
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProduction of furans from C5 and C6 sugars in the presence of polar organic solvents
Luca
Ricciardi
a,
Willem
Verboom
 *a,
Jean-Paul
Lange
*bc and
Jurriaan
Huskens
*a
*a,
Jean-Paul
Lange
*bc and
Jurriaan
Huskens
*a
aMolecular NanoFabrication Group, Department of Molecules & Materials, MESA+ Institute, University of Twente, P.O. Box 217, 7500 AE, Enschede, The Netherlands. E-mail: w.verboom@utwente.nl; j.huskens@utwente.nl; Tel: +31-53-489-2977 Tel: +31-53-489-2995
bSustainable Process Technology Group, University of Twente, P.O. Box 217, 7500 AE, Enschede, The Netherlands. E-mail: j.p.lange@utwente.nl; Tel: +31-20-630-3428
cShell Global Solutions International B.V., Grasweg 31, 1031 HW Amsterdam, The Netherlands
First published on 17th November 2021
Abstract
Furfural and hydroxymethylfurfural are promising platform molecules for manufacturing chemicals and fuel components. These furanic compounds are the product of the acid-catalyzed dehydration of sugars (e.g., xylose and glucose), components obtained from lignocellulosic biomass. Manufacturing furans employs the use of mineral acid catalysts (e.g., H2SO4) in an aqueous medium. This approach limits the selectivity towards furans to approx. 45 mol%, mainly by the formation of solid by-products (humins). The use of aqueous–organic biphasic conditions raises the selectivity to approx. 60–70 mol%. However, even higher selectivities (>80 mol%) can be achieved by switching to organic solvent systems. Specifically, aprotic polar organic solvents (e.g., DMSO) can improve both the conversion and the selectivity from sugars to platform molecules. The presence of aprotic polar organic solvents has an influence on the solvent shell of the sugar and on the activity of the catalyst. Studying these two effects is crucially important to understand improvements of the selectivity. The aim of this review is to explore the use of polar organic solvents in the process of the manufacture of furans, while addressing the challenges of its industrial application, particularly in solvent recovery and recycling.
 Luca Ricciardi | Luca Ricciardi was born in Naples, Italy in 1993. He recently obtained his PhD (2021) at the University of Twente under the supervision of Prof. Dr Ir. Jurriaan Huskens, Prof. Dr Jean-Paul Lange and Dr Willem Verboom. He achieved his BSc in Chemical Science and Technology, under the supervision of Prof. Dr Osvaldo Lanzalunga, and his MSc in Biophysical Chemistry, under the supervision of Prof. Dr Maria Enrica Di Cocco and Prof. Dr Maurizio Delfini, both at the University of Rome “la Sapienza”. His PhD project focused on the development of cost-effective approaches to selectively obtain furfural from xylose. |
 Willem Verboom | Willem Verboom studied chemistry at Utrecht University, where he also obtained his PhD (1980). Subsequently, he went to the University of Twente, where he now is an associate professor of organic chemistry. His current research interests involve the design of specific ligands for actinides and lanthanides, the use of microreactors, and green chemistry. He is the (co-)author of about 450 scientific publications. |
 Jean-Paul Lange | Jean-Paul Lange is principal research scientist at Shell Projects & Technology in Amsterdam, the Netherlands, where he has been exploring novel catalytic processes for producing fuels and chemicals, initially from natural gas and oil and, since more than twenty years from biomass and plastic wastes. Jean-Paul Lange is also Professor in Chemical Biorefining at the University of Twente, the Netherlands, where he is investigating thermo-chemical and -catalytic routes to convert biomass to fuels and chemicals and to recycle plastic wastes. Jean-Paul Lange is co-author of >100 patent series, >65 scientific publications, 10 book chapters and co-editor of one scientific book (in press). |
 Jurriaan Huskens | Jurriaan Huskens (1968) obtained his PhD (1994) at the Delft University of Technology with Herman van Bekkum. After postdoctoral stays with Dean Sherry and Manfred Reetz, he became assistant professor with David Reinhoudt at the University of Twente in 1998, and full professor in 2005. He received the Unilever Research Award (1990), a Marie Curie fellowship (1997), the Gold Medal 2007 of the Royal Netherlands Chemical Society, and a Fellowship from the Institute of Advanced Study, Durham University, UK (2019). Present research interests encompass: supramolecular chemistry at interfaces, supramolecular materials, multivalency, nanofabrication, and green chemistry. He is (co)author of over 400 refereed research papers and five patents. |
Introduction
Research over the last few decades has been devoted to the exploration and development of new, non-fossil sources such as biomass, to produce fuels and chemicals.1–5 Biomass can be converted by a series of chemical processes referred to as biorefinery, which is an alternative to fossil-based oil refinery and the related chemical complexes.3–7 The application of biorefinery technologies is key to upgrade complex biological resources, such as non-edible biomass (e.g., forestry and agricultural waste), into valuable chemicals, some of which can also be used as bio-based alternatives to fossil fuels.3–18 In this scenario, lignocellulosic biomass serves as an important renewable starting material for biorefinery.8–11 It is, in fact, the most abundant plant dry matter and can be obtained from a wide range of sources, e.g., sugarcane bagasse, maple wood, corncob, corn stover, pinewood, eucalyptus, wheat straw, and barley husk.8–11Lignocellulosic biomass is composed mainly of three biopolymers: lignin, cellulose and hemicellulose (Fig. 1).1,12–14 Cellulose is a homopolymer of glucose.1 Cellulose can be hydrolyzed to obtain glucose, which can be isomerized to fructose and other C6 sugars, e.g., mannose, through an ene-diol intermediate.1 Depending on the crop, the hemicellulose fraction of lignocellulosic biomass may contain C5 sugars, e.g., xylose and arabinose, and/or C6 sugars, e.g., glucose, mannose and galactose, as well as uronic acids, e.g., glucuronic acid.12 In hardwoods, xylose and arabinose are present mainly as glucuronoxylan and minor quantities of xyloglucan, while arabinoxylan occurs in grasses and, in minor quantities, in softwood.1 The average xylan contents is up to 5 w% in softwoods, 15 w% in hardwoods, and 20 w% in grass straw.1,12
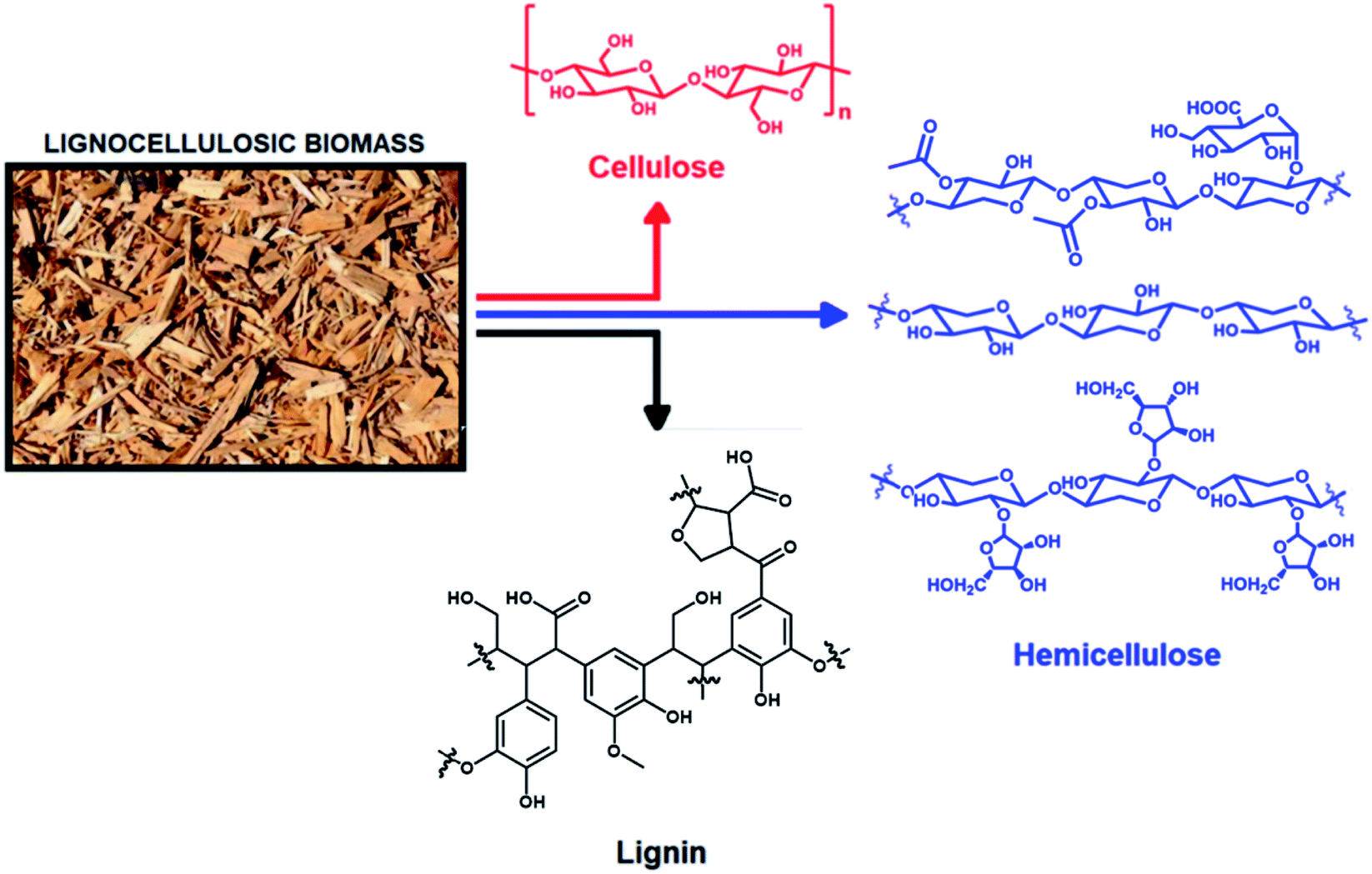 | ||
| Fig. 1 Constituents of lignocellulosic biomass in various biopolymers. Specifically, hemicellulose (in blue) is represented, from top to bottom, by glucuronoxylan, xylan and arabinoxylan. | ||
The conversion of biomass is much more challenging than that of model carbohydrates, as the decomposition behavior of the feedstock depends also on the interactions between its components (i.e., cellulose, hemicellulose, and lignin).8–14 Various methodologies of fractionation are used to separate the different components of biomass.3,5,15,16 Typically, the aim is to extract the hemicellulose and/or the lignin to deliver a cellulose-rich pulp.8–16
The fractionation of biomass is followed by the conversion of its constituents into a variety of high-value products.17,18 Amongst them, furfural and 5-hydroxymethylfurfural (HMF) stand out as top added-value platform molecules for chemicals and fuels.1–3 In particular, these two furanic compounds and their rich tree of derivatives offer many opportunities for fuel and chemical manufacture.1,2,4–7 HMF can be upgraded to several added-value intermediates like 2,5-furandicarboxylic acid (FDCA), 2,5-dimethylfuran, various furan-derived molecules, higher alkanes and aromatic gasoline.1,3–7,17,19 Furfural can be upgraded to THF, furan, butane and pentane diols, esters (e.g., furfuryl acetate, esters of levulinic acid and dimethyl pentanoate), and diesel alkanes.1–3,6,7,17,19–22
Furfural and HMF are generally produced by an acid-catalyzed dehydration of C5 and C6 sugars, respectively.1–3,23 The most common substrates for the production of HMF are fructose and glucose.3,4 While the production of HMF from fructose is efficient and direct, using glucose as a starting material requires the use of an isomerization catalyst.3,4 However, producing HMF from glucose could take advantage of the low feedstock costs if it can proceed directly, without isomerization to fructose.3,4 In the production of furfural from xylose, this additional isomerization step is not implemented, as no specific preferential isomer has been detected yet.1,2 Today, the industrial production of furans still largely relies on the batch dehydration of biomass using sulfuric acid, with yields of furfural and HMF of typically around 45–50%.1,2,23
There are several inherent differences between HMF and furfural in terms of stability and water solubility.1 HMF easily suffers from a rehydration reaction to form levulinic acid.24 Its hydroxymethyl group easily undergoes alkylation and triggers the production of humins.25 Furfural is much more stable intrinsically, as its degradation arises from a subsequent condensation reaction with sugar molecules and acid-catalyzed resinification reactions, both of which lead to the formation of solid humic by-products.1,2 Additionally, due to its hydroxymethyl group, HMF is highly water soluble, while furfural has a solubility limit of 8 w% in water.1,26
Several strategies have been employed to improve the sugar-to-furan selectivity.1–3,27–33 The parameters used for optimizing this reaction are the choice of the catalyst and possible additives, such as halide salts or metal ions, but also changing the characteristics of the solvent system.27–33 Most of the strategies applied to optimize this selectivity have been recently reviewed.27–34 Shuai and Luterbacher reviewed the effect of solvents on general biomass processing, focusing on the solvent effect on the behavior of the biopolymers lignin and cellulose.30 Lee and Wu reviewed all the solvent systems used in furfural production, including ionic liquids and deep eutectic solvents.33 Zhao et al. offered a comprehensive review for the production of furfural and HMF through the hydrothermal conversion of biomass, focusing on homogeneous catalysis in different solvent environments.34
The present review focuses on the use of polar organic solvents (e.g., DMSO and ethanol) as a promising reaction environment or as additives to the solvent system, with the aim to boost reactivity and selectivity. Recent developments in the understanding of their beneficial effects on the sugar-to-furan selectivity and their effect on catalyst activity are discussed, both in homogeneous and heterogeneous catalysis. Moreover, the feasibility of the possible application of such systems in industrial processes is discussed.
Chemistry of sugar dehydration
The key step in the conversion of a sugar to a furanic compound is an acid-catalyzed dehydration reaction.1 Several mechanisms have been proposed in different studies under various reaction conditions.23 Various aspects seem to influence the pathway of the reaction, such as catalyst type, salt concentration, presence of specific metal ions and the solvent system.23,34–37The reaction can be performed both in monophasic and biphasic environments, using both homogeneous and heterogeneous catalysis.1,31 In industrial setups, homogeneous catalysis from mineral acids, such as H2SO4 and HCl, is the preferred choice for such reactions because of the high catalyst and regeneration costs that result from using heterogeneous catalysts instead.1,38 Whenever possible, the homogeneous acid is recycled together with the solvent after recovery of the product.38,39
There are, however, other examples in literature in which supercritical CO2 and carboxylic acids have been used to catalyze the production of both furfural and HMF.37 Various heterogeneous catalysts, such as zeolites, sulfonated resins, and other acidic solids, have been successfully applied to produce furanic compounds from sugars.1,31,33,34 Moreover, due to the formation of acidic by-products, the dehydration of a sugar shows autocatalytic behavior.1,40
Heterogeneous catalysis potentially offers the benefit of ease of separation of solvent and catalyst, aiding the recyclability of the catalyst.41 However, heterogeneous catalysts are not beneficial over homogeneous catalysts in terms of sugar-to-furan selectivity, which strongly depends on the reaction conditions and the solvent system.1,31,40,41 More importantly, heterogeneous catalysts are prone to fouling and deactivation by deposition of humins as well as irreversible degradation by hydrothermal conditions, which remains a problem in their industrial application.41–43
Ionic liquids (ILs) and deep eutectic solvents (DESs) have also been used to successfully upgrade sugars to furans.33,34 ILs show low volatility, high stability (thermal and chemical), and a high degree of designable characteristics.33,34 However, ILs are often costly and composed of chemicals with a high toxicity.33,34 For this reason, DESs, which are generally less costly and toxic, appear to be a more viable alternative to ILs.33,34 However, DESs rarely show the same chemical and thermal stability of ILs, which currently impairs their industrial application.33,34 Additionally, product and catalyst recovery from such solvent systems remains an issue for their industrial application as well.33,34
Non-traditional heating methods, such as microwave heating, have also been used for the conversion of sugars to furans, both in heterogeneous and homogeneous catalysis.1,2,8,35,44–49 The presence of specific, non-thermal microwave effects has been excluded, and the rate enhancement observed at microwave heating conditions has been attributed to local overheating.48,50
Reaction towards furans
Rationalizing the mechanism of these reactions is an important aspect to optimize the reaction conditions to avoid the formation of side products.23,34 From a mechanistic point of view, several intermediate steps, such as isomerization, ring opening and closing, have been hypothesized in the dehydration process of both C5 and C6 sugars (see Schemes 1 and 2, respectively).23,34–42,51,52 As previously mentioned, an intermediate isomerization step to fructose most likely occurs in the production of HMF from glucose (Scheme 2a and b), even if a closed-ring pathway, with no isomerization, is theoretically possible (Scheme 2c).1,3,4,27 In the case of furfural manufacture from xylose, several groups have studied the importance of a C5 ketose analogous to fructose, i.e., xylulose.23,51–53 While xylulose can be detected in various concentrations depending on the reaction conditions, its key relevance as an intermediate in the process of furfural formation has been ruled out by Ershova et al., who showed, using both experiments and kinetic modeling, the role of xylulose to be along a parallel reaction pathway.51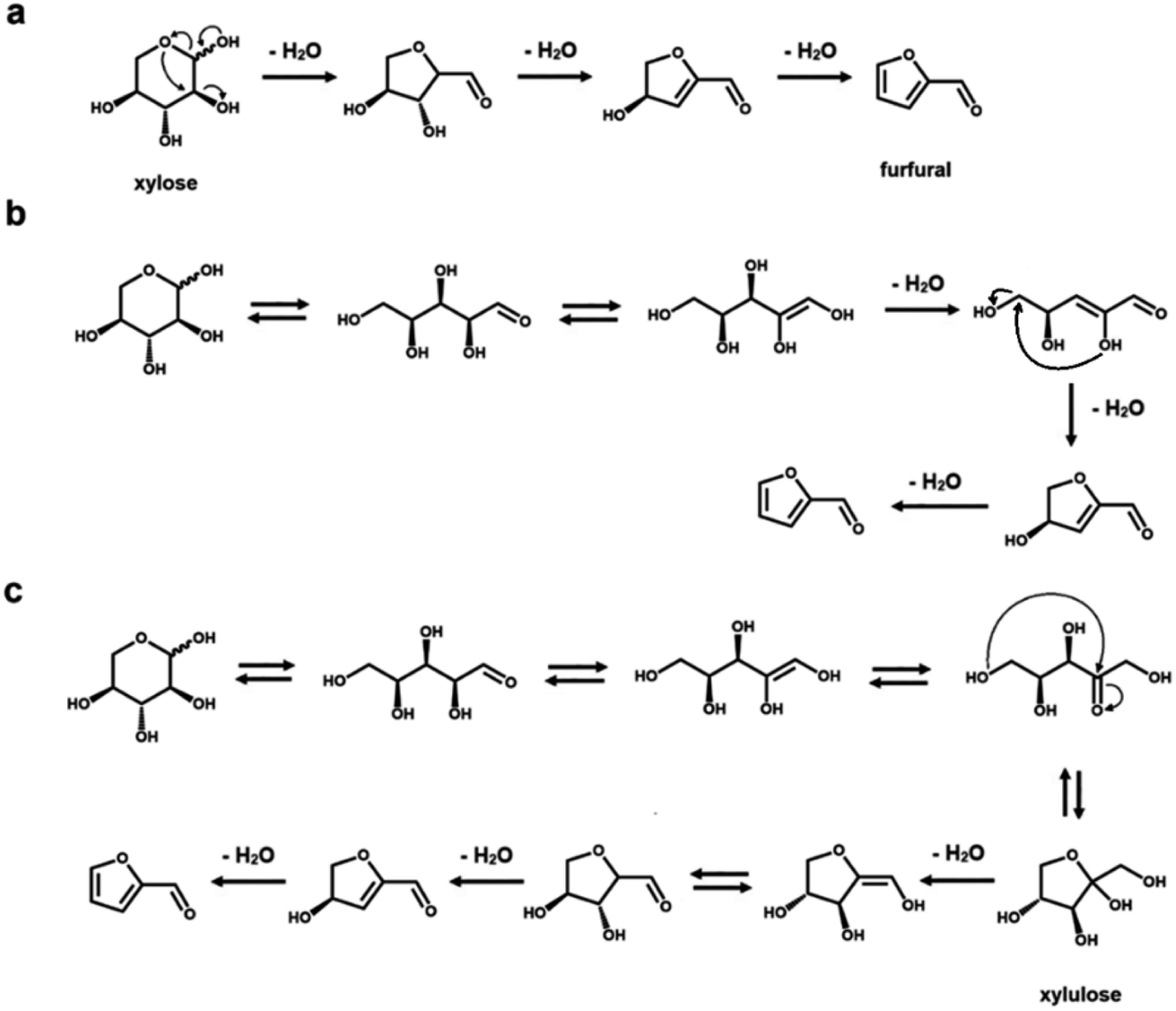 | ||
| Scheme 1 Mechanistic pathways from xylose to furfural. (a) Closed-ring pathway, dehydration in three steps with no isomerization. (b) Open-ring pathway, including a retro-Michael elimination. (c) Closed-ring mechanistic pathway inclusive of xylose-to-xylulose isomerization. Back-reactions in shown equilibria do not necessarily proceed with retention of configuration. | ||
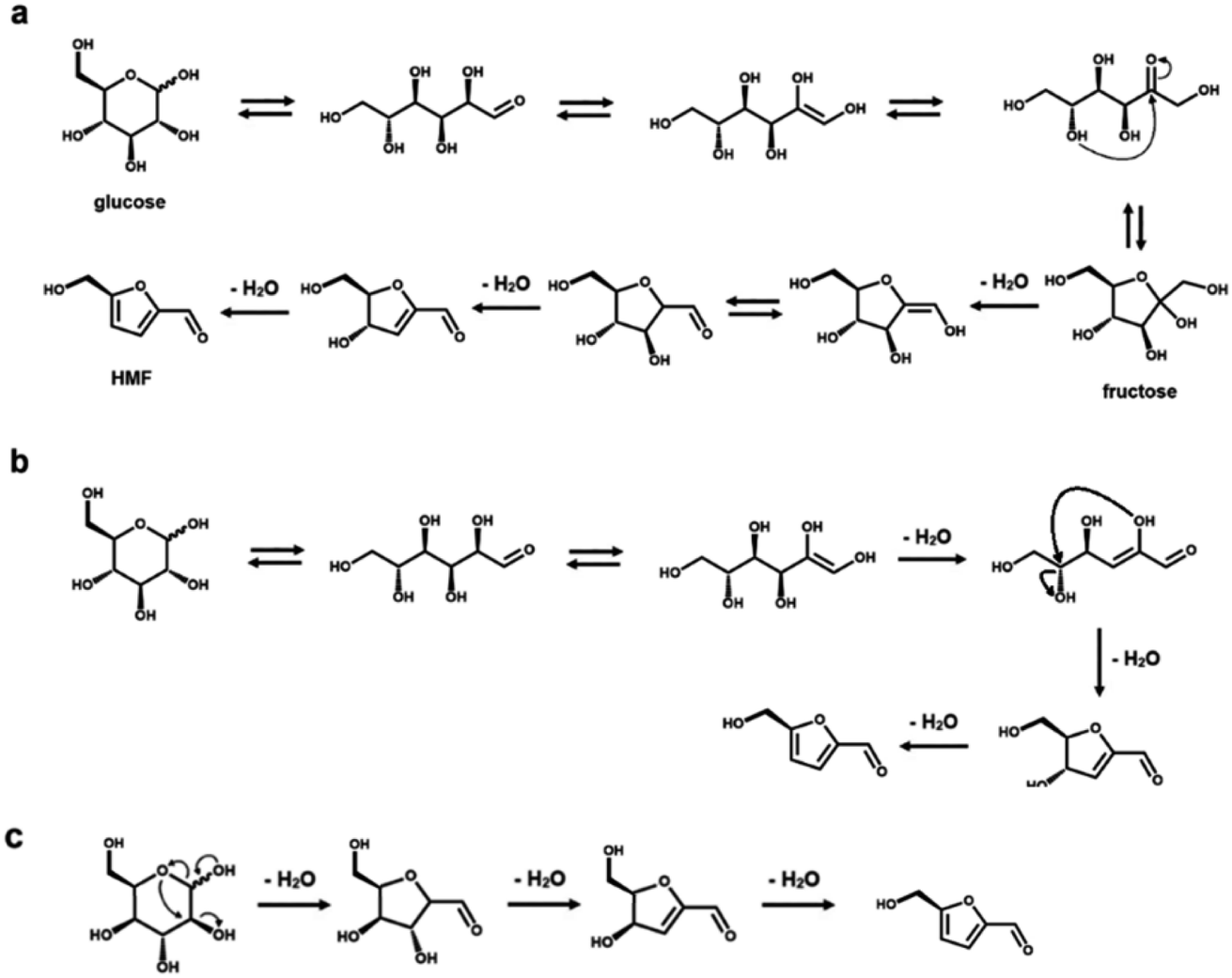 | ||
| Scheme 2 Mechanistic pathways from glucose and fructose to HMF. (a) Closed-ring pathway, after glucose-to-fructose isomerization. (b) Open-ring pathway, including a retro-Michael elimination. (c) Closed-ring pathway, without isomerization. Back-reactions in shown equilibria do not necessarily proceed with retention of configuration. | ||
Reaction towards humins and other by-products
As previously mentioned, the sugar-to-furan selectivity for the reaction of dehydration is limited by the formation of several by-products.1,2,24,25,54–58 Some can be both water- or organic-soluble molecules of low molecular weight, e.g., acetic acid, furanoic acid, γ-valerolactone (GVL). Others can be solid by-products, i.e., humins.1,2,24,25,54–58While the low-molecular weight compounds are produced by the degradation of both sugars and furans, via reactions of fragmentation, decomposition, and rehydration, humins are formed by progressive sugar–sugar, furan–furan and sugar–furan condensation.24,25,54–58 It has to be noted that some of the low molecular weight by-products and intermediates of dehydration can also participate in the formation of humins.24,57–60 Additionally, as previously mentioned, HMF is more reactive than furfural and more prone to polymerization, hence a comparison between humin formation from C6 and C5 sugars is not necessarily straightforward.25,60 Conversely, humic by-products obtained from the same sugar feed, but from reactions performed in different solvent systems (e.g., aqueous monophasic vs. aqueous–organic biphasic) do not show differences in structure and composition.48,49
When performing the acid-catalyzed dehydration of sugars in the presence of water, rehydration of the newly formed carbon–carbon double bonds leads to the formation of low-molecular weight by-products (e.g., carboxylic acids and lactones).55,56 Nimlos et al. propose, based on quantum-mechanical modelling, the significance of the hydroxyl group of xylose, which is protonated first, in steering the selectivity.55 Specifically, simulating the protonation of 2-OH leads preferentially to furfural formation, while the protonation of 3-OH leads to decomposition of the sugar based on C–C cleavage.55 It is worth mentioning, however, that the model proposed by Nimlos et al. does not successfully implement the closed-ring mechanism (Scheme 1a), in contradiction with most literature.34–37,40–42 Qian et al. have performed ab initio simulations, both in gas phase and simulated aqueous environment, that suggest that mechanisms that include ring-opening (Schemes 1b and 2b) result in a higher number of reaction paths to low-molecular weight side-products (e.g., formic acid and acetic acid), in the case of dehydration of both C5 and C6 sugars.56 This can be rationalized by the deprotection of the very reactive aldehyde functionality of the sugar. The deprotected aldehyde could also bind with another sugar molecule through a cyclic ketal or dioxolane link, opening thereby the way towards the formation of humins.
Horvat et al. propose a direct connection between rehydration of furans or partially dehydrated sugars and formation of humins.59,60 Specifically, the products of rehydration are thought to trigger the polymerization to humins, reacting with sugars in solution. However, this is only inferred from the structure of isolated intermediates in reactions analogous to HMF rehydration to form levulinic acid.59,60 IR analyses indicate that humins retain furan rings in their structure, an evidence which is consistent with the formation of humic by-products also via aldol addition/condensation of furans with partial rehydration by-products, e.g., 2,5-dioxo-6-hydroxyhexanal.59 The addition of HMF to a glucose feed did not significantly affect the yield of humins.61 On the contrary, adding a molecule that can act as a crosslinker, i.e., 1,2,4-trihydroxybenzene, increased the production of humins during acid-catalyzed dehydration of glucose.61 It has to be noted that 1,2,4-trihydroxybenzene is also formed, in minor amounts, from HMF during carbohydrate dehydration, and its inclusion in the humic by-products is an indication that humins are not only formed through aldol condensation.61 Based on all this, the stabilization of furans and partially dehydrated intermediates appears to be a key aspect of preventing the formation of humins and, consequently, reaching high sugar-to-furan selectivity.57,59,60
Some of the side-products of the dehydration of carbohydrates, such as carboxylic acids (e.g., levulinic acid, acetic acid and formic acid), lactones (e.g., GVL and angelica lactone) and cyclohexanones, can be reutilized and valorized.1–3,31,40,62–69 Humic by-products can also be processed and applied as platform materials such as graphene oxide.70–72 However, their high heterogeneity limits their possible applications in industry.70–72
Aqueous–organic biphasic solvent systems
A common, and widely applied, strategy to improve the selectivity toward furfural and HMF is the add a water-immiscible organic solvents, such as toluene, methylisobutylketone, diethyl ether, or p-xylene, to form an aqueous–organic biphasic system.1,31,40,73–77 Such aqueous–organic biphasic systems can be low-cost, relatively non-toxic and environmentally friendly. The concept of biphasic operation, based on the process of reactive extraction, exploits the continuous liquid–liquid extraction of the formed HMF or furfural into the organic phase.78–82 The characteristics of these biphasic systems are variable in terms of catalysis and composition.31,40,73,75While homogeneous catalysis by mineral acids remains the preferred source, in several studies heterogeneous catalysis has also been successfully applied at biphasic conditions.31,40 The effect of adding a water-immiscible layer affects the sugar-to-furan selectivity by continuous extraction of the newly formed HMF or furfural into the organic phase.1–3,31,40 In both cases the sugar-to-furan selectivity increases, on average, to 60–70 mol% because of the suppression of the furan-sugar condensation reaction towards solid by-products.1,31,40,63,64
Biphasic operation has been applied to sugar dehydration at several aqueous–organic volume ratios, with mostly organic biphasic systems being reported to be the most beneficial in terms of sugar-to-furan selectivity.1,20,40,73–75 Under microwave heating, biphasic systems were shown to deliver higher yields when the organic solvent in use is a non-polar, microwave-silent solvent.49 In such systems, the selective heating of the aqueous phase, while the organic phase stays relatively ‘cold’, has resulted in xylose-to-furfural selectivities up to 90 mol%.49 Large fractions of organic solvent, however, can be a limit for the efficiency of the process, as the feed for such a reaction comes as an aqueous solution and limiting the volume of the aqueous phase limits the final furan production.1,76,77
Aqueous–organic monophasic solvent systems
Several examples of monophasic solvent systems composed of water and a polar organic solvent have been investigated for the acid-catalyzed dehydration of sugars.30,33,34,83–85 Specifically, both solvents that fully mix with water at room temperature, e.g., GVL, DMSO or dioxane, and solvents that mix with water at operating conditions, e.g., n-butanol, are widely used in this field.30,33,34,83–89 The so-called ‘solvent effect’ resulting from adding such a co-solvent refers to changes in reaction rate, reaction pathway, product distribution, and yield.30,33,34 Contrary to biphasic operation, the use of this type of co-solvents will affect the interaction between the solute (i.e., the C5 and C6 sugars) and the solvent (i.e., water).30,33,34 These changes occur because of the influence of the co-solvent on hydrogen bonding and the overall thermodynamic behavior of the solvent system.30,33,34 Below, the effects of such miscible co-solvents on the production of HMF and furfural will be reviewed.Protic organic solvents
In the realm of water-miscible organic solvents, there is a distinction between protic solvents, such as ethanol, and aprotic polar solvents, such as DMSO.30 Protic solvents can both accept and donate hydrogen bonds, whereas the aprotic ones can only accept hydrogen bonding; additionally, protic solvents can react as nucleophiles.30 Protic solvents, like isopropanol and ethanol, have been successfully used both in HMF and furfural synthesis.89–94 The use of a protic solvent is justified because of the beneficial suppressing effect on the formation of humic by-products by sugar–furan condensation.89–94 In fact, the in situ formation of an alkyl-glucoside has been hypothesized to protect the sugar, suppressing parallel condensation reactions that result in the formation of humic by-products (Scheme 3).89 Köchermann et al. have shown that, while side reactions seem to be suppressed, the presence of ethanol below 50 vol% does not influence the rate of xylose consumption.89 This is explained by considering differences in the activation energy for the reaction of dehydration and degradation, which are strongly temperature-dependent.89 Hu et al., in fact, show that at low temperatures, the presence of an alcohol (i.e., methanol) results in a decreased rate of xylose consumption.89,90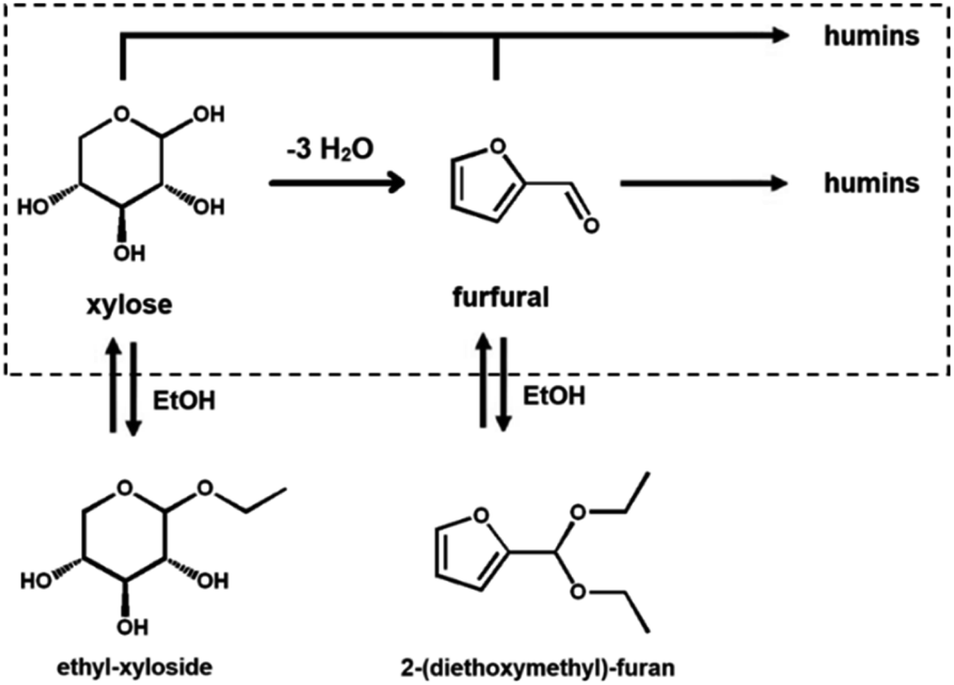 | ||
| Scheme 3 Possible reaction pathway for the formation of the ethyl-xyloside and 2-(diethoxymethyl)furan in a water–ethanol solvent system. The dashed box indicates the species present in a water-only system. | ||
This approach was also successfully applied using heterogeneous and homogeneous catalysis conditions, but the most promising results in terms of sugar-to-furan selectivity have been observed under homogeneous catalysis conditions.89–94 Specifically, about 75 mol% xylose-to-furfural selectivity was obtained using a water–ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) mixture, using H2SO4 as acid catalyst.89 Among the alcohols, n-butanol has the specific characteristic of forming a biphasic system with water at T < 80 °C, transitioning into a monophasic system at T > 80 °C.95
:1 v/v) mixture, using H2SO4 as acid catalyst.89 Among the alcohols, n-butanol has the specific characteristic of forming a biphasic system with water at T < 80 °C, transitioning into a monophasic system at T > 80 °C.95
The use of different organic solvents, like formic acid, as additives to the aqueous phase, has also been reported.96 Specifically, the addition of increasing non-catalytic amounts of formic acid (>0.5 M) to a water/n-butanol 1:3 v/v solvent system resulted in a consistent increase in the selectivity of the fructose dehydration to HMF, with an ultimate selectivity of about 80 mol%.96
Using formic acid as a co-catalyst allows the production of furfural combined in tandem with that of other derivatives, e.g., furfuryl alcohol (with an overall yield of approx. 70 mol%).97 The same effect was obtained using other carboxylic acids (e.g., oxalic acid), which are also routinely used for the formation of deep eutectic solvents (DES).98,99 A related combined approach has also been reported for isopropanol.100,101 In this case furfural is reduced to furfuryl alcohol through catalytic hydrogen transfer (CHT) over metal–organic frameworks (MOFs).100 CHT in alcohol media has also been reported in a tandem process for the production of furfural, and minor percentages of furfuryl alcohol and levulinic acid, from a xylose feed.101 A similar facilitative effect has been observed for tert-butanol.102 Specifically, Peng et al. have reported that tert-butanol dehydrates to isobutylene, which then undergoes oxygenolysis to acetone and formic acid.102 Over metallic surfaces, formic acid decomposes to CO2/H2, while in presence of strong acids it decomposes to CO/H2O.102 In such a systems, however, the regeneration of consumed H-donors is an expense which negatively affects application in industry.
Much like sugars, however, alcohol can also undergo dehydration under the same reaction conditions, i.e., high temperature and acid catalysis.103–106 Different alcohols result in different products of dehydration (e.g., ethanol to ethylene or isopropanol to isobutylene).103–106 These reactions will negatively affect the costs of possible industrial applications. Additional alcohol will need to be constantly added to the solvent system, to balance the losses. Moreover, as the product feed will contain multiple new components, the product yield and isolation might also be influenced.
Aprotic polar solvents
As previously discussed, aprotic solvents behave in a fundamentally different way than protic ones.30,33,34 This results in differences in the solvent–solute interaction, which also reflects on the reaction parameters, mechanism, and kinetics, with no possibility of forming glucosides.107–111 Specifically, the presence of aprotic solvents in the mixture has been reported to promote the glucose-to-fructose isomerization (Scheme 2) and the formation of furanose-aldehyde intermediates (Schemes 1a and 2a).107,111 The product/by-product distribution is also affected, largely increasing the selectivity of the dehydration of glucose and fructose towards HMF.107 This specific behavior of aprotic solvents can be generalized to all dehydration reactions, with a direct effect on its mechanism, owing to competition between water and the polar organic solvent to form the solvent shell for the sugar, by interacting with its hydroxyl groups.108 This has been analyzed through DFT simulations of the spatial distribution of the solvents.108 Specifically, Chew et al. modeled the reactant with a small number of explicit solvent molecules in the direct vicinity, while the rest of the solvent was modeled implicitly.108 This allowed to extract the configuration of the solvent molecules close to the reactant, obtaining the energy-minimized structures for several solvent mixtures.108Aprotic solvents alter the relative stability of the starting materials, transition states and products in the dehydration reactions.109,110 This opens the possibility of predicting and rationalizing the effect of a specific solvent in this type of acid-catalyzed processes98 through computational work.109,110 Several aprotic solvents have been used for the dehydration of sugars both in water and in water-free environments, providing a wide variety of sugar-to- furan selectivities depending on the reaction conditions.35,112–148
For illustration purpose, the presence of an aprotic organic solvent in a solvent system has a direct effect on the mutarotation of sugars.118 Specifically, in a mainly aqueous environment at room temperature, the β-pyranose form of a sugar is dominant, whereas in mainly organic environments (i.e., in presence of organic solvents, such as DMSO, THF, γ-butyrolactone (GBL) and DMF), the α-pyranose form is favored (Fig. 2).118 The preference for the β-pyranose form in aqueous environment can be rationalized by the fact that the sugar has a more hydrophilic surface area compared to the one of the α-pyranose, due to the spatial arrangement of the OH groups in the β-pyranose form.118 This influences the reactivity of the sugar, as it depends strongly on the conformation of the functional groups (e.g., in reaction of dehydration and hydrogenation).118
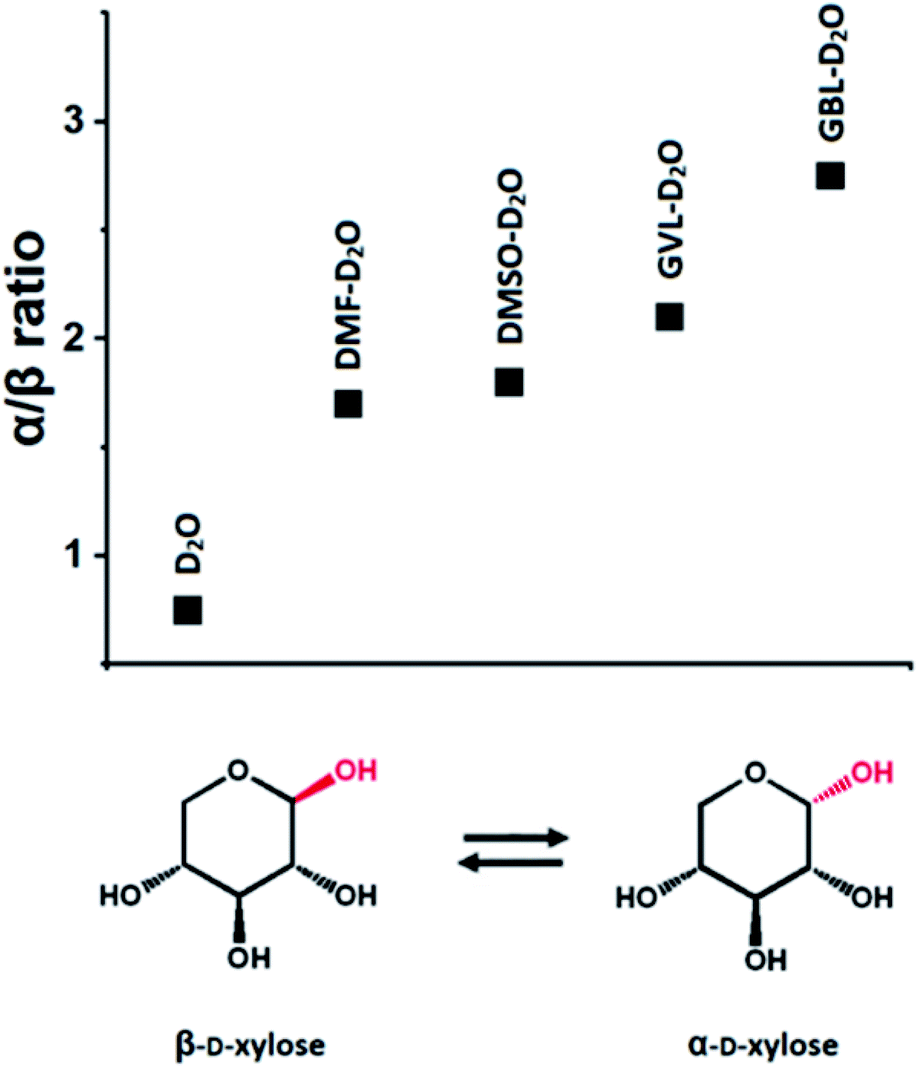 | ||
| Fig. 2 α/β-Anomer ratios in different solvent systems, determined by 1H-NMR spectroscopy. Data from ref. 118. | ||
:3 v/v water–DMSO monophasic systems), paired with microwave heating and a Lewis acid catalyst (i.e., CrCl3 or Al(NO3)3), resulted in a relatively high fructose-to-HMF selectivity (approx. 65 mol%), with a strong influence on conversion and selectivity also coming from the catalyst loading.113
| DMSO:H2O (v/v) |
Starting material | Starting conc. (mM) | Product | Catalyst | Sel. (mol%) | Ref. |
|---|---|---|---|---|---|---|
| a Hierarchically porous carbon-based catalyst. b Sulfonated carbon catalyst based on Eucalyptus Kraft lignin. | ||||||
| 1:1 |
Fructose | 380 | HMF | Maleic acid–SnCl4 | 54 | 115 |
| 1:1 |
Xylose | 33 | Furfural | Sn0.625Cs0.5PW | 61 | 119 |
| 3:1 |
Cellulose formate | 250 | HMF | HCl/AlCl3 | 52 | 115 |
| 1:0 |
Fructose | 550 | HMF | H2SO4 | 81 | 116 |
| 1:0 |
Xylose | 33 | Furfural | H3PW12O40 | 67 | 119 |
| 1:0 |
Fructose | 180 | HMF | SiO2–SO3H | 91 | 122 |
| 1:0 |
Fructose | 50 | HMF | HPC-P25–Sa | 98 | 124 |
| 1:0 |
Fructose | 110 | HMF | Nb2O5 | 86 | 125 |
| 1:0 |
Fructose | 720 | HMF | EKLSCb | 85 | 126 |
The use of DMSO enables the effective solubilization of rather recalcitrant starting materials (e.g., coffee grounds, bread waste or cellulose formate).112–125 Moving from a fully aqueous monophasic system to a 1:1 water–DMSO mixture, with HCl and AlCl3 as cocatalysts, has resulted into a 35 mol% selectivity improvement in the conversion of cellulose formate to HMF, with a concomitant reduction of the production of levulinic acid.115 This selectivity improvement is explained by specific interactions between the reactant (i.e., formylated sugars) and the aprotic solvent (see above).115 However, it has to be noted that some of the mentioned side reactions (e.g., HMF to levulinic acid) are the result of rehydration, so operating in a water-lean environment will also have a non-negligible effect.1,2
Experimental evidence supports that the effect of DMSO on the sugar-to-furan selectivity is mainly the result of solvation of the sugar, and not of catalysis by acidic species generated by the degradation of DMSO.116,117 Moreover, solvation of the product can also be a contributor, as adding aprotic organic solvents will result in a stabilization of HMF and furfural.118 Molecular dynamics simulations on the interaction of glucose in progressively more organic solvent mixtures of water with DMSO, THF and DMF, exhibited that these solvents compete with water in forming the first solvation shell around the sugar, even upon the addition of relatively low amounts (<40–50 vol%).117 Vasudevan et al. modeled a small number of sugar molecules within a solvent environment constituted of a large number of explicitly modeled solvent molecules, with no constraints on bonds and angles.117
A 20–25 mol% xylose-to-furfural selectivity improvement is observed when moving from aqueous systems to 1:1 v/v water–DMSO conditions, using bi-metallic salts of tungstophosphoric acid as catalysts.119 It was demonstrated that the presence of DMSO improved the catalytic activity and stability, with the catalyst retaining >90% of its activity after six reaction cycles.119 Similar results have been obtained using Preyssler heteropolyacids (e.g., H14NaP5W30O110) as catalysts in a water-free DMSO monophasic system, with xylose-to-furfural selectivities as high as 80 mol%; a value that can compete with biphasic operations.120 In this study, which has employed significantly higher xylose concentrations (i.e., approx. 200 mM compared to the approx. 33 mM used by Guo et al.119), DMSO was also combined with other organic solvents (namely, dichloromethane and methylisobutylketone), with the aim of further stabilizing the produced furfural and limiting the subsequent resinification reactions.120
This effect was also supported by the analysis of the Gibbs free energy of different tautomers of fructose in different solvent systems, performed by Fu et al., who state that the presence of DMSO has a direct effect on the mechanism of the reaction.120 Specifically, the presence of DMSO (as well as dioxane and NMP) stabilizes the α-furanose form of fructose, favoring, as also previously mentioned, the closed-ring dehydration mechanism (Scheme 2a). Moreover, the presence of aprotic solvents suppresses the formation of fructose–HMF oligomers, thereby improving the fructose-to-HMF selectivity.121
Using DMSO as the sole component of a monophasic system is rather common in the heterogeneously catalyzed production of furans from xylose, glucose and fructose.120,122–126 Among heterogeneous catalysts, sulfonated solids (e.g., silica, carbon and palygorskite) are specifically used, always with sugar-to-furan selectivities >85 mol%.122–126 Acidic metal oxide catalysts (e.g., Nb2O5) have also been successfully applied, specifically for the dehydration of fructose, with HMF production of approx. 85–90 mol% selectivity.126
:1 v/v THF–water or dioxane–water), combined with catalytic amounts of Cl− anions, results in the stabilization of protonated transition states of the acid-catalyzed process, leading to significant increases in reactivity with an approx. 80 mol% fructose-to-HMF selectivity.127 Mellmer et al. have modelled the molecular interactions of the sugar and the solvent using an all-atom simulation of the molecules.127
Increasing the THF content of the solvent system resulted in an increased xylose-to-furfural selectivity, catalyzed by pressurized CO2, leading to a selectivity improvement of approx. 30–40 mol% when moving from mainly aqueous to mainly organic systems.129 A similar pattern has been observed when producing furfural from alginic acid, in a reaction catalyzed by heteropolyacids in a 95:5 THF–water system.130
Extremely high fructose-to-HMF selectivities (>95 mol%) were obtained when performing fructose dehydration in water-free THF.131 As in the case of DMSO, the absence of water is generally combined with a heterogeneous catalyst. In this case, the best performing catalyst is a functionalized poly(styrene sulfonate) (PSS), which bears ionic liquid moieties (PSS-30IL-SO3H), yielding an almost quantitative HMF production from fructose.131
:1 v/v organic–aqueous system.124 Among different solvents, the best performing one is sulfolane, which is structurally similar to DMSO, yielding a xylose-to-furfural selectivity up to approx. 70 mol%, with an enhancement from 10 to 25 mol% compared with other organic solvents, e.g., THF, GVL, and GBL (Table 3).133–138
| Sulfolane:H2O (v/v) |
Starting material | Starting conc. (mM) | Product | Catalyst | Sel. (mol%) | Ref. |
|---|---|---|---|---|---|---|
| a The pre-hydrolysate liquor composes 20 w% of the mixture. b The bamboo biomass composes approx. 5 w% of the mixture. c In this specific case, furfural and HMF are produced concomitantly. | ||||||
| 1:1 |
Xylose | 140 | Furfural | — | 68 | 132 |
| 4:1 |
Pre-hydrolysate liquora | n.a. | Furfural | H-β zeolite | 70 | 134 |
| 9:1 |
Bamboo biomassb | n.a. | Furfuralc | AlCl3 | 25 | 135 |
| 9:1 |
Bamboo biomassb | n.a. | HMFc | AlCl3 | 39 | 135 |
| 1:0 |
Fructose | 360 | HMF | LiCl | 67 | 136 |
| 1:0 |
Fructose | 360 | HMF | HBr | 91 | 136 |
However, the high boiling point of sulfolane (285 °C), compared with that of other aprotic organic solvents (e.g., DMSO 189 °C and THF 66 °C), allows for more complex reaction setups, such as reactive distillation.134 Continuous furfural removal by means of distillation would allow to avoid subsequent side-reactions that limit the selectivity.134 However, this stays a challenge in such systems, and the balance between the various system components is yet to be determined.134
In reaction systems that don't exploit reactive distillation, xylose-to-furfural selectivities up to 70 mol% were reached when using heterogeneous catalysis in mainly organic environments.134 Additionally, the use of highly organic sulfolane–water mixtures (9:1 v/v) in combination with metal chlorides (e.g., SnCl4 and FeCl3) resulted in 25–30 mol% fructose-to-HMF selectivity improvements, with an absolute yield of approx. 40 mol%, when compared with fully aqueous systems.135
This is rationalized by the fact that the SnCl4 catalyst is homogeneous in the presence of sulfolane, whereas it is largely precipitated as a solid in aqueous system, impairing its activity.135 SnCl4 has been reported to hydrolyze, and Liu et al. have hypothesized, based on ESI-MS spectrometry, about a possible synergic effect of the presence of tin hydroxides in catalyzing a glucose-to-fructose isomerization.135 The effect of the solvent system on the activity of metal halides has been confirmed by Caes and Raines, who studied several metal halides to catalyze the dehydration of fructose to HMF in a water-free sulfolane environment.136 Compared to catalyst-free conditions, they showed fructose-to-HMF improvements up to 60–70 mol%, with absolute HMF yields of approx. 67 mol% when using LiCl and approx. 91 mol% when using HBr.136
In the case of fructose dehydration catalyzed by phosphate-functionalized porous organic polymers and performed in fully organic environment, moving from DMSO to dioxane resulted in a 20 mol% fructose-to-HMF selectivity increase, up to 97 mol%.139 This can be rationalized by specific interactions between solvent and sugars, solvent and solvent, and solvent and catalysts.116–118 In fact, it is proven, that no acidic species are formed due to DMSO degradation.116
:1 v/v dioxane–water mixture, the conversion of glucose to fructose drops from > 90 mol% to approx. 50 mol% (after 2 h of reaction).140 Jeong et al. reported the use of thick corn syrup with high fructose content (water content less than 25 w%) as a starting material.141 Using Amberlyst-15 as the acid catalyst and dioxane as the solvent an ultimate fructose-to-HMF selectivity of approx. 80 mol% was reached.141 Heterogeneous catalysis was also successfully applied using Amberlyst-131 in mainly organic dioxane–water solvent systems to catalyze the production of HMF from glucose/fructose mixtures, reaching an overall selectivity towards HMF of approx. 75 mol%.140
| Dioxane:H2O (v/v) |
Starting material | Starting conc. (mM) | Product | Catalyst | Sel. (mol%) | Ref. |
|---|---|---|---|---|---|---|
| a Fructose syrup contains approx. 67 w% of fructose and approx. 8 w% of other sugars. b Phosphate-functionalized polymer catalyst. c Calcium gluconate-derived sulfonated carbon catalyst. d The bamboo biomass composes approx. 10 w% of the mixture. | ||||||
| 9:1 |
Fructose | 50 | HMF | HCl | 73 | 127 |
| 9:1 |
Glucose/fructose | 55 | HMF | Sn-β/Amberlyst-131 | 75 | 140 |
| 9:1 |
Fructose syrupa | n.a. | HMF | Amberlyst-15 | 72 | 141 |
| 1:0 |
Fructose | 280 | HMF | B-POPb | 91 | 139 |
| 1:0 |
Xylose | 130 | Furfural | SC-GCa-800c | 76 | 142 |
| 1:0 |
Bamboo biomassd | n.a. | Furfural | HCl | 83 | 143 |
Water-free dioxane was also used to produce furfural from xylose with approx. 75–80 mol% selectivity in the presence of a solid carbon-based acid catalyst, derived from calcium gluconate.142 Like DMSO, dioxane has also been employed in the treatment of more recalcitrant starting materials (e.g., corncob and bamboo lignocellulosic biomass), with a very efficient biomass fractionation, >90 mol% hydrolysis yield and 80–90 mol% conversion yield of furfural.143,144 Specifically, a fully organic dioxane environment gave rise to >90 mol% yield of furfural starting from bamboo biomass, in an HCl-catalyzed process.144 A more complex solvent system, composed of a mixture of ethanol, dioxane and formic acid, was successfully used for lignocellulosic biomass liquefaction, which yielded furans such as furfural and HMF.144
:5 v/v DES–acetone mixture) gave furfural starting from xylose with approx. 70 mol% selectivity, using H2SO4 as a catalyst.145 However, Chen and Wan do not discuss the recovery of the product from such a solvent mixture.145 Wang et al. reported that using a 7:3 v/v acetone–water mixture and pressurized phosphoric acid as a catalyst resulted in a xylose-to-furfural selectivity of 45–50 mol%, i.e., an improvement of approx. 20 mol%, in comparison with pure water.146
| Solvent | Solvent:H2O (v/v) |
Starting material | Starting conc. (mM) | Product | Catalyst | Sel. (mol%) | Ref. |
|---|---|---|---|---|---|---|---|
| a This solvent system also includes a DES. b Pressurized phosphoric acid. c 1-Butyl-3-methylimidazolium chloroferrate. d Calcium gluconate-derived sulfonated carbon catalyst. | |||||||
| Acetone | 5:3a |
Xylose | 130 | Furfural | H2SO4 | 70 | 145 |
| Acetone | 7:3 |
Sugarcane | 100 | Furfural | PPAb | 45 | 146 |
| Acetone | 4:1 |
Fructose | 55 | HMF | HCl | 98 | 147 |
| Acetone | 4:1 |
Fructose | 55 | HMF | H2SO4 | 97 | 147 |
| Acetone | 4:1 |
Fructose | 55 | HMF | Amberlyst-15 | 95 | 147 |
| Butanone | 2:1 |
Xylose | 200 | Furfural | [bmim]Cl/FeCl3c | 53 | 150 |
| GBL | 1:1 |
Xylose | 140 | Furfural | — | 57 | 132 |
| GBL | 9:1 |
Fructose | 290 | HMF | HY zeolite | 67 | 148 |
| GBL | 1:0 |
Xylose | 130 | Furfural | SC-GCa-800d | 82 | 142 |
Motagamwala et al. demonstrated that a 4:1 v/v acetone–water system, using HCl as catalyst, gave rise to an almost quantitative HMF production from fructose (>95 mol% selectivity).147 GBL has been successfully used, in combination with water, to produce both furfural and HMF using heterogeneous catalysts, e.g., zeolites or Amberlyst-15 as solid acid catalysts, giving sugar-to-furan selectivities up to 70 mol%.148,149 Butanone, although it is only partially miscible with water, has been used, combined with water, in highly organic mixtures with xylose-to-furfural selectivity improvements up to 40 mol% when compared with pure water.150
It has to be noted that polar aprotic solvents are not interchangeable and using different solvents will result in different sugar-to-furan selectivities.112,123,137–139,142,152 Molecular dynamics indicate major differences in specific solvent–sugar interactions between different organic solvents, which might be the source of such asymmetry.56,117,118 Combining various solvents also results in specific difference in behavior.138 Specifically, when operating in solvent systems composed of equal volumes of three different solvents (e.g., water, toluene and dioxane) and phenylboronic acid as an additive, switching from dioxane to sulfolane or DMSO resulted in a xylose-to-furfural selectivity increase from approx. 75 to 90–95 mol%. This selectivity improvement is related to a change in its phase behavior.138 Specifically, while the system is biphasic at room temperature, it transitions to monophasic at the reaction temperature, resulting in a selectivity increase of approx. 20 mol%.137
Outlook on the effects of polar aprotic solvents
Effect on starting materials and products
As shown in Tables 1–5, the sugar concentration in the feed is almost always in the mM range. Low concentration of sugar in the feed can be connected to high sugar-to-sugar selectivity.124,125,131,147 However, in some cases, relatively high sugar concentrations in the feed result in sugar-to-furan selectivities >80 mol%.116,126,136 This shows that the sugar concentration in the feed does contribute to the final selectivity, but the catalyst and the solvent system do as well.1,2Different solvent–sugar interactions will lead to different sugar solubility, depending on the solvent system and the water content.153,154 Every organic solvent can be described by empiric solubility parameters, which are influenced by the water content and different for each compound under analysis, which will result in a very system-specific behavior.153,154 For example, adding acetone to the aqueous phase improved the rate of fructose dehydration, but reduced the sugar solubility.155,156 While a high reaction rate is beneficial, lowering the solubility of the sugar will result in a feed at a lower sugar concentration, which can be a disadvantage for an industrial-scale process design.38
When the starting material is not a solution of sugar in water but solid biomass (e.g., lignocellulose), its impregnation with the solvent will affect the hydrolysis process.135,137,143 Specifically, higher polarity of the solvent will lead to more efficient swelling of the starting material and improve the hydrolysis.135,137,143 Switching from sulfolane to DMSO in a 9:1 v/v organic–aqueous system in the acid-catalyzed degradation of cellulose, has resulted in a selectivity increase towards HMF from approx. 25 to 70 mol%.137
The amount of water present in the system can affect the behavior of the sugar and the mechanism of dehydration (Schemes 1 and 2).107,109,110 Specifically, the ring opening is triggered by the protonation of the pyranose (or furanose) oxygen, and the site of protonation is dependent on the structure of the solvent, as solvent–solute interaction can influence the pKa of OH groups, through the formation of hydrogen bonds.157,158 Performing the reaction in a mostly, or fully, organic system will influence the site of protonation, directly affecting the reaction mechanism.
As previously mentioned, the effect of the solvent system on the stability of products and intermediates cannot be neglected.30,33,34,133,138 Specifically, increasing the stability of furans and impairing the rehydration process will result in a lower production of humic by-products.57,59,60 Fu et al. show that THF inhibits oligomer formation in the context of glucose dehydration to HMF, thereby regulating the formation of solid by-products.133 The inhibited reactions are the cross-condensation of HMF and a rehydration product (i.e., levulinic acid) and the self-condensation of HMF.133 In such system, the catalyst, i.e., compressed CO2, had also a crucial role as a phase splitting agent.133
Effect on the catalyst
When the water content of the system is limited or even absent, the characterization of the behavior of the acid catalyst and that of the sugar is more complex.116–118,122–126,131,136,138,141–144 The acid-catalyzed process of sugar dehydration involves a proton transfer from the catalyst onto the sugar, which affects the β-elimination of the hydroxyl groups.1,23,34–37,40–42,51 Proton transfer can happen even in the complete absence of water.159–166 In fact, the equilibria of the dissociation of acids and bases are strongly influenced by the properties of the solvent, e.g., its basicity.159,160It is assumed that proton transfer reactions generally take place along hydrogen bonds, with the proton forming a full bond to a base as it breaks its full bond to another.157 Several mechanistic aspects may influence the rate of proton transfer, hence affecting the catalyst activity: steric factors, polarity and basicity of the solvent, possible delocalization of the charges and stabilization of transition states.162 Protic solvents (e.g., alcohols) can be protonated by the acid and serve as an intermediary between the acid and the sugar, similar to water.167 In aprotic solvents, formation of ion pairs in solution (e.g., in the case of DMSO) can be an important factor to take into account, both in mainly organic and water-free environments,161 since it affects the formation of hydrogen bonds and hence proton transfer.162 Furthermore, molecular dynamics simulations confirm that solvent–water interactions influence the stability of H3O+ cations in mixed water–organic solvent environments, while organic solvents successfully stabilize H+ in purely organic solution, based on their basicity.163 Notably, the dissociation constants of several acids vary quite significantly even when moving from one aprotic solvent to another, mirroring well the behavior of sugar dehydration observed in literature (see above).137,139,161
With the decrease of the water content of the solvent system, the catalyst activity generally increases, resulting in a faster sugar conversion.120,122–126,136,140–146 Direct interactions between the catalyst and the solvent have been reported to affect the strength of heterogeneous acid catalysts, as well as the homogeneous ones, with higher acid strengths in organic solvents compared to aqueous conditions.164 The nature of the solvent system influences strongly the activity of solid catalysts (i.e., supported sulfonic acid catalysts) as well.164 Depending on the solvent system, in particular the accessibility of the catalyst to the sugar will be affected, e.g., by swelling of ion exchange resins, differences in pore sizes or a poor wetting of the catalyst.164 In some instances, this increase of the catalytic activity can be explained by a complete solubilization of the catalyst in the solvent system, or by a stabilization of the catalyst, which reduces its progressive deactivation.118,135 Specifically, Liu et al. have indicated that the presence of sulfolane in the solvent system successfully suppresses the hydrolysis of SnCl4, which acts as the catalyst, to SnO2, which has a poor catalytic activity for sugar dehydration.136 All this information is used to rationalize and design solvent environments beneficial to biomass processing.165–168
Beyond the effects discussed above, the choice of solvent can also have a determining impact on the deactivation of solid acid catalysts.43 Firstly, polar organic solvents may reduce the catalyst fouling by reducing the formation of humins and increasing their solubility in the medium.43 Secondly, the (near) absence of water could severely reduce the propensity of solid acid catalysts to degrade under hydrothermal conditions.43 However, the deactivation challenge remains significant and largely underestimated. To remain affordable, catalysts are expected to produce 1000× their weight of product before being discarded.38 Solid catalysts are readily deactivated by deposition of <10 w% of fouling agents. A non-selective formation of only 1 w% of fouling humins reaches this deactivation level with a productivity of only 10 g product per g catalyst and, thereby, requires some 100 regeneration cycles over the catalyst lifetime, i.e., every 3 days for 1 year of operation. The batch experiments that are reported in literature cannot provide significant information, as they rarely demonstrate productivities above approx. 10 g product per g catalyst, even after a few consecutive runs.38
Application perspectives
In industrial settings, furfural is generally produced in aqueous monophasic solvent systems.1,31,40,73–77 However, biphasic operations, which are still in a development phase for industrial application, generally surpass the sugar-to-furan selectivity of aqueous monophasic operations (40–50 mol%), resulting in a selectivity of 60–70 mol%, which is still limited.1,31,40,73–77 As previously mentioned, moving from aqueous–organic biphasic systems to monophasic ones, using protic and aprotic polar organic solvents, can lead to sugar-to-furan selectivities of >90 mol%.30,33,34 However, the application of polar organic solvents can cause logistic problems in industrial set-ups. Specifically, the major shortcoming of the application of polar organic solvents is the loss of solvent in the waste disposal step.1 When sugars are separated from the other products of biomass processing, all the leftovers need to be processed to remove the polar organic solvents, to limit the dispersal in the environment, which is a problem from both environmental and economical points of view.1,168–170Additionally, it is reported that the use of some aprotic organic solvents (e.g., dioxane) negatively affects the solubility of sugars in the feed.155,156 This could lower the overall production rate, if it forces to operate at sugar concentrations that are below the desired level, e.g., as dictated for good selectivity.156 Typically, a general industrial criterion for reactor productivity indicates a minimum rate of 100 g L−1 h−1.171 A lower reactor productivity would affect the applicability of such solvent systems, e.g., resulting in prohibitively large reactors.171
Selection of the solvent system
Highly organic and fully organic polar monophasic solvent systems result in high sugar-to-furan selectivities, both for the processing of xylose and of glucose and fructose.120,122–131,134–144,146,147 In this scenario, Walker et al. rationalized the choice of a suitable solvent system for biomass conversion processes based on molecular dynamics and machine learning tools, combined with a specific experimental workflow based on two case studies (i.e., the dehydration of cyclohexanol to cyclohexene and that of fructose to HMF).166Predictive tools were developed for the selection of preferred solvent mixtures, depending on their desired properties.166 Walker et al. show that a computational screening method efficiently allows to select the best-performing candidates among a library of solvent systems, with the aim of minimizing experimental screening.166 The efficiency of the specific reaction, however, is not the only important factor to be considered in the choice of the solvent system.167
Most of the common polar organic solvents (e.g., DMSO, DMF, THF) have detrimental effects both on the environment and on human health, hence green and safer alternatives are needed.170 Specifically, green alternatives for conventional dipolar aprotic solvents mainly include more task-specific replacements, formulated by rational design.170 Biobased solvents (e.g., GVL and GBL) can be seen as possible alternatives because of their renewability, biodegradability, and also their commercial availability.170 However, applications are limited due to their instability toward strong acids and the already mentioned challenge of solvent–product separation.170
Product recovery is generally operated by means of distillation and, therefore, requires the solvent and solute to have suitable boiling points and chemical/thermal stabilities.169,170,172,173 To minimize the energy demand, the product is preferably recovered from the product stream by distillation.1,167 Therefore, the boiling point of the solvents, either at atmospheric pressure or at reduced pressure, needs to be sufficiently high with respect to that of HMF or furfural to minimize the distillation resistance.169,170 For this reason, sulfolane (bp 285 °C) can be more suitable for industrial application in the case of furfural (bp 162 °C) than DMSO (bp 189 °C), which also suffers thermal degradation.169,170
An alternative to non-green polar organic solvents is cyrene (i.e., dihydrolevoglucosenone), which is a water-miscible, high-boiling (bp 226 °C), biobased and fully biodegradable aprotic polar organic solvent (Scheme 4).174–176 Its properties are comparable with the most common non-biobased polar organic solvents and, for this reason, it has been used as a replacement of its ‘less green’ counterparts in several chemical processes.173–177
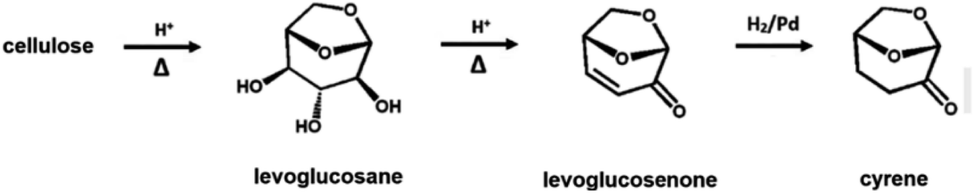 | ||
| Scheme 4 Cyrene production from cellulose. | ||
Cyrene has been successfully used as a solvent for fluorination reactions, the synthesis of ureas and SN2 substitution reactions, which are usually performed in DMF or NMP.174–178 Recently, cyrene was successfully employed in the pretreatment of biomass, in combination with water and other aprotic solvents (e.g., dioxane) at mild temperature (120 °C), to obtain free carbohydrates through enzymatic hydrolysis and removing lignin.179 However, the application for sugar dehydration reactions requires every component of the solvent system to be stable at low pH and high temperature, hence, the analysis stability of cyrene at the reaction conditions is of high importance.
Water removal and sugar extraction
Isolating the sugar from a sugar-rich feed in water and taking it into and reacting it an organic solvent is another viable option for implementing polar organic solvents in an industrial setup. Typically, the industrial feed for the production of furans is an acidic aqueous stream containing a few percent (e.g., 5 w%) of sugar, which is derived by pretreatment of biomass.1,2,167 Removal of water from this stream is highly costly and ineffective, because water distillation is characterized by a prohibitive distillation resistance.169,170There are alternative approaches to remove the desired sugars from this aqueous stream, e.g., by membrane enrichment, sugar crystallization, and liquid–liquid extraction.180–184 The latter has been successfully performed employing the formation of an organic-soluble boronate ester (here, phenylboronic acid; PBA), and this method has already been used successfully to produce sugar alcohols selectively and to isolate solid sugars from a sugar-rich hydrolysate (Scheme 5).180–183 This promising approach is challenging due to the high solubility of sugar in water and the operation parameters (mainly the high pH, ≥6), which are not compatible with those of an industrial application as the aqueous stream has a pH around 3.182,183
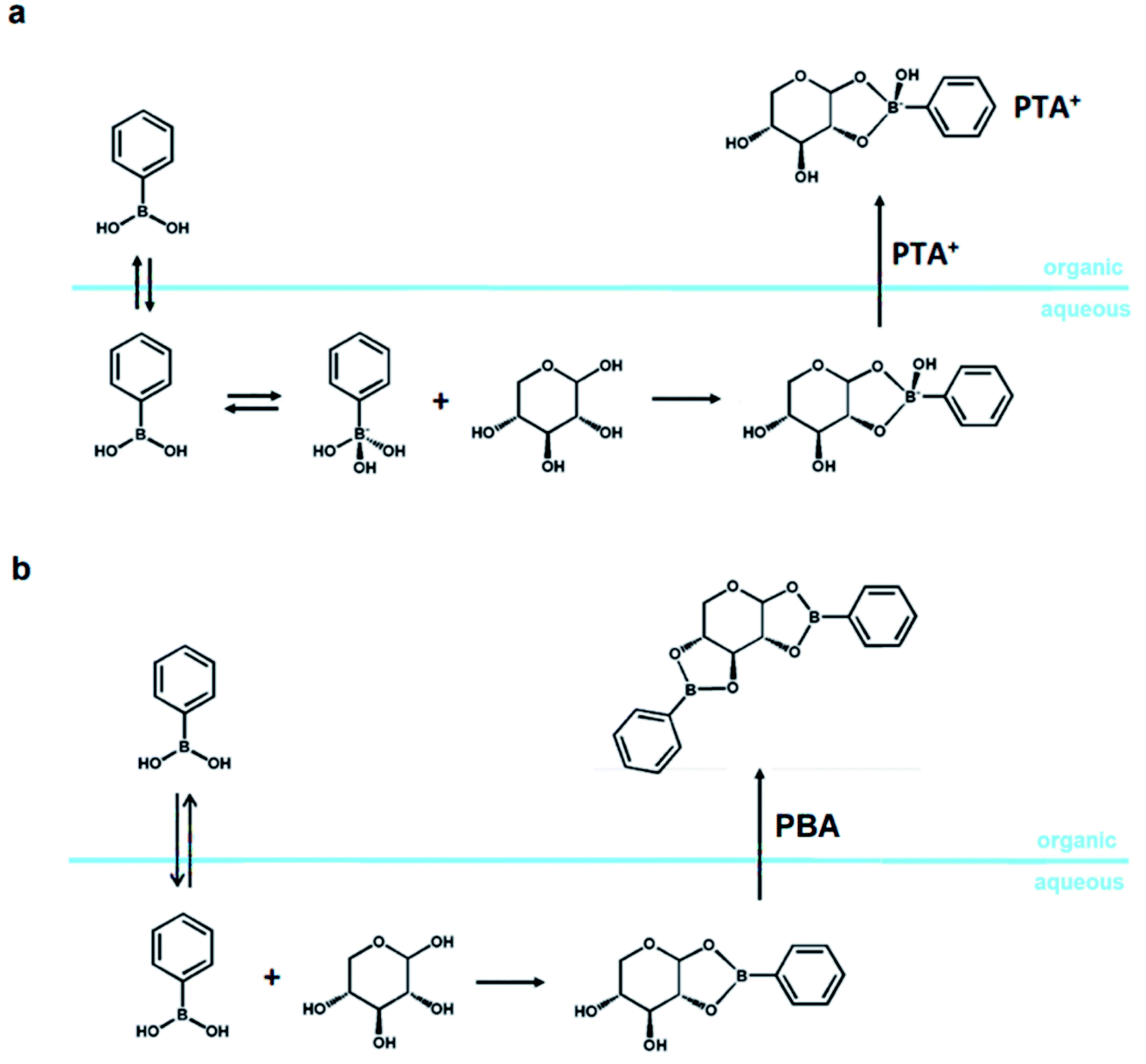 | ||
| Scheme 5 Liquid–liquid xylose extraction via formation of (a) a negatively charged PBA monoester, mediated by a phase transfer agent (PTA), (b) an uncharged PBA diester, without any phase-transfer agent. | ||
When performed at alkaline pH, the extraction relies on the phase transfer of a negatively charged monoester, formed by condensation of the sugar and the negatively charged tetragonal boronate anion (Scheme 5a), operated by a phase-transfer agent (PTA), e.g., halide salts of organic-soluble quaternary ammonium salts like Aliquat 366.182 However, xylose and, to a lesser extent, also glucose and fructose, can be extracted from an aqueous feed into toluene at pH < pKa of the boronic acid of choice (i.e., PBA).180 The extraction relies on the formation of a diester between the sugar and the neutral trigonal form of the PBA (PBA2X; Scheme 5b).184
Extracting the sugar into an organic phase opens the way for more control on the solvent system in which the dehydration of sugars is performed.138,181,183 Specifically, the process of boronate-mediated sugar extraction described in Scheme 5b has been successfully combined with the dehydration reaction in a three-solvent system. This results in a highly selective process of xylose dehydration which uses a xylose-rich hydrolysate as feed.138 In this specific case, the process concept dictates for the furfural to be isolated from the product stream by means of distillation.138 When operating with a three-solvent system composed of equal volumes of water (pH = 1, H2SO4), sulfolane and 1-methylnaphthalene (MN), the system partitions at room temperature into two phases, i.e., an apolar phase (MN) and a polar phase (water–sulfolane).138 The apolar catalyst-free phase contains most of the furfural, which can be isolated by means of distillation.138 The polar phase contains a minor portion of the furfural, which can be extracted through liquid–liquid extraction using clean MN.138 However, degradation of sulfur-containing solvents could be an issue for this process concept, and additional research towards industrial application is necessary.
When a xylose extraction step precedes the dehydration, a number of biomass hydrolysis by-products are left in the hydrolysate feed, resulting in the production of furfural with a much higher purity.183 Additionally, the polar organic solvent does not contact the aqueous waste feed, reducing the possibility of spillage, hence reducing the environmental impact.138,168–170
Summary and future prospects
The application of polar organic solvents in the production of HMF and furfural from C5 and C6 sugars leads to sugar-to-furan selectivities up to 90–95 mol%, depending on the reaction conditions. This is an improvement compared to fully aqueous and aqueous–organic biphasic conditions, for which the sugar-to-furan selectivity is limited to approx. 45–50 mol% and 60–70 mol%, respectively. This improvement can be rationalized by solvent effects interacting with both the starting material and the catalyst, leading to effects such as: (i) the stabilization of specific sugar conformers, which are favorable to a selective dehydration, (ii) a change in the catalyst activity, possibly related to enhanced intrinsic acidity and, in the case of solid acids, improved accessibility and reduced fouling, and (iii) generally, a higher rate of dehydration.Each aprotic organic solvent shows a specific behavior, depending on the specific solvent–sugar and solvent–catalyst interactions, leading to different outcomes for each solvent at different reaction conditions.
However, this beneficial effect of polar solvents on the reaction parameters of sugar dehydration, though promising, comes with an increased cost, due to solvent recycling and environmental impact, which impairs their commercial availability. The sugar feed is generally delivered in water, as sugar syrup extracted from sugar beet or cane, or produced by hydrolysis of starch, cellulose or hemicellulose. To be recycled, the polar solvent needs to be separated from the water that the sugar is delivered in, or the sugar needs to be delivered dry. Moreover, additional techniques for ensuring the full compatibility with highly organic operations and the industrial feed, e.g., liquid–liquid sugar extraction, are to be taken into consideration. All these additional steps will inevitably increase the process costs and complexity, requiring the sugar-to-furfural yield to be sufficiently high to justify such a trade-off. Additional research and investigation of the upscaling of viable process concepts are needed. Organic–aqueous monophasic conditions remain promising to improve the selectivity in the production of furans from biomass. Moreover, increasing the sugar concentration in the feed and recycling the organic solvents are also important factors to take into consideration, as they can improve the production rate of furans, reducing the operation costs.
Author contributions
L. R. proposed the outline of the review and analysed the literature in depth. Manuscript preparation and subsequent editing/improvement of the text was performed by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from Shell Global Solutions International B.V. is gratefully acknowledged.Notes and references
- J.-P. Lange, E. van der Heide, J. van Buijtenen and R. Price, ChemSusChem, 2012, 5, 150–166 CrossRef CAS.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. López Granadosa, Energy Environ. Sci., 2016, 9, 1144–1189 RSC.
- P. L. Arias, J. A. Cecilia, I. Gandarias, J. Iglesias, M. López Granados, R. Mariscal, G. Morales, R. Moreno-Tost and P. Maireles-Torres, Catal. Sci. Technol., 2020, 10, 2721–2757 RSC.
- Y. Luo, Z. Li, X. Li, X. Liu, J. Fan, J. H. Clark and C. Hu, Catal. Today, 2019, 319, 14–24 CrossRef CAS.
- H. Wang, C. Zhu, D. Li, Q. Liu, J. Tan, C. Wang, C. Cai and L. Ma, Renewable Sustainable Energy Rev., 2019, 103, 227–247 CrossRef CAS.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- K. Gupta, R. K. Rai and S. K. Singh, ChemCatChem, 2018, 10, 2326–2349 CrossRef CAS.
- I. K. M. Yu and D. C. W. Tsang, Bioresour. Technol., 2017, 238, 716–732 CrossRef CAS.
- B. Ward, Bacterial Energy Metabolism in Molecular Medical Microbiology, Academic Press, Cambridge, Massachusetts, 2nd edn, 2014, pp. 201–233 Search PubMed.
- J. Cai, Y. He, X. Yu, S. W. Banks, Y. Yang, X. Zhang, Y. Yu, R. Liu and A. V. Bridgwater, Renewable Sustainable Energy Rev., 2017, 76, 309–322 CrossRef CAS.
- A. Zoghlami and G. Paës, Front. Chem., 2019, 7, 874 CrossRef CAS.
- X. Li, Y. Chen and J. Nielsen, Curr. Opin. Biotechnol., 2019, 57, 56–65 CrossRef CAS.
- S. K. Bhatia, S. S. Jagtap, A. A. Bedekar, R. K. Bhatia, A. K. Patel, D. Pant, J. R. Banu, C. V. Rao, Y.-G. Kim and Y.-H. Yang, Bioresour. Technol., 2020, 300, 122724 CrossRef CAS.
- J. Baruah, B. K. Nath, R. Sharma, S. Kumar, R. C. Deka, D. C. Baruah and E. Kalita, Front. Energy Res., 2018, 6, 141 CrossRef.
- Y. Kawamata, T. Yoshikawa, Y. Koyama, H. Ishimaru, S. Ohtsuki, E. Fumoto, S. Sato, Y. Nakasaka and T. Masuda, Ind. Crops Prod., 2021, 159, 113078 CrossRef CAS.
- B. Song, R. Lin, C. H. Lam, H. Wu, T.-H. Tsui and Y. Yu, Renewable Sustainable Energy Rev., 2021, 135, 110370 CrossRef CAS.
- Y. Jing, Y. Guo, Q. Xia, X. Liu and Y. Wang, Chem, 2019, 5, 2520–2546 CAS.
- R. Calvo-Serrano, M. Guo, C. Pozo, Á. Galán-Martín and G. Guillén-Gosálbez, ACS Sustainable Chem. Eng., 2019, 7, 10570–10582 CrossRef CAS.
- J. M. J. M. Ravasco, C. M. Monteiro, F. Siopa, A. F. Trindade, J. Oble, G. Poli, S. P. Simeonov and C. A. M. Afonso, ChemSusChem, 2019, 12, 4629–4635 CrossRef CAS PubMed.
- S. Jiang, W. Ramdani, E. Muller, C. Ma, M. Pera-Titus, F. Jerôme and K. De Oliveira Vigiera, ChemCatChem, 2020, 13, 1699–1704 CrossRef CAS.
- J. Xu, N. Li, X. Yang, G. Li, A. Wang, Y. Cong, X. Wang and T. Zhang, ACS Catal., 2017, 7, 5880–5886 CrossRef.
- J.-P. Lange and S. H. Wadman, ChemSusChem, 2020, 13, 5329–5337 CrossRef CAS PubMed.
- B. Danon, G. Marcotullio and W. de Jong, Green Chem., 2014, 16, 39–54 RSC.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Green Chem., 2006, 8, 701–709 RSC.
- G. Tsilomelekis, M. J. Orella, Z. Lin, Z. Cheng, W. Zheng, V. Nikolakis and D. G. Vlachos, Green Chem., 2016, 18, 1983–1993 RSC.
- I. Agirrezabal-Telleria, I. Gandarias and P. L. Arias, Bioresour. Technol., 2013, 143, 258–264 CrossRef CAS PubMed.
- A. Mukherjee, M.-J. Dumont and V. Raghavan, Biomass Bioenergy, 2015, 72, 143–183 CrossRef CAS.
- S. Peleteiro, S. Rivas, J. L. A. V. Santos and J. C. Parajó, Bioresour. Technol., 2016, 202, 181–191 CrossRef CAS PubMed.
- Z. Xue, M.-G. Ma, Z. Li and T. Mu, RSC Adv., 2016, 6, 98874–98892 RSC.
- L. Shuai and J. Luterbacher, ChemSusChem, 2016, 9, 133–155 CrossRef CAS PubMed.
- J. E. Romo, N. V. Bollar, C. J. Zimmermann and S. G. Wettstein, ChemCatChem, 2018, 10, 4805–4816 CrossRef PubMed.
- U. Tyagi, N. Anand and D. Kumar, Bioresour. Technol., 2019, 289, 121675 CrossRef PubMed.
- C. B. T. L. Lee and T. Y. Wu, Renewable Sustainable Energy Rev., 2021, 137, 110172 CrossRef CAS.
- Y. Zhao, K. Lu, H. Xu, L. Zhu and S. Wang, Renewable Sustainable Energy Rev., 2021, 139, 110172 CrossRef.
- O. Ershova, K. Nieminen and H. Sixta, ChemCatChem, 2017, 9, 3031–3040 CrossRef CAS.
- Y. Kim, A. Mittal, D. J. Robichaud, H. M. Pilath, B. D. Etz, P. C. St. John, D. K. Johnson and S. Kim, ACS Catal., 2020, 10, 14707–14721 CrossRef CAS.
- M. Sajid, M. R. Dilshad, M. S. U. Rehman, D. Liu and X. Zhao, Molecules, 2021, 26, 2208 CrossRef CAS PubMed.
- J.-P. Lange, Catal. Sci. Technol., 2016, 6, 4759–4767 RSC.
- I. B. Elkina, A. B. Gilman, V. V. Ugrozov and V. V. Volkov, Ind. Eng. Chem. Res., 2013, 52, 8856–8863 CrossRef CAS.
- M. Peters, M. F. Eckstein, G. Hartjen, A. C. Spiess, W. Leitner and L. Greiner, Ind. Eng. Chem. Res., 2007, 46, 7073–7078 CrossRef CAS.
- I. Agirrezabal-Telleria, I. Gandarias and P. L. Arias, Catal. Today, 2014, 234, 42–58 CrossRef CAS.
- S. Shrestha, X. Fonoll, S. K. Khanal and L. Raskin, Bioresour. Technol., 2017, 245, 1245–1257 CrossRef CAS PubMed.
- J.-P. Lange, Angew. Chem., Int. Ed., 2015, 54, 13186–13197 CrossRef CAS.
- J. Asomaning, S. Haupt, M. Chae and D. C. Bressler, Renewable Sustainable Energy Rev., 2018, 92, 642–657 CrossRef CAS.
- P. Priecel, J. E. Perez Mejia, P. D. Carà and J. A. Lopez-Sanchez, Green Chem., 2018, 56, 243–299 CAS.
- K. R. Enslow and A. T. Bell, ChemCatChem, 2015, 7, 479–489 CrossRef CAS.
- O. Yemiş and G. Mazza, Waste Biomass Valorization, 2019, 10, 1343–1353 CrossRef.
- L. Ricciardi, W. Verboom, J.-P. Lange and J. Huskens, ACS Sustainable Chem. Eng., 2019, 7, 14273–14279 CrossRef CAS.
- L. Ricciardi, W. Verboom, J.-P. Lange and J. Huskens, ChemSusChem, 2020, 13, 3589–3593 CrossRef CAS PubMed.
- C. Xiouras, N. Radacsi, G. Sturm and G. D. Stefanidis, ChemSusChem, 2016, 9, 2159–2166 CrossRef CAS PubMed.
- O. Ershova, J. Kanervo, S. Hellsten and H. Sixta, RSC Adv., 2015, 5, 66727–66737 RSC.
- L. Zhu, X. Fu, Y. Hu and C. Hu, ChemSusChem, 2020, 18, 4812–4832 CrossRef PubMed.
- E. Nikolla, Y. Román-Leshkov, M. Moliner and M. E. Davis, ACS Catal., 2011, 1, 1724–1728 CrossRef.
- K. L. Baugh and P. L. McCarty, Biotechnol. Bioeng., 1988, 31, 50–61 CrossRef CAS.
- M. R. Nimlos, X. Qian, M. Davis, M. E. Himmel and D. K. Johnson, J. Phys. Chem. A, 2006, 110, 11824–11838 CrossRef CAS.
- X. Qian, M. R. Nimlos, M. Davis, D. K. Johnson and M. E. Himmel, Carbohydr. Res., 2005, 340, 2319–2327 CrossRef CAS PubMed.
- S. K. R. Patil and C. R. F. Lund, Energy Fuels, 2011, 25, 4745–4755 CrossRef CAS.
- S. K. R. Patil, J. Heltzel and C. R. F. Lund, Energy Fuels, 2012, 26, 5281–5293 CrossRef CAS.
- J. Horvat, B. Klaić, B. Metelko and V. Šunjić, Tetrahedron Lett., 1985, 26, 2111–2114 CrossRef CAS.
- J. Horvat, B. Klaić, B. Metelko and V. Šunjić, Croat. Chem. Acta, 1986, 59, 429–438 CAS.
- I. van Zandvoort, Y. Wang, C. B. Rasrendra, E. R. H. van Eck, P. C. A. Bruijnincx, H. J. Heeres and B. M. Weckhuysen, ChemSusChem, 2013, 6, 1745–1758 CrossRef CAS PubMed.
- J. M. R. Gallo, D. M. Alonso, M. A. Mellmera and J. A. Dumesic, Green Chem., 2013, 15, 85–90 RSC.
- H. Shen, H. Shan and L. Liu, ChemSusChem, 2020, 13, 513–519 CrossRef CAS.
- N. Shi, Q. Liu, H. Cen, R. Ju, X. He and L. Ma, Biomass Convers. Biorefin., 2020, 10, 277–287 CrossRef CAS.
- A. T. Pedersen, R. Ringborg, T. Grotkjær, S. Pedersen and J. M. Woodley, Chem. Eng. J., 2015, 273, 455–464 CrossRef.
- Y. Zhao, H. Xu, K. Lu, Y. Qu, L. Zhu and S. Wang, Energy Sci. Eng., 2019, 7, 2237–2246 CrossRef CAS.
- Z. Chen, X. Bai, A. Lusi, W. A. Jacoby and C. Wan, Bioresour. Technol., 2019, 289, 121708 CrossRef PubMed.
- R. Xing, W. Qi and G. W. Huber, Energy Environ. Sci., 2011, 4, 2193–2205 RSC.
- A. Mitta, S. K. Black, T. B. Vinzant, M. O'Brien, M. P. Tucker and D. K. Johnson, ACS Sustainable Chem. Eng., 2017, 5, 5694–5701 CrossRef.
- T. M. C. Hoang, L. Lefferts and K. Seshan, ChemSusChem, 2013, 6, 1651–1658 CrossRef CAS.
- J.-M. Pin, N. Guigo, A. Mija, L. Vincent, N. Sbirrazzuoli, J. C. van der Waal and E. de Jong, ACS Sustainable Chem. Eng., 2014, 2, 2182–2190 CrossRef CAS.
- R. M. Abdilla-Santes, S. Agarwal, X. Xi, H. Heeres, P. J. Deuss and H. J. Heeres, J. Anal. Appl. Pyrolysis, 2020, 152, 104963 CrossRef CAS.
- B. Saha and M. M. Abu-Omar, Green Chem., 2014, 16, 24–38 RSC.
- R. Weingarten, J. Cho, W. C. Conner Jr and G. W. Huber, Green Chem., 2010, 12, 1423–1429 RSC.
- Z. Wang, S. Bhattacharyya and D. G. Vlachos, Green Chem., 2020, 22, 8699–8712 RSC.
- J. Esteban, A. J. Vorholt and W. Leitner, Green Chem., 2020, 22, 2097–2128 RSC.
- F. Delbecq, Y. Takahashi, T. Kondo, C. Cara Corbas, E. Ruiz Ramos and C. Len, Catal. Commun., 2018, 110, 74–78 CrossRef CAS.
- M. Papaioannou, R. J. T. Kleijwegt, J. van der Schaaf and M. F. N. d'Angelo, Ind. Eng. Chem. Res., 2019, 58, 16106–16115 CrossRef CAS.
- G.-T. Jeong, S.-K. Kim and D.-H. Park, Biotechnol. Bioprocess Eng., 2013, 18, 88–93 CrossRef CAS.
- A. A. Kiss, J.-P. Lange, B. Schuur, D. W. F. Brilman, A. G. J. van der Ham and S. R. A. Kersten, Biomass Bioenergy, 2016, 95, 296–309 CrossRef CAS.
- S. Kumar, S. Pandey, K. L. Wasewar, N. Ak and H. Uslu, J. Chem. Eng. Data, 2021, 66, 1557–1573 CrossRef CAS.
- T.-Y. Chen, P. Desir, M. Bracconi, B. Saha, M. Maestri and D. G. Vlachos, Ind. Eng. Chem. Res., 2021, 60, 3723–3735 CrossRef CAS.
- L. Zhang, H. Yu, P. Wang and Y. Li, Bioresour. Technol., 2014, 151, 355–360 CrossRef CAS PubMed.
- Q. Zhang, C. Wang, J. Mao, S. Ramaswamy, X. Zhang and F. Xu, Ind. Crops Prod., 2019, 138, 111454 CrossRef CAS.
- E. S. Morais, M. G. Freire, C. S. R. Freire, J. A. P. Coutinho and A. J. D. Silvestre, ChemSusChem, 2020, 13, 784–790 CrossRef CAS PubMed.
- C. Liu, L. Wei, X. Yin, X. Pan, J. Hu, N. Li, J. Xu, J. Jiang and K. Wang, Chem. Eng. J., 2021, 425, 130608 CrossRef CAS.
- X. Wang, M. Qiu, Y. Tang, J. Yang, F. Shen, X. Qia and Y. Yu, Int. J. Biol. Macromol., 2021, 187, 232–239 CrossRef CAS PubMed.
- A. Cañada-Barcala, D. Rodríguez-Llorente, L. López, P. Navarro, E. Hernández, V. I. Águeda, S. Álvarez-Torrellas, J. C. Parajó, S. Rivas and M. Larriba, ACS Sustainable Chem. Eng., 2021, 9, 10266–10275 CrossRef.
- J. Köchermann, J. Schreiber and M. Klemm, ACS Sustainable Chem. Eng., 2019, 7, 12323–12330 Search PubMed.
- X. Hu, C. Lievens and C.-Z. Li, ChemSusChem, 2012, 5, 1427–1434 CrossRef CAS PubMed.
- X. Zhang, H. Lu, K. Wu, Y. Liu, C. Liu, Y. Zhu and B. Liang, Chin. J. Chem. Eng., 2020, 28, 136–142 CrossRef CAS.
- J. K. C. N. Agutaya, R. Inoue, S. Siew Vin Tsie, A. T. Quitain, J. de la Peña-García, H. Perez-Sànchez, M. Sasaki and T. Kida, Ind. Eng. Chem. Res., 2020, 59, 16527–16538 CrossRef CAS.
- K. Sun, Q. Li, L. Zhang, Y. Shao, Z. Zhang, S. Zhang, Q. Liu, Y. Wang and X. Hu, Bioresour. Technol. Rep., 2020, 10, 100419 CrossRef.
- X. Cheng, D. Jiang, X. Hu, B. Barati, Y. Hu, L. Qian, Z. He, S. Wang and H. Li, J. Anal. Appl. Pyrolysis, 2021, 154, 104996 CrossRef CAS.
- Y. Romàn-Leshkov and J. A. Dumesic, Top. Catal., 2009, 52, 297–303 CrossRef.
- N. Jiang, R. Huang, W. Qi, R. Su and Z. He, BioEnergy Res., 2012, 5, 380–386 CrossRef CAS.
- L. Xu, R. Nie, H. Xu, X. Chen, Y. Li and X. Lu, Ind. Eng. Chem. Res., 2020, 59, 2754–2760 CrossRef CAS.
- X.-X. Xue, C.-L. Ma, J.-H. Di, X.-Y. Huo and Y.-C. He, Bioresour. Technol., 2018, 268, 292–299 CrossRef CAS PubMed.
- M. Zdanowicz, K. Wilpiszewska and T. Spychaj, Carbohydr. Polym., 2018, 200, 361–380 CrossRef CAS PubMed.
- J. Sittiwong, S. Boonmark, W. Nunthakitgoson, T. Maihom, C. Wattanakit and J. Limtrakul, Inorg. Chem., 2021, 60, 4860–4868 CrossRef CAS PubMed.
- A. Gupta, S. U. Nandanwar, P. Niphadkar, I. Simakova and V. Bokade, Biomass Bioenergy, 2020, 139, 105646 CrossRef CAS.
- L. Peng, M. Wang, H. Li, J. Wang, J. Zhang and L. He, Green Chem., 2020, 22, 5656–5665 RSC.
- S. Roy, G. Mpourmpakis, D.-Y. Hong, D. G. Vlachos, A. Bhan and R. J. Gorte, ACS Catal., 2012, 2, 1846–1853 CrossRef CAS.
- S. Arifin and I.-L. Chien, Ind. Eng. Chem. Res., 2008, 47, 790–803 CrossRef CAS.
- M. Zhang and Y. Yu, Ind. Eng. Chem. Res., 2013, 52, 9505–9514 CrossRef CAS.
- J. Bedia, R. Barrionuevo, J. Rodríguez-Mirasol and T. Cordero, Appl. Catal., B, 2011, 103, 302–310 CrossRef CAS.
- B. Song, Z. Wu, Y. Yu and H. Wu, Ind. Eng. Chem. Res., 2020, 59, 7336–7345 CrossRef CAS.
- A. K. Chew, T. W. Walker, Z. Shen, B. Demir, L. Witteman, J. Euclide, G. W. Huber, J. A. Dumesic and R. C. Van Lehn, ACS Catal., 2020, 10, 1679–1691 CrossRef CAS.
- M. A. Mellmer, C. Sanpitakseree, B. Demir, P. Bai, K. Ma, M. Neurock and J. A. Dumesic, Nat. Catal., 2018, 1, 199–207 CrossRef CAS.
- J. J. Varghese and S. H. Mushrif, React. Chem. Eng., 2019, 4, 165–206 RSC.
- K. M. Vilonen, A. Vuolanto, J. Jokela, M. S. A. Leisola and A. O. I. Krause, Biotechnol. Prog., 2004, 20, 1555–1560 CrossRef CAS PubMed.
- J. Chen, B. Chan, Y. Shao and J. Ho, Phys. Chem. Chem. Phys., 2020, 22, 3855–3866 RSC.
- R. J. J. Ganado, D. E. C. Yu and F. C. Franco, Ind. Eng. Chem. Res., 2019, 58, 14621–14631 CrossRef.
- I. K. M. Yu, D. C. W. Tsang, S. S. Chen, L. Wang, A. J. Hunt, J. Sherwood, K. De Oliveira Vigier, F. Jérôme, Y. Sik Ok and C. Sun Poon, Bioresour. Technol., 2017, 245, 456–462 CrossRef CAS PubMed.
- C. Jin, N. Xiang, X. Zhu, E. Shuang, K. Sheng and X. Zhang, Appl. Catal., B, 2021, 285, 119799 CrossRef CAS.
- M. R. Whitaker, A. Parulkar, P. Ranadive, R. Joshi and N. A. Brunelli, ChemSusChem, 2019, 12, 2211–2219 CrossRef CAS PubMed.
- V. Vasudevan and S. H. Mushrif, RSC Adv., 2015, 5, 20756–20763 RSC.
- Q. Lin, S. Liao, L. Li, W. Li, F. Yue, F. Peng and J. Ren, Green Chem., 2020, 22, 532–539 RSC.
- X. Guo, F. Guo, Y. Li, Z. Zheng, Z. Xing, Z. Zhu, T. Liu, X. Zhang and Y. Jin, Appl. Catal., A, 2018, 558, 18–25 CrossRef CAS.
- O. H. Pardo Cuervo, G. P. Romanelli, J. A. Cubillos, H. A. Rojas and J. J. Martínez, ChemistrySelect, 2020, 5, 4186–4193 CrossRef CAS.
- X. Fu, Y. Hu, Y. Zhang, Y. Zhang, D. Tang, L. Zhu and C. Hu, ChemSusChem, 2020, 13, 501–512 CrossRef CAS PubMed.
- S. L. Barbosa, M. d. S. Freitas, W. T. P. dos Santos, D. Lee Nelson, S. I. Klein, G. Cesar Clososki, F. J. Caires, A. C. M. Baroni and A. P. Wentz, Sci. Rep., 2021, 11, 1919 CrossRef CAS PubMed.
- R. Wang, X. Liang, F. Shen, M. Qiu, J. Yang and X. Qi, ACS Sustainable Chem. Eng., 2020, 8, 1163–1170 CrossRef CAS.
- S. Luan, W. Li, Z. Guo, W. Li, X. Hou, Y. Song, R. Wang and Q. Wang, Green Energy Environ., 2021 DOI:10.1016/j.gee.2021.01.005.
- F. Wang, H. Z. Wu, C. L. Liu, R. Z. Yang and W. S. Dong, Carbohydr. Res., 2013, 368, 78–83 CrossRef CAS PubMed.
- R. S. Nunes, G. M. Reis, L. M. Vieira, D. Mandelli and W. A. Carvalho, Catal. Lett., 2021, 151, 398–408 CrossRef CAS.
- M. A. Mellmer, C. Sanpitakseree, B. Demir, K. Ma, W. A. Elliott, P. Bai, R. L. Johnson, T. W. Walker, B. H. Shanks, R. M. Rioux, M. Neurock and J. A. Dumesic, Nat. Commun., 2019, 10, 1132 CrossRef PubMed.
- Q. Xia, H. Peng, Y. Zhang, G. Fu, Y. Liu, Z. Xiao, L. Huang and H. Bi, Biomass Convers. Biorefin., 2021, 11, 1–13 CrossRef.
- A. R. C. Morais and R. Bogel-Lukasik, Green Chem., 2016, 18, 2331–2334 RSC.
- G. Park, W. Jeon, C. Ban, H. C. Woo and D. H. Kim, Energy Convers. Manage., 2016, 118, 135–141 CrossRef CAS.
- Q. Sun, S. Wang, B. Aguila, X. Meng, S. Ma and F.-S. Xiao, Nat. Commun., 2018, 9, 3236 CrossRef PubMed.
- J. E. Romo, K. C. Miller, T. L. Sundsted, A. L. Job, K. A. Hoo and S. G. Wettstein, ChemCatChem, 2019, 11, 4715–4719 CrossRef CAS.
- X. Fu, J. Dai, X. Guo, J. Tang, L. Zhu and C. Hu, Green Chem., 2017, 19, 3334–3343 RSC.
- P. S. Metkar, E. J. Till, D. R. Corbin, C. J. Pereira, K. W. Hutchenson and S. K. Sengupta, Green Chem., 2015, 17, 1453–1466 RSC.
- C. Liu, M. Wei, F. Wang, L. Wei, X. Yin, J. Jiang and K. Wang, J. Energy Inst., 2020, 93, 1642–1650 CrossRef CAS.
- B. R. Caes and R. T. Raines, ChemSusChem, 2011, 4, 353–356 CrossRef CAS PubMed.
- K. Wang, J. Ye, M. Zhou, P. Liu, X. Liang, J. Xu and J. Jiang, Cellulose, 2017, 24, 1383–1394 CrossRef CAS.
- L. Ricciardi, W. Verboom, J.-P. Lange and J. Huskens, Green Chem., 2021, 23, 8079–8088 RSC.
- S. Ravi, Y. Choi and J. K. Choe, Appl. Catal., B, 2020, 271, 118942 CrossRef CAS.
- Q. Guo, L. Ren, S. M. Alhassan and M. Tsapatsis, Chem. Commun., 2019, 55, 14942–14945 RSC.
- J. Jeong, C. A. Antonyraj, S. Shin, S. Kim, B. Kim, K.-Y. Lee and J. K. Cho, J. Ind. Eng. Chem., 2013, 19, 1106–1111 CrossRef CAS.
- T. Yang, W. Li, M. Su, Y. Liu and M. Liu, New J. Chem., 2020, 44, 7968–7975 RSC.
- L. Ji, P. Li, F. Lei, X. Song, J. Jiang and K. Wang, Energies, 2020, 13, 5294 CrossRef CAS.
- Z. Wu, F. Wang, L. Hu, Y. Jiang, X. Wang and J. Xu, J. Chem. Technol. Biotechnol., 2021, 96, 971–979 CrossRef CAS.
- Z. Chen and C. Wan, Bioresour. Technol. Rep., 2019, 8, 100318 CrossRef.
- Q. Wang, X. Zhuang, W. Wang, X. Tan, Q. Yu, W. Qi and Z. Yuan, Chem. Eng. J., 2018, 334, 698–706 CrossRef CAS.
- A. H. Motagamwala, K. Huang, C. T. Maravelias and J. A. Dumesic, Energy Environ. Sci., 2019, 12, 2212–2222 RSC.
- L. Wang, H. Guo, Q. Xie, J. Wang, B. Hou, L. Jia, J. Cui and D. Li, Appl. Catal., A, 2019, 572, 51–60 CrossRef CAS.
- H. Kim, S. Yang and D. H. Kim, Environ. Res., 2020, 187, 1–7 CrossRef PubMed.
- Y. Zhao, H. Xu, K. Wang, K. Lu, Y. Qu, L. Zhu and S. Wang, Sustainable Energy Fuels, 2019, 3, 3208–3218 RSC.
- K. Sun, Y. Shao, P. Liu, L. Zhang, G. Gao, D. Dong, S. Zhang, G. Hu, L. Xu and X. Hu, Fuel, 2021, 300, 120199 Search PubMed.
- R. Li, Q. Lin, Y. Wang, W. Yang, X. Liu, W. Li, X. Wang, X. Wang, C. Liu and J. Ren, Appl. Catal., B, 2021, 286, 119862 CrossRef CAS.
- M. Saha, P. Bernstein Saynik, A. Borah, R. S. Malani, P. Arya, Shivangi and V. S. Moholkar, Bioresour. Technol. Rep., 2019, 5, 206–211 CrossRef.
- A. Ghosh, R. C. Brown and X. Bai, Green Chem., 2016, 18, 1023–1031 RSC.
- H. Chang, I. Bajaj, G. W. Huber, C. T. Maravelias and J. A. Dumesic, Green Chem., 2020, 22, 5285–5295 RSC.
- H. Chang, I. Bajaj, A. H. Motagamwala, A. Somasundaram, G. W. Huber, C. T. Maravelias and J. A. Dumesic, Green Chem., 2021, 23, 3277–3288 RSC.
- H. Rasmussen, H. R. Sørensen and A. S. Meyer, Carbohydr. Res., 2014, 385, 45–57 CrossRef CAS PubMed.
- S. Feng, C. Bagia and G. Mpourmpakis, J. Phys. Chem. A, 2013, 117, 5211–5219 CrossRef CAS PubMed.
- I. M. Kolthoff, Anal. Chem., 1974, 46, 1992–2003 CrossRef CAS.
- W. C. Barrette Jr, H. W. Johnson Jr and D. T. Sawyer, Anal. Chem., 1984, 56, 1898–1902 CrossRef.
- A. Jarczewski and C. D. Hubbard, J. Mol. Struct., 2003, 649, 287–307 CrossRef CAS.
- S. Kong, I. G. Shenderovich and M. V. Vener, J. Phys. Chem. A, 2010, 114, 2393–2399 CrossRef CAS PubMed.
- A. K. Chew and R. C. Van Lehn, Front. Chem., 2019, 7, 439 CrossRef CAS PubMed.
- S. Koujout and D. R. Brown, Catal. Lett., 2004, 98, 195–202 CrossRef CAS.
- A. K. Chew, T. W. Walker, Z. Shen, B. Demir, L. Witteman, J. Euclide, G. W. Huber, J. A. Dumesic and R. C. Van Lehn, ACS Catal., 2020, 10, 1679–1691 CrossRef CAS.
- T. W. Walker, A. K. Chew, R. C. Van Lehn, J. A. Dumesic and G. W. Huber, Top. Catal., 2020, 63, 649–663 CrossRef CAS.
- I. M. Kolthoff and K. Chantooni Jr, J. Am. Chem. Soc., 1968, 90, 3320–3326 CrossRef CAS.
- X. Li, Y. Chen and J. Nielsen, Curr. Opin. Biotechnol., 2019, 57, 56–65 CrossRef CAS PubMed.
- S. Ramaswamy, H. J. Huang and B. V. Ramarao, Separation and Purification Technologies in Biorefineries, John Wiley & Sons, Hoboken, New Jersey, 2013, pp. 233–258 Search PubMed.
- J.-P. Lange, ChemSusChem, 2017, 10, 245–252 CrossRef CAS PubMed.
- J.-P. Lange, Nat. Catal., 2021, 4, 186–192 CrossRef CAS.
- F. Gao, R. Bai, F. Ferlin, L. Vaccaro, M. Li and Y. Gu, Green Chem., 2020, 22, 6240–6257 RSC.
- D. L. Head and C. G. McCarty, Tetrahedron, 1973, 16, 1405–1408 CrossRef.
- B. F. M. Kuster, Starch, 1900, 42, 314–321 CrossRef.
- J. Sherwood, M. De Bruyn, A. Constantinou, L. Moity, C. R. McElroy, T. J. Farmer, T. Duncan, W. Raverty, A. J. Hunt and J. H. Clark, Chem. Commun., 2014, 50, 9650–9652 RSC.
- J. E. Camp, ChemSusChem, 2018, 11, 3048–3055 CrossRef CAS PubMed.
- A. O. Komarova, G. R. Dick and J. S. Luterbacher, Green Chem., 2021, 23, 4790–4799 RSC.
- L. Mistry, K. Mapesa, T. W. Bousfield and J. E. Camp, Green Chem., 2017, 19, 2123–2128 RSC.
- X. Meng, Y. Pu, M. Li and A. J. Ragauskas, Green Chem., 2020, 22, 2862–2872 RSC.
- F. Cao, T. J. Schwartz, D. J. McClelland, S. H. Krishna, J. A. Dumesic and G. W. Huber, Energy Environ. Sci., 2015, 8, 1808–1815 RSC.
- G. J. Griffin and L. Shu, J. Chem. Technol. Biotechnol., 2004, 79, 505–511 CrossRef CAS.
- N. Sánchez-Bastardo, I. Delidovich and E. Alonso, ACS Sustainable Chem. Eng., 2018, 6, 11930–11938 CrossRef.
- S. S. Gori, M. V. Ramakrishnam Raju, D. A. Fonseca, J. Satyavolu, C. T. Burns and M. H. Nantz, ACS Sustainable Chem. Eng., 2015, 3, 2452–2457 CrossRef CAS.
- L. Ricciardi, W. Verboom, J.-P. Lange and J. Huskens, ACS Sustainable Chem. Eng., 2021, 9, 6632–6638 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |