the reverse process called mesenchymal to epithelial transition (MET), the cytoskeletal components are very differently expressed. These different compositions lead to different mechanical and motile behaviours. Besides actin-related structures, crucial elements are keratin IFs, which, with their 37 cytoplasmic members, constitute the largest group of the IF protein-family.5,6 They are typically expressed in epithelial cells in various combinations and provide crucial cell type-specific structural support upon mechanical stresses. These diverse mechanical properties are realized by expressing various specific keratin pairs as keratins obligate heterodimeric complexes representing parallel coiled coils made from two alpha-helical molecules of two sequence-related classes each.7 They are also involved in other non-mechanical functions, including regulation of cell growth, migration, and protection from apoptosis.8,9 Several studies have highlighted the potential inhibitory role of keratins for cell mobility and thus the need to downregulate them for EMT to increase cellular mobility. For example, it was found that keratinocytes with all keratins deleted by gene-targeting are softer, more deformable, and more invasive compared to the small overall effect generated after actin depolymerization.10 They are also able to migrate twice as fast as wild type cells.11 Conversely, keratin re-expression in these studies has the opposite effect. This indicates a direct link between downregulation of keratins during EMT and loss of stiffness with increased migration and invasion ability of tumor cells. Although F-actin networks dramatically reorganize as a prerequisite for morphological and migratory changes during EMT and MET, it is indeed remarkable that the entire IF system is rebuilt by a switch between keratin and vimentin expressions.12 As demonstrated for numerous physiological situations, the cytoskeletal systems act synergistically.13 By coordinating their functions, they affect each other's mechanics. However, it is challenging to investigate this mechanical interplay in cells due to their inherent complexity, which makes it difficult to disentangle mechanical crosstalk and regulatory biochemical interactions. In this respect, reconstituted composite cytoskeletal filament networks have been investigated in vitro by quantitative rheological measurements and theoretical modelling. The results have often revealed unexpected mechanical responses. For instance, the presence of microtubules was found to induce unexpected local compressibility14 and non-linear strain stiffening in actin networks.15,16 Concomitantly, actin reinforces microtubules against compressive loads.17When designing and interpreting such in vitro studies, it is important to choose the system parameters carefully as observed when measuring the mechanical properties of composite filament networks of actin and vimentin. Holding the molecular content constant for different mixing ratios of actin and vimentin has yielded contradictory results for their mechanical response, namely a synergistic increase in the linear elasticity18 and formation of stiffer or softer networks depending on the investigation technique.19,20 In this case, however, vimentin filaments carry roughly twice as many monomers per unit length than actin filaments, i.e. total filament length is half compared to that of F-actin. Consequently, it has been shown that actin–vimentin filament composites with the same total polymer length and mesh size, can be described by a superposition of the mechanical properties of the underlying constituents.21
Due to their intrinsic ability to self-assemble in vitro, the co-polymerization of actin and keratin filaments is possible without additional accessory proteins. Hence, it has been recently demonstrated that encapsulation of actin and keratin within vesicles leads to the formation of composite filament networks, where F-actin acts as steric resistance for keratin IFs preventing their collapse to bundled clusters.22
Here, we investigate the mechanical properties of in vitro reconstituted F-actin and keratin 8/18 (K8–K18) composite networks by bulk shear rheology measurements. We aim to characterize networks with similar total filament lengths. By systematically varying the relative mass ratios of actin and keratin, we investigate how their relative presence impacts the overall mechanical response of the composites. We complement these rheology measurements with confocal microscopy of the composites.
Experimental
Polymer lengths
The mass per unit length (mL) for actin is 2.66 × 10−11 g m−1,23 and for K8–K18 in Tris-based assembly buffer is 3.16 × 10−11 g m−1.24 At 0.5 mg ml−1 actin monomers (11.9 μM) and 0.6 mg ml−1 K8–18 tetramers (11.34 μM), both actin and keratin filament networks will have similar total filament length. Using these concentrations as our boundary conditions, we can select actin–keratin mixtures with the same total polymer length per unit volume.Protein preparation and co-polymerization
G-actin was prepared from rabbit muscle and stored at −80 °C in G-Buffer (2 mM Tris–HCl pH 7.5, 0.2 mM ATP, 0.1 mM CaCl2, 1 mM DTT, 0.01% NaN3, pH 7.8) as described previously.25 Vectors containing keratin genes (pET 24a–K8 and pET 23a–K18) were transformed into E. coli BL21 for protein expression. Recombinant K8 and K18 were isolated and purified as previously described.26For reconstitution, purified K8 and K18 proteins were mixed in equimolar amounts and renatured by stepwise dialysis against 8 M urea, 2 mM Tris–HCl, 1 mM DTT, pH 9.0 with stepwise reduction of urea concentration (6 M, 4 M, 2 M, 1 M, and 0 M). Each dialysis step was done for 20 minutes at room temperature, then the dialysis was continued overnight against 2 mM Tris–HCl, 1 mM DTT, pH 9.0 at 4 °C. The final protein concentration was determined by measuring the absorption at 280 nm using a DU 530 UV/vis Spectrophotometer (Beckman Coulter Inc., USA). Composite networks were prepared by mixing actin monomers at 0.5 mg ml−1 and K8–K18 tetramers at 0.6 mg ml−1 in varying mixing ratios. Assembly of pure and mixed networks was initiated by adding 1/10 volume of 10× F-buffer (20 mM Tris–HCl, 1 M KCl, 10 mM MgCl2, 2 mM ATP, 10 mM DTT, pH 7.5) to the protein sample.
Shear rheology
Rheology measurements were conducted using a strain-controlled ARES (TA Instruments, USA) and a MCR 502 WESP (Anton Paar, Austria) rheometer using 40 mm plate–plate geometry with 140 μm gap width at 20 °C. In all measurements, F-buffer was mixed with the protein mixture on ice, and a sample volume of 200 μl was loaded quickly between the two plates. F-Buffer with the same conditions as in the sample was distributed around the sample to prevent artefacts from interfacial elasticity and a solvent trap was placed around the sample to prevent evaporation. The sample was given two hours to equilibrate between the rheometer plates as the filament assembly was monitored with a dynamic time sweep for 2 hours, one data point per minute at a frequency (ω) of 1 Hz and a strain (γ) of 2%. The linear viscoelastic response of equilibrated networks was measured with dynamic frequency sweeps ranging from 0.01 Hz to 80 Hz at a strain of 2% and 20 points per decade. Data points plotted in Fig. 2 are the mean of at least five independent rheology measurements. A transient step rate test with a strain rate of 0.1 s−1 was applied to measure the strain-dependent stress in the non-linear strain regime. The differential shear modulus (K) was determined with a self-written Python script, calculating the gradient of the smoothed stress data divided by the strain step width. The linear differential shear modulus Klin is given by the first non-negative value of the smoothed stress data, whereas negative stress is measured due to technical limitations, particularly for small strains, lacking every physical relevance. The linear frequency sweeps as well as the nonlinear step rate measurements were evaluated in the frame of the GWLC model with a self-written Python script. At least six independent rheology measurements were taken into account for the evaluation and comparison with the model predictions. Details about the model are presented in the ESI.†Protein labelling, sample preparation and imaging
We have developed a new method for direct labelling of the wild type K8–K18 without mutation, unlike the techniques used in previous studies.22,27 The labelling is based on the coupling of Atto-488-NHS-ester (ATTO-TEC, Siegen, Germany) to the free amine (NH2) residues (lysine or terminus) in K8 tetramers. K8 was renatured by stepwise dialysis against the labelling buffer (2 mM sodium phosphate, pH 8.5) as described above. K8 tetramers were mixed with Atto-488-NHS-ester solubilized in DMSO at 10 mM at a molar ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The mixture was incubated in the dark for 1 hour at room temperature. In one step, the free dye was removed, and labelled K8 was denatured into monomers by overnight dialysis against 8 M urea, 2 mM sodium phosphate buffer pH 8.5 at 4 °C. On the second day, the labelling buffer was exchanged by the dialysis buffer (8 M urea, 2 mM Tris–HCl, pH 9.0) by dialysis for 2 hours at room temperature. The concentration of labelled K8 was determined by measuring the absorbance at 280 and 500 nm using a DU 530 UV/vis Spectrophotometer (Beckman Coulter Inc., USA). Labelled K8 was then distributed to appropriate aliquots and stored at −80 °C. One day before imaging, equal amounts of 20% labelled K8 were mixed with unlabelled K8 and K18 in 8 M urea, 2 mM Tris–HCl, pH 9.0 to have a final sample labelling of 10% by concentration. This mixture was renatured into tetramers by stepwise dialysis against 2 mM Tris–HCl, pH 9.0.
:1. The mixture was incubated in the dark for 1 hour at room temperature. In one step, the free dye was removed, and labelled K8 was denatured into monomers by overnight dialysis against 8 M urea, 2 mM sodium phosphate buffer pH 8.5 at 4 °C. On the second day, the labelling buffer was exchanged by the dialysis buffer (8 M urea, 2 mM Tris–HCl, pH 9.0) by dialysis for 2 hours at room temperature. The concentration of labelled K8 was determined by measuring the absorbance at 280 and 500 nm using a DU 530 UV/vis Spectrophotometer (Beckman Coulter Inc., USA). Labelled K8 was then distributed to appropriate aliquots and stored at −80 °C. One day before imaging, equal amounts of 20% labelled K8 were mixed with unlabelled K8 and K18 in 8 M urea, 2 mM Tris–HCl, pH 9.0 to have a final sample labelling of 10% by concentration. This mixture was renatured into tetramers by stepwise dialysis against 2 mM Tris–HCl, pH 9.0.
For network visualization, labelled K8–K18 prepared in the previous step was mixed with unlabelled K8–K18 tetramers in a ratio of 1:10, with a final protein concentration of 0.6 mg ml−1. Fluorescently labelled actin was prepared by mixing G-actin at 5 μM with phalloidin–tetramethylrhodamine B isothio-cyanate (phalloidin–TRITC – Sigma-Aldrich Co.) in a molar ratio of 1:1. For composite networks, 10% labelled keratin samples were mixed with labelled actin samples in varying ratios. Networks were created by adding 1/10 volume fraction of 10× F-buffer to the sample and immediately pipetting it into an experimental sample chamber to rest for 2 hours at room temperature before imaging as described in ref. 28. Images were captured using a spinning disc confocal microscope (inverted Axio Observer.Z1/Yokogawa CSU-X1A 5000, Carl Zeiss Microscopy GmbH, Germany), 100× oil immersion objective NA 1.40 with a Hamamatsu camera at an exposure time of 100 ms.
Results and discussion
Co-polymerization and filament length
In order to investigate the mechanical properties of composite filament networks of F-actin and K8–K18, we chose the initial concentrations of actin monomers and keratin tetramers that resulted in comparable total filament lengths of actin and keratin as depicted in Fig. 1. | ||
| Fig. 1 Illustration of F-actin–keratin composite filament networks with varying mixing ratios. By co-polymerizing actin monomers at 0.5 mg ml−1 and keratin tetramers at 0.6 mg ml−1, an intermixed network of actin (red) and keratin (green) filaments is formed. At the noted concentrations, networks with the total polymer lengths of the composite will be approximately the same. The ability of actin to pervade the keratin networks, depending on its content, impacts the architecture and mechanics of the composites. As depicted, actin and keratin differ in their persistence length with Lp ≈ 10 μm and 0.5 μm respectively. | ||
We reconstituted composite networks of actin and keratin by co-polymerization of actin monomers and tetrameric complexes of K8 and K18. It is well known that the ionic requirements for polymerization of actin and keratin are quite different. Keratin can assemble in vitro into networks in an unusually low ionic strength buffer24 and addition of ions such as magnesium (Mg2+) or potassium (K+) at physiological concentrations is enough to induce immediate bundling, resulting in heterogeneous networks.29–31 In contrast, for actin the addition of mono- and divalent ions to physiological concentrations is essential to initiate the polymerization from globular actin (G-actin) to filamentous actin (F-actin) to form entangled networks.32 A striking difference between actin and keratin filaments is the filament persistence length, which is the length scale for the decay of the tangent–tangent correlation along the filament contour, and is proportional to the stiffness of the polymer.33 Keratins have a significantly smaller persistence length (Lp) than actin, with Lp ≈ 0.5 μm and 10 μm, respectively.34 Here, we assessed the polymerization efficiency of keratin and actin in different concentrations of MgCl2 (1, 0.5, 0.1 and 0 mM) by high-speed sedimentation of protein assemblies followed by SDS-PAGE of pellet and supernatant fractions (data not shown). We found that a lack of Mg2+ ions had no effect on the polymerization efficiency of keratin, and soluble keratin tetramers were polymerized completely into filaments, whereas for actin at least 1 mM MgCl2 was required to ensure complete polymerization. Thus, we had to adjust the polymerization buffer (see Materials and methods) to include a concentration of 1 mM MgCl2 for the co-polymerization in order to form actin–keratin filament networks.
Linear viscoelasticity of pure actin and keratin filament networks
Using bulk shear rheology, the linear viscoelastic properties of polymer solutions can be quantified by the frequency (ω) dependent complex shear modulus G*(ω) = G′(ω) + iG′′(ω), where G′ and G′′ are the elastic and viscous moduli, respectively. Under the selected assembly conditions, pure K8–K18 filament networks at 0.6 mg ml−1 exhibit predominantly elastic behaviour with G′ being consistently larger than G′′ by nearly one order of magnitude (Fig. 2A). Accordingly, the loss factor tan(ϕ) = G′′/G′ has a small value (tan(ϕ) at 1 Hz = 0.13), indicative of highly elastic networks. Over the entire frequency range, G′ and G′′ show a very weak frequency dependence. A weak power law was obtained by a linear fit of G′ in the log–log-plot yielding a power-law exponent of α(G′) = 0.07. We observed no G′′/G′ crossover in the measurable frequency range for pure keratin. This is consistent with results previously reported for K8–K18 network bulk rheological behaviour.24,35,36 By comparison, G′ and G′′ of pure F-actin filament networks showed a more pronounced frequency dependence with α(G′) = 0.23, (Fig. 2E). Accordingly, the crossover between G′ and G′′ regimes was observed at low frequency. Actin networks have a larger loss factor (tan(ϕ) at 1 Hz = 0.52) and, consequently, the elastic contributions are less dominant than in pure keratin networks. These mechanical features are similar to those obtained for entangled actin networks under comparable conditions.21,37 | ||
| Fig. 2 Linear viscoelasticity of reconstituted filemant networks. By comparison, the keratin network (A) shows more predominant elasticity with a weaker frequency dependence of both elastic G′ (filled symbols) and viscous G′′ (open symbols) moduli than the F-actin network (E); the keratin network is more elastic (tanϕker = 0.13) than the F-actin network (tanϕact= 0.52) with no observed G′/G′′ crossover, which, in contrast, is a signature of actin networks in this frequency range. Actin–keratin composite filament networks with varying mixing ratios of actin and keratin (B–D) show intermediate viscoelastic properties. With increasing actin/decreasing keratin content, the frequency dependence of G′ and G′′ moduli increases gradually, and the networks become less elastic as the loss factor values increase. Consequently, the G′/G′′ crossover appears at high frequency in keratin-dominated networks and shifts gradually to lower frequencies with increasing actin content. α represents the slope between 0.1 and 10 Hz of G′. Data points in all curves represent the mean of at least five independent measurements and error bars represent the standard deviation from the mean. | ||
Composite filament networks exhibit an intermediate linear viscoelastic behaviour
To date, previous in vitro studies have investigated the mechanical properties of only one-component systems of either actin filaments as entangled38–40 and crosslinked28,41–44 networks or keratin single filaments27 and networks.24,29,35,36,45In our study, composite networks of F-actin and K8–K18 filaments with varying mixing ratios revealed an intermediate linear viscoelastic behaviour with regard to their composite-specific protein content. With increasing actin/decreasing keratin contents as shown in Fig. 2B–D, the dependence of G′ and G′′ on the frequency increased gradually as indicated by the gradual increase in α values; α = 0.09 in keratin-dominated networks (0.45 mg ml−1 K8–K18–0.125 mg ml−1 actin), 0.16 in equal ratio-networks (0.3 mg ml−1 K8–K18–0.25 mg ml−1 actin) and 0.21 in F-actin-dominated networks (0.15 mg ml−1 K8–K18–0.375 mg ml−1 actin). Furthermore, the crossover to the predominant viscous regime, observed only in F-actin networks, started to appear in keratin-dominated networks at high frequencies. With increasing actin content, actin contributes more to the composite's behaviour, shifting the crossover point gradually to lower frequencies. The loss factor tan(ϕ) increased with increasing actin/decreasing keratin content, which indicates a smooth transition to less elastic networks, as shown in Fig. 3A. We used a Mann–Whitney U test to investigate the statistical significance of the loss factor values with respect to each other. We found p-values less than 0.05, indicating significant difference, for all compared distributions except for the pure networks compared to composites with a respective division of 0.75/0.25 (Table S1, ESI†). In Fig. 3B, we show that the elastic moduli G′ for all networks exhibit only minor variations.
 | ||
| Fig. 3 Mechanical properties of pure and composite filament networks in the linear regime. (A) Mean values of the loss factor tan(ϕ) at f = 1 Hz of composite networks showing a gradual increase in the loss factor values, i.e. the network's viscosity, with increasing actin/decreasing keratin content. We found the distributions to be significantly different except for the constellations of pure actin compared to 0.15 mg ml−1 keratin–0.375 mg ml−1 actin and pure keratin compared to 0.45 mg ml−1 keratin–0.125 mg ml−1 actin respectively. (B) The plateau modulus G0 = G′ (f = 1 Hz) for all networks shows only minor variations between networks in the linear regime while in (C) the slopes α show a gradual increase with actin content where we found distributions differing significantly except for the constellation of pure actin compared to 0.15 mg ml−1 keratin–0.375 mg ml−1 actin (p-value = 0.5). In (A)–(C) dots represent single measurements and error bars represent the standard deviation from the mean. | ||
Filament–filament interactions constitute a fundamental cause for mechanical responses
Inter-filament interactions are directly related to microscopic details of polymers constituting a network, which, however, are mostly neglected in theoretical approaches. Thus, we have limited our argumentations to points we could directly control in the experiments or which were predefined by the polymer type. The transition from keratin-rich to actin-rich networks involves a gradual increase of the frequency dependence of the elastic modulus as illustrated in Fig. 3C, showing the slopes of G′ between 0.1 and 10 Hz for respective composites.The weak power law behaviour exhibited in actin filament networks can be described by the glassy worm-like chain model (GWLC), where the exponent of this power law depends on the level of pre-stress and the interaction strength (ε).46 This interaction strength is a phenomenological parameter that is typically neglected in models of entangled networks such as the tube model.47 The fundamental idea of the GWLC is an exponential stretching of a wormlike chain's mode relaxation times (τn) according to:

 | ||
| Fig. 4 Attractive filament–filament interactions (captured in the stickiness parameter ε) determine the mechanical response of composite filament networks in linear rheology. Normalized storage moduli (G′) versus frequency for different actin/keratin compositions evaluated with the glassy wormlike chain model. Solid lines represent the measured data and dashed lines represent the fitting curves, yielding values for stickiness ε as shown. For pure keratin, the model could not be fitted to the data. | ||
Fitting the GWLC model to the experimental data yields values for ε that indicate stronger attractive filament–filament interactions for increasing keratin contents in composite networks (Fig. 4). While actin networks are considered a model system for entangled networks, keratin networks form sticky clusters due to pronounced hydrophobic interactions36,46 and, therefore, pure keratin networks are not readily accessible to the GWLC model.
As the GWLC model is based on the assumption of isotropic networks, it cannot capture pure keratin systems due to the inherent bundling under the investigated conditions (Fig. S4, ESI†). Cluster-forming networks may be better described by models for crosslinked networks such as an affine model.48 Recently, a detailed model for bundling of keratin was suggested based on the interplay between inter-filament electrostatic and hydrophobic interactions. This model predicts that the process of keratin bundling is determined by the electric charge of the filaments, the number of hydrophobic residues, and the exclusion of the ions from the bundle interior.49 Compared with actin, the attractive interactions between filaments in pure keratin structures are much stronger than those in entangled actin networks.37 Consequently, the viscous loss modulus in keratin networks during oscillatory shear is significantly reduced compared to the viscous dissipation in actin networks.50 Therefore, the viscous modulus G′′ of actin networks was observed steeply increasing towards the elastic modulus G′, and the crossover point was observed in the linear regime.
This denotes the transition from a regime dominated by filament interactions within the network to a regime dominated by single filament behaviour, which is a signature of actin networks.51 The resulting parameters for the stickiness parameter ε are shown in Fig. 4. For increasing keratin content, we observed a stronger attractive interaction between individual filaments, reflected in higher ε values.
Keratin induces strain stiffening in the non-linear regime
Strain stiffening of biopolymer networks is a feature shared by various intermediate filament proteins.52 This physical response is of particular physiological significance for keratins, which make up a major portion of structural proteins found in epithelial cells, and provide protection against large-scale deformation. Interestingly, keratin filament networks exhibit strain stiffening even in the absence of crosslinkers or divalent cations,24,36 while in actin networks the strain stiffening depends on the crosslinks.51,53These features of keratin and actin filament assemblies are reflected in the differential shear modulus K for the different composite networks. For networks with lower keratin content, we see weak strain-stiffening effects comparable to that of pure actin networks. This is expressed in a maximum value K that is two orders of magnitude lower than for pure keratin and keratin-dominated networks (Fig. 5, inset) appearing at stresses shifted to values more than one order of magnitude lower (Fig. 5). Besides distinctly higher values for the differential modulus, we see a clear increase in yield stresses for networks of high keratin content. This becomes evident from the shift of maxima in stress–strain relations towards higher strains, translating to higher stresses where the differential modulus intersects the x-axis as illustrated in Fig. 5. The onset of the differential modulus occurs at stresses/strains that are comparable for all networks except for the pure actin, where the onset stress is shifted an order of magnitude to higher stresses.
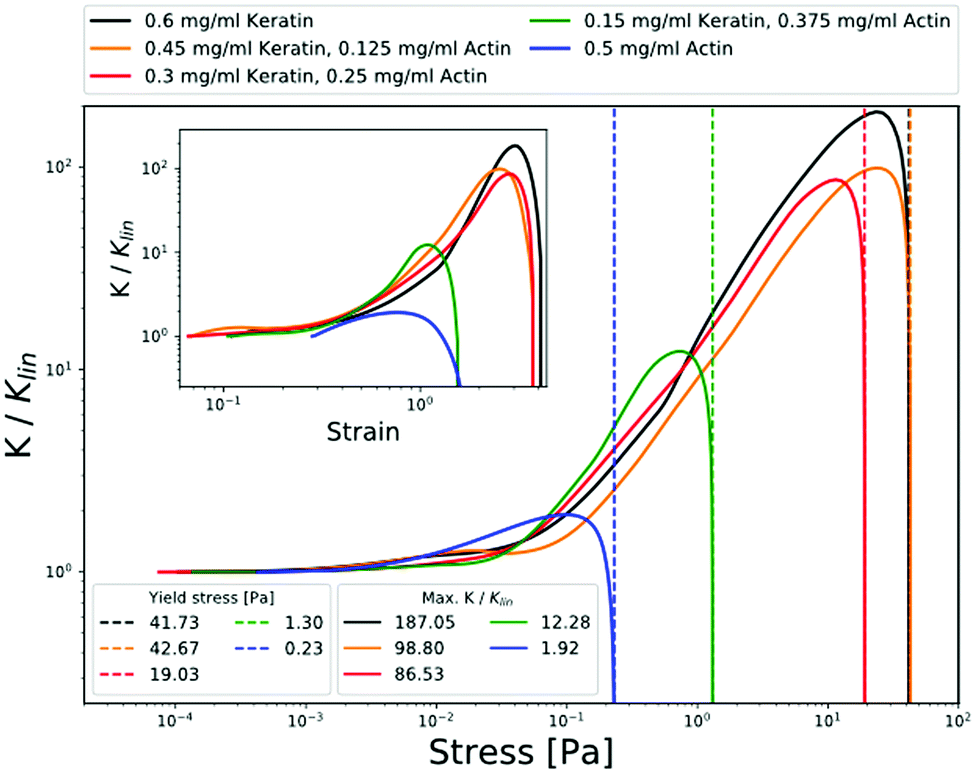 | ||
| Fig. 5 Actin–keratin filament composite networks exhibit strain stiffening increasing in manifestation with increasing keratin content. This is expressed in the differential shear modulus K = dσ/dγ rescaled by its value in the linear regime Klin and plotted versus stress σ. Solid lines are measurement curves and dashed vertical lines indicate the yield stresses. The inset shows K/Klinversus strain. | ||
Network architectures
Using spinning disc confocal microscopy, we examined the architecture of all filament networks (10% labeled sample) under these assembly conditions. As mentioned above, keratin is known to quickly form networks due to its high self-affinity, and bundled networks if positively charged ions are present.24,31 We initiated the assembly at low temperature by mixing the protein solution with the assembly buffer on ice in order to slow down the assembly process. A dense, heterogeneous network was obtained as shown in Fig. 6A. Interestingly, a bundled keratin network was also formed even at a very low protein concentration (e.g. as low as 0.1 mg ml−1) (Fig. S1A, ESI†). In the absence of salts, keratin can assemble into isotropic networks even at high protein concentrations (Fig. S1B, ESI†). By contrast, actin at 0.5 mg ml−1 assembled solely into highly isotropic entangled networks under selected ionic conditions (Fig. 6B). Actin filaments often associate into bundles and networks with diverse structures only in the presence of “bundling factors” such as high salt conditions or actin-binding proteins.54–56 Consequently, bundling of actin was not an issue, and actin filaments completely arranged into isotropic networks under the investigated conditions. | ||
| Fig. 6 Confocal micrographs of in situ formed filmanet networks. Keratin assembles into a dense heterogeneous network immediately after the addition of F-buffer (A), actin assembles solely into an isotropic entangled network as no accessory proteins or crosslinking factors have been used (B). In keratin-dominated networks, a complete co-localization of F-actin and keratin filament networks culminates in a large cluster (C). In equal ratio and F-actin-dominated networks, the isotropic actin networks provide steric hindrance against tight bundling of keratin, and thus keratin appears as filaments and small clusters in these composites (D and E respectively). Scale bar for all images: 10 μm. | ||
In composite networks, actin provides steric hindrance, creating “obstacles” in between keratin filaments, thereby preventing or reducing their tight bundling. This observation is consistent with a previous in vitro study on actin–keratin composites encapsulated in vesicles.22 However, we observed that this steric effect became less pronounced in increasingly keratin-dominated networks (Fig. 6C). Under these conditions, actin and keratin were observed completely co-localized, but mostly appeared as a large cluster. In actin-dominated regimes as well as equal ratio networks, keratin filaments appeared as extended networks or as very small clusters surrounded by homogenous actin filament networks (Fig. 6D and E). In all composites, both keratin and actin networks did not demix, but appeared as interdependent elements.
To visualize keratin networks, we developed a new method for labeling the wild type keratin without mutations (described in Materials and methods). As a control, we tested the bulk mechanical properties of the 10% labeled keratin samples. We found that these labeled samples behave as unlabeled keratin, indicating that the filaments and network properties were not affected by the presence of 10% labeled keratin (Fig. S2, ESI†).
Conclusions
We investigated the bulk rheology of composite networks made from F-actin and K8–K18 filaments at varying relative concentrations. We show that for small deformations, i.e., the linear deformation regime, the composites revealed an intermediate linear viscoelastic behaviour compared to that of the pure networks. This provides important new information about the mechanical coupling between networks of these two structural proteins within the cell environment. Similar to mixed F-actin/vimentin IF networks,21 the mechanics of composite F-actin/K8–K18 filament networks can be described from their respective substructures as a superposition in the frame of the GWLC model.21 This suggests that cells can tune their network mechanics by varying the relative ratios of actin and keratin. They may likewise utilize this tunability when changing their viscoelastic properties by varying cytoskeletal network components to meet the mechanical demands in various different simple epithelia such as found in lung and small intestine. These changes are especially important for large deformations, i.e. non-linear deformations. The F-actin/K8–K18 filament composites show drastic strain stiffening under larger deformations, which is induced and dominated by the keratin content. Strain stiffening can be especially used to absorb large external forces, ensuring that tissue structures such as the epithelium remain stable. This may provide insights into the mechanical interplay they might display in a physiological situation, for instance, for cells integrated into an epithelial cell layer when starting to turn into a migrating cell during and after EMT. In the latter situation, cells build up a vimentin IF system, which provides completely different mechanical properties.57–59 Hence, this change of the IF system is probably of direct importance to the process as vimentin IF provides a much softer counterpart system to F-actin with strain stiffening more than one order of magnitude lower than K8–K18 IF.21Here, our rheological measurements are in the same line with the concept that the downregulation of K8–K18 may contribute to the loss of cell stiffness. This is supported by several knockout experiments showing that cells lacking keratins are more deformable and invasive,10 migrating faster than wild type cells.11 These effects are also much more pronounced than the softening effects resulting from actin depolymerization.10 In particular, loss of K8–K18 in epithelial cancer cells was found to increase collective cell migration.60
To study how actin and keratins affect each other's organization, we have developed a suitable labelling procedure for K8–K18, which enabled us to find that actin sterically hinders and effectively reduces the tight bundling of keratin in some composites. This observation highlights the supportive role of actin within the cell as it enables keratin networks to extend in order to provide protection to the entire cell. Previous reports demonstrate that the downregulation of keratins can provide space in the cell periphery to enable F-actin networks to reorganize more freely to form protrusions for migration, while the presence of an intact keratin network in the cell periphery, however, slows down actin reorganization.61,62 These results support our observation that this hindrance effect provided by actin diminishes in keratin-dominated networks.
The behaviour of composite cytoskeletal networks in cells is also highly affected by different crosslinkers. For example, many actin crosslinkers mediate the formation of actin filament bundles in cells63 and induce strain stiffening in in vitro reconstituted actin filament networks.51,53 In the case of keratin, depletion of keratin-associated plectin, which is able to crossbridge individual IFs and to connect them to other cytoskeletal components, alters the organization and dynamics of keratin, but does not affect the overall mechanical properties of the cell.64,65 Further studies centering on authentic cellular cytolinkers such as plectin that crossbridge keratin and actin filaments are necessary to begin to understand the complex and dynamic behaviour these composite networks exhibit in a living cell.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Dr Josef Käs for fruitful discussions. We acknowledge funding by the ESF: European Social Fund for I. E. (ESF—100327895), P. M. (ESF—100316844), and C. T. (ESF—100380880). Furthermore, we acknowledge funding by the European Research Council (ERC-741350) and the German Research Foundation (INST 268/296-1 FUGG & HE 1853/11-1).References
- D. A. Fletcher and R. D. Mullins, Nature, 2010, 463, 485–492 CrossRef CAS PubMed.
- T. Hohmann and F. Dehghani, Cells, 2019, 8, 362 CrossRef CAS PubMed.
- F. Huber, J. Schnauss, S. Ronicke, P. Rauch, K. Muller, C. Futterer and J. Kas, Adv. Phys., 2013, 62, 1–112 CrossRef CAS PubMed.
- D. H. Kim, T. Xing, Z. Yang, R. Dudek, Q. Lu and Y.-H. Chen, J. Clin. Med., 2018, 7, 1 CAS.
- H. Herrmann and U. Aebi, Cold Spring Harbor Perspect. Biol., 2016, 8, a018242 CrossRef PubMed.
- J.-F. Nolting and S. Köster, New J. Phys., 2013, 15, 045025 CrossRef CAS.
- J. T. Jacob, P. A. Coulombe, R. Kwan and M. B. Omary, Cold Spring Harbor Perspect. Biol., 2018, 10, a018275 CrossRef.
- S. Yoon and R. E. Leube, Essays Biochem., 2019, 63, 521–533 CrossRef CAS.
- X. Pan, R. P. Hobbs and P. A. Coulombe, Curr. Opin. Cell Biol., 2013, 25, 47–56 CrossRef CAS PubMed.
- K. Seltmann, A. W. Fritsch, J. A. Kas and T. M. Magin, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18507–18512 CrossRef CAS PubMed.
- K. Seltmann, W. Roth, C. Kroger, F. Loschke, M. Lederer, S. Huttelmaier and T. M. Magin, J. Invest. Dermatol., 2013, 133, 181–190 CrossRef CAS PubMed.
- B. O. Sun, Y. Fang, Z. Li, Z. Chen and J. Xiang, Biomed. Rep., 2015, 3, 603–610 CrossRef CAS PubMed.
- M. Schoumacher, R. D. Goldman, D. Louvard and D. M. Vignjevic, J. Cell Biol., 2010, 189, 541–556 CrossRef CAS PubMed.
- V. Pelletier, N. Gal, P. Fournier and M. L. Kilfoil, Phys. Rev. Lett., 2009, 102, 188303 CrossRef PubMed.
- Y.-C. Lin, G. H. Koenderink, F. C. MacKintosh and D. A. Weitz, Soft Matter, 2011, 7, 902–906 RSC.
- S. N. Ricketts, J. L. Ross and R. M. Robertson-Anderson, Biophys. J., 2018, 115, 1055–1067 CrossRef CAS PubMed.
- C. P. Brangwynne, F. C. MacKintosh, S. Kumar, N. A. Geisse, J. Talbot, L. Mahadevan, K. K. Parker, D. E. Ingber and D. A. Weitz, J. Cell Biol., 2006, 173, 733–741 CrossRef CAS PubMed.
- O. Esue, A. A. Carson, Y. Tseng and D. Wirtz, J. Biol. Chem., 2006, 281, 30393–30399 CrossRef CAS PubMed.
- M. H. Jensen, E. J. Morris, R. D. Goldman and D. A. Weitz, Bioarchitecture, 2014, 4, 138–143 CrossRef PubMed.
- H. Lopez-Menendez and L. Gonzalez-Torres, J. Mech. Phys. Solids, 2019, 127, 208–220 CrossRef CAS.
- T. Golde, C. Huster, M. Glaser, T. Handler, H. Herrmann, J. A. Kas and J. Schnauss, Soft Matter, 2018, 14, 7970–7978 RSC.
- J. Deek, R. Maan, E. Loiseau and A. R. Bausch, Soft Matter, 2018, 14, 1897–1902 RSC.
- U. Aebi, R. Millonig, H. Salvo and A. Engel, Ann. N. Y. Acad. Sci., 1986, 483, 100–119 CrossRef CAS PubMed.
- P. Pawelzyk, H. Herrmann and N. Willenbacher, Soft Matter, 2013, 9, 8871 RSC.
- B. Gentry, D. Smith and J. Kas, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2009, 79, 031916 CrossRef PubMed.
- K. L. A. U. Herrmann Hh, Methods Cell. Biol., 2004, 78, 3–24 CrossRef.
- C. Lorenz, J. Forsting, A. V. Schepers, J. Kraxner, S. Bauch, H. Witt, S. Klumpp and S. Koster, Phys. Rev. Lett., 2019, 123, 188102 CrossRef CAS PubMed.
- D. Strehle, J. Schnauss, C. Heussinger, J. Alvarado, M. Bathe, J. Kas and B. Gentry, Eur. Biophys. J., 2011, 40, 93–101 CrossRef CAS PubMed.
- A. Leitner, T. Paust, O. Marti, P. Walther, H. Herrmann and M. Beil, Biophys. J., 2012, 103, 195–201 CrossRef CAS PubMed.
- I. Martin, M. Moch, T. Neckernuss, S. Paschke, H. Herrmann and O. Marti, Soft Matter, 2016, 12, 6964–6974 RSC.
- C. Y. Hemonnot, M. Mauermann, H. Herrmann and S. Koster, Biomacromolecules, 2015, 16, 3313–3321 CrossRef CAS PubMed.
- H. Lodish, A. Berk, S. L. Zipursky, P. Matsudaira, D. Baltimore and J. Darnell, Cell Motility and Shape I: Microfilaments, Molecular Cell Biology, W. H. Freeman, 2000 Search PubMed.
- Q. Wen and P. A. Janmey, Curr. Opin. Solid State Mater. Sci., 2011, 15, 177–182 CrossRef CAS PubMed.
- A. F. Pegoraro, P. Janmey and D. A. Weitz, Cold Spring Harbor Perspect. Biol., 2017, 9, a022038 CrossRef PubMed.
- S. Yamada, D. Wirtz and P. A. Coulombe, Mol. Biol. Cell, 2002, 13, 382–391 CrossRef CAS PubMed.
- P. Pawelzyk, N. Mücke, H. Herrmann and N. Willenbacher, PLoS One, 2014, 9 Search PubMed.
- T. Golde, M. Glaser, C. Tutmarc, I. Elbalasy, C. Huster, G. Busteros, D. M. Smith, H. Herrmann, J. A. Käs and J. Schnauß, Soft Matter, 2019, 15, 4865–4872 RSC.
- B. J. Gurmessa, N. Bitten, D. T. Nguyen, O. A. Saleh, J. L. Ross, M. Das and R. M. Robertson-Anderson, Soft Matter, 2019, 15, 1335–1344 RSC.
- M. L. Gardel, M. T. Valentine, J. C. Crocker, A. R. Bausch and D. A. Weitz, Phys. Rev. Lett., 2003, 91, 158302 CrossRef CAS PubMed.
- T. T. Falzone and R. M. Robertson-Anderson, ACS Macro Lett., 2015, 4, 1194–1199 CrossRef CAS.
- B. Gurmessa, S. Ricketts and R. M. Robertson-Anderson, Biophys. J., 2017, 113, 1540–1550 CrossRef CAS PubMed.
- M. L. Gardel, J. H. Shin, F. C. MacKintosh, L. Mahadevan, P. Matsudaira and D. A. Weitz, Science, 2004, 304, 1301–1305 CrossRef CAS PubMed.
- D. Strehle, P. Mollenkopf, M. Glaser, T. Golde, C. Schuldt, J. A. Kas and J. Schnauss, Molecules, 2017, 22, 1–11 CrossRef PubMed.
- R. Tharmann, M. M. Claessens and A. R. Bausch, Phys. Rev. Lett., 2007, 98, 088103 CrossRef CAS PubMed.
- S. Yamada, D. Wirtz and P. A. Coulombe, J. Struct. Biol., 2003, 143, 45–55 CrossRef CAS PubMed.
- K. Kroy and J. Glaser, New J. Phys., 2007, 9, 416 CrossRef.
- H. Isambert and A. C. Maggs, Macromolecules, 1996, 29, 1036–1040 CrossRef CAS.
- F. C. MacKintosh, J. Kas and P. A. Janmey, Phys. Rev. Lett., 1995, 75, 4425–4428 CrossRef CAS PubMed.
- E. Haimov, R. Windoffer, R. E. Leube, M. Urbakh and M. M. Kozlov, Biophys. J., 2020, 119, 65–74 CrossRef CAS PubMed.
- O. Lieleg, M. M. Claessens, Y. Luan and A. R. Bausch, Phys. Rev. Lett., 2008, 101, 108101 CrossRef CAS PubMed.
- C. Semmrich, T. Storz, J. Glaser, R. Merkel, A. R. Bausch and K. Kroy, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20199–20203 CrossRef CAS PubMed.
- C. Storm, J. J. Pastore, F. C. MacKintosh, T. C. Lubensky and P. A. Janmey, Nature, 2005, 435, 191–194 CrossRef CAS PubMed.
- M. L. Gardel, J. H. Shin, F. C. MacKintosh, L. Mahadevan, P. Matsudaira and D. A. Weitz, Science, 2004, 304, 1301–1305 CrossRef CAS PubMed.
- J. X. Tang and P. A. Janmey, Biol. Bull., 1998, 194, 406–408 CrossRef CAS PubMed.
- C. Schuldt, J. Schnauss, T. Handler, M. Glaser, J. Lorenz, T. Golde, J. A. Kas and D. M. Smith, Phys. Rev. Lett., 2016, 117, 197801 CrossRef PubMed.
- J. Schnauß, T. Golde, C. Schuldt, B. S. Schmidt, M. Glaser, D. Strehle, T. Händler, C. Heussinger and J. A. Käs, Phys. Rev. Lett., 2016, 116, 108102 CrossRef PubMed.
- A. E. Patteson, A. Vahabikashi, K. Pogoda, S. A. Adam, K. Mandal, M. Kittisopikul, S. Sivagurunathan, A. Goldman, R. D. Goldman and P. A. Janmey, J. Cell Biol., 2019, 218, 4079–4092 CrossRef CAS PubMed.
- A. Aufderhorst-Roberts and G. H. Koenderink, Soft Matter, 2019, 15, 7127–7136 RSC.
- J. Block, H. Witt, A. Candelli, E. J. Peterman, G. J. Wuite, A. Janshoff and S. Koster, Phys. Rev. Lett., 2017, 118, 048101 CrossRef PubMed.
- A.-M. Fortier, E. Asselin and M. Cadrin, J. Biol. Chem., 2013, 288, 11555–11571 CrossRef CAS PubMed.
- A. W. Holle, M. Kalafat, A. S. Ramos, T. Seufferlein, R. Kemkemer and J. P. Spatz, Sci. Rep., 2017, 7, 45152 CrossRef CAS PubMed.
- S. Karsch, F. Buchau, T. M. Magin and A. Janshoff, Cell. Mol. Life Sci., 2020, 77, 4397–4411 CrossRef CAS PubMed.
- S. J. Winder and K. R. Ayscough, J. Cell Sci., 2005, 118, 651–654 CrossRef CAS PubMed.
- M. Moch, R. Windoffer, N. Schwarz, R. Pohl, A. Omenzetter, U. Schnakenberg, F. Herb, K. Chaisaowong, D. Merhof, L. Ramms, G. Fabris, B. Hoffmann, R. Merkel and R. E. Leube, PLoS One, 2016, 11, e0149106 CrossRef PubMed.
- N. Bonakdar, A. Schilling, M. Sporrer, P. Lennert, A. Mainka, L. Winter, G. Walko, G. Wiche, B. Fabry and W. H. Goldmann, Exp. Cell Res., 2015, 331, 331–337 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sm02261f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |