
A facile approach to construct organic D–π–A dyes via sequential condensation reactions for dye-sensitized solar cells†
Hongjin
Chen
a,
Ashraful
Islam
 *b,
Towhid H.
Chowdhury
b,
Idriss
Bedja
c,
Hamid M.
Ghaithan
c,
Rui
Zhang
a and
Jian
Liu
*a
*b,
Towhid H.
Chowdhury
b,
Idriss
Bedja
c,
Hamid M.
Ghaithan
c,
Rui
Zhang
a and
Jian
Liu
*a
aJiangsu Co-Innovation Center of Efficient Processing and Utilization of Forest Resources, College of Chemical Engineering, Nanjing Forestry University, Jiangsu Key Lab of Biomass-based Green Fuels and Chemicals, Nanjing 210037, China. E-mail: liu.jian@njfu.edu.cn
bPhotovoltaic Materials Group, National Institute for Materials Science (NIMS), 1-2-1 Sengen, Tsukuba, Ibaraki 305-0047, Japan. E-mail: Islam.Ashraful@nims.go.jp
cCornea Research Chair, Optometry Department, College of Applied Medical Sciences, King Saud University, Riyadh 11433, Saudi Arabia
First published on 18th November 2020
Abstract
We developed a new strategy for the molecular engineering of organic D–π–A sensitizers for dye-sensitized solar cell (DSSC) applications. Different from the synthesis of organic D–π–A dyes by conventional C–C coupling method, the present approach highlights the construction of D–π–A backbone via sequential condensation reactions. This approach avoids noble metal catalysis as well as harsh reaction conditions. Based on this concept, four organic D–π–A dyes have been developed as sensitizers for DSSCs, which produced high IPCE values and photovoltage. Moreover, expansion of the π-conjugated spacer in this type of dyes resulted in enhanced photovoltaic performance, which provided a new pathway for designing D–π–A dyes.
Introduction
Solar cells, producing electric energy from solar energy via the photovoltaic effect, have attracted great attention in the field of clean renewable energy.1 As a promising alternative to conventional inorganic photovoltaic devices, solar cells employing molecule-based photovoltaic materials have received considerable research interest owing to their low cost and high efficiency for practical applications.2 Among them, dye-sensitized solar cells, pioneered by Grätzel in 1991,2b have been considered as a promising photovoltaic technology due to their simple preparation process.3 Besides device engineering, continuous breakthroughs in achieving the highest efficiency in DSSCs have been reported, which is due to the meticulous molecular engineering of new sensitizers toward efficient light harvesting which play a crucial role in DSSCs.4Ruthenium(II)-polypyridyl complexes (such as N3, N719, black dye, and so on) have been demonstrated to be a kind of efficient light harvesters for high-efficiency DSSCs thanks to their inherent broad absorption spectra that mainly originate from the intrinsic metal-to-ligand charge transfer (MLCT) transitions.5 However, the shortage of noble metal resources and environmental issues greatly limit their application prospects. On the other hand, organic sensitizers, typically consisting of donor–π–acceptor (D–π–A) conjugated systems, have been gradually considered as more promising candidates for DSSCs.6 The main absorption bands of organic D–π–A dyes attributed to π–π* and intramolecular charge transfer (ICT) transitions can be easily tuned through variation of the molecular structure. So far, many successful organic sensitizers have been developed via meticulous molecular design, and yield comparative or even superior performance in comparison with ruthenium(II)-based complexes.7 However, the synthesis of organic D–π–A dyes was necessarily carried out using C–C coupling reactions to link the donor and/or acceptor parts via precious metal catalysis in an inert atmosphere, which increase the difficulty and cost of the industrial process.8
Quinoxaline, a kind of π-conjugated electron deficient moiety, has received considerable attention in the molecular engineering of D–π–A sensitizers, in which the electron donor and acceptor were frequently introduced in the 5- or 8- position successively via metal catalysis coupling.9 Recently, quinoxaline was employed as an acceptor in D–π–A sensitizers for DSSCs, but their synthesis procedure still needs Pd-catalysis coupling reactions.10 Indeed, triarylamine derivatives, which are classic electron donors, can be easily converted to benzil derivatives by Friedel–Crafts acylation, which gives an opportunity to install electron donors on the conjugated backbone via condensation with aryldiamine derivatives.11 However, this simple strategy has been rarely employed to construct organic D–π–A sensitizers.
Herein, we report a novel approach involving sequential condensation reactions for the construction of organic D–π–A dyes, in which two triphenylamine groups acted as electron donors and cyanoacetic acid acted as an electron acceptor as well as anchoring group. Briefly, triphenylamine was Friedel–Crafts acylated by oxalyl chloride to give an intermediate diketone for subsequent condensation with a 1,2-diamine substituted π-conjugated spacer with a terminal aldehyde group. The acceptor moiety was further introduced by Knoevenagel condensation. As a result, the present strategy can avoid noble metal catalysis throughout the synthesis of the organic D–π–A dyes.
Results and discussion
The molecular structures and synthesis routes of the four D–π–A dyes LJ-10, LJ-11, LJ-12 and LJ-13 are shown in Scheme 1. Compound 2a was commercially available, compounds 1 and 2b were prepared according to the reported methods.12 The two dyes LJ-10 and LJ-12 with terminal carboxy groups were synthesized directly from triphenylamine diketone 1 by a condensation reaction with 1,2-diamine substituted aryl acids 2a and 2b, respectively. To obtain dyes LJ-11 and LJ-13 with a terminal cyanoacetic acid moiety, the corresponding intermediate aldehydes 3a and 3b were prepared by the reduction of LJ-10 and LJ-12 and subsequent oxidation, and were further reacted with cyanoacetic acid via Knoevenagel condensation, respectively. The structures of the desired organic D–π–A dyes were characterized by NMR spectroscopy as well as mass spectrometry. | ||
| Scheme 1 Molecular structures and synthetic routes of D–π–A dyes LJ-10, LJ-11, LJ-12 and LJ-13. | ||
The absorption spectra of the four D–π–A dyes LJ-10, LJ-11, LJ-12 and LJ-13 in CH2Cl2 are shown in Fig. 1, and the corresponding data have been summarized in Table 1. These new dyes LJ-10, LJ-11, LJ-12 and LJ-13 exhibited two distinct absorption bands: the shorter wavelength absorption band in the UV region (300–380 nm) mainly originates from the π–π* electron transitions of the conjugated molecules; the longer wavelength absorption band in the visible region (420–500 nm) can be assigned to an intramolecular charge transfer (ICT) from the triphenylamine donating groups to the electron accepting moiety.13 All the four new D–π–A dyes exhibited emission in CH2Cl2 solutions (Fig. S1, ESI†). The UV-vis absorption spectra of dyes LJ-10, LJ-11, LJ-12 and LJ-13 adsorbed on TiO2 films are shown in Fig. S2.† The λmax values for the dye-loaded TiO2 films are also listed in Table 1. After being adsorbed on TiO2 films, all these four dyes exhibited fine absorption peaks without significant breadth in comparision to those in solutions respectively. Probably the steric hindrance of the two triphenylamine groups was effective to suppress the intermolecular interactions. On the other hand, the maximum absorption of these dyes anchored on TiO2 film exhibited a slight hypochromatic shift in comparison with that of the measurement in solution, respectively. Such a blue shift would be assigned to the solvent effect and/or deprotonation of the carboxylic acid in the anchoring process.14
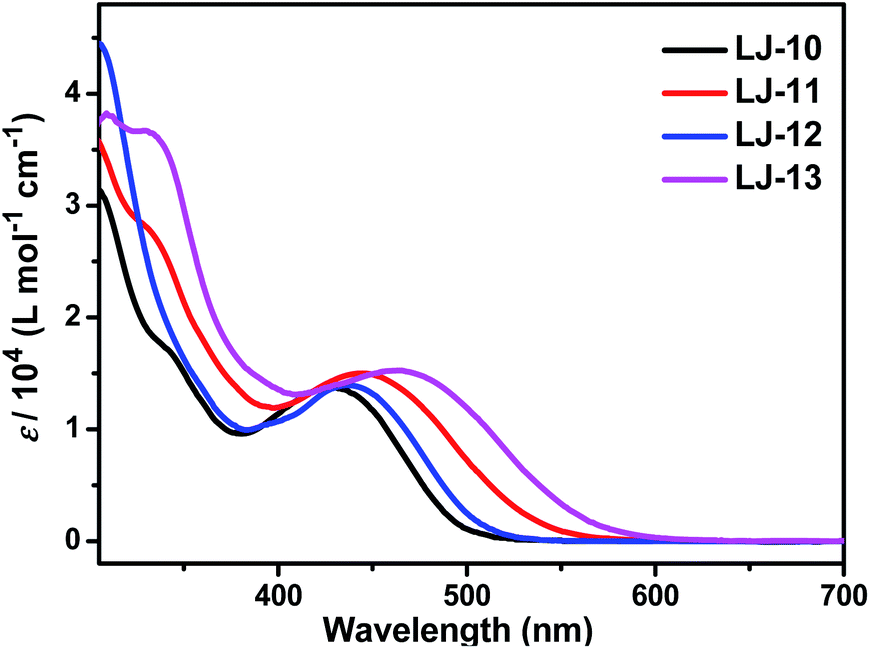 | ||
| Fig. 1 Absorption spectra of dyes LJ-10, LJ-11, LJ-12 and LJ-13 in CH2Cl2. | ||
| Dye | λ max/nm (ε/L mol−1 cm−1) | λ max/nm | S +/0/eV (vs. NHE)c | S +/*/eV (vs. NHE)d | E 0–0/eV |
|---|---|---|---|---|---|
| a The absorption spectra were measured in CH2Cl2 solutions (2 × 10−5 M). b Measured on TiO2 films dipped in 0.3 mM solution of the dye in CH3CN/n-BuOH (1/1, v/v). c The S+/0 corresponding to the ground-state oxidation potential (vs. NHE) in CH2Cl2 internally calibrated with ferrocene (Fig. S5). d S +/* = S+/0 − E0–0, where E0–0 is the zero–zero transition energy. e E 0–0 values were estimated from the intersection between the normalized absorption and emission spectra in CH2Cl2 (Fig. S1, ESI). | |||||
| LJ-10 | 431 (13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700) 700) |
429 | 0.94 | −1.54 | 2.48 |
| LJ-11 | 447 (15000) |
432 | 1.03 | −1.36 | 2.39 |
| LJ-12 | 439 (13930) |
436 | 0.90 | −1.54 | 2.44 |
| LJ-13 | 466 (15250) |
456 | 1.01 | −1.26 | 2.27 |
Cyclic voltammetry (CV) was carried out in CH2Cl2 containing 0.1 M tetra-n-butylammonium hexafluorophosphate (TBAHFP) as a supporting electrolyte to investigate the electrochemical properties of the four D–π–A dyes LJ-10, LJ-11, LJ-12 and LJ-13. As shown in Fig. 2, the present dyes exhibited two reversible oxidative waves, respectively. As the four dyes have the same donor part, their HOMO levels corresponding to the first oxidation potentials exhibited little difference, and increase in the order of LJ-12 (0.90 eV) < LJ-10 (0.94 eV) < LJ-13 (1.01 eV) < LJ-11 (1.03 eV). The variation tendency was in accordance with the result determined by differential pulse voltammetry (Fig. S6†). The result indicated that the introduction of the electron-donating thiophene moiety in the conjugated backbone decreases the oxidation potential of the dye. They are all more positive than the Nernst potential of the I−/I3− redox couple (0.4 V vs. NHE), ensuring regeneration of the oxidized dyes by I− after electron injection, with a driving force of 0.50–0.64 V, respectively. The excited state redox potentials of these four dyes (−1.26 to −1.54 eV vs. NHE) are sufficiently more negative than the conduction band of TiO2 (−0.5 eV vs. NHE), ensuring sufficient driving force (0.76–1.04 eV) for electron injection from the excited dyes to the conduction band of TiO2. These results suggested the potential application of these new dyes in dye-sensitized solar cells.15
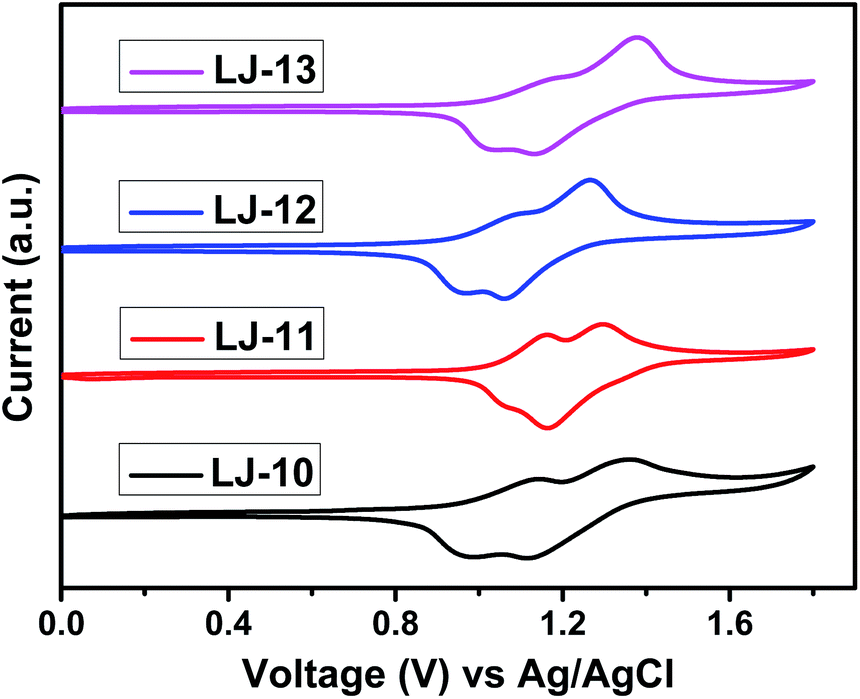 | ||
| Fig. 2 Cyclic voltammograms of dyes LJ-10, LJ-11, LJ-12 and LJ-13 in CH2Cl2/TBAHFP (0.1 M), [c] = 1 × 10−4 mol L−1, 293 K, scan rate = 100 mV s−1. | ||
The optimized configurations of dyes LJ-10, LJ-11, LJ-12 and LJ-13 were investigated by density functional theory (DFT) calculations. The optimized ground-state geometries indicated that all these four dyes have similar dihedral angles around 35° between their electron-donating triphenylamine groups and π-conjugated units, respectively (Fig. S3, ESI†). The frontier molecular orbitals of these four dyes were investigated by DFT calculations at the B3LYP/6-31G** level. As shown in Fig. S4 (ESI†), all these four dyes showed similar electron cloud distribution in HOMOs, which are mainly localized at two triphenylamine groups. Their LUMOs show localized electron distributions through the anchoring group and its adjacent quinoxaline spacer, respectively. Moreover, the variation tendencies of both HOMO and LUMO levels are in accord with the order estimated from optical and electrochemical characterizations, respectively.
DSSCs based on these dyes were fabricated according to our previous method,16 which is detailed in the ESI.† The dye-loading amount of each dye in the device was estimated by an adsorption/desorption method. The dye-loading amounts of all the four dyes are around 1 × 10−7 mol cm−2. Photovoltaic characteristics of the DSSCs based on dyes LJ-10, LJ-11, LJ-12 and LJ-13 have been investigated systematically.17 As a reference, we also investigated the device based on N719, which yielded a power conversion efficiency around 7.9% (Fig. S7†). As shown in Fig. 3, the highest value for the IPCE spectra of DSSCs based on all the four dyes reaches up to 88% in the visible region, which implied the efficient electron injection from the LUMO level of the dye molecules into the conduction band of TiO2, respectively. The onset wavelength of IPCE spectra shifts toward the red region in the order LJ-10 < LJ-12 < LJ-11 < LJ-13 devices (Fig. 3), in agreement with the absorption spectra observed for the dye-loaded TiO2 films (Fig. S2†). The result was consistent with the short circuit current (Jsc) of their corresponding devices measured by J–V characterizations under 100 mW cm−2 and simulated AM1.5G conditions (Table 2 and Fig. 4).
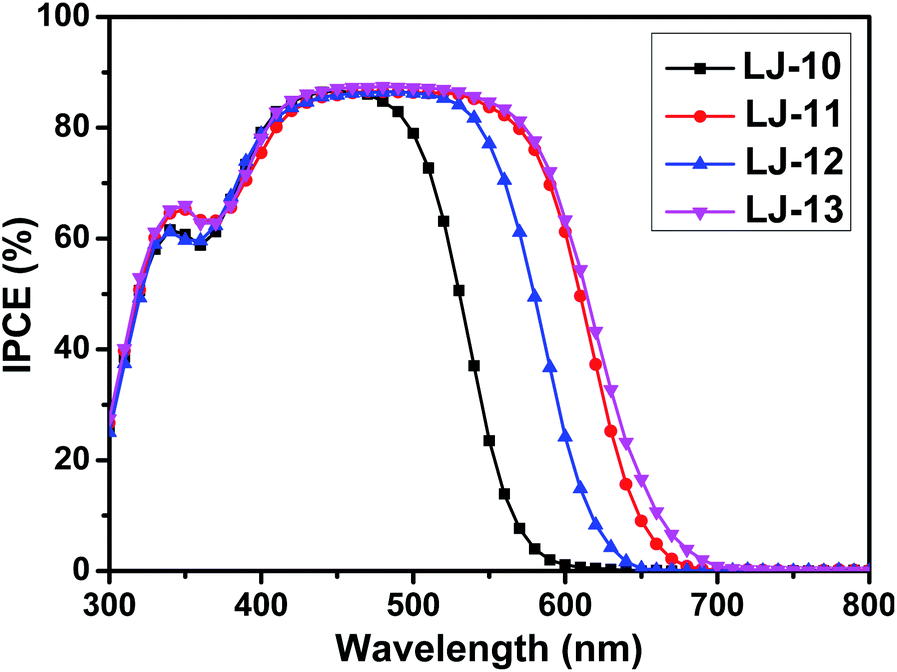 | ||
| Fig. 3 IPCE spectra of DSSCs based on the dyes LJ-10, LJ-11, LJ-12 and LJ-13. | ||
| Dyes | J SC [mA cm−2] | V OC [V] | FF | η [%] | Dye-loading amount [mol cm−2] |
|---|---|---|---|---|---|
| a Measurements were performed under AM 1.5 irradiation on the DSSC devices with 0.2304 cm2 active surface. JSC, short circuit current; VOC, open circuit voltage; FF, fill factor; η, conversion efficiency. | |||||
| LJ-10 | 7.00 | 0.706 | 0.741 | 3.66 | 9.93 × 10−8 |
| LJ-11 | 11.47 | 0.756 | 0.734 | 6.36 | 1.10 × 10−7 |
| LJ-12 | 9.74 | 0.728 | 0.741 | 5.25 | 9.88 × 10−8 |
| LJ-13 | 12.28 | 0.764 | 0.745 | 7.00 | 1.15 × 10−7 |
| N719 | 15.01 | 0.735 | 0.719 | 7.92 | 1.20 × 10−7 |
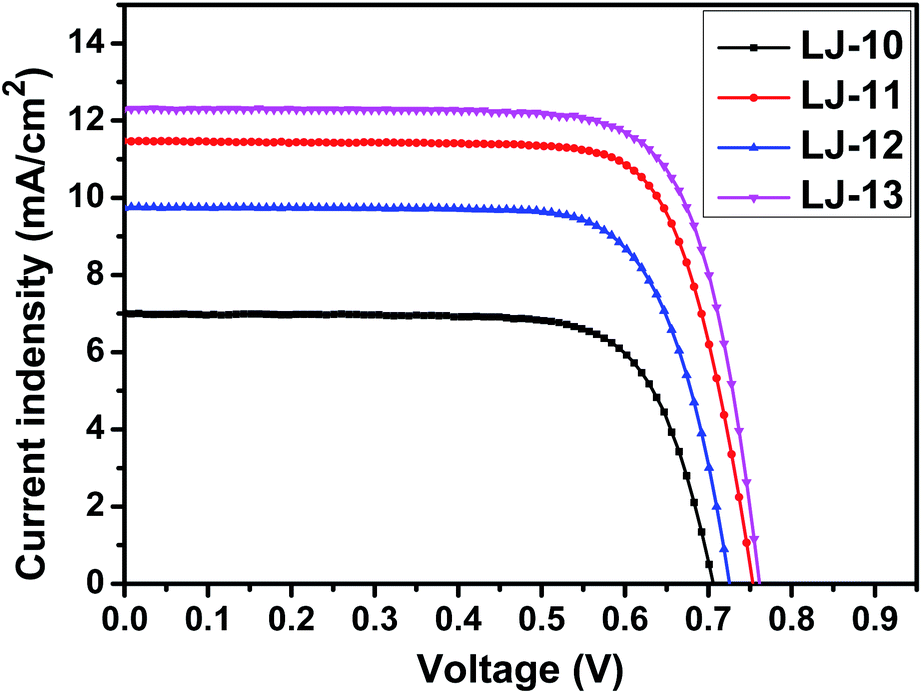 | ||
| Fig. 4 J–V curves of DSSCs based on dyes LJ-10, LJ-11, LJ-12 and LJ-13. | ||
As shown in Fig. 4, the open-circuit voltages (VOC) of DSSCs based on the pristine dyes LJ-10, LJ-11, LJ-12 and LJ-13 are 0.706 V, 0.756 V, 0.728 V and 0.764 V, respectively. DSSCs based on dyes LJ-11 and LJ-13 with a terminal cyanoacetic acid group exhibited higher photovoltage than their analogues with a terminal aryl acid moiety LJ-10 and LJ-12, respectively. To gain insight into the difference between VOC obtained for devices based on these four dyes, the VOC dependence on electron density was firstly characterized by means of the charge extraction method (CEM). As shown in Fig. S8,† DSSCs based on LJ-10, LJ-11, LJ-12 and LJ-13 showed a similar linear increase in electron density as a function of VOC. This result suggests that all these DSSCs had a similar level of conduction band edge of TiO2 regardless of the dye structure. Then, we further investigated the electron lifetimes (τ) in the conduction band of TiO2 by means of intensity-modulated photovoltage spectroscopy (IMVS). As shown in Fig. 5, the τ values of the DSSCs based on these dyes varied in the order of LJ-13 > LJ-11 > LJ-12 > LJ-10, which was at par with the increasing order of the corresponding device photovoltage.
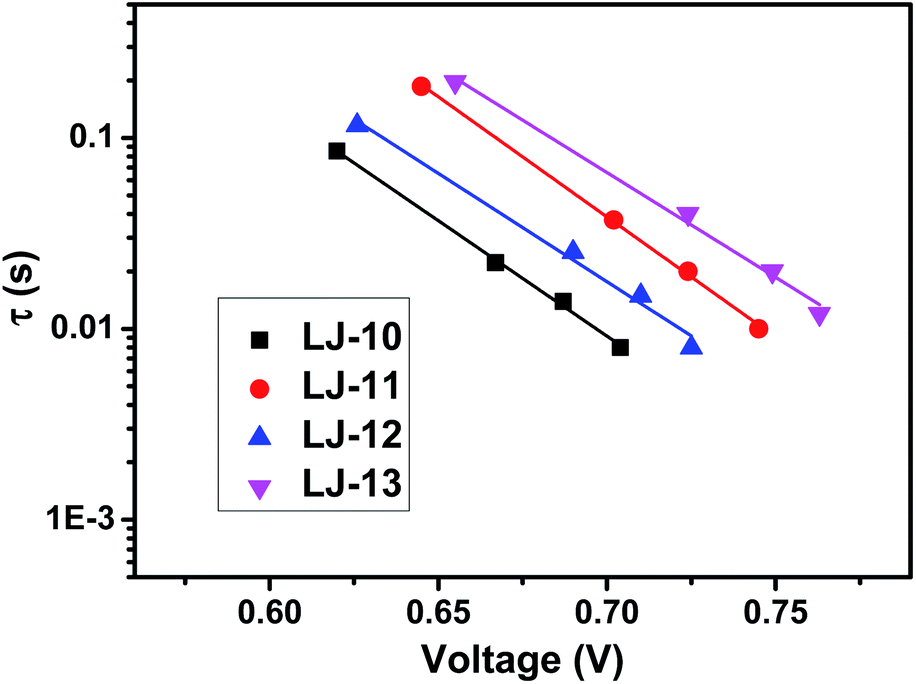 | ||
| Fig. 5 Electron lifetime (τ) as a function of VOC for DSSCs based on dyes LJ-10, LJ-11, LJ-12 and LJ-13, respectively. | ||
The degree of orbital overlap between the π*-orbitals of the anchoring group in dyes LJ-10, LJ-11, LJ-12 and LJ-13 and titanium 3d-orbitals on the photoanode surface can be investigated by the measurement of decay lifetimes of charge transfer kinetics between the dye and TiO2 surface. Nanosecond laser flash photolysis was used as a reference tool for investigating such interfacial kinetics in DSSCs, which is considered to be one of the crucial steps for the device performance.18 We employed the LP920 laser flash photolysis system to investigate and collaborate the device performances with the regeneration kinetics of dyes.
On account of the very low signals detected, sufficiently higher laser energy has been used to record a suitable transient absorbance signal. According to the results reported by Grätzel et al.,18a this should result in no significant change in the decay rate. Due to the very poor signals measured for the bleaching of the ground state of dyes, we measured the transient optical signals (Fig. 6) representing the concentration of the oxidized dye (positive ΔOD) (both have the same decay lifetime), produced after electron injection from the excited dye to the conduction band of TiO2. All decay lifetimes are listed in Table 3. Without the inert electrolyte (Fig. 6a), in acetonitrile, the absorption decays recorded half-times (τ1/2) of 6.47, 7.26, 8.87 and 1.88 μs for LJ-10, LJ-11, LJ-12 and LJ-13, respectively, which implied the unwanted recombination kinetics between injected electrons into TiO2 with the oxidative dyes. In the presence of the electrolyte, the absorption decays (τ1/2) (Fig. 6b) in acetonitrile are 3.61, 2.39, 2.17, and 0.86 μs for LJ-10, LJ-11, LJ-12 and LJ-13, respectively. These short time decays suggest that the electrolyte has regenerated at the respective dyes with suitable efficiency, which proved again the thermodynamical feasibility for dye reduction (note that the respective dyes are all more positive than the Nernst potential of the I−/I3− redox couple (0.4 eV vs. NHE), as mentioned above for Fig. 2). Once again the above-measured decay lifetimes happen to be within the order of rates mentioned by Booschloo et al.19 Usually the higher current density collected at the photoanode is a consequence of a faster dye regeneration by the electrolyte.
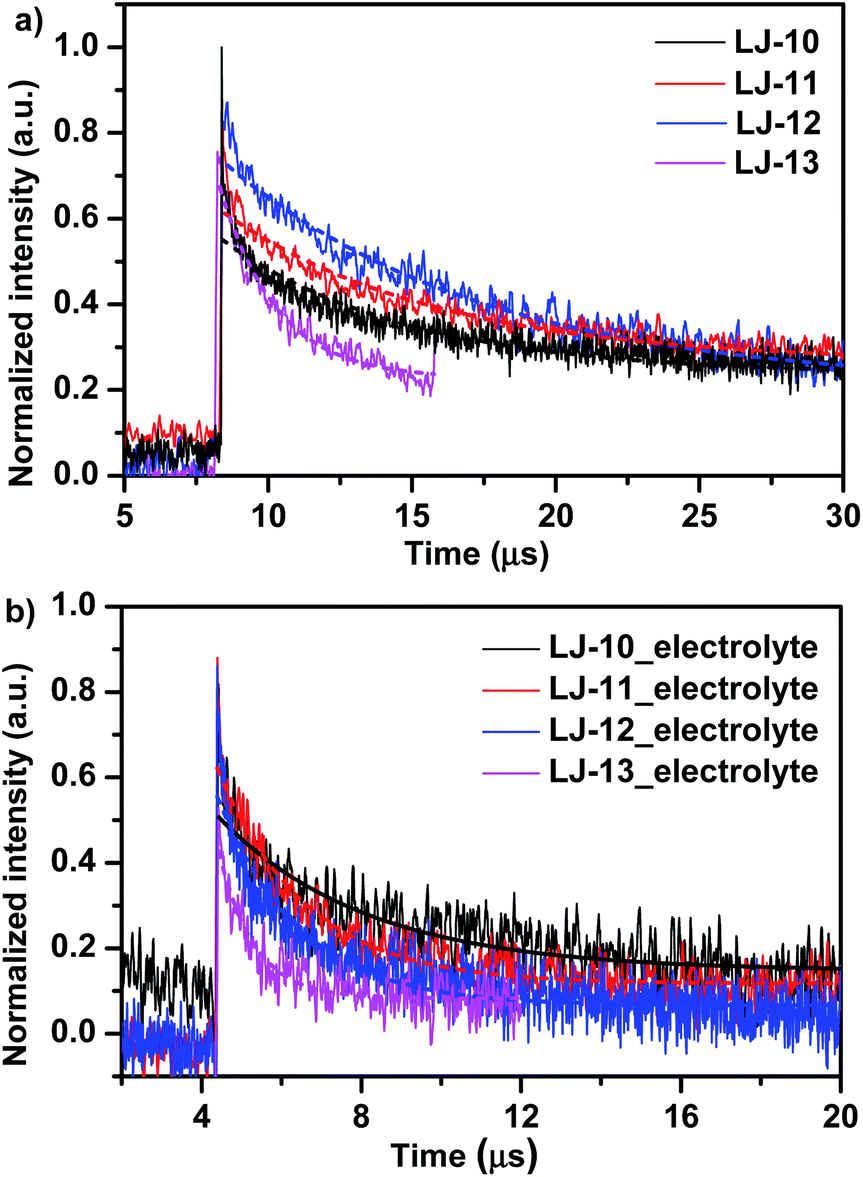 | ||
| Fig. 6 Transient absorbance decay profiles obtained upon nanosecond pulsed laser excitations on mesoporous TiO2 films sensitized with LJ-10, LJ-11, LJ-12 and LJ-13 dyes at a laser excitation of 430–470 nm, in the absence (a) and in the presence (b) of the LiI/I2 electrolyte, respectively. | ||
| Sample | No electrolyte (μs) | With electrolyte (μs) |
|---|---|---|
| LJ-10 | 6.47 | 3.61 |
| LJ-11 | 7.26 | 2.39 |
| LJ-12 | 8.87 | 2.17 |
| LJ-13 | 1.88 | 0.86 |
Generally, a good corroboration (within bar errors) was found here, where faster regeneration decay has allowed higher current density collected. LJ-13 showed the fastest decay lifetime and produced the highest current density. Although LJ-12 exhibited faster decay and dye regeneration lifetime than LJ-11, its current density collected is lower than that of the latter. The difference in the short circuit currents collected originated mostly from the overall total IPCE spectra covered (Fig. 3). In addition, based on the cyclic voltammograms of LJ-10, LJ-11, LJ-12 and LJ-13 in Fig. 3, all dyes presented driving forces greater than 0.5 V, higher than the redox potential of I−/I3− which is 0.4 V versus NHE. According to the study of Daeneke et al.,20 quantitative dye regeneration (theoretical regeneration yield 99.9%) can be achieved with a driving force of 20–25 kJ mol−1 (ΔE ≈ 0.20–0.25 V). Hence, a much higher driving force may not necessarily make a difference since the dye regeneration process is diffusion controlled if it exceeds 0.25 V. Therefore, in view of this, dye regeneration times measured in the presence of an electrolyte do corroborate with the driving forces and the recorded photocurrents, respectively.
Conclusion
In summary, we have developed a facile strategy to construct organic D–π–A dyes via sequential condensation reactions for DSSCs. The present approach does not employ the conventional C–C coupling method that is habitually used in the construction of a D–π–A backbone, and avoids the conventional noble metal catalysis as well as harsh reaction conditions. DSSCs based on the present four sensitizers produced high IPCE values and photovoltage, which suggested the successful molecular design for efficient electron injection, dye-regeneration, and retardation of charge recombination in DSSCs. Moreover, investigation of the relationship between the molecular structure and device performances may place hopes on further improvement of efficiency by expanding the conjugation length of the π-spacer in the future.Experimental
Synthesis and characterization
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the Natural Science Foundation of Jiangsu Province (BK20191385), Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX20_0860), the High Level Talent Project of Nanjing Forestry University (GXL2018003). A. Islam acknowledges the support from JSPS KAKENHI grant no. 18H02079. I. Bedja extends his appreciation to the Deanship of Scientific Research at King Saud University for funding this work through research group no. RG-1438-041.Notes and references
- (a) W. Kraut and R. von Baltz, Phys. Rev. B, 1979, 19, 1548 CrossRef CAS; (b) A. M. Cook, B. M. Fregoso, F. de Juan, S. Coh and J. E. Moore, Nat. Commun., 2017, 8, 14176 CrossRef CAS; (c) D. J. Norris and E. S. Aydil, Science, 2012, 338, 625 CrossRef CAS.
- (a) W. C. Sinke and M. M. Wienk, Nature, 1998, 395, 544 CrossRef CAS; (b) B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef; (c) A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050 CrossRef CAS; (d) H.-S. Kim, C.-R. Lee, J.-H. Im, K.-B. Lee, T. Moehl, A. Marchioro, S.-J. Moon, R. Humphry-Baker, J.-H. Yum, J. E. Moser, M. Grätzel and N.-G. Park, Sci. Rep., 2012, 2, 591 CrossRef.
- (a) Z. H. Kafafi, Science, 2015, 348, 644 CrossRef; (b) I. Chung, B. Lee, J. He, R. P. H. Chang and M. G. Kanatzidis, Nature, 2012, 485, 486 CrossRef CAS; (c) M. Grätzel, Nature, 2003, 421, 586 CrossRef.
- (a) M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Müller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382 CrossRef CAS; (b) A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS; (c) N. Duvva, U. Chilakamarthi and L. Giribabu, Sustainable Energy Fuels, 2017, 1, 678 RSC; (d) Y.-C. Chen and J. T. Lin, Sustainable Energy Fuels, 2017, 1, 969 RSC; (e) X. Li, Y. Hu, I. Sanchez-Molina, Y. Zhou, F. Yu, S. A. Haque, W. Wu, J. Hua, H. Tian and N. Robertson, J. Mater. Chem. A, 2015, 3, 21733 RSC; (f) M. F. Shah, A. Mirloup, T. H. Chowdhury, S. Alexandra, A. S. Hanbazazah, A. Ahmed, J.-J. Lee, N. Leclerc, M. Abdel-Shakour and A. Islam, Sustainable Energy Fuels, 2019, 3, 2983 RSC; (g) J. Luo, J. Zhang, K.-W. Huang, Q. Qi, S. Dong, J. Zhang, P. Wang and J. Wu, J. Mater. Chem. A, 2016, 4, 8428 RSC; (h) Y. S. Tingare, N. S. Vinh, H.-H. Chou, Y.-C. Liu, Y.-S. Long, T.-C. Wu, T.-C. Wei and C.-Y. Yeh, Adv. Energy Mater., 2017, 18, 1700032 CrossRef; (i) Y. Huang, W.-C. Chen, X.-X. Zhang, R. Ghadari, X.-Q. Fang, T. Yu and F.-T. Kong, J. Mater. Chem. C, 2018, 6, 9445 RSC; (j) K. S. K. Reddy, Y.-C. Liu, H.-H. Chou, K. Kala, T.-C. Wei and C.-Y. Yeh, ACS Appl. Mater. Interfaces, 2018, 10, 39970 CrossRef CAS; (k) Y. Kurumisawa, T. Higashino, S. Nimura, Y. Tsuji, H. Iiyama and H. Imahori, J. Am. Chem. Soc., 2019, 141, 9910 CrossRef CAS; (l) J.-M. Ji, S. H. Kim, H. Zhou, C. H. Kim and H. K. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 24067 CrossRef CAS; (m) Y. Wu, Q. Zhang, J. Li, X. Tian, D. Li, X. Lu, B. Xu, Y. Wu and K. Guo, J. Mater. Chem. C, 2019, 7, 1974 RSC; (n) A. Slodek, D. Zych, S. Golba, S. Zimosz, P. Gnida and E. Schab-Balcerzak, J. Mater. Chem. C, 2019, 7, 5830 RSC; (o) A. Baumann, C. Curiac and J. H. Delcamp, ChemSusChem, 2020, 13, 2503 CrossRef CAS.
- (a) A. S. Polo, M. K. Itokazu and N. Y. M. Iha, Coord. Chem. Rev., 2004, 248, 1343 CrossRef CAS; (b) W. M. Campbell, A. K. Burrell, D. L. Officer and K. W. Jolley, Coord. Chem. Rev., 2004, 248, 1363 CrossRef CAS; (c) P. Wang, C. Klein, R. Humphry-Baker, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 808 CrossRef CAS; (d) N. Robertson, Angew. Chem., Int. Ed., 2006, 45, 2338 CrossRef CAS; (e) L. Y. Han, A. Islam, H. Chen, C. Malapaka, B. Chiranjeevi, S. Zhang, X. Yang and M. Yanagida, Energy Environ. Sci., 2012, 5, 6057 RSC.
- (a) S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242 CrossRef CAS; (b) M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453 RSC; (c) M. Urbani, M. Grätzel, M. K. Nazeeruddin and T. Torres, Chem. Rev., 2014, 114, 12330 CrossRef CAS; (d) Y. Wu and W. Zhu, Chem. Soc. Rev., 2013, 42, 2039 RSC; (e) Y. Cheng, G. Yang, H. Jiang, S. Zhao, Q. Liu and Y. Xie, ACS Appl. Mater. Interfaces, 2018, 10, 38880 CrossRef CAS; (f) J. Wang, S. Liu, Z. Chai, K. Chang, M. Fang, M. Han, Y. Wang, S. Li, H. Han, Q. Li and Z. Li, J. Mater. Chem. A, 2018, 6, 22256 RSC; (g) A. F. Buene, E. E. Ose, A. G. Zakariassen, A. Hagfeldt and B. H. Hoff, J. Mater. Chem. A, 2019, 7, 7581 RSC; (h) H. Zhou, J.-M. Ji, S. H. Kang, M. S. Kim, H. S. Lee, C. H. Kim and H. K. Kim, J. Mater. Chem. A, 2019, 7, 2843 CAS; (i) T. Hua, K. Zhang, Z.-S. Huang, L. Wang, H. Tang, H. Meier and D. Cao, J. Mater. Chem. C, 2019, 7, 10379 RSC; (j) S. H. Lee, A. J. Matula, G. Hu, J. L. Troiano, C. J. Karpovich, R. H. Crabtree, V. S. Batista and G. W. Brudvig, ACS Appl. Mater. Interfaces, 2019, 11, 8000–8008 CrossRef CAS; (k) P. Heng, L. Mao, X. Guo, L. Wang and J. Zhang, J. Mater. Chem. C, 2020, 8, 2388 RSC; (l) P. Ferdowsi, Y. Saygili, F. Jazaeri, T. Edvinsson, J. Mokhtari, S. M. Zakeeruddin, Y. Liu, M. Grätzel and A. Hagfeldt, ChemSusChem, 2020, 13, 212 CrossRef CAS; (m) Y. Ezhumalai, F.-S. Lin, M.-S. Fan, K. Prabakaran, J.-S. Ni, Y.-C. Wu, G.-H. Lee, M.-C. Chen and K.-C. Ho, ACS Appl. Mater. Interfaces, 2020, 12, 15071 CrossRef CAS; (n) M. C. Sil, L.-S. Chen, C.-W. Lai, C.-C. Chang and C.-M. Chen, J. Mater. Chem. C, 2020, 8, 11407–11416 RSC; (o) Y. Ren, Y. Cao, D. Zhang, S. M. Zakeeruddin, A. Hagfeldt, P. Wang and M. Grätzel, Adv. Mater., 2020, 2000193 CrossRef CAS.
- (a) A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Gratzel, Science, 2011, 334, 629 CrossRef CAS; (b) Y. Xie, Y. Tang, W. Wu, Y. Wang, J. Liu, X. Li, H. Tian and W.-H. Zhu, J. Am. Chem. Soc., 2015, 137, 14055 CrossRef CAS; (c) J. Wang, H. Wu, L. Jin, J. Zhang, Y. Yuan and P. Wang, ChemSusChem, 2017, 10, 2962 CrossRef CAS; (d) Y. Ren, D. Sun, Y. Cao, H. N. Tsao, Y. Yuan, S. M. Zakeeruddin, P. Wang and M. Grätzel, J. Am. Chem. Soc., 2018, 140, 2405 CrossRef CAS; (e) W. Zhang, Y. Wu, H. W. Bahng, Y. Cao, C. Yi, Y. Saygili, J. Luo, Y. Liu, L. Kavan, b.-E. Moser, A. Hagfeldt, H. Tian, S. M. Zakeeruddin, W.-H. Zhu and M. Grätzel, Energy Environ. Sci., 2018, 11, 1779 RSC; (f) Y. Mu, H. Wu, G. Dong, Z. Shen, S. Li and M. Zhang, J. Mater. Chem. A, 2018, 6, 21493 RSC; (g) C.-T. Li, Y.-L. Kuo, C. P. Kumar, P.-T. Huang and J. T. Lin, J. Mater. Chem. A, 2019, 7, 23225 RSC; (h) J.-M. Ji, H. Zhou, Y. K. Eom, C. H. Kim and H. K. Kim, Adv. Energy Mater., 2020, 2000124 CrossRef CAS; (i) H. Jiang, Y. Ren, W. Zhang, Y. Wu, E. C. Socie, B. I. Carlsen, J.-E. Moser, H. Tian, S. M. Zakeeruddin, W.-H. Zhu and M. Grätzel, Angew. Chem., Int. Ed., 2020, 59, 9324 CrossRef CAS; (j) K. Zeng, Y. Chen, W.-H. Zhu, H. Tian and Y. Xie, J. Am. Chem. Soc., 2020, 142, 5154 CrossRef CAS.
- (a) N. Koumura, Z.-S. Wang, S. Mori, M. Miyashita, E. Suzuki and K. Hara, J. Am. Chem. Soc., 2006, 128, 14256 CrossRef CAS; (b) S. Chaurasia, C.-J. Liang, Y.-S. Yen and J. T. Lin, J. Mater. Chem. C, 2015, 3, 9765 RSC; (c) Z. Wang, M. Liang, Y. Tan, L. Ouyang, Z. Sun and S. Xue, J. Mater. Chem. A, 2015, 3, 4865 RSC; (d) Z.-S. Huang, H. Meier and D. Cao, J. Mater. Chem. C, 2016, 4, 2404 RSC; (e) P.-Y. Ho, C.-H. Siu, W.-H. Yu, P. Zhou, T. Chen, C.-L. Ho, L. T. L. Lee, Y.-H. Feng, J. Liu, K. Han, Y. H. Lo and W.-Y. Wong, J. Mater. Chem. C, 2016, 4, 713 RSC; (f) H. Klfout, A. Stewart, M. Elkhalifa and H. He, ACS Appl. Mater. Interfaces, 2017, 9, 39873 CrossRef CAS; (g) P. Brogdon, H. Cheema and J. H. Delcamp, Chemsuschem, 2018, 11, 86 CrossRef CAS; (h) A. Dessì, M. Calamante, A. Sinicropi, M. L. Parisi, L. Vesce, P. Mariani, B. Taheri, M. Ciocca, A. D. Carlo, L. Zani, A. Mordini and G. Reginato, Sustainable Energy Fuels, 2020, 4, 2309 RSC; (i) M. L. Parisi, A. Dessì, L. Zani, S. Maranghi, S. Mohammadpouras, M. Calamante, A. M. R. Basosi, G. Reginato and A. Sinicropi, Front. Chem., 2020, 8, 214 CrossRef CAS.
- (a) K. Pei, Y. Wu, A. Islam, Q. Zhang, L. Han, H. Tian and W. Zhu, ACS Appl. Mater. Interfaces, 2013, 5, 4986–4995 CrossRef CAS; (b) J.-S. Ni, W.-S. Kao, H.-J. Chou and J. T. Lin, ChemSusChem, 2015, 8, 2932–2939 CrossRef CAS; (c) K. Demirak, M. Can, C. Ozsoy, M. Z. Yigit, B. Gultekin, S. Demic and C. Zafer, Turk. J. Chem., 2017, 41, 309–322 CrossRef CAS; (d) L. Lyu, R. Su, S. Y. Al-Qaradawi, K. A. Al-Saad and A. El-Shafei, Dyes Pigm., 2019, 171, 107683 CrossRef CAS; (e) H. D. Weldekirstos, M.-C. Kuo, S.-R. Li, W.-L. Su, M. A. Desta, W.-T. Wu, C.-H. Kuo and S.-S. Sun, Dyes Pigm., 2019, 163, 761–774 CrossRef CAS.
- (a) J. M. Park, C. Y. Jung, Y. Wang, H. D. Choi, S. J. Park, P. Ou, W.-D. Jang and J. Y. Jaung, Dyes Pigm., 2019, 170, 107568 CrossRef CAS; (b) R. Su, L. Lyu, M. R. Elmorsy and A. El-Shafei, Sol. Energy, 2019, 194, 400–414 CrossRef CAS; (c) J. M. Park, C. Y. Jung, Y. Wang, H. D. Choi, S. J. Park, P. Ou, W.-D. Jang and J. Y. Jaung, Electrochim. Acta, 2019, 298, 650e662 CrossRef.
- S. Bijesh and R. Misra, Asian J. Org. Chem., 2018, 7, 1882–1892 CrossRef CAS.
- (a) C. Wang, B. Hu, J. Wang, J. Gao, G. Li, W.-W. Xiong, B. Zou, M. Suzuki, N. Aratani, H. Yamada, F. Huo, P. S. Lee and Q. Zhang, Chem.–Asian J., 2015, 10, 116 CrossRef; (b) X. Jiang, Y. Yang, J. Zhu, T.-K. Lau, P. Cheng, X. Lu, X. Zhan and X. Chen, J. Mater. Chem. C, 2017, 5, 8179 RSC.
- (a) J.-M. Ji, H. Zhou and H. K. Kim, J. Mater. Chem. A, 2018, 6, 14518 RSC; (b) M. B. Desta, N. S. Vinh, C. P. Kumar, S. Chaurasia, W.-T. Wu, J. T. Lin, T.-C. Wei and E. W.-G. Diau, J. Mater. Chem. A, 2018, 6, 13778 RSC.
- Y. Cui, Y. Wu, X. Lu, X. Zhang, G. Zhou, F. B. Miapeh, W. Zhu and Z.-S. Wang, Chem. Mater., 2011, 23, 4394 CrossRef CAS.
- G. S. Selopal, H.-P. Wu, J. Lu, Y.-C. Chang, M. Wang, A. Vomiero, I. Concina and E. W.-G. Diau, Sci. Rep., 2016, 6, 18756 CrossRef CAS.
- (a) J. Liu, Y. Numata, C. Qin, A. Islam, X. Yang and L. Han, Chem. Commun., 2013, 49, 7587 RSC; (b) J. Liu, X. Yang, A. Islam, Y. Numata, S. Zhang, N. T. Salim, H. Chen and L. Han, J. Mater. Chem. A, 2013, 1, 10889 RSC; (c) H. Chen, G. Lyu, Y. Yue, T. Wang, D.-P. Li, H. Shi, J. Xing, J. Shao, R. Zhang and J. Liu, J. Mater. Chem. C, 2019, 7, 7249 RSC.
- (a) X. Yang, M. Yanagida and L. Han, Energy Environ. Sci., 2013, 6, 54 RSC; (b) M. Pazoki, U. B. Cappel, E. M. J. Johansson, A. Hagfeldt and G. Boschloo, Energy Environ. Sci., 2017, 10, 672 RSC.
- (a) S. Pelet, J.-E. Moser and M. Gratzel, J. Phys. Chem. B, 2000, 104, 1791 CrossRef CAS; (b) M. Freitag, J. Teuscher, Y. Saygili, X. Zhang, F. Giordano, P. Liska, J. Hua, S. M. Zakeeruddin, J.-E. Moser, M. Grätzel and A. Hagfeldt, Nat. Photonics, 2017, 11, 372–378 CrossRef CAS; (c) J. V. S. Krishna, D. Koteshwar, T. H. Chowdhury, S. P. Singh, I. Bedja, A. Islam and L. Giribabu, J. Mater. Chem. C, 2019, 7, 13594 RSC.
- G. Boschloo and A. Hagfeldt, Acc. Chem. Res., 2009, 42, 1819 CrossRef CAS.
- T. Daeneke, A. J. Mozer, Y. Uemura, S. Makuta, M. Fekete, Y. Tachibana, N. Koumura, U. Bach and L. Spiccia, J. Am. Chem. Soc., 2012, 134, 16925 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectra characterization, DFT calculations of dyes LJ-10, LJ-11, LJ-12 and LJ-13. See DOI: 10.1039/d0se01519a |
| This journal is © The Royal Society of Chemistry 2021 |