
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanism of formaldehyde and formic acid formation on (101)-TiO2@Cu4 systems through CO2 hydrogenation†
Deobrat
Singh
 *af,
Sanjeev K.
Gupta
*b,
Nicola
Seriani
c,
Igor
Lukačević
d,
Yogesh
Sonvane
a,
P. N.
Gajjar
e and
Rajeev
Ahuja
fg
*af,
Sanjeev K.
Gupta
*b,
Nicola
Seriani
c,
Igor
Lukačević
d,
Yogesh
Sonvane
a,
P. N.
Gajjar
e and
Rajeev
Ahuja
fg
aApplied Materials Lab, Department of Physics, S. V. National Institute of Technology, Surat, 395007, India. E-mail: deobrat.singh@physics.uu.se
bComputational Materials and Nanoscience Group, Department of Physics and Electronics, St. Xavier's College, Ahmedabad, 380009, India. E-mail: sanjeev.gupta@sxca.edu.in
cThe Abdus Salam International Centre for Theoretical Physics, Strada Costiera 11, 34151 Trieste, Italy
dDepartment of Physics, Josip Juraj Strossmayer University of Osijek, 31000 Osijek, Croatia
eDepartment of Physics, University School of Sciences, Gujarat University, Ahmedabad, 380009, India
fCondensed Matter Theory Group, Materials Theory Division, Department of Physics and Astronomy, Uppsala University, Box 516, 75120 Uppsala, Sweden
gApplied Materials Physics, Department of Materials Science and Engineering, Royal Institute of Technology (KTH), S-100 44 Stockholm, Sweden
First published on 23rd November 2020
Abstract
The decoration of a copper cluster on the anatase phase of a (101)-TiO2 surface to increase the reduction of CO2 has gained significant interest and potential to trigger sustainable solar-fuel-based economy. In the present work, we studied a heterogeneous surface for the reduction of CO2, which can produce various organic compounds such as formic acid, formaldehyde, methanol, ethanol, and methane. The density functional theory calculations were employed to study the formation of formaldehyde and methanol from CO2via hydrogenation by H2 on a Cu catalyst. The copper cluster is a unique catalyst for charge separation and conversion into important organic compounds. Theoretical investigations suggest that these organic compounds can be used as feedstock or be converted into solar fuel.
I. Introduction
Nowadays, capture of carbon dioxide (CO2), its conversion into solar fuels and the development of non-fossil fuel energy sources are critical to minimize the effects of CO2 as a greenhouse gas for both environmental protection and industrial activities, along with solving the energy crises.1–10 The potential application of the photocatalytic reduction of CO2 emissions and the development of non-fossil fuel energy sources are crucial for the repression of greenhouse effects in the environment and for the human health.1–6 The main problems of removing some acidic gases from the atmosphere are a global challenge. Traditional methods to capture acidic gases and electrochemically reduce CO2 can produce a variety of hydrocarbon compounds together with some other useful organic compounds such as formaldehyde, formic acid, carbon monoxide, methane, ethanol, methanol and ethylene with high current efficiency.7–9 Olah et al. have studied the electrolysis of CO2 to carbon monoxide and hydrogen in aqueous media for the production of important organic chemicals like methanol.10 This gives a motivation to study various nanomaterials such as CeO2-(110) surface,11 Rh-(211) surface,12 binary metal oxide (ZnO–ZrO2) solid solution catalyst13 and several phases of titanium dioxide (TiO2) such as anatase, rutile and brookite14–19 in the presence of different active catalysts such as platinum (Pt), palladium (Pd), rhodium (Rh) and copper (Cu). They can work as fuel producers from the reduction of carbon dioxide (CO2) with water or hydrogen. These studies explain that the photocatalytic reduction of CO2 has recently gained such significant interest, as it has the potential to trigger a sustainable solar fuel-based economy. Organic products such as hydrocarbons and other compounds can be used as feedstocks for conversion into valuable carbon-neutral fuels. This process is also of interest for the recycling of CO2 as an energy carrier, thereby reducing its accumulation in the atmosphere. Furthermore, the same process is interesting for the production of renewable solar fuels from CO2, and the production of water and hydrogen for usage as transport fuels. Moreover, the by-product of this reaction mechanism is oxygen, which is an essential component for life on earth.TiO2 has been proven to be one of the most suitable materials for photocatalysis and solar energy conversion because of its suitable valence and conduction band alignment, long-term stability, non-toxicity and cost-effectiveness.20–25 According to Zhang et. al., the anatase phase of TiO2 displays much higher photocatalytic activity than rutile and brookite phases, because it has an indirect band gap, leading to longer charge carrier lifetimes.26,27 Generally, the photocatalytic activity of TiO2 is mainly dependent on its structure, crystallite size and distinguished surface areas. Additionally, the lifetime of photogenerated electrons and holes in anatase is about an order of magnitude larger than that in rutile, thus greatly enhancing the probability of photoexcited electrons and holes to participate in surface chemical reactions.27 Recently, a copper cluster decorated on the anatase phase of TiO2 at different surfaces has been successfully investigated by first-principles calculations.28 The (101)-TiO2 surface with a copper cluster is more stable than the (110)-TiO2 surface with the same copper cluster, and this increases the activity of TiO2 toward CO2 reduction. Furthermore, it is responsible for the photoproduction of hydrocarbons. The copper cluster decorated on the titania surface is very important for charge separation, with electrons moving toward the copper cluster and holes toward Ti3+ polaron sites. Moreover, Seriani et al. also suggested that the presence of Cu4 cluster enhances the photoresponse of TiO2 and contributes to the increased lifetime of photogenerated electron–hole pairs and to the increased activity of CO2 reduction, a key step in the photoproduction of hydrocarbons.28 Another study suggested that the Cu/TiO2 catalytic system is responsible for enhancing the CO2 photoreduction efficiency.29 Yang et al. have systematically investigated the structural and adsorption properties of metal impurities adsorbed on anatase TiO2 to enhance the photocatalytic CO2 reduction.30 Additionally, a lot of other experimental and theoretical investigations suggest that the (101)-TiO2 surface of anatase performs better in the photocatalytic CO2 reduction to produce important organic compounds useful for fuels in real life.30,31,31–36 It was also reported that Cu is one of the extraordinary metals that exists stably as a metallic form, also as metallic oxides under atmospheric conditions. Furthermore, it is expected that decorated/loaded Cu on the TiO2 system can significantly enhance the photocatalytic activity of the TiO2 system since the work function of Cu is quite similar to the conduction band of TiO2 and standard reduction potential of the oxygen molecule, which is considered a probable electron acceptor in an aqueous suspension of TiO2.37 From this prospective, several studies demonstrated that the loading of Cu clusters on TiO2 surfaces is promising for photocatalytic applications such as CO2 photoreduction, H2 production, antimicrobial coatings, solar water splitting, gas sensing and photodetection.37–43,43–48 These findings open up new possibilities for the production of organic compounds such as hydrocarbons using copper cluster-based catalysts.
Motivated by the studies on TiO2, we have taken the TiO2 anatase (101)-surface with a Cu4 cluster (denoted as (101)-TiO2@Cu4) model for the CO2 reduction process. According to previous studies, the (101)-TiO2@Cu4) system is more stable than other titania surfaces.38 For this reason, we investigated the copper cluster decorated on the (101)-TiO2 anatase phase using the first-principles calculations. The main aim of this work is to discuss the reaction mechanism of the chemical reduction, the capturing of CO2 gas molecules and the charge transfer from the theoretical point of view. Moreover, we discuss the dissociation process of CO2 molecules and the reaction mechanism of CO2 conversion into important organic compounds such as formaldehyde and formic acid.
II. Methodology
The computational method employed in the present study is based on density functional theory. A three-dimensional model was used to consider the periodic nature of the surface. In this model, supercells were repeatedly used periodically in all three directions, creating an infinite slab in the plane of the slab and an infinite stack of slabs in the direction perpendicular to the slab. Moreover, a vacuum layer of 15 Å was created to separate each slab from its neighbors to make the unphysical interaction between the slabs negligible. Spin-polarized DFT-based ground-state properties were calculated using projector augmented-wave (PAW) datasets to represent the electronic wave functions. For exchange-correlation functionals, the generalized-gradient approximation (GGA) within the Perdew–Burke–Ernzerhof (PBE) parametrization was employed.49 In a limited number of cases, DFT estimations for the on-site coulombic interactions can be corrected using the DFT+U technique.50 Specifically, a Dudarev51U parameter as implemented in the Quantum Espresso (QE) code52 with the values of 4.2 eV and 5.2 eV for titanium53–55 and copper atoms,28,56 respectively, was employed. In this case, only valence electrons were represented explicitly in the calculations, with the ionic potential described by ultrasoft pseudopotentials similar to those introduced by Vanderbilt.57 We have also considered van der Waals (vdW) interactions by including a rectification to the GGA functionals associated with the vdW-DF2 functionals.58 All the calculations were done using the QE code.59 The cutoff energy was chosen to ensure the convergence of the total energy with respect to the size of plane-wave basis set. In our calculations, we used a cutoff energy of 40 Ry and 480 Ry for the wave functions and charge densities, respectively. A series of tests converged a k-point grid in the Brillouin zone to (4 × 4 × 2) according to the Monkhorst–Pack scheme.60 The positions of all atoms in the unit cell were fully relaxed using the conjugated gradient algorithm until the forces on each atom were less than 10−3 a.u. In the periodic slab model, the anatase-(101) surface was represented using a rectangular (1 × 2) supercell with the surface cell size of 10.27 Å × 7.57 Å. The charge transfer trends between the anatase-(101) surface, Cu4 cluster and CO2 and CO gas molecules was obtained by the Bader analysis.61,62The adsorption energies of CO2 and CO on the (101)-TiO2@Cu4 anatase surface were calculated as follows:
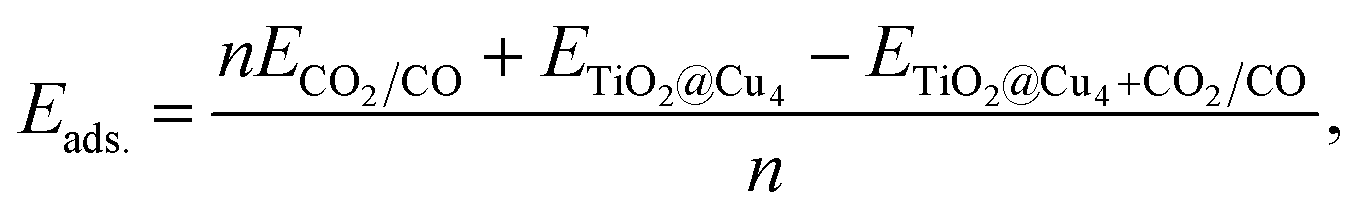 | (1) |
III. Results and discussion
First, we discuss the results concerning the molecular adsorption and dissociation on the anatase (101)-surface with copper cluster decoration. Later on, we will discuss the enhancement of photoresponse of the anatase (101)-surface in the presence of empty states of copper cluster, which contribute to the lifetime of photogenerated electron–hole pairs and to the increase in the CO2 reduction activity. The CO2 adsorption and dissociation process on the (101)-TiO2 anatase was discussed previously, and it was found that the maximum adsorption energy is 1.35 eV, while the maximum dissociation barrier equals 0.90 eV.17 However, according to another study, defective TiO2 materials exhibit better performance than pristine TiO2 for CO2 photoreduction as well as enhance sunlight absorption efficiency.64 The reduced (101)-surface of TiO2 is much more favorable for CO2 binding with the accompanying charge transfer to CO2.65 The correlation of photo-activity with material property suggests that the high activity of He or H2-pretreated Cu/TiO2 is probably attributed to the synergy of the Cu species and the surface defect sites such as oxygen vacancies, which facilitate the separation of electron–hole pairs and promote electron transfer to adsorbed CO2.29A. Structural properties
The Cu4 cluster was deposited on the surface of (101)-TiO2 (Fig. 1), where Cu atoms were placed just above the oxygen and titanium atoms in a form of triangular pyramidal (tetrahedron) structure with two copper atoms covalently bonded with oxygen atoms. Each copper atom was tightly bonded with the surrounding three copper atoms. The tetrahedron structure of Cu4 cluster was introducing more active sites that might play a significant role for the photocatalytic activity of (101)-TiO2 surface. The bond length between the two Cu atoms was slightly changed due to the interaction with the (101)-TiO2 surface and the local environment of copper tetrahedron structure. The bond lengths of two Cu atoms varied from 2.37 Å to 2.54 Å, being slightly shorter than the bond length in Cu bulk (2.55 Å). The Cu–Ti and Cu–O bond lengths were 2.71 Å and 1.89 Å, respectively. Cu–O is slightly shorter than the standard bond length of 1.94 Å, while the Cu–Ti bond length varies according to the cluster size deposited on the (101)-TiO2 surface. Previously reported values of the Cu–Ti bond length vary from 2.59 Å to 2.92 Å.28 To understand the stability of Cu4 deposited on the (101)-TiO2 surface, we calculated the binding energy of (101)-TiO2@Cu4 (2.41 eV), which shows strong interaction between the copper cluster and the atoms at the topmost layer of the (101)-TiO2 surface, in a good agreement with the previously reported work.23 The optimized structure of (101)-TiO2@Cu4 has two different types of coordinated atoms. First one is a 6-fold (6f) coordinated Ti-atom positioned just below the copper cluster and the second one is a 5-fold (5f) coordinated Ti-atom just below the first copper atom. Actually, the Ti1(5-fold coordinated Ti atom) atom is presented just below the copper cluster for the charge transport in the system, as shown in Fig. 1.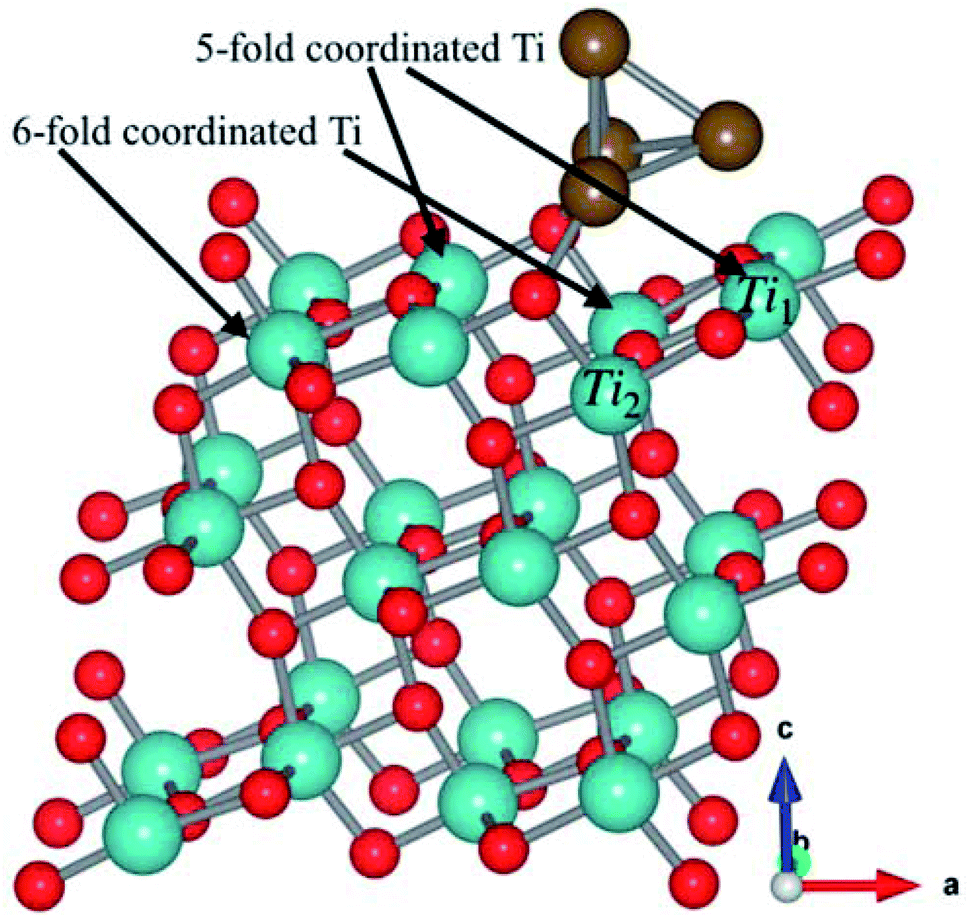 | ||
| Fig. 1 Fully optimized structure of the (110)-plane of anatase TiO2 with a tetrahedron copper cluster (Cu4). The red color shows oxygen atoms, dark cyan color shows titanium atoms and wine color shows copper atoms. The 5-fold (Ti1) and 6-fold (Ti2) coordinated Ti atoms are indicated with black arrows. | ||
B. CO2 photoreduction on the (101)-TiO2@Cu4 interface: charge transfer mechanism
In a (1 × 2) supercell containing 48 O atoms, 24 Ti atoms and the copper cluster of 4 Cu atoms distributed in trilayers of six O–Ti–O, three different binding configurations of CO2 and CO were identified on the (101)-TiO2@Cu4 surface (Fig. 2). In two configurations, CO2 and CO molecules bind to the surface Ti(5f) site in a linear geometry relative to the surface (Fig. 2a and c), with O–C–O angles nearly linear (179.29° and 178.81°). The remaining adsorption configuration identified on this surface has a bent CO2 structure, with the O–C–O angle of 135.59° (Fig. 2b). All configurations indicate a strong interaction between the (101)-TiO2@Cu4 and CO2 molecules, bonded to Ti(5f) and 3-fold O(3f) atom (or Vo–Ti3+) sites, as the adsorption energies are 2.94, 3.00 and 3.03 eV (Table S2†), respectively. Due to the presence of polaron sites of Ti3+, the electrons drift towards the Ti3+ site and the holes travel towards the tetramer copper cluster site. Using the Bader charge analysis, we found the total charge donated by the tetramer copper cluster to the titania surface to be 0.78|e|. Furthermore, the presence of Ti3+ near the strong binding Cu–Ti interface could contribute to the formation of active sites for CO2 reduction. Subsequently, the CO2 molecule can react with the (101)-TiO2@Cu4 surface (see Fig. 2b) receiving an excess of 0.17|e| to make CO2δ−.18 During this process the Cu4 cluster losses 0.19|e|, most of which transfer to CO2 molecules, while the rest of electrons are delocalized. This means that the charge transfer occurs between active sites (101)-TiO2 and the copper cluster, which is responsible for CO2 reduction. Thus, titania has an important role in charge separation along with the tetramer copper cluster deposited on its surface.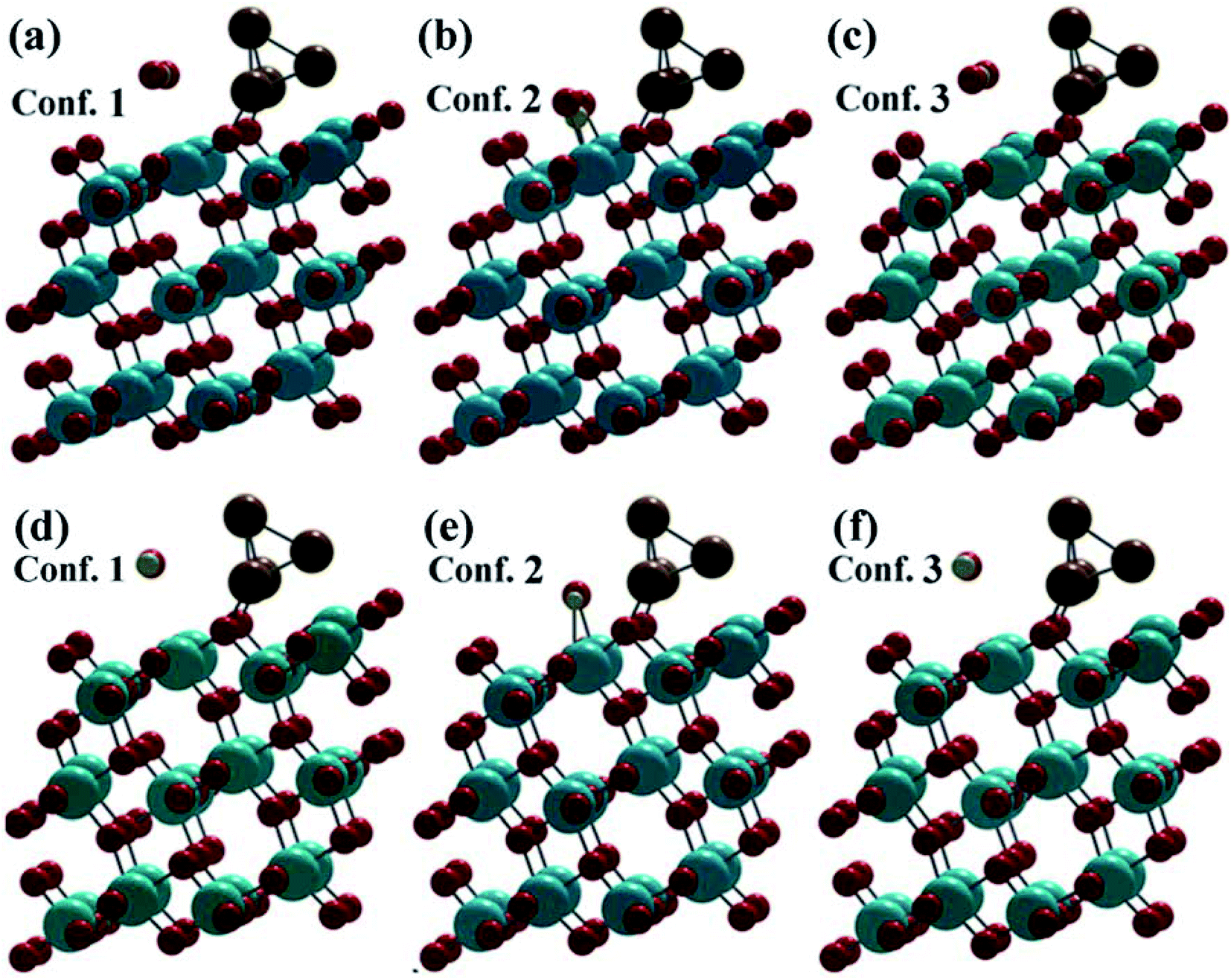 | ||
| Fig. 2 Fully optimized structures after CO2 and CO adsorption on (101)-TiO2@Cu4. (a–c) represent the CO2 configurations and (d–f) represent the CO configurations. | ||
After the dissociation of O–C–O bonds, carbon monoxide interacts strongly with the copper cluster and the active sites on (101)-TiO2. Due to the strong bonding between the active sites and CO2 molecule, the bond dissociation energy was found to be 516.70 kJ mol−1. This was slightly lower than its standard values (532 kJ mol−1).66,67 However, in case of configurations 1 and 3 (Fig. 2a and c), the CO2 molecule losses 0.30|e| and 0.34|e|, respectively. This means that the (101)-TiO2@Cu4 surface accumulates the charge. According to the analysis, Cu4 cluster gains 0.01|e| and 0.09|e| from CO2 molecules and the rest of electrons go to the titanium surface in configurations 1 and 3, respectively. The adsorption process of the CO molecule on the same surface is presented in Fig. 2d–f. In the case of configuration 2, the CO molecule is weakly bonded to the active sites and the corresponding adsorption energy is 0.34 eV. During this process, the CO molecule losses a charge of 0.08|e|, with the Cu4 cluster gaining 0.06|e| from the CO molecule, while the remaining electrons are delocalised between them.
The adsorption energies of configurations 1 and 3 are 0.30 and 0.24 eV, respectively, much less than those in the case of CO2 adsorption. The reason lies in the smaller charge transfer from the CO molecule to the (101)-TiO2@Cu4 surface, which is 0.20|e| and 0.19|e| for configurations 1 and 3, respectively, compared to CO2 (0.30|e| and 0.34|e|). In the same cases, a part of the transferred charge (0.15|e| and 0.17|e|) is stored by the Cu4 cluster, while the rest of electrons go to the titanium surface or are delocalized between the surface and the adsorbant. On the contrary, CO2 and CO molecules gain/loss 0.17|e| and 0.08|e| to the (101)-TiO2@Cu4 surface in configuration 2 (Fig. 2b and e), as shown in Table S3.† According to that, the charge transfer in configurations 1 and 3 is higher than that in configuration 2, and contributes to the stronger interaction between the atoms in those cases. This finally results in bond length differences (C–O, Cu–O), as shown in Table S1.† Other bond lengths (Cu–Ti, Cu–Cu) are not much influenced by the charge gain/transfer from CO2/CO to the (101)-TiO2@Cu4 surface. The Cu–O bond length (O from CO2 molecule) varies from 1.86 to 1.92 Å, the C–O bond length in CO2 from 1.14 to 1.27 Å, the Cu–Cu bond length from 2.34 to 2.55 Å and the Cu–Ti bond length from 2.71 to 2.94 Å. Configuration 2 describes a chemisorption process while the remaining two are physisorption processes. Now, when we dissociated the O–C–O bonds, the carbon monoxide (CO) strongly interacted with active sites of 5- and 6-fold coordinated systems of the (101)-TiO2@Cu4 surface. The dissociation energy (CO2 to CO and O) of this bond was found to be 324.88 kJ mol−1 and 463.37 kJ mol−1 for configurations 1 and 3, respectively.
From the electronic band structures, the (101)-TiO2 surface of anatase shows the semiconducting behaviour with a band gap of 1.81 eV, which is in excellent agreement with previous work (Eg = 1.77 eV).68 While the Cu4 cluster is deposited on the surface of (101)-TiO2, the band gap suddenly reduces from 1.81 eV to 0.44 eV, which is presented (see Fig. S1 in ESI†). This result is consistent with the previous work.28 The Cu4 cluster doping induced a significant reduction in the band gap of the anatase (101)-TiO2 surface. The band gap of the (101)-TiO2@Cu4 surface (0.44 eV) was narrower than that of the (101)-TiO2 surface (1.81 eV), and it would play a significant role for enhancing the photocatalytic activity and increasing absorption in the visible region. The Cu4 cluster-doped (101)-TiO2 surface is presented in Fig. S1(b).† For a semiconductor photocatalyst, effective absorption is one of the most important properties. This kind of transformation will be a significant advantage for the modified material by doping to perform better in the photocatalytic activity. It has been argued that the electronic transition under visible light irradiation must involve localized state transition to the conduction band.69 The electronic band structure calculated for the Cu4 cluster-doped system indicated the formation of impurity states in the electronic band gap (see Fig. S1(b)†). Furthermore, we computed the electronic band structure of gas molecules CO2 and CO absorbed on the surface of (101)-TiO2@Cu4 as shown in Fig. S2.† Excitation from the impurity states (valence band) of the Cu4 cluster to the conduction band could account for the optical absorption edge shift toward the lower photon energies, i.e. in the visible region compared with the (101)-TiO2 surface (see Fig. S3†). The optical absorption spectra of the (101)-TiO2@Cu4 system with CO2 and CO gas molecules reveal the absorption shifted more in the visible region, which is the evidence of the optical response starts in the visible range. To understand the contribution of atomic orbitals in the process of CO2 reduction, we have calculated projected density of states (PDOS), as shown in Fig. 3. The PDOS of 3d and 4p orbitals of the Cu4 cluster and 3d orbitals of its neighboring Ti3+ polaron state appear near the Fermi level (configuration 1 – Fig. 2a). The Ti3+ polaron state appears toward the majority down-spin states. This polaronic state is characterized by the magnetic moment of the Ti atom and by its geometric distortion due to the strong interaction between the Cu4 cluster and (101)-TiO2 surface.28 The gas molecules were located at different positions, therefore, the change in the magnetic moment for the Ti atom was between 0.82 μB and 1.42 μB, as shown in Table S2.† The maximum change of 1.42 μB in the magnetic moment (for both 5-fold and 6-fold coordinated Ti atoms below the copper cluster) of Ti for CO2 reduction was found in configuration 2 (Fig. 2b), while for pure forms it was 0.86 μB (for 5-fold coordinated atom) by the Ti1 atom (see Fig. 1). After the dissociation of oxygen atoms from the CO2 molecule, the Ti3+ polaron state appears and the polaronic state is shifted towards the up-spin channel side for configuration 2, which is presented in the valence band maximum (VBM). The formation of polaronic states comes from the 6f-Ti atom below the Cu4 cluster. In case of configuration 2 of the (101)-TiO2@Cu4 surface, the polaronic state was found in a wide energy range between −1.0 eV and 0 eV due to the strong hybridization of the Ti-d state and the Cu-p state and, in this case, both 5-fold and 6-fold coordinated Ti atoms presented below the copper cluster contributed to form polaronic states.
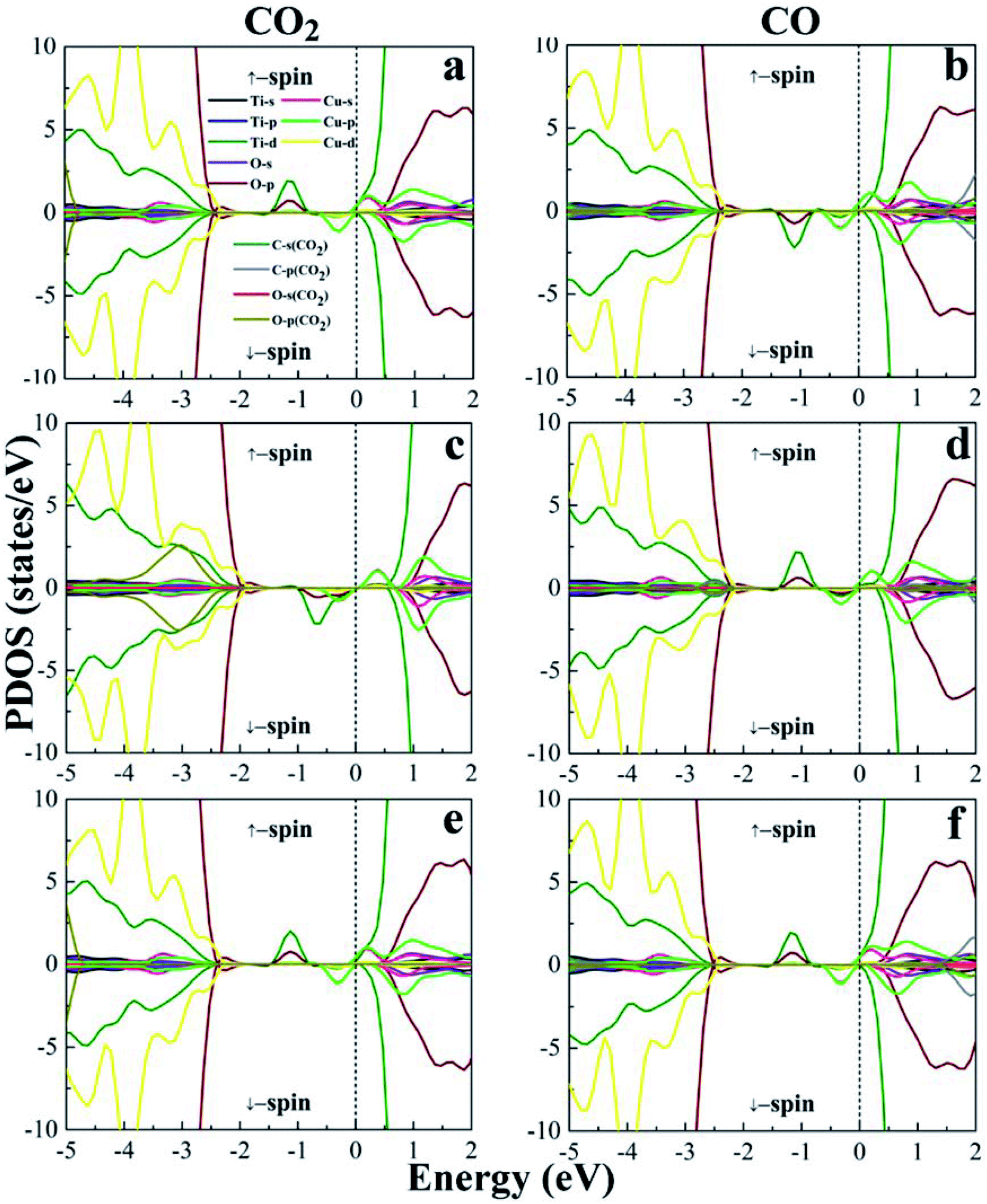 | ||
| Fig. 3 Projected density of states (PDOS) of configuration 1, 2 and 3 of CO2 and CO adsorption on (101)-TiO2@Cu4. (a and b) PDOS of CO2 and CO for configuration 1, (c and d) PDOS of CO2 and CO for configuration 1 and (e and f) PDOS of CO2 and CO for configuration 1, respectively. Black line: Ti-s states; blue line: Ti-p states; olive line: Ti-d states; violet line: O-s states; wine line: O-p states; pink line: Cu-s states; green line: Cu-p states; yellow line: Cu-d states; dark gray line: C-s states from CO2/CO molecules; gray line: C-p states CO2/CO molecules; red line: O-s states from CO2/CO molecules; dark yellow line: O-p states CO2/CO molecules. The zero of energy has been set to the Fermi energy. | ||
In case of configuration 2, the gas molecules strongly interacted with the titania surface. That is why the change in the magnetic moment for the Ti atom was maximally 1.42 μB. It was calculated through the Löwdin analysis.70 However, after the dissociation of the oxygen atom from CO2 molecules, the polaronic state will be compacted in the range between −1.38 eV and −0.73 eV. A change in the magnetic moment (for both 5-fold and 6-fold coordinated Ti atoms below the copper cluster) is almost the same (1.41 μB) due to the large geometric distortion by the CO molecule. Polaron formation has been found for both molecule adsorptions, but in the case of CO2, it shifted toward the down-spin side, while for the CO case, it shifted in the opposite spin channel side.
Moreover, in the case of configurations 1 and 3, the 3d and 4p orbitals of the Cu4 cluster and the 3d orbitals of its neighboring Ti3+ polaronic states showed the same behaviour near the Fermi level. Due to the presence of the Cu4 cluster, the change in the magnetic moment (for 5-fold coordinated Ti atoms below the copper cluster) was 0.82 μB, for both configurations. The majority spin states of the Ti3+ polaron state appeared towards the up-spin states. At the Fermi levels, more contribution came from the p and d states of Cu atoms and d states of Ti atoms. The polaronic states of Ti3+ were found to be in the range of −1.48 eV to −0.80 eV and −1.46 eV to −0.79 eV for configurations 1 and 3, respectively. The PDOS of CO adsorption on the (101)-TiO2@Cu4 in configurations 1 and 3 is shown in Fig. 3b and f. The majority of charge carriers shift towards the down-spin and the magnetic moment comes from these states. The magnetic moment of Ti atoms due to the Cu4 cluster of configuration 1 was 0.84 μB (for 5-fold coordinated Ti atoms below the copper cluster). For configuration 3, the polaronic states Ti3+ appeared on up-spin side and the change in polarization in it was 0.83 μB (for 5-fold coordinated Ti atoms below the copper cluster), due to the Cu4 cluster decorated on the (101)-TiO2 surface.
The isosurface of the electron charge density shows the charge dynamics from the active sites on the (101)-TiO2@Cu4 surface to the CO2/CO system and vice versa. The computed charge density difference is defined by the following equation:
| Δρ = ρ(101-TiO2@Cu4+CO2/CO) − ρ(101-TiO2@Cu4) − ρ(CO2/CO), | (2) |
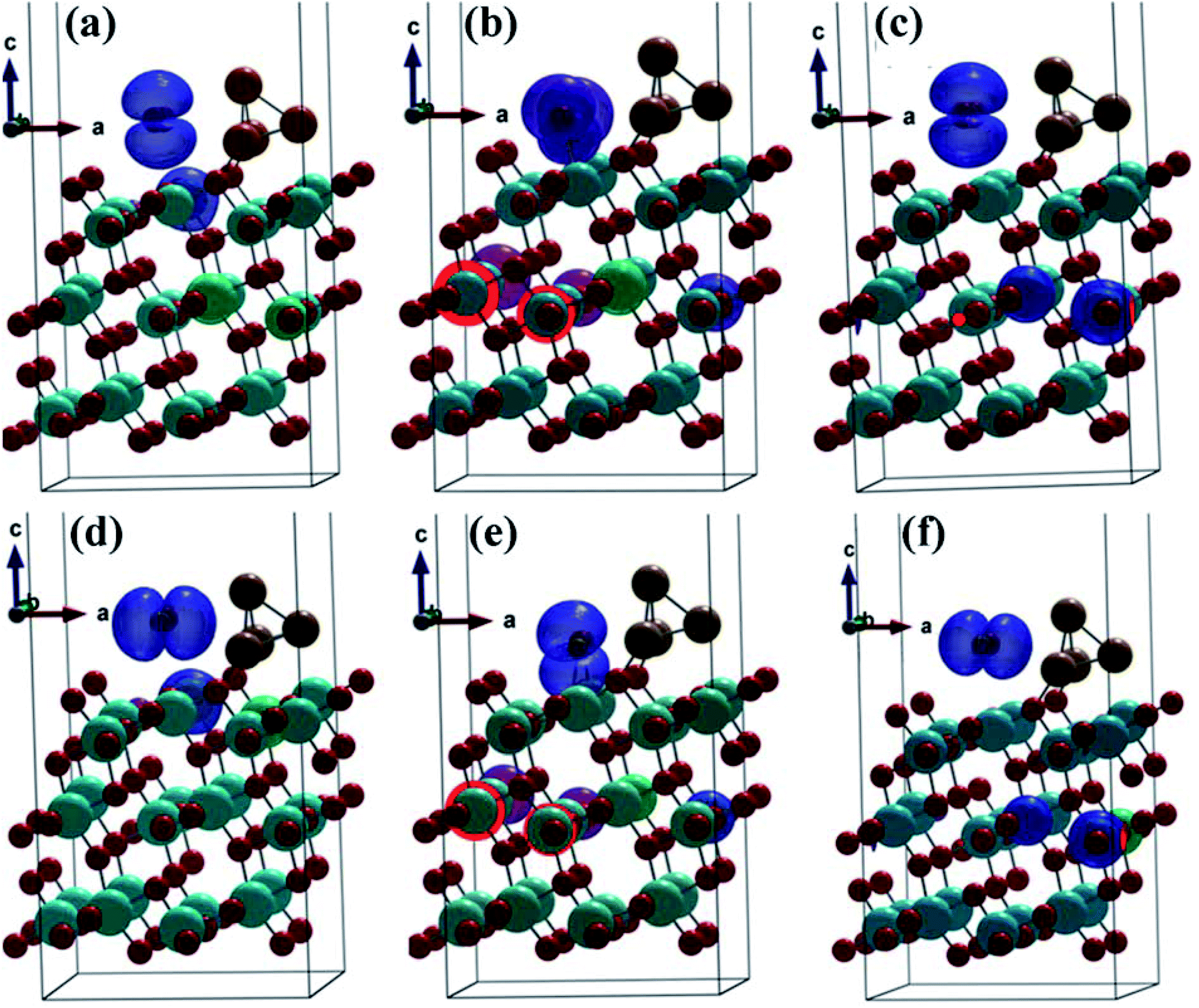 | ||
| Fig. 4 A charge density map for each of configuration 1, 2 and 3 of CO2 and CO absorption on the surface of (101)-TiO2@Cu4. Here (a–c) regard CO2 adsorption and (d–f) regard CO adsorption on (101)-TiO2@Cu4. The observed interstitial electron charge accumulation points to the active sites on (101)-TiO2@Cu4. The blue color shows the charge accumulation. | ||
C. Dissociation mechanism of CO2 on TiO2@Cu4
For the dissociation process, we chose the most energy preferable sites of CO2 with (101)-TiO2@Cu4 from configurations 1, 2 and 3 for the co-adsorption of CO and O. In order to compare the co-adsorption behaviour of CO adsorption on the (101)-TiO2@Cu4 system, we have calculated the co-adsorption configurations of the CO fragment and O atom from CO2 (Fig. 5). Carbon and oxygen atoms of CO were shifted slightly above (101)-TiO2@Cu4 in each configuration with different orientations of the CO molecule. The O atom was bonded to the Ti atom of the (101)-TiO2 surface for configurations 1 and 3, while for configuration 2, it was bonded to Ti and Cu atoms of the (101)-TiO2@Cu4 system. After the dissociation of CO2 molecule, the symmetry of the Cu4 cluster changed for configurations 2 and 3; it was slightly deformed via the bridge-like bonds, as shown in Fig. 5B and C. The structures and bonding characteristics between the fragments of CO2 and the (101)-TiO2@Cu4 system are presented in Fig. 5, along with the reaction energy barriers of the acidic gas dissociation process. In case of configuration 1, the dissociation process of CO2 (CO2 → CO + O) is as follows: the O atom moves toward the Ti atom (Ti–O bond is formed) as the C–O bond is extended from 1.18 Å to 1.20 Å. However, in the case of configurations 2 and 3, the activated O atom moves toward Ti, as well as Cu atoms, while the activated C–O bond varies from 1.14 Å to 1.23 Å and 1.14 Å to 1.18 Å for configurations 2 and 3, respectively. Ti–O and Cu–O bonds were formed in configuration 2 and the Ti–O bond was formed in configuration 3. The energy barriers (Ea) for the CO2 dissociation process (1.72 eV, 1.59 eV and 1.55 eV for configurations 1, 2 and 3, respectively) can be compared to other theoretical values for the activation energy of CO2 dissociation reaction: 1.53 eV and 1.73 eV on the Fe(111) and Pt(111) surfaces, respectively.71 Other theoretical as well as experimental data on the energy barrier/activation energy of CO2 dissociation vary up to 2.10 eV (theoretical)15,71–73 and 1.50 eV (experimental).74,75 During the fragmentation of carbon dioxide to carbon monoxide, we found the energy differences of 1.01 eV, 0.93 eV and 0.01 eV between IS and FS for configurations 1, 2 and 3, respectively. Therefore, the dissociation process of CO2 is an endothermic reaction in each case. Additionally, the optical absorption spectra calculated for the CO2 and CO with (101)-TiO2@Cu4 from configurations 1, 2 and 3 systems are depicted in Fig. S3.† It is evident from the optical spectra that all these systems show the first absorption peaks start at low photon energy. In the next part, the formation of formic acid and formaldehyde is discussed.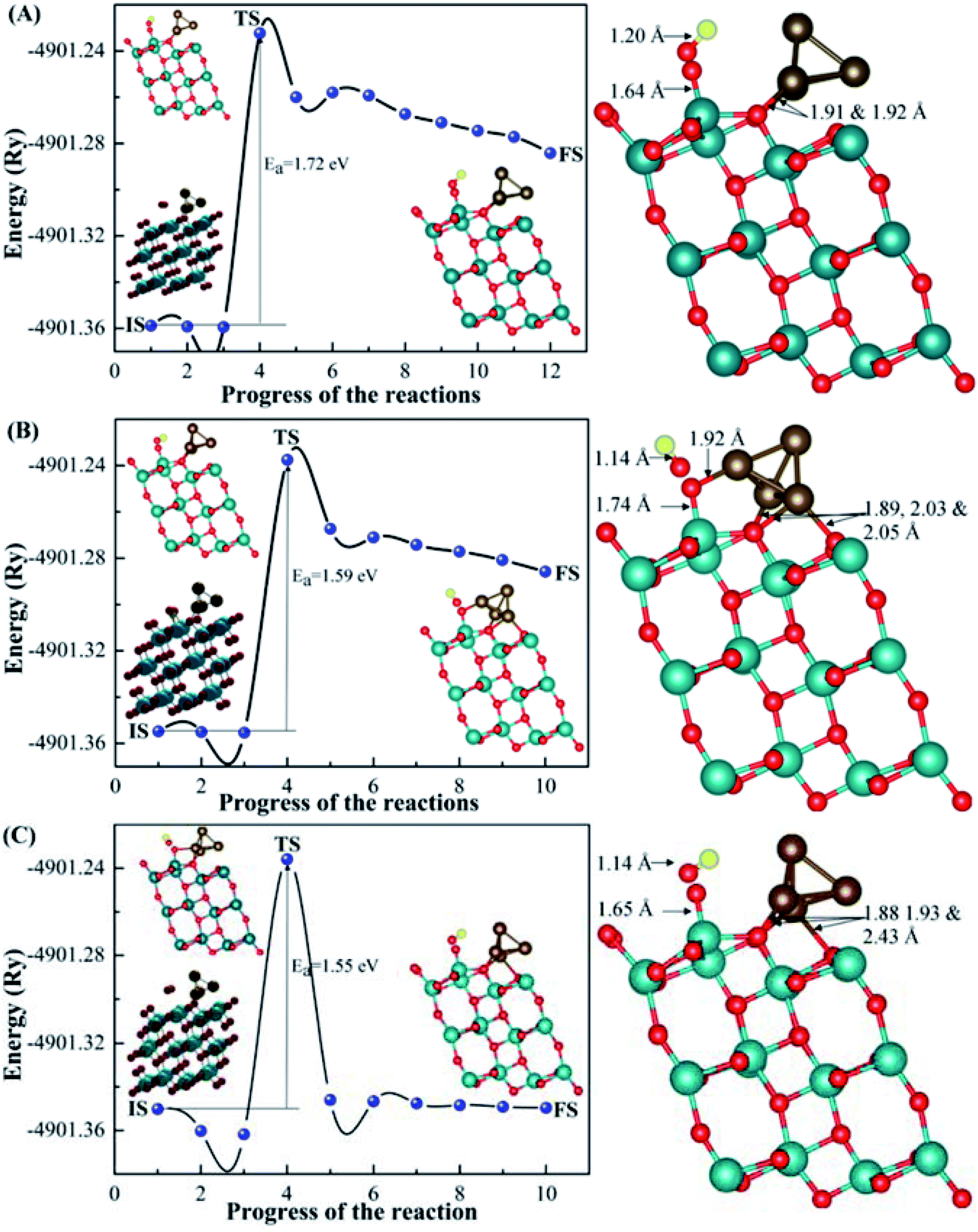 | ||
| Fig. 5 Optimized structures and bond lengths of final states and energetic profiles of the CO2 gas molecule dissociated from the active sites of the (101)-TiO2@Cu4 system for configuration 1 (A), configuration 2 (B) and configuration 3 (C). (101)-TiO2@Cu4 with CO2 configurations are chosen as the initial state (IS), while co-adsorption configurations of (101)-TiO2@Cu4 with CO and O are chosen as the final state (FS). The transition state (TS) is chosen between the IS and the FS. | ||
D. Photocatalytic reduction of CO2 to form formic acid and formaldehyde
When co-adsorbed with hydrogen molecules (H2) between 5-fold and 6-fold coordinated titanium sites of the (101)-TiO2@Cu4 system, CO2 tends to bind at the bridge site, while one H atom is at the top site (Fig. 6). Here, we have taken two hydrogen molecules for configuration-2 as carbon monoxide can also react with hydrogen molecules to yield formic acid and formaldehyde.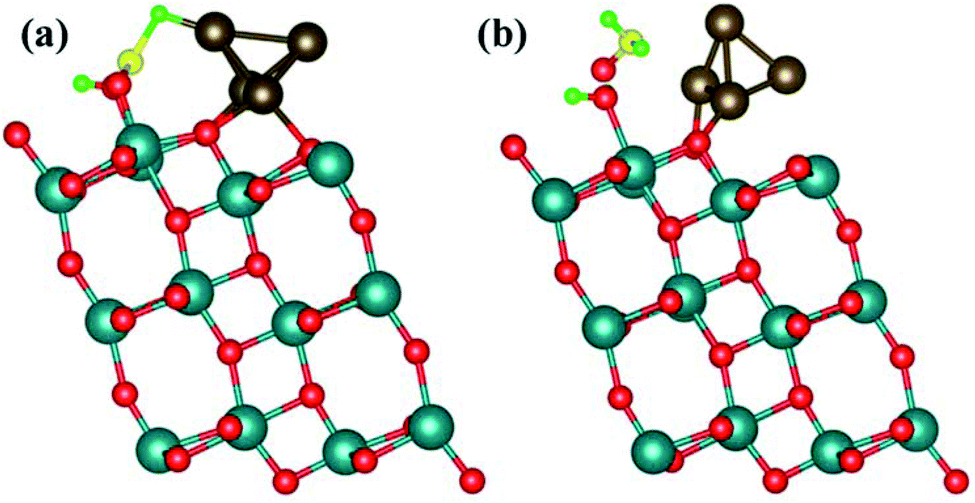 | ||
| Fig. 6 Formation reactions of (a) formic acid and (b) formaldehyde from CO2, 1H2 and 2H2 on the active sites of the (101)-TiO2@Cu4 system represented as the optimized structures from the final relaxation state. The red color shows oxygen, yellow carbon, green hydrogen, dark cyan titanium and wine color copper atoms. | ||
Initially, during the formation of formic acid, CO2 molecule fragments into CO and O. Remaining oxygen from CO2 bonds with the Ti atom of the TiO2 surface and oxygen from CO molecule are bonded to the Ti atom of the same surface. H atoms of the hydrogen molecule bond at appropriate places mean just top of the TiO2 surface and near to the copper cluster (Fig. 6a). The reaction path for the formation of HCOOH considers hydrogenation as the first possibility, with a sequence of elementary steps as follows:
| CO2 → CO + O → CO + O + H2 → CH2O + O → HCOOH. |
Both oxygen atoms are bonded with Ti atoms, while the H atom is bonded with Cu atoms; the bond lengths H–C, C–O, O–H were found to be 1.60 Å, 1.85 ± 0.05 Å and 0.98 Å, respectively, which is larger than the standard bond length 1.06, 1.16 and 0.96 Å, respectively. The bond dissociation energy (D0) is defined as the standard-state enthalpy change for the reaction at a specified temperature.66,67,76,77 That is,
| D0 = ΔH = ∑H(bonds broken) − ∑H(bonds formed), | (3) |
Second, we discuss the formation of formaldehyde and water molecule from CO2 and 2H2 molecules (Fig. 6b). The reaction path for the formation of formaldehyde is as follows:
| CO2 → CO + O → CO + O + 2H2 → OH + H2 + HCO → H2O + CH2O. |
The bond lengths of H–C, C–O, and O–H, are 1.36 ± 0.31 Å, 1.67 Å, and 0.99 ± 0.01 Å, respectively. During the reaction mechanism, the change in enthalpy is −61.12 kJ mol−1. Again, the enthalpy change shows a negative sign, meaning that the reaction is exothermic and indicating that the reaction is favorable.
As we know that CO2 is abundant in our environment and it has many hazards on our health as well as on our environment, there is a need to utilize this toxic gas to obtain something useful in practical applications. Therefore, we propose the theoretical model in which CO2 dissociates into CO and O, which after the reaction with hydrogen molecules gives useful organic compounds. This theoretical model concerns two big issues in our environment: absorption of hazardous CO2 gas and the resulting energy fuel.
IV. Conclusions
In summary, we systematically investigated the adsorption and dissociation processes of CO2 and CO gas molecules on the anatase (101)-TiO2@Cu4 system using ab initio simulations. The copper cluster decorated on the (101)-TiO2 surface increases the catalytic activity regarding the charge separation. Further studies could be directed toward finding alternative cluster formations or atomic species, which would increase even more the charge separation. It also shows the strong ability to capture the CO2 and CO gas molecules: the adsorption energy of studied three configurations, at the active sites of (101)-TiO2@Cu4 system, are 2.94, 3.00 and 3.03 eV for CO2 gas molecules and 0.30, 0.34 and 0.24 eV for CO gas molecules, respectively. These results indicated that the copper cluster used as a catalyst on the top of the (101) surface of TiO2 can enhance the adsorption energy of CO2 and CO molecules compared to the undecorated TiO2 surface. It was found to significantly modify the adsorption properties of CO2 on the surface of (101)-TiO2 anatase, and hence, it is expected to play an important role for even more complex problems related to CO2 photoreduction and selective conversion of CO2 into carbon-natural fuels. Moreover, the dissociation process of the CO2 molecule on these configurations shows an endothermic character. The corresponding energy barriers range between 1.72 eV and 1.55 eV. The formation of valuable carbon-neutral fuels for practical applications in real life is also studied. CO2 gas molecules react with several hydrogen gas molecules to form important organic compounds such as formic acid and formaldehyde. The changes in enthalpy for these reaction mechanisms are negative, meaning that the processes are significantly exothermic (359.90 kJ mol−1 and 61.12 kJ mol−1). These results indicated that, during the formation of formic acid and formaldehyde, the system will release sufficient amounts of heat. According to this, it is very useful for non-fossil fuel energy sources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. K. G. acknowledges award of the Fulbright-Nehru Postdoctoral Research Fellowship by United States-India Educational Foundation (USIEF) and Grant no. 1638/FNPDR/2012. S. K. G. acknowledges the use of high-performance computing clusters at Michigan Technological University, Houghton, USA, and IUAC, New Delhi to obtain the partial results presented in this paper. S. K. G thank the Science and Engineering Research Board (SERB), Department of Science and Technology (India) and the Russian Foundation for Basic Research (Russia) for the financial support grant numbers: YSS/2015/001269 and INT/RUS/RFBR/IDIR/P-6/2016. D. S. and R. A. also thanks Olle Engkvists stiftelse (198-0390), Carl Tryggers Stiftelse for Vetenskaplig Forskning (CTS: 18:4) and Swedish Research Council (VR-2016-06014) for financial support. P. N. G. is thankful to the Department of Science and Technology, India for the support under DST-FIST and the University Grants Commission, India for the support under DRS-SAP. The authors gratefully acknowledge computational resources from the Swedish National Infrastructure (SNIC) and HPC2N. I. L. acknowledges the financial support from the ZUP2018-51 project funded by Josip Juraj Strossmayer University of Osijek, Croatia. I. L. also acknowledges the usage of computational resources CRO-NGI and Isabella at SRCE, Zagreb, Croatia.References
- A. Kuang, M. Kuang, H. Yuan, G. Wang, H. Chen and X. Yang, Appl. Surf. Sci., 2017, 410, 505–512 CrossRef CAS.
- G. Shi, L. Yu, X. Ba, X. Zhang, J. Zhou and Y. Yu, Dalt. Trans., 2017, 46, 10569–10577 RSC.
- N. Nakaten, R. Schlüter, R. Azzam and T. Kempka, Energy, 2014, 66, 779–790 CrossRef.
- Y. Sun, J. Chen and Z. Zhang, Energy, 2019, 167, 688–697 CrossRef CAS.
- O. de Queiroz Fernandes Araújo, J. Luiz de Medeiros, L. Yokoyama and C. do Rosário Vaz Morgado, Energy, 2015, 92, 556–568 CrossRef.
- L. Liu, J. Jin, Y. Lin, F. Hou and S. Li, Energy, 2016, 106, 212–220 CrossRef CAS.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS.
- D. T. Whipple and P. J. A. Kenis, J. Phys. Chem. Lett., 2010, 1, 3451–3458 CrossRef CAS.
- N. Li, X. Chen, W.-J. Ong, D. R. MacFarlane, X. Zhao, A. K. Cheetham and C. Sun, ACS Nano, 2017, 11, 10825–10833 CrossRef CAS.
- G. A. Olah and G. S. Prakash, US Pat., 7704369B2, 2010 Search PubMed.
- N. Kumari, N. Sinha, M. A. Haider and S. Basu, Electrochim. Acta, 2015, 177, 21–29 CrossRef CAS.
- N. Kapur, J. Hyun, B. Shan, J. B. Nicholas and K. Cho, J. Phys. Chem. C, 2010, 114, 10171–10182 CrossRef CAS.
- J. Wang, G. Li, Z. Li, C. Tang, Z. Feng, H. An, H. Liu, T. Liu and C. Li, Sci. Adv., 2017, 3, e1701290 CrossRef.
- H.-Y. T. Chen, S. Livraghi, E. Giamello and G. Pacchioni, Chempluschem, 2016, 81, 64–72 CrossRef CAS.
- W.-J. Yin, B. Wen, S. Bandaru, M. Krack, M. Lau and L.-M. Liu, Sci. Rep., 2016, 6, 23298 CrossRef CAS.
- M. M. Rodriguez, X. Peng, L. Liu, Y. Li and J. M. Andino, J. Phys. Chem. C, 2012, 116, 19755–19764 CrossRef CAS.
- D. C. Sorescu, W. A. Al-Saidi and K. D. Jordan, J. Chem. Phys., 2011, 135, 124701 CrossRef.
- L. Liu and Y. Li, Aerosol Air Qual. Res., 2014, 14, 453–469 CrossRef CAS.
- A. Sarkar, E. Gracia-Espino, T. Wågberg, A. Shchukarev, M. Mohl, A.-R. Rautio, O. Pitkänen, T. Sharifi, K. Kordas and J.-P. Mikkola, Nano Res., 2016, 9, 1956–1968 CrossRef CAS.
- N. Shehzad, M. Tahir, K. Johari, T. Murugesan and M. Hussain, J. CO2 Util., 2018, 26, 98–122 CrossRef CAS.
- N. Ambrožová, M. Reli, M. Šihor, P. Kuśtrowski, J. C. S. Wu and K. Kočí, Appl. Surf. Sci., 2018, 430, 475–487 CrossRef.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS.
- A. Meng, L. Zhang, B. Cheng and J. Yu, ACS Appl. Mater. Interfaces, 2019, 11, 5581–5589 CrossRef CAS.
- J. Low, B. Dai, T. Tong, C. Jiang and J. Yu, Adv. Mater., 2019, 31, 1802981 CrossRef.
- F. Xu, K. Meng, B. Cheng, J. Yu and W. Ho, ChemCatChem, 2019, 11, 465–472 CrossRef CAS.
- J. Zhang, P. Zhou, J. Liu and J. Yu, Phys. Chem. Chem. Phys., 2014, 16, 20382–20386 RSC.
- M. Xu, Y. Gao, E. M. Moreno, M. Kunst, M. Muhler, Y. Wang, H. Idriss and C. Wöll, Phys. Rev. Lett., 2011, 106, 138302 CrossRef.
- N. Seriani, C. Pinilla and Y. Crespo, J. Phys. Chem. C, 2015, 119, 6696–6702 CrossRef CAS.
- L. Liu, F. Gao, H. Zhao and Y. Li, Appl. Catal. B Environ., 2013, 134–135, 349–358 CrossRef CAS.
- C.-T. Yang, B. C. Wood, V. R. Bhethanabotla and B. Joseph, J. Phys. Chem. C, 2014, 118, 26236–26248 CrossRef CAS.
- H. Zhao, F. Pan and Y. Li, J. Mater., 2017, 3, 17–32 Search PubMed.
- J. Yu, J. Low, W. Xiao, P. Zhou and M. Jaroniec, J. Am. Chem. Soc., 2014, 136, 8839–8842 CrossRef CAS.
- M. J. Jackman, A. G. Thomas and C. Muryn, J. Phys. Chem. C, 2015, 119, 13682–13690 CrossRef CAS.
- X. Wang, Y. Zhao, K. Mølhave and H. Sun, Nanomaterials, 2017, 7, 382 CrossRef.
- P. Kar, S. Farsinezhad, N. Mahdi, Y. Zhang, U. Obuekwe, H. Sharma, J. Shen, N. Semagina and K. Shankar, Nano Res., 2016, 9, 3478–3493 CrossRef CAS.
- Y. Cao, M. Yu, S. Qi, T. Wang, S. Huang, Z. Ren, S. Yan, S. Hu and M. Xu, Phys. Chem. Chem. Phys., 2017, 19, 31267–31273 RSC.
- K. Y. Song, Y. T. Kwon, G. J. Choi and W. I. Lee, Bull. Korean Chem. Soc., 1999, 20, 957–960 CAS.
- S. Obregón, M. J. Muñoz-Batista, M. Fernández-García, A. Kubacka and G. Colón, Appl. Catal. B Environ., 2015, 179, 468–478 CrossRef.
- S. Sagadevan, S. Vennila, P. Singh, J. A. Lett, W. C. Oh, S. Paiman, F. Mohammad, H. A. Al-Lohedan, I. Fatimah, M. M. Shahid and P. K. Obulapuram, J. Exp. Nanosci., 2020, 15, 337–349 CrossRef CAS.
- M. A. Gondal, S. G. Rashid, M. A. Dastageer, S. M. Zubair, M. A. Ali, J. H. Lienhard, G. H. McKinley and K. K. Varanasi, IEEE Photonics J, 2013, 5, 2201908 Search PubMed.
- V. Meng’wa, N. Makau, G. Amolo, S. Scandolo and N. Seriani, J. Phys. Chem. C, 2018, 122, 16765–16771 CrossRef.
- A. Heidarpour, Y. Mazaheri, M. Roknian and S. Ghasemi, J. Alloys Compd., 2019, 783, 886–897 CrossRef CAS.
- B. Xin, P. Wang, D. Ding, J. Liu, Z. Ren and H. Fu, Appl. Surf. Sci., 2008, 254, 2569–2574 CrossRef CAS.
- D. Liu, Y. Fernández, O. Ola, S. Mackintosh, M. Maroto-Valer, C. M. A. Parlett, A. F. Lee and J. C. S. Wu, Catal. Commun., 2012, 25, 78–82 CrossRef CAS.
- T. Koklic, I. Urbančič, I. Zdovc, M. Golob, P. Umek, Z. Arsov, G. Dražić, Š. Pintarič, M. Dobeic and J. Štrancar, PLoS One, 2018, 13, e0201490 CrossRef.
- X. Wei and W. Gao, in Proceedings of the 8th Pacific Rim International Congress on Advanced Materials and Processing, Springer International Publishing, Cham, 2013, pp. 1663–1669 Search PubMed.
- M. Janczarek, A. Zielińska-Jurek, I. Markowska and J. Hupka, Photochem. Photobiol. Sci., 2015, 14, 591–596 RSC.
- C. Liu, S. L. Nauert, M. A. Alsina, D. Wang, A. Grant, K. He, E. Weitz, M. Nolan, K. A. Gray and J. M. Notestein, Appl. Catal. B Environ., 2019, 255, 117754 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- V. I. Anisimov, M. A. Korotin, A. S. Mylnikova, A. V. Kozhevnikov, D. M. Korotin and J. Lorenzana, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 172501 CrossRef.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- M. Cococcioni and S. de Gironcoli, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 035105 CrossRef.
- B. J. Morgan and G. W. Watson, Surf. Sci., 2007, 601, 5034–5041 CrossRef CAS.
- M. Farnesi Camellone, P. M. Kowalski and D. Marx, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 035413 CrossRef.
- P. M. Kowalski, M. F. Camellone, N. N. Nair, B. Meyer and D. Marx, Phys. Rev. Lett., 2010, 105, 146405 CrossRef.
- D. O. Scanlon, A. Walsh, B. J. Morgan, G. W. Watson, D. J. Payne and R. G. Egdell, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 035101 CrossRef.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef.
- K. Lee, É. D. Murray, L. Kong, B. I. Lundqvist and D. C. Langreth, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 081101 CrossRef.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys. Condens. Matter, 2009, 21, 395502 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- R. F. W. Bader and T. T. Nguyen-Dang, Adv. Quantum Chem., 1981, 14, 63–124 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- J. Zhang, M. Zhang, Y. Han, W. Li, X. Meng and B. Zong, J. Phys. Chem. C, 2008, 112, 19506–19515 CrossRef CAS.
- H. He, P. Zapol and L. A. Curtiss, J. Phys. Chem. C, 2010, 114, 21474–21481 CrossRef CAS.
- J. A. Dean, Properties of atoms, radicals, and bonds, Lange's handbook of chemistry, McGraw-Hill, Inc.New York, 1999, vol. 15, pp. 4–1 Search PubMed.
- B. de Darwent, Bond Dissociation Energies in Simple Molecules, NBS Publication, 1970, https://nvlpubs.nist.gov/nistpubs/Legacy/NSRDS/nbsnsrds31.pdfhttps://doi.org/10.6028/NBS.NSRDS.31 Search PubMed.
- M. Kinga Bruska, K. Szaciłowski and J. Piechota, Mol. Simul., 2009, 35, 567–576 CrossRef CAS.
- C. Di Valentin, G. Pacchioni, A. Selloni, S. Livraghi and E. Giamello, J. Phys. Chem. B, 2005, 109, 11414–11419 CrossRef CAS.
- P.-O. Löwdin, Phys. Rev., 1955, 97, 1474–1489 CrossRef.
- D. S. H. S. J. Jo and D. H. Park, Bull. Korean Chem. Soc., 2000, 21(8), 779–784 Search PubMed.
- M. J. Roberts, R. C. Everson, G. Domazetis, H. W. J. P. Neomagus, J. M. Jones, C. G. C. E. Van Sittert, G. N. Okolo, D. Van Niekerk and J. P. Mathews, Carbon, 2015, 93, 295–314 CrossRef CAS.
- A. I. Fergusson, Identifying CO2 dissociation pathways on stepped and kinked copper surfaces using first principles calculations, Doctoral dissertation, Georgia Institute of Technology, 2012, http://hdl.handle.net/1853/43652.
- E. C. Neyts, K. Ostrikov, M. K. Sunkara and A. Bogaerts, Chem. Rev., 2015, 115, 13408–13446 CrossRef CAS.
- Y. Toda, H. Hirayama, N. Kuganathan, A. Torrisi, P. V. Sushko and H. Hosono, Nat. Commun., 2013, 4, 2378 CrossRef.
- S. Eckle, H.-G. Anfang and R. J. Behm, J. Phys. Chem. C, 2011, 115, 1361–1367 CrossRef CAS.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: See the figures in the supplementary material for the structural parameters, adsorption energy, magnetic moments, Bader charges, band structures and optical absorption of CO2 and CO gas molecules on anatase (101)-TiO2@Cu4 system. See DOI: 10.1039/d0se01587c |
| This journal is © The Royal Society of Chemistry 2021 |