A perylene-based aromatic polyimide with multiple carbonyls enabling high-capacity and stable organic lithium and sodium ion batteries†
Received
21st August 2020
, Accepted 28th October 2020
First published on 29th October 2020
Abstract
Redox-active organic electrode materials are considered promising alternatives to inorganic intercalation analogs in organic metal-ion batteries. However, their poor cycling stability owing to high solubility in organic electrolytes and poor electronic conductivity remain a challenge for all-organic battery applications. Constructing new conjugated aromatic polyimides (PIs) or polymers with characteristics of improved electronic conductivity and abundant redox-active units (i.e., dual redox units containing multiple carbonyl groups) is an effective method of preventing these battery problems. In this study, we synthesized a perylene-3,4:9,10-tetracarboxylic dianhydride (perylene)-based aromatic PI as the cathode material, which is incorporated with two distinct types of redox-active units through the polymerization of perylene-3,4:9,10-tetracarboxylic acid with a 2,6-diaminoanthraquinone moiety, which has multiple redox-active carbonyl sites. The as-prepared PI exhibited significantly lower stability problems and enhanced the performance of electrochemical kinetics in both organic lithium and sodium ion batteries owing to improved electronic conductivity via a unique π-conjugation structure and the PI's multiple redox kinetics. The battery cells with the PI cathode exhibited initial discharge capacities of 209 mA h g−1 (for Li+/Li) and 207 mA h g−1 (for Na+/Na) at a high current rate of 200 mA g−1. The PI exhibited a better long-life cycling stability with a high-rate discharge ability (15 mA h g−1 for Li+/Li) with a capacity retention of 14%; and 78 mA h g−1 for Na+/Na with 54% capacity retention at a current density of 1C over 1000 cycles. These values are among the best when the delivered high specific capacities and stable cycle performance of both Li+/Na+ ion storage are compared with the previously reported similar PIs used for Li+-ion storage. This demonstrates the promising potential application of multiple redox-active units (i.e., dual redox-active units) in the design of sustainable cathodic materials for next-generation electrochemical energy storage devices.
1. Introduction
Lithium-ion batteries (LIBs) have been employed as the dominant power source for portable electronic devices and electric vehicles (EVs) owing to their high energy densities, long cycle life, and negligible self-discharging.1–3 Meanwhile, sodium-ion batteries (SIBs) have also emerged as some of the most promising battery technologies owing to their comparable theoretical energy density, large natural abundance of sodium (Na)-based intercalation materials, low cost of Na metal compared with lithium (Li), and intercalation chemistries similar to those of LIBs.4 However, most redox-active electrode materials utilized in today's conventional LIBs and SIBs generally rely on inorganic Li/Na-based intercalation compounds, which are frequently constrained by limited mineral resources, low utilization efficiencies even at high rates, irreversibility of the redox reactions, sluggish kinetics owing to large Na ions inserting and de-inserting into their rigid lattices, and energy-intensive processes required during the production of these materials.5–8 Therefore, these constraints have motivated research interest in the development of alternative electrode materials that are naturally abundant, more cost-effective, have negligible environmental effects, and excellent capability to store more Li+/Na+ ions to compete with inorganic intercalation materials.
Organic electrode materials are highly promising alternatives to inorganic intercalation materials because they have the potential to create high migration kinetics of different metal ions (i.e., Li+, Na+, K+, Mg2+, Zn2+, Al3+, and Ca2+), a high theoretical storage capacity, structural flexibility, low weight, large resources, and easy functionalization.9–17 Recent publications highlighted four promising families of redox-active organic electrode materials utilized in various metal-ion batteries depending on their electrochemically active functional groups involved in redox reaction kinetics. These materials include (i) conducting polymers or conjugated polymers,18,19 (ii) organic free radical polymers and organosulfides,20,21 (iii) covalent organic frameworks and macromolecules/porous organic polymers,22–28 and (iv) conjugated carbonyl compounds or conjugated carbonyl-based polyimides (PIs)/polymers.29–32 Among them, the conjugated carbonyl compounds have been considered promising electrode materials for batteries because of their potentially tailored high theoretical capacity, fast redox reaction kinetics, and high structural diversity.33,34 However, the high solubility of these carbonyl compounds in organic electrolytes during cycling, low discharge voltage, low electronic conductivity, and low active carbonyl utilization have severely inhibited their further applications. To resolve these intrinsic problems, scholars have extensively investigated conjugated carbonyl-based PIs/polymers as some of the most promising high-capacity electrode materials utilized in various metal-ion batteries owing to their exceptional mechanical strength, high chemical resistance, and higher electronic conductivity through an extended π-conjugation system, excellent structural stability that can significantly enhance their potentially tailored high specific capacity, and prolonged cycle performance over 5000 cycles.28,35–38
To date, considerable research effort has been adopted to prevent the dissolution problems of organic electrodes in electrolytes and increase the electronic conductivity and cycling performance of conjugated carbonyl-based battery electrode materials by synthesizing structurally stable and conjugated aromatic multi-carbonyl PIs/polymers (i.e., dual redox-active units with multiple carbonyls) using polymerization reactions.39–42 Among the electrochemically active aromatic PIs, the aromatic dianhydride of perylene-3,4:9,10-tetracarboxylic acid (perylene)-based PIs combined with electrochemically active and inactive linker groups have been extensively explored as the most promising high-capacity materials for battery electrodes with prolonged stable cycle performance.17,35,43–47 In practice, the perylene-based aromatic PIs exhibit the structural characteristics of redox-active carbonyl conjugated systems with five condensed aromatic rings and the ring closure of imide groups, which can accommodate more Li+/Na+ ions and enable fast multiple redox kinetics. Thus, they can deliver an exceptionally high specific capacity of over 200 mA h g−1 (theoretical capacity density up to 274 mA h g−1), a high reversible capacity while maintaining long-cycling stability because they have high theoretical storage capacity and ultrafast charge–discharge kinetics.48,49 Several studies demonstrated that the electrochemically active redox sites of the aromatic anhydride/imide groups primarily originate from the enolization of carbonyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and their theoretical charge transfer interactions (capacity density) associated with the number of redox-active carbonyl groups.50–56 The theoretical specific capacity (Cspec) of redox-active PIs or polymers can be calculated using the following well-known formula.57
O) and their theoretical charge transfer interactions (capacity density) associated with the number of redox-active carbonyl groups.50–56 The theoretical specific capacity (Cspec) of redox-active PIs or polymers can be calculated using the following well-known formula.57
Cspec = nF/(3.6 × Mw) = 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 801 × n/Mw (mA h g−1) 801 × n/Mw (mA h g−1) |
where
n is the number of transferred electrons (C
O groups) in each structural unit,
F is the Faraday constant (96
485.332 C mol
−1), and
Mw is the molecular weight of the structural unit (PI/polymer). Increasing the ratio of
n to
Mw is more beneficial to obtaining abundant Li
+/Na
+ ion storage redox sites and a higher theoretical capacity.
58 Therefore, the theoretical specific capacities of PIs are expected to increase by increasing the rate of C
O groups (higher content of carbonyl activity) in the design of perylene-based aromatic PI chains with low molecular weights of the units. For instance, Sharma
et al. demonstrated that perylene-tetracarboxylic dianhydride (PTCDA)-based PIs containing an additional C
O group from the low molecular weight of urea polymerization resulted in increased Li
+-ion storage and better cycling stability.
59 Our group recently reported PTCDA-based aromatic PIs combining PTCDA units and electrochemically redox-active groups; these polyimides exhibited high discharge capacities (over 119 mA h g
−1 at 25 mA g
−1) and good cycling stability in both LIBs and SIBs.
47 Jung
et al. also fabricated PTCDA-based PI containing PTCDA unit and 2,6-diaminoanthraquinone (2,6-DAAQ) as an additional redox-active unit (tricarbonyl moiety) for Li
+-ion storage, and it exhibited a specific capacity of 128.43 mA h g
−1 over 20 cycles at a current density of 50 mA g
−1, and an excellent rate capability retention of 78% at 200 mA g
−1 in LIBs.
46
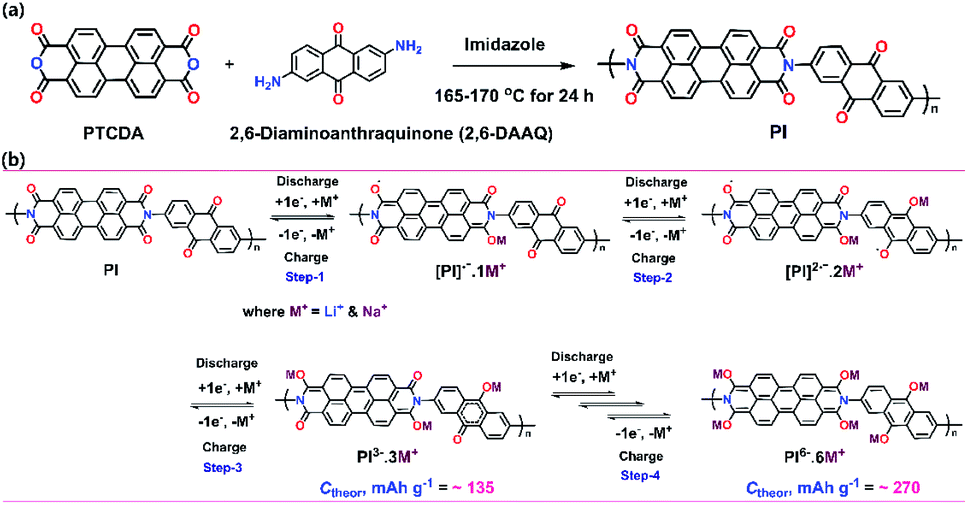
Herein, we report a PTCDA (perylene)-based aromatic PI that incorporates two distinct types of redox-active units by combining a PTCDA unit and a 2,6-DAAQ moiety (Scheme 1a). This PI served as a high-capacity cathode material with improved cycling stability and high-rate performance when compared with a previously reported similar PI.46 This cathode material was prepared through a simple one-pot solution polymerization reaction of a PTCDA unit and 2,6-DAAQ. To achieve a high theoretical storage capacity and higher electronic conductivity of the PI, we selected 2,6-DAAQ as an additional carbonyl moiety as it has a high carbonyl rate to facilitate new Li+/Na+ ion storage sites through its carbonyl groups. We synthesized this cathode material for use in both LIBs and SIBs because SIBs using PIs have not been reported until now. The PI exhibited initial specific capacities of 209 mA h g−1 (for Li+/Li) and 207 mA h g−1 (for Na+/Na) at high discharge voltage plateaus (1.9–2.4 V for Li+/Li and Na+/Na). Moreover, the cell delivered a high-rate performance with average reversible capacities of 58 mA h g−1 (for Li+/Li) and 77 mA h g−1 (for Na+/Na) over 20 cycles at 50 mA g−1. These discharge capacities are comparable to or even higher than the typical specific capacities of a previous study.46
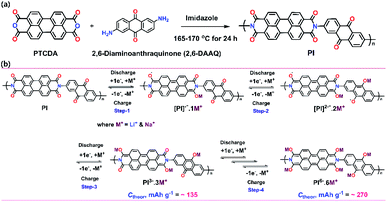 |
| | Scheme 1 (a) A synthetic method to synthesize the perylene-based aromatic PI through the polymerization of a PTCDA unit and 2,6-DAAQ in a single polycondensation step. (b) Representative scheme of the PI-based cathode indicating the proposed mechanism of the Li+/Na+-ion storage redox reaction during CV (one-step reaction of three electrons was equally applicable for the PI through three steps via multiple carbonyl rates). The calculated theoretical capacity (Ctheor) of PI and its electrogenerated species are indicated in each step. | |
2. Experimental section
Material synthesis
The procedure shown in Scheme 1a was used to synthesize the perylene-based aromatic PI at a 95.7% yield from a PTCDA unit and 2,6-DAAQ through a condensation polymerization reaction. The ESI† provides detailed synthetic procedures and characterization using standard spectroscopic techniques for the perylene-based aromatic PI with multiple carbonyls.
Characterization
All reagents, chemicals, organic solvents, and electrolytes were purchased from Tokyo Chemical Industry Co., Ltd and Sigma-Aldrich and were used as received. All electrochemical measurements and the construction of LIBs and SIBs were performed at room temperature in an argon-filled glovebox with oxygen and moisture levels below 1 ppm. All battery capacities and current densities were calculated based on the amount of the material in the electrode. Fourier transform infrared (FT-IR) spectra for the prepared final PI were measured using a Smart iTR NICOLET iS10 FT-IR spectrometer (Thermo Scientific). Thermogravimetric analysis (TGA) was performed using a TGA Q50 thermal analyzer at a heating ramp rate of 10 °C min−1 from 0 to 900 °C under a nitrogen atmosphere at the Core Research Support Center for Natural Products and Medical Materials in Yeungnam University. Solid state 13C CP/MAS NMR measurements were performed on a Bruker Avance 500 (500 MHz) spectrometer with a CPTOSS 5 kHz spinning. The morphologies of the samples were determined using a S-4800 (Hitachi) field-emission scanning electron microscope (FE-SEM). X-ray diffraction (XRD) analysis was used to investigate the crystal structures of the bulk and electrode samples using Cu-Kα radiation (operation voltage: 40 kV, current: 30 mA, λ = 1.5418 Å). X-ray photoelectron spectroscopy (XPS) measurements were recorded using K-Alpha (ThermoScientific) as an X-ray source to analyze the elemental surface composition of the samples and electrodes. Electrochemical impedance spectroscopy (EIS) was conducted to estimate the charge transfer resistance of the fabricated battery cells. The frequency was varied from 100 kHz to 0.1 Hz with an alternating current signal amplitude of 10 mV.
Electrochemical measurements
The PI cathode was obtained by mixing the cathode material (PI), conductive carbon black (ENSACO, MMM Super P, 230 m2 g−1, Sigma-Aldrich), and a polymer binder (Kynar 2801, polyvinylidene difluoride (PVDF)) at a weight ratio of 60:30:10 to form a well-dispersed slurry in N-methylpyrrolidone (NMP), and was stirred for 6 h. The slurry was uniformly spread over an aluminum foil (Shenzhen Vanlead Tech. Co. Ltd, China) as a current collector, using the doctor blade technique at a thickness of 20 μm. Subsequently, the coated aluminum foil was immediately dried in a blast oven at 100 °C for 12 h to produce electrode films. A twin roller machine was used to press the coated electrodes to increase the contact between the PI and the aluminum foil. Finally, the electrodes were cut into small circular discs with diameters of 12 mm. The mass of the active material coated on the current collector was 0.3 ± 0.18 mg cm−2. After the electrodes were weighed, they were placed in an argon-filled glove box with the moisture and oxygen/water levels below 1 ppm and fabricated into half-cell LIBs using the 2032 coin cell assembly. The pure lithium foil was used as the counter electrode, 1 M LiPF6 in an ethylene carbonate (EC)/dimethylcarbonate (DMC) mixture (EC/DMC; 1:1 molar ratio) as the electrolyte (1 mL of electrolyte was used to make a cell), and a Celgard 2500 microporous polypropylene membrane as the separator. The battery cells were assembled and left for approximately 12 h at room temperature. The electrochemical characterization of the PIs as the cathode in SIBs was evaluated similarly as the aforementioned lithium battery tests, but we used sodium foil as the counter electrode, glass fiber as the separator (GF/D, Whatman), and approximately 1 mL of EC/DMC (1:1 molar ratio) solution of NaPF6 as the electrolyte. Subsequently, a galvanostatic charge–discharge cycle was performed on the fabricated battery cells using a Neware multi-channel battery tester (model BTS-7.6.0, Shenzhen). Cyclic voltammetry (CV) was performed at a scan rate of 0.1 mV s−1 in a voltage range between 1.5 and 3.5 V for Li+/Li and Na+/Na using a WonATech WBCS3000 battery test unit. EIS was conducted to estimate the charge transfer resistance of both fresh and cycled battery cells using a Solartron impedance/gain phase analyzer (model SI 1255) coupled with a potentiostat (SI 1268). The frequency was varied from 100 kHz to 0.1 Hz with an alternating current signal amplitude of 10 mV.
Ex situ methods
For ex situ analysis, the samples in different states were obtained by discharging and charging at a current density of 200 mA g−1. The electrodes were rinsed with dimethyl carbonate. After washing, all the samples were dried in a vacuum at room temperature. All the samples were sealed in an argon-filled glove box before the ex situ experiments to prevent exposure to air.
3. Results and discussion
Materials synthesis and characterization of the PI
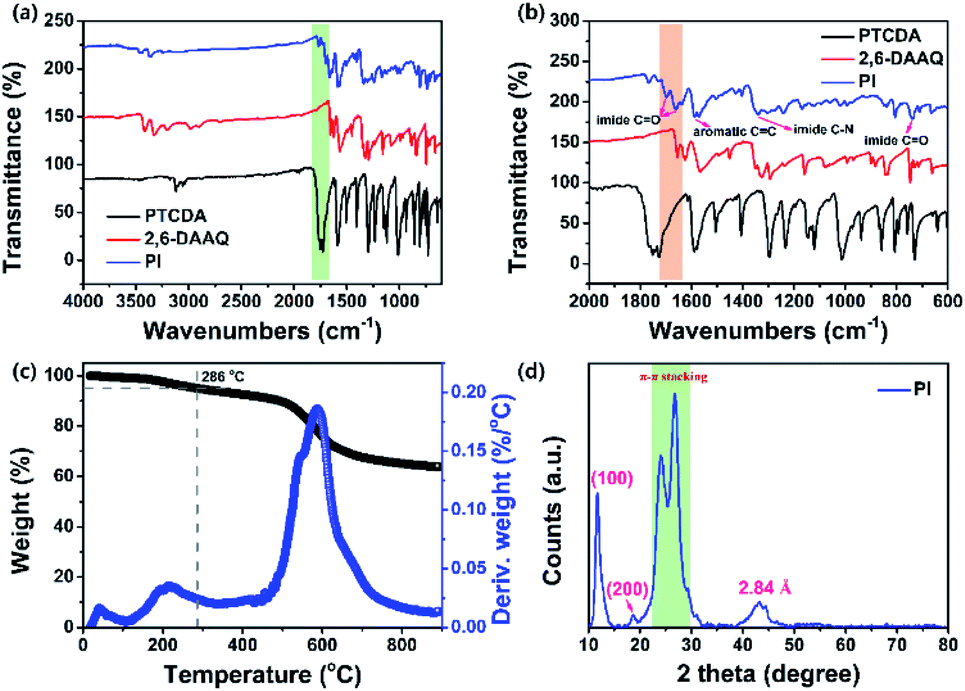
A perylene-based aromatic multi-carbonyl PI (Scheme 1a) was synthesized through the polymerization of a PTCDA unit and 2,6-DAAQ (which has multiple Li+/Na+ ion storage redox sites) in a 95.7% yield. The FT-IR spectrum of the PTCDA indicated two sharp vibrational bands at 1766 and 1727 cm−1 (Fig. 1a and b), which were associated with the asymmetric and symmetric stretching vibration modes of carbonyl groups (CO bonds), respectively. These vibrational bands shifted to 1698 and 1663 cm−1 in the PI. The strong vibrational bands at 1297 and 938 cm−1 of the PTCDA were assigned to C–O–C, which clearly disappeared in the PI. Meanwhile, a broad new vibrational band at 1340 cm−1 corresponding to the stretching vibration of imide C–N bonds and a sharp vibration band at 723 cm−1 assigned to imide CO bond symmetric in-plane bending of the PI were observed, indicating the complete conversion of the anhydride groups into the imide in the PI.45–47,59,60 Two vibrational bands were observed at 3428 and 3328 cm−1 in the 2,6-DAAQ, and they were primarily attributed to the asymmetric and symmetric stretching vibration modes of –NH2, while these two vibrational bands clearly disappeared in the PI. Two vibrational bands were located at 1655 and 1626 cm−1 in the 2,6-DAAQ, corresponding to the signals of CO bonds; they shifted to 1662 cm−1 in the PI. In addition, the FT-IR spectrum of the 2,6-DAAQ indicated a strong vibrational band located at 1567 cm−1 corresponding to the aromatic CC stretching vibration mode, which shifted and was located at 1571 and 1587 cm−1 in the PI. These results indicated that the PTCDA and 2,6-DAAQ coupled well in the molecular structure of the PI. The synthesized PI exhibited a high thermal stability with 5% and 10% weight loss of the decomposition temperatures (Td) above 280 and 490 °C (i.e., the major product decomposition temperature was in the range of 400 to 650 °C), respectively, under a nitrogen atmosphere (Fig. 1c). This suggested a potential candidate for the fabrication of safe battery cells using this PI. The molecular structure of PI was confirmed by using a solid-state 13C CP/MAS NMR spectroscopic method (see the Experimental section and Fig. S1†). The structure of the PI was further confirmed using CHNS (elemental) analysis, which has a formula composition of (C38H14N2O6)n with the content of C, H, N, and O as 76.77%, 2.37%, 4.70%, and 16.09%, respectively (see the Experimental section in the ESI†). The CHNS analysis indicated that the contents of C, H, and N were 76.25%, 2.41%, and 4.78% respectively, which corresponded with the calculated values of the expected composition of the PI; moreover, these values matched well with elemental values for a PI reported by Jung et al.46 Furthermore, the high-resolution XPS indicated characteristic deconvoluted peaks of the corresponding binding energies for the C, N, and O elements (Fig. S2†) and the emergence of π–π* satellite peaks in the local-scan XPS spectra of both C, N, and O 1s signals, implying that the PTCDA and 2,6-DAAQ units were strongly coupled in the successfully synthesized PI.
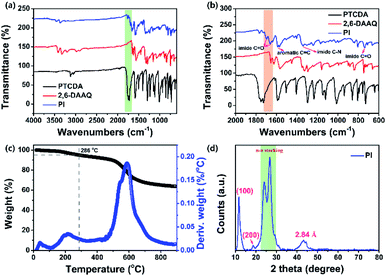 |
| | Fig. 1 (a) FT-IR spectra of PTCDA, 2,6-DAAQ, and the PI; (b) extended FT-IR spectra of the PTCDA, 2,6-DAAQ, and PI; (c) thermogravimetric analysis curve for the PI and the corresponding derivative weight change curve; (d) powder XRD patterns of the PI. | |
The XRD peaks for the PI exhibited good crystallinity (Fig. 1d). The strong reflection peak at approximately 2θ = 11.72° indicated intermolecular π–π stacking distances (d-spacing) of 7.6 Å and was indexed as the d100 reflection, which corresponded to the molecular length and irregularly ordered packing along the N–N direction of the PI aggregates. The reflection peak at approximately 2θ = 18.69° had a d-spacing of 4.75 Å and was indexed as the d200 reflection. The diffraction peaks were detected in the range of 2θ = 23.99° and 26.77°, corresponding to the d-spacing in the range of 3.3–3.7 Å, which are the general characteristic patterns of the intermolecular π–π stacking distances of PI aggregates.47 As a result, the XRD pattern of the PI exhibited up to second-order diffractions, indicating that the PI tended to self-organize into a few orders of the disordered aggregated structures by fewer intermolecular π–π stacking interactions between conjugated perylene cores in the PI in cooperation with hydrophilic interactions of the imide-functional groups (carbonyl groups). Thus, micro/nano-particle aggregated structures with good crystallinity were formed. This result was further supported by the FE-SEM images of the surface morphology of the PI (Fig. S3†). Furthermore, the broad reflection peak was detected in the range 2θ = 38–48°, which belonged to the d-spacing of 2.84 Å in the meridional direction, which was related to the intercolumnar disordered aggregated structures of the PI.61 Note that the observed diffraction patterns for PI differed in the degree of structural order (crystallinity) from the report by Jung et al.46 The as-prepared PI was more beneficial for boosting electronic conductions and ionic conductance (fast redox kinetics of Li+/Na+ ions), indicating that the as-prepared PI material may have a better rate performance in a battery system.
The morphology of the PI was inspected using FE-SEM (Fig. 2). As Fig. 2a–c, PI exhibited microparticle sizes in the range 500 nm to 2 μm, which indicated a morphology of disordered aggregates (irregular edges of microparticle structures) with good crystallinity, and the formation of a π-stacked arrangement of the PI aggregated morphology between the conjugated perylene cores in the PI (Fig. 2d and S3†), which was consistent with the powder XRD analysis of the PI (Fig. 1d). Remarkably, this morphology differed from the similar PI electrode reported by Jung et al., which was clearly amorphous.46
 |
| | Fig. 2 (a–c) Typical FE-SEM micrographs for the PI; (d) FE-SEM images depicting the formation of π-stacked PI aggregated morphology between conjugated perylene cores in the PI. | |
Electrochemical performance evaluation of the PI
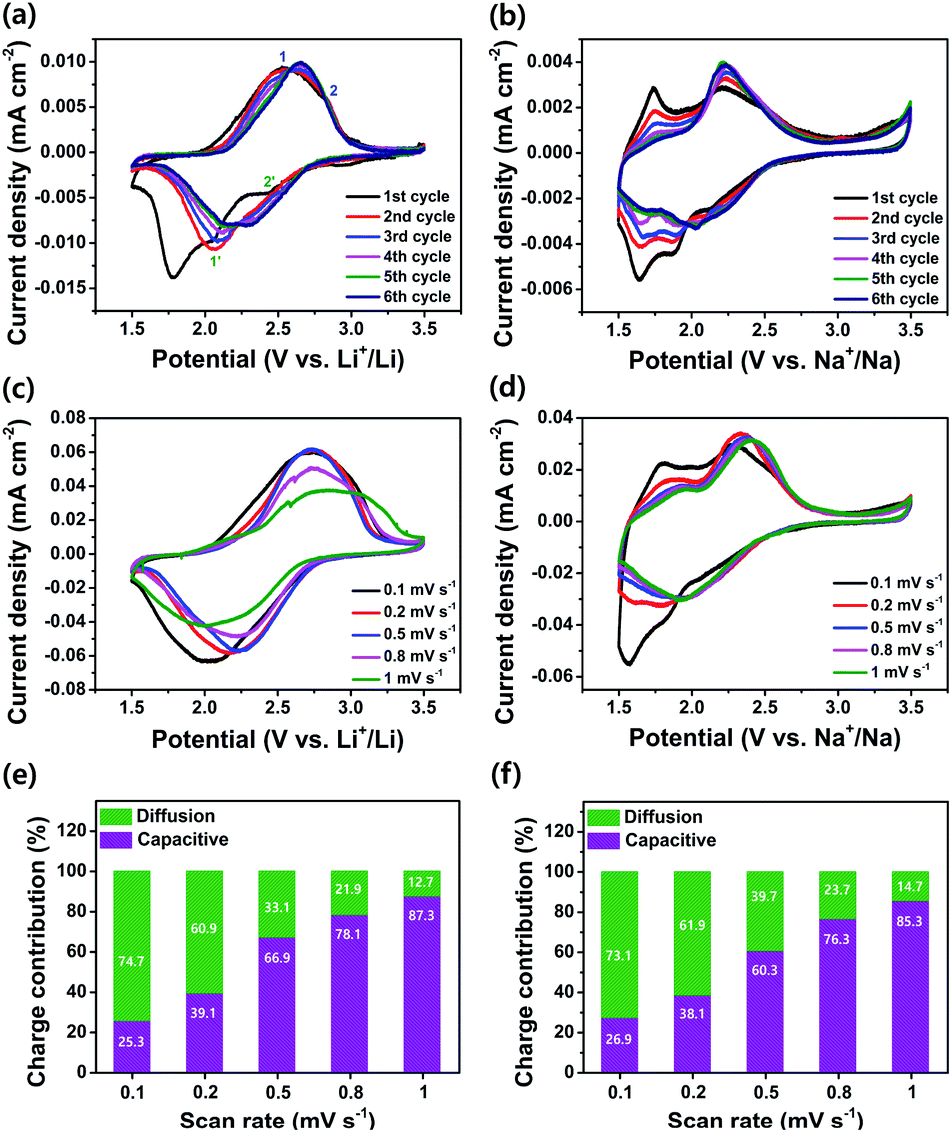
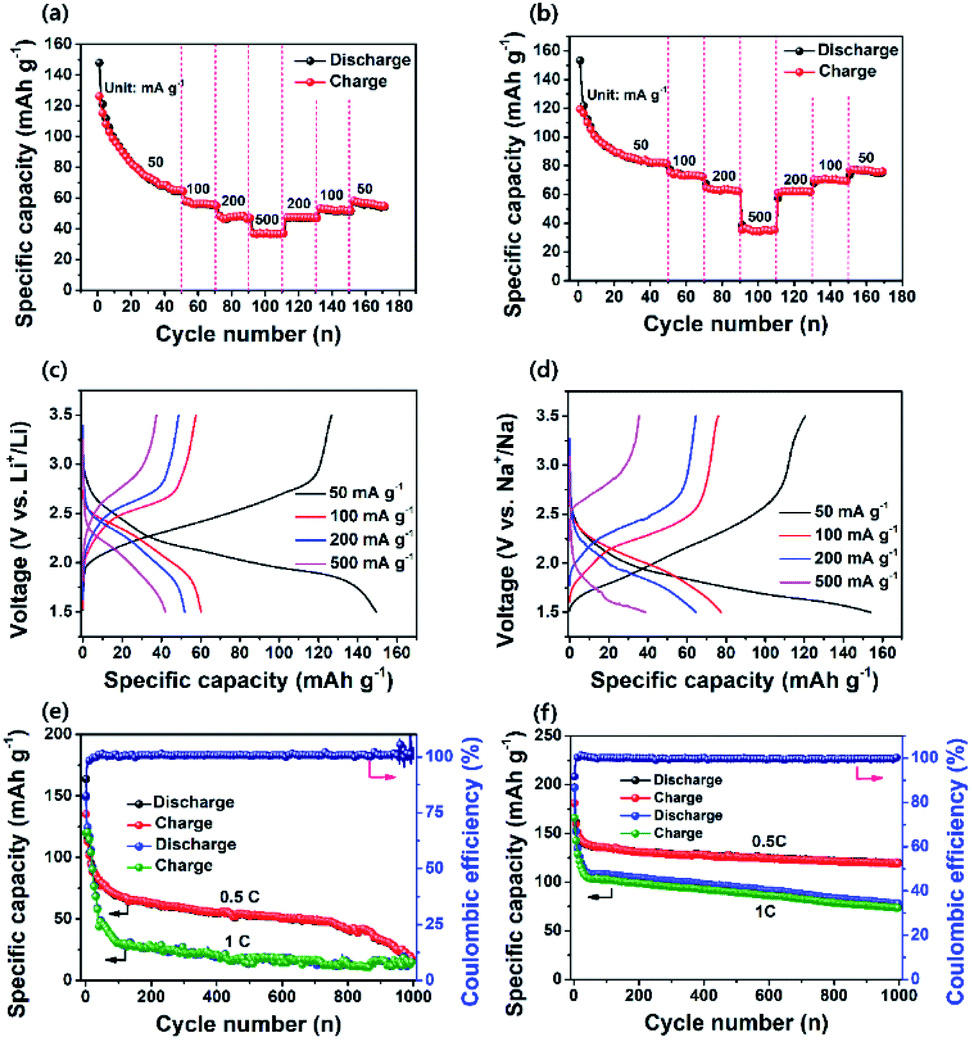
The electrochemical performance of the as-prepared PI as a cathode was investigated using coin-type cells. Fig. 3a and b show the CV curves for the LIB and SIB, including the reversible process of both Li+/Na+ ions into the PI at a scan rate of 0.1 mV s−1 in a potential range of 1.5–3.5 V for Li+/Li and Na+/Na, respectively. In the LIB (Fig. 3a), the CV curves of the PI were broad with two pairs of reversible redox peaks for Li+/Li (i.e., the oxidation peaks centered at 2.52 & 2.83 V, and the reduction peaks centered at 2.44 V & 2.06 V), and were red-shifted to higher potential regions from 2.52 to 2.65 V (oxidation peaks) and 2.06 to 2.15 V (reduction peaks), with the appearance of a new peak located at 2.28 V in the subsequent cycles (second → sixth cycle). By contrast, the CV curves of the PI in the SIB exhibited two pairs of reversible redox peaks (i.e., the oxidation peaks centered at 1.74 & 2.21 V, and the reduction peaks located at 1.87 & 2.13 V) with broadening peaks for Na+/Na (Fig. 3b). In addition, the PI exhibited redox curves at 2.21 and 2.13 V, and altered peak positions with increasing intensities in the subsequent cycles (second → sixth cycle). The observed broad curves in the CV involved fast multiple redox kinetics by sequential rearrangements of the PI structure via the enolization reaction between CO and Li+/Na+ ions, as intermediate compounds illustrated in steps 1 and 2 of Scheme 1b. These results were primarily attributed to the three-electron redox reaction in the three conjugated carbonyl groups (CO) of the PI through the direct formation of trianion species via fast multiple redox kinetics between the radical anion and trianion species, which in turn resulted in two reversible redox peaks (i.e., two broader peaks involving a three electron transfer process) rather than three well-resolved redox peaks. This result indicated that the high degree of delocalization of the perylene cores, which favoured the fast electron transformation between the radical anions and trianion species (as illustrated in Scheme 1b) resulted in two broader peaks in cycling curves involving three electrons. In addition, the cycling curves became broader as the cycle number increased owing to fast multiple reaction kinetics for Li/Na-ion storage redox behaviour through sequential structural rearrangements of PI via the enolization reaction following a multiple Li/Na-ion storage.62–64 These phenomena corresponded with the CV results by Jung et al.46 A two-electron transfer occurred from the conjugated carbonyl groups presented in the perylene core and one-electron transfer from the DAAQ unit attached in the imide region of the PI, which could reversibly exchange three electrons; thus, this was considered the three-electron transfer redox process involved in the PI, as indicated in step 3 of Scheme 1b. The redox process in the PI can be explained in three steps (Scheme 1b). The aromatic anhydride of the PTCDA and DAAQ units in the PI could reversibly exchange three electrons at the carbonyl groups through an enolization reaction (Li+/Na+ ions enolate, as shown in step 3 of Scheme 1b) and was then theoretically associated with 3 mol of Li+/Na+ ions (metal ions). During the first step of the discharge (step 1), one electron was initially captured from the one-carbonyl group of the PTCDA unit in PI to form a radical anion ([PI]˙−) as an intermediate and was then associated with 1 mol of metal ions, as indicated by the appearance of reduction peaks at 2.44 V for Li+/Li (Fig. 3a) and 2.13 V for Na+/Na (Fig. 3b). In the second step of discharge (step 2), another electron was captured from the opposite carbonyl group of the DAAQ unit to form the dianion species ([PI]2˙−) of the PI and was then associated with another 1 mol of metal ions to form ([PI]2˙−·2M+) as the intermediate compound. This process is ascribed to the CV peaks centered at 2.06 V (for Li+/Li) and 1.87 V (for Na+/Na). In the third step of discharge (step 3), another electron was gained from the opposite CO group of the PTCDA unit to form trianion (PI3−) species. This species could be accompanied by the insertion of another 1 mol of metal ions to finally generate the trianion species (PI3−·3M+) in the subsequent step. These phenomena were attributed to the CV peaks detected at 1.78 V (for Li+/Li) and 1.64 V (for Na+/Na). The CO groups were rebuilt and returned to the original state (de-insertion of metal ions) through the charge process. These results corresponded with previously reported CV results by Wu et al.51 and Jung et al.46 Therefore, the theoretical capacity of the PI was calculated to be 135 mA h g−1, according to the three-electron transfer redox process. Moreover, the broadened peaks of the PI in the LIB and SIB were attributed to the less diffusion-controlled process of insertion/de-insertion of metal ions. Therefore, a high degree of polarization is required to insert the metal ions into the uncoordinated PI molecules during electrochemical kinetics. The CVs of the PI electrode at different scan rates were conducted to investigate the charge storage mechanism and reaction kinetics (Fig. 3c and d). It can be seen that the anodic peaks remained unchanged from the scan rate of 0.1 to 0.8 mV s−1, and then shifted to high potentials at 1 mV s−1. The cathodic peaks shifted to higher potentials from 0.1 to 0.8 mV s−1 and then shifted to lower potentials at the scan rate of 1 mV s−1. The peak current density reveals a gradual decrease with the increase of the scanning rate for Li+/Li (Fig. 3c). By contrast, both the anodic and cathodic peaks shifted to higher potentials with the increase in the scanning rate from 0.1 to 1 mV s−1 for Na+/Na (Fig. 3d). At the anodic scan, the peak current density reveals a gradual increase with the increase of the scanning rate, yet a gradual decrease in the cathodic peak current intensity regardless of the scanning rate. The ideal voltammetry response of a PI electrode at various sweep rates can be estimated according to the equation:65where i is the current, v is the sweep rate, and a and b are the constant parameters, which is an important indicator of the charge storage kinetics. The b-value can be estimated from the slope of log(V) vs. log(i) plots. The b-value is close to 1, the process is predominantly controlled by a capacitive effect, i.e., a surface-controlled capacitive process at the surface of the electrode (capacitive-controlled process), and if b-value equals to 0.5, the process is mainly dominated by a solid-state diffusion-controlled process.66 As Fig. 3c and d show, the redox reaction of the PI electrode has a b value of 0.14 for Li+/Li (Fig. S4a†) and 2.2 for Na+/Na (Fig. S4b†), which demonstrates predominantly the characteristic of a diffusion-controlled charge process in LIB cells and a surface-controlled capacitive process in SIB cells, respectively. By using these b-values, the diffusion coefficient (Do) of PI (i.e., the insertion of Li+/Li and Na+/Na ions) can be calculated as expressed by using the modified Randles–Sevick equation.67 The PI showing the highest Do ≈ 2.80 × 10−5 cm2 s−1 for Na+/Na compared to Li+/Li (Do ≈ 1.20 × 10−7 cm2 s−1), assuming one-electron transfer at each step. These results demonstrated that the PI has faster reaction kinetics and rate-independent behaviour during the electrochemical redox process for Na+/Na compared to Li+/Li. Further, the contribution from diffusion-controlled processes and surface-controlled capacitive behaviour is quantified to further elucidate the energy storage mechanism using the following equation with the two parts, i.e., the surface capacitive effects (k1v) and diffusion-controlled insertion (k2v1/2).68where k1v and k2v1/2 are the capacitive contribution and diffusion-controlled contribution, respectively. The CV results at different scan rates as shown in Fig. 3c and d, the values of k1 (the slope of each line) and k2 (the intercept of each line) can be calculated at a certain potential. The capacitive contributed ratio can be estimated at the scan rate of 0.1 to 1.0 mV s−1 by using eqn (2) and from the shadow region in the CV profiles of the charge contribution ratio for the capacitive- and diffusion-controlled behaviours vs. Li+/Li (Fig. S5†) and Na+/Na (Fig. S6†). As Fig. 3e and f show, the capacitive contribution becomes higher as the scanning rates increase and moreover, reaches the maximum values of 87.3% for Li+/Li and 85.3% for Na+/Na at a high scan rate of 1.0 mV s−1, respectively. This high capacitive contribution are attributed to the short ion-diffusion length and efficient ion/electron transfer at the high current densities, which are further justified by the better high-rate capability of PI in both battery systems (Fig. 5a and b). With the increase in scanning rates, the fraction of the diffusion-controlled process contribution decreased due to the insufficient time for the Li+/Na+ ion insertion into the PI electrode during the high-rate electrochemical charge–discharge cycles. Furthermore, the step 4 of Scheme 1b shows that a further six-electron transfer redox process was possible in a lower potential range of 1.2–0.01 V. However, a low-charge capacity could be obtained during the following charge process (i.e., the charge capacity realized approximately half of the deep-discharge capacity), suggesting that the electrochemical redox kinetics in the PI was irreversible in both battery systems in the potential range of 0.01–3.0 V (at a very low voltage of <1.2 V). Our recent study confirmed this phenomenon by both deep-discharge/charge curves and CV results obtained for a perylene-based aromatic polyimide electrode in the potential range of 0.01–3.0 V.60 Moreover, when deep-discharging to below 1.2 V, the structural degradation of the electrogenerated PI species could also be realized in both battery systems owing to the formation of repulsive forces between the injected charges on the conjugated PI structural units.57,62–64,69
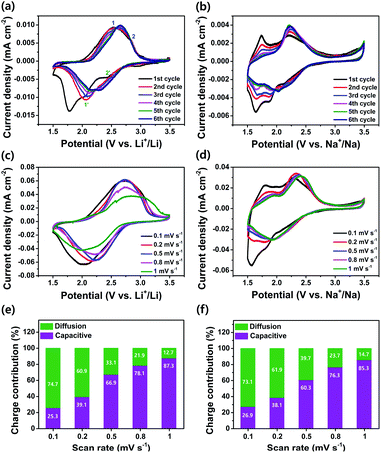 |
| | Fig. 3 CV curves for (a) PI vs. Li+/Li; (b) PI vs. Na+/Na, within a potential range of 1.5–3.5 V. CV curves of (c) PI vs. Li+/Li and (d) PI vs. Na+/Na at different scan rates ranging from 0.1 to 1.0 mV s−1. The total capacitive- and diffusion charge-storage contributions of (e) the PI electrode for Li+/Li, and (f) the PI electrode for Na+/Na under the different scan rates ranging 0.1 to 1.0 mV s−1. | |
Evaluation of the Li/PI and Na/PI battery cells
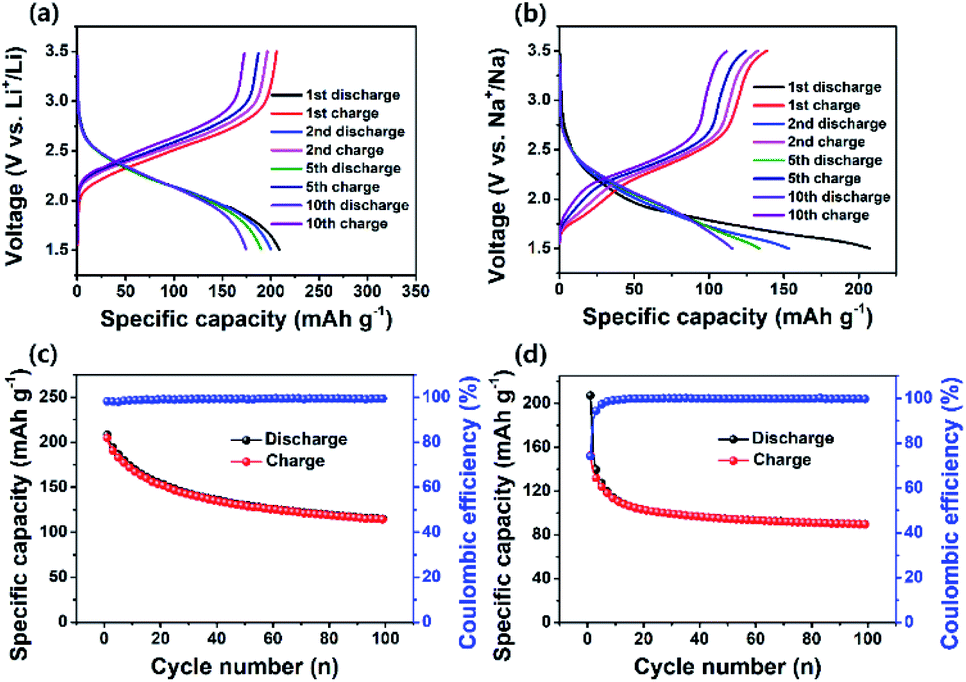
Fig. 4a and b show the charge–discharge profiles of PI in the LIB and SIB at a current density of 200 mA g−1. The PI in the LIB exhibited noticeable sloping charge and discharge voltage plateaus during the charge and discharge process of the first cycle (Fig. 4a), whereas the PI in the SIB exhibited almost four discharge voltage plateaus at the first cycle (i.e., one major and three minor discharge voltage plateaus) with two charge voltage plateaus (Fig. 4b), as strongly evident from their corresponding differential capacity–voltage (dQ/dV) profiles (Fig. S7†). These results are in good accordance with their corresponding CV profiles (Fig. 3a and b). The PI in the LIB exhibited a high initial discharge capacity of ∼209 mA h g−1 at a high current rate of 200 mA g−1 and decreased to ∼115 mA h g−1 over 100 cycles, and it exhibited a good capacity retention of ∼45% with a high coulombic efficiency of ∼100% (Fig. 4c). The PI in the SIB delivered an initial discharge capacity of ∼207 mA h g−1 and decreased to ∼90 mA h g−1; ∼43% of the initial discharge capacity was retained over 100 cycles (Fig. 4d). This is owing to the minimal numbers of Li+ and Na+ ions extracted from the backbone structure of PI. It is not possible to extract fully Li+/Na+ ions from the structure of PI during the charging and discharging process, because it may generate an unstable PI structure, which can affect the overall stability of battery cells. In addition, we observed that the PI exhibited twice the high discharge capacities compared with that of a similar PI reported by Jung et al. (∼128.43 mA h g−1 at a current density of 50 mA g−1 in 20 cycles).46 Both PI-based cells delivered initial discharge capacities over ∼65% efficiencies of their theoretical values (∼135 mA h g−1), assuming a three-electron transfer redox process occurred, as depicted in step 3 of Scheme 1b. It is noticeable that the initial discharge capacities of PI in both battery systems is larger than their theoretical capacity, which is attributed to the relative low first cycle coulombic efficiency (CE) of 98% for Li+/Li and 74.2% for Na+/Na, as illustrated in Fig. 4c and d. The observed low first cycle CE for Na+/Na suggested that a large portion of Na+ ions are immobilized on the surface or inside the bulk phases of the PI electrode due to the large size of Na+ ions as compared to Li+ ions. Such low initial CE values have already been reported on the graphitic carbon materials for the large size Na-ion storage.70,71 In addition, the significant amount of conductive carbon (Super P, 30%) can also be attributed to the little capacity from Super P in the initial few cycles (about 10–15 mA h g−1 on the Al foil as cathode or ∼293 mA h g−1 on the copper foil as anode). Therefore, this initial capacity surpasses the theoretical capacity (∼135 mA h g−1) of PI calculated based on the three-lithium (sodium)-insertion model, as indicated in Scheme 1b. However, the revealed rapid capacity decays after the first few turns as revealed in Fig. 4c and d suggested that the insertion of Li+/Na+ ions into the PI is not fully reversible and also potentially slow metal-ion diffusion resulting from irreversible side reactions between Li+/Na+ ions and CO groups or the absorbed molecules present on the surface of the amorphous Super P due to differences in Li+/Na+ ion kinetics, which resulted in a significant amount of irreversible capacity.63 This phenomenon is further well-supported with the CV curves (Fig. 3a and b) and cycling performance of PI in both the battery cells (Fig. 4c and d). Consequently, the revealed irreversible side reactions of Li+/Na+ ions on the surface of the amorphous carbon is owing to differences in Li+/Na+ ion kinetics and raised interfacial resistance due to the surface side reactions with CO groups during the charge–discharge cycling results in a significant shift in the higher voltage region (1st cycle → 10th cycle),47 as strongly further evidenced from the differential capacity–voltage profile (Fig. S7†). In addition, the rapid capacity fading observed in Fig. 4c and d assigned to the relative dissolution of the electrogenerated Li+/Na+ enolate species of PI (i.e., discharged PI2˙−·2M+ or PI3−·3M+ species) in the electrolytes during the repeated high-rate electrochemical charging/discharging process. Moreover, the PI attained a CE of approximately 100% across 100 cycles in both battery cells, suggesting the efficient suppression of PI electrode loss during the discharge–charge cycling. The rate performance of the PI in both the LIB and SIB cells was also measured at different current densities for 20 consecutive discharge–charge cycles (Fig. 5a and b). Initially, the number of test cycles was increased to 50 cycles to attain the stable capacity at the rate of 50 mA g−1. The PI in the LIB was observed to exhibit discharge capacities of ∼148 mA h g−1 at 50 mA g−1, ∼60 mA h g−1 at 100 mA g−1, ∼51 mA h g−1 at 200 mA g−1, and ∼42 mA h g−1 at 500 mA g−1. The PI in the SIB exhibited ∼154 mA h g−1 at 50 mA g−1 ∼77 mA h g−1 at 100 mA g−1, ∼65 mA h g−1 at 200 mA g−1, and ∼39 mA h g−1 at 500 mA g−1. When the current density reverted to 50 mA g−1 in both cells, the PI exhibited the average reversible capacities of ∼58 mA h g−1 (for Li+/Li) and 77 mA h g−1 (for Na+/Na). The PI was observed to exhibit a better rate capability in both battery cells and attained approximately 57% (for Li+/Li) and 42% (for Na+/Na) of 50 mA g−1 at 500 mA g−1, respectively. In addition, we observed that there is no distinct difference in the recovered capacity in Li-based and Na-based systems except for the current density of 50 mA g−1, demonstrating excellent rate performance with a better electrochemical stability. Some differences are observed in the recovered/reversible capacity of PI at 50 mA g−1 for both battery cells owing to the fact that many factors including the side reactions, which chemically degraded the electrodes after many cycles at high rates and the changes in the geometry of the PI during the charge–discharge process. This phenomenon caused the PI to isolate itself from the Super P network or an overall lack of crystallinity of the PI in the electrodes for Li+/Li and Na+/Na systems. This factor could lead to inferior ion/electron transfer and electrochemical stability, which is reflected in the reversible capacity when the current density goes back to 50 mA g−1. Fig. 5c and d show the voltage profiles of the PI at different current rates, indicating that a large polarization effect occurred at a high charging rate of 500 mA g−1, and this result indicated a good consistency with the report by Jung et al.46 To further investigate the long-life cycling stability of the PI in both battery cells, we performed the galvanostatic discharge–charge tests at 0.5 and 1C (where 1C = 135 mA h g−1, corresponding to a three-electron transfer redox process) for approximately 1000 cycles. As Fig. 5e shows, the specific capacity of the PI in the LIB decreased steeply to 106 mA h g−1 during the initial 10 cycles at current densities of 0.5 and 1C, respectively. After 1000 cycles, only 17% of capacity was retained at a 0.5C rate with a discharge capacity of ∼18 mA h g−1, which was comparable to the report by Jung et al.46 At a 1C rate, the PI in the LIB delivered an initial discharge capacity of ∼149 mA h g−1, which decreased to 106 mA h g−1 over 10 cycles and exhibited a dramatic decrease in the discharge capacity to ∼15 mA h g−1 with a capacity retention of ∼14% after 1000 cycles, although the cycling stability of the PI deteriorated owing to its increasing large polarization effect during Li-ion insertion/de-insertion kinetics, as
strongly revealed from Fig. 3c and e. By contrast, the PI in the SIB delivered an initial discharge capacity of ∼196 mA h g−1, which rapidly decreased to ∼156 mA h g−1 during the initial 10 cycles at a 0.5C rate (Fig. 5f). Thereafter, the capacity was maintained at ∼77% based on the capacity of 120 mA h g−1 after 1000 cycles. The PI delivered an initial discharge capacity of 197 mA h g−1 at a 1C rate, which abruptly decreased to 144 mA h g−1 over 10 cycles (Fig. 5f). The PI electrode in the SIB retained a capacity of 78 mA h g−1 with a capacity retention of approximately 54% after 1000 cycles. At a 1C rate, the capacity of Li/PI and Na/PI battery cells reached 149 mA h g−1 and 197 mA h g−1, respectively, which are obviously larger than the theoretical capacity of PI. This is attributed to the low first cycle CE, as illustrated in Fig. 5e and f. The observed low first cycle CE indicating that a large portion of Li+/Na+ ions are immobilized on the surface or inside the bulk phases of the PI electrode due to their ionic size. In addition, the CE values of both cells approached ∼99%. These results indicated that the long-life cycling stability of the PI in the LIB deteriorated owing to the significantly large polarization effect for Li+ storage compared with that of the SIB. This factor can be associated with the inferior electrochemical activity (all the redox units) in the PI after many cycles, in agreement with the CV results (Fig. 3). The unique aromatic conjugated structure and the intrinsic high electrical conductivity of the PI significantly facilitated the strong π–π stacking interaction (π-stacked structure) and efficient electron transfer in the PI electrode for Na+/Na, which resulted in a better long-life cycling stability in the SIB.
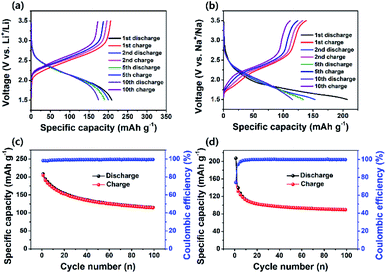 |
| | Fig. 4 Discharge–charge curves of (a) PI for Li+/Li and (b) PI for Na+/Na. Cycling performance of (c) PI for Li+/Li and (d) PI for Na+/Na at a current density of 200 mA h g−1. | |
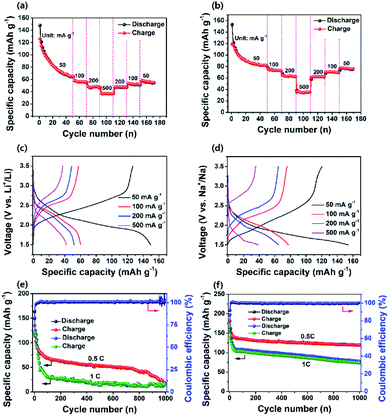 |
| | Fig. 5 Rate performance of (a) PI for Li+/Li and (b) PI for Na+/Na. Discharge–charge voltage profiles of (c) PI for Li+/Li and (d) PI for Na+/Na at different current rates. Long-life cycling stability of (e) PI for Li+/Li and (f) PI for Na+/Na at the different rates (0.5 and 1C) over 1000 cycles. | |
For an in-depth understanding of the formation of the solid electrolyte interface (SEI) composition/film on the cycled electrodes, the PI electrodes were disassembled from the Li/PI and Na/PI battery cells after over 1000 cycles and ex situ morphologies of both electrodes were analyzed using FE-SEM analysis (Fig. S8†). The pristine PI electrodes exhibited abundant PI nanoparticles with π-stacked aggregated structures, which resulted in well-distributed surface morphologies (Fig. S8a and b†). Remarkably, the post-mortem analysis of the PI electrodes revealed that the morphology of the PI accumulated with a significant amount of SEI films on the electrode surface during the cycling in both the LIB (Fig. S8c and d†) and SIB cells (Fig. S8e and f†), although the aggregate size of the PI particles did not seem to change. These results indicated that the PI electrodes maintained their structural integrity and were almost completely insoluble in electrolytes during the reversible redox process. Studies have already demonstrated that the high performance in perylene-based aromatic PI electrodes can be explained by the crystallinity of the active material, intrinsic high electrical conductivity of the active material, and electrode morphology.28 This excellent battery performance of the as-prepared PI was realized by its retained crystallinity after 1000 cycles and its favorable aggregated morphology of electrodes, as revealed by ex situ SEM images (Fig. S8c–f†). These are crucial factors for short ion-diffusion pathways and extremely short pathways to the conductivity carbon material leading to an increase in the short range-order of interchain electrons (charge transfer) hopping between the π-stacked PI aggregates (i.e., the interconnection between PI aggregates, which enabled the efficient hopping of local interchain electrons).72,73 The high degree of redox activity was facilitated by the higher rate of carbonyl groups in the PI and the high structural integrity of the PI electrodes after over 1000 cycles (for Li+/Li and Na+/Na), as strongly evident from SEM images (Fig. S8c–f†). These enabled the PI to have a higher discharge capacity during the charge–discharge cycling than in the report by Jung et al. owing to the multiple three-carbonyl-group accessibility.46
Ex situ FT-IR studies for cycled PI electrodes
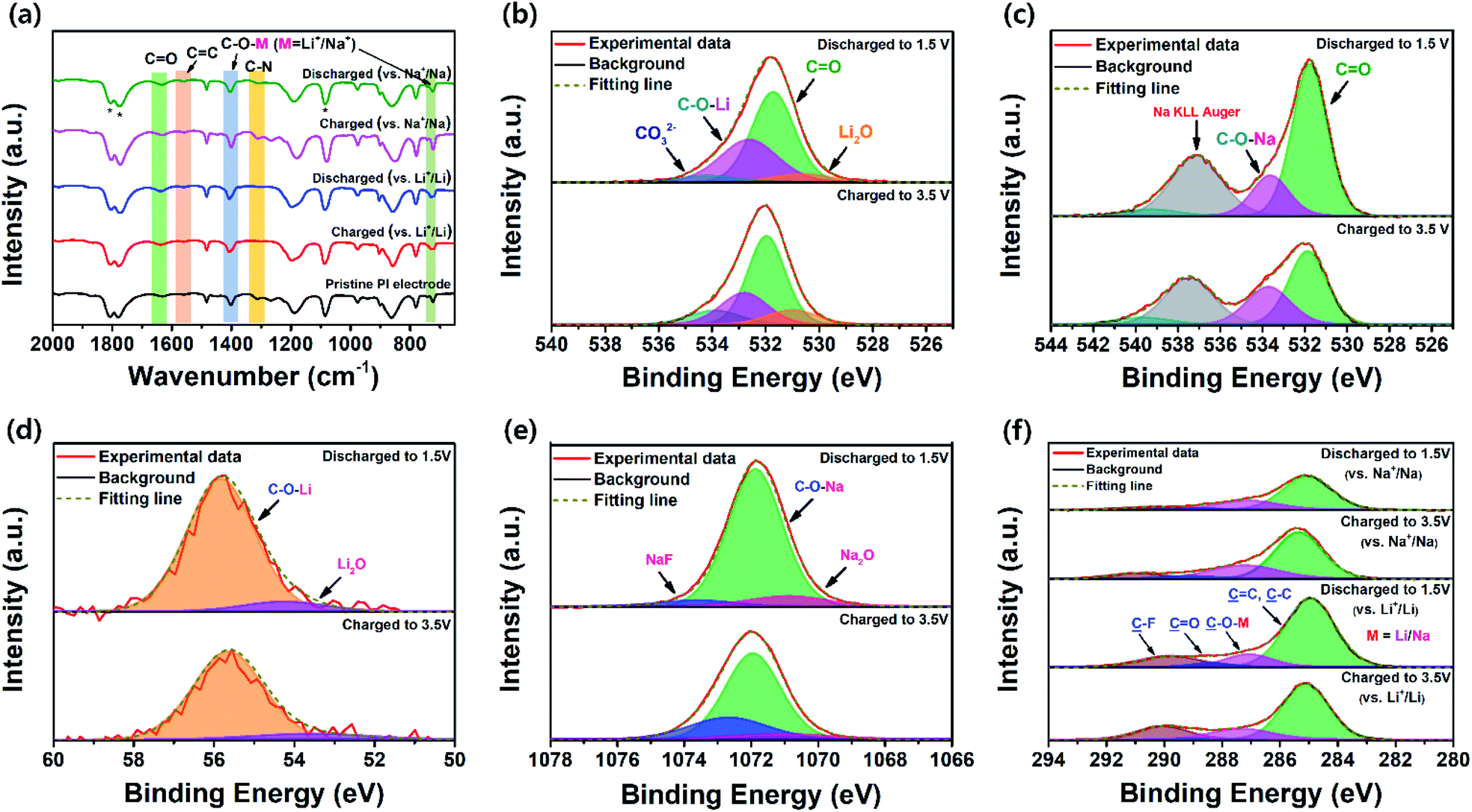
The chemical changes in the PI electrode during the electrochemical redox process were investigated using ex situ FT-IR spectra (Fig. 6a). The spectra for the cycled PI electrodes were captured in two different states to identify the metal-ion (Li+/Na+ ions) storage sites during the discharge–charge process of the first cycle. As Fig. 6a depicts, the vibrational bands at 1560 and 1315 cm−1 from the CC stretching vibration mode of the perylene core and the C–N stretching vibration of the imide, respectively, were clearly unchanged during the electrochemical charge–discharge process. This result indicated that the CC bonds of the perylene core and C–N bonds of the imide functionality did not participate in the redox process. However, the two main vibrational bands were at approximately 1698 and 1662 cm−1, corresponding to the asymmetric and symmetric stretching frequencies of carbonyl (CO) groups, respectively. These vibration bands were red-shifted and exhibited two broad bands at 1635 cm−1 and 1405 cm−1 in the electrode, respectively; these were attributed to the vibration of the electrochemically active CO bonds during charge and discharge processes.43 When the PI electrode was fully discharged to 1.5 V in both battery cells, the CO bands gradually weakened with the insertion of Li+/Na+ ions, which corresponded with the symmetric and asymmetric stretching vibrations of lithium/sodium enolate groups, such as C–O–Li/C–O–Na bonds (C–O–M bonds, where M = Li+/Na+), appearing at 1400 cm−1 (for Li+/Li and Na+/Na). The sharp vibrational band located at 723 cm−1 was attributed to the symmetric in-plane bending of imide CO groups of the PI, which were seemingly weakened in the PI electrodes during a galvanostatic discharge voltage to 1.5 V and recharged to 3.5 V in the PI, indicating the formation of a new bond between the symmetric in-plane bending of imide CO groups and insertion of metal ions to form metal-ion enolate groups (i.e., C–O–Li and C–O–Na bonds). By considering the highly reversible electrochemical redox behaviour of the PTCDA units, even after being recharged to 3.5 V in both states, the PI exhibited the remaining broad bands located at 1635, 1405, and 723 cm−1; this was attributed to the formation of the irreversible SEI layer on the surface of the PI electrodes in both cells.60 Therefore, these results demonstrated that only the CO bonds participated in the reversible redox processes. These results corresponded with the report by Tian et al.74 and the redox mechanism of the reversible insertion and de-insertion of Li+/Na+ ions through enolate formation, as depicted by the chemical equations in Scheme 1b.
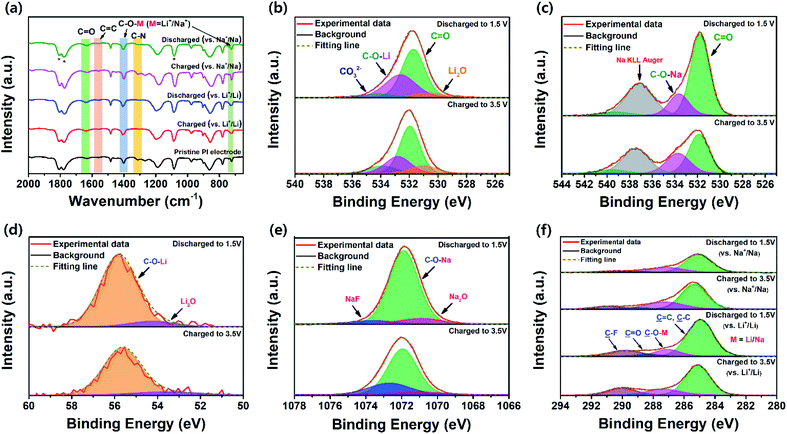 |
| | Fig. 6 (a) Ex situ FT-IR spectra of the pristine PI and cycled PI electrodes in different states for Li+/Li and Na+/Na (discharged to 1.5 V and recharged to 3.5 V; the peaks marked with a star represent the vibration of electrolytes). Ex situ XPS local-scan spectra of the PI electrodes for (b) O 1s regions in different states for Li+/Li, (c) O 1s regions in different states for Na+/Na, (d) Li 1s regions in different states, and (e) Na 1s regions in different states. (f) C 1s regions for the different states of PI electrodes (for Li+/Li and Na+/Na). | |
Ex situ XPS studies for the cycled PI electrodes
The ex situ XPS measurements verified the evidence for the participation or consumption of only carbonyl groups during the charge–discharge process (Fig. 6b–e). The local-scan XPS spectra of the O 1s signal of the PI electrodes under different charge and discharge states (for Li+/Li and Na+/Na) could be deconvoluted into two main peaks (Fig. 6b and c), which strongly indicated that the Li+/Na+ ions were inserted into the PI electrodes at two different redox-active carbonyl sites. In the LIB, a Li2O peak at 530.5 eV was detected and may have originated from the SEI film (Fig. 6b).75 The ratio of the C–O (C–O–Li) peak at 532.6 eV increased when the ratio of the CO peak at 531.6 eV decreased in the discharged voltage of 1.5 V. However, these tendencies were seemingly different in the local-scan spectra of O 1s regions for Na+/Na (Fig. 6c). This was because the sodium Auger overlapped with the local-scan spectra of the O 1s signals since the sodium Auger peak (Na KLL) was occasionally observed in the range of 535–537 eV.76 Meanwhile, the carbonyl binding energies were visibly red-shifted (i.e., shifted to lower binding energy) by approximately 0.16 eV (for Li+/Li) and 0.32 eV (for Na+/Na) because the carbonyl groups gained electrons to form C–O–Li and C–O–Na bonds, respectively, during the discharging process.56 These results strongly indicated that the carbonyl groups primarily participated in the reversible insertion and de-insertion of Li+/Na+ ions. Fig. 6d and e depicts the XPS spectra of Li 1s and Na 1s signals for the PI electrodes after discharging to 1.5 V and followed by recharging to 3.5 V in the first cycle, respectively. As Fig. 6d shows, the Li2O peak at 54.14 eV could have originated from the SEI film. The peak at 55.87 eV was attributed to the Li+ ions in C–O–Li groups originating from the reduction of the PI. The ratio of these peaks increased upon being discharged to 1.5 V compared with that when charged to 3.5 V. Fig. 6e indicates that the peaks appeared at 1071 and 1071.87 eV, corresponding to NaF and C–O–Na peaks, respectively.77 The peak at 1071.87 eV could be attributed to the Na+ ions in C–O–Na groups originating from the reduction of the PI during the discharge to 1.5 V for Na+/Na.78 The peak located at 1072.66 eV was attributed to the Na2O peak, which was formed in the SEI film after charge–discharge cycling. Note that the intensities of both the C–O–Li and C–O–Na peaks increased at the discharged voltage of 1.5 V, compared with that of the charged voltage of 3.5 V. These results strongly indicated that metal-ion storage sites were only the carbonyl groups. In the NaPF6-based electrolytes, NaF formation could be possible in the PI electrode after charge–discharge cycling. The formation of such an F-containing SEI film on the surface of the PI electrode after cycling is known to induce superior mechanical strength, which would be beneficial to the excellent long cycle life of PI electrodes, particularly in SIB cells. This phenomenon indicated good consistency with an excellent long-term cycling stability of PI at higher charging rates for Na+/Na (Fig. 5f).
In addition, the local-scan spectra of C 1s and N 1s signals were analyzed to identify any chemical changes in the PI molecular structures during the electrochemical redox process vs. Li+/Li and Na+/Na (Fig. 6f and S9†). The peak appeared at 284.98 eV in the local-scan spectra of the C 1s signal during the discharged voltage of 1.5 V, which corresponds with CC and C–C peaks (Fig. 6f). The intensity of the CC and C–C peaks increased; this was further supported by the results of ex situ FT-IR analysis (Fig. 6a). The peak located at 399.50 eV in the local-scan spectra of N 1s when discharging to 1.5 V, which was assigned to the C–N peak (Fig. S9†). Note that both CC/C–C peaks (Fig. 6f) and C–N peaks (Fig. S9†) shifted to lower binding energies when the discharged voltage decreased to 1.5 V with respect to those of recharged to 3.5 V, indicating a good π–π stacking aggregated morphology in the PI electrodes and excellent electron transfer between the PTCDA and 2,6-DAAQ units in the PI.53 These results strongly confirmed that the carbonyl groups (CO bonds) were the main binding sites for metal-ion storage rather than the aromatic benzene rings of the PI (i.e., the CC of the perylene core and C–N of the imide functionality did not participate in the electrochemical redox process). Thus, the reversible binding and release of metal ions from the CO bonds occurred in the PI during the charge and discharge states.
Ex situ XRD evaluation for cycled PI electrodes
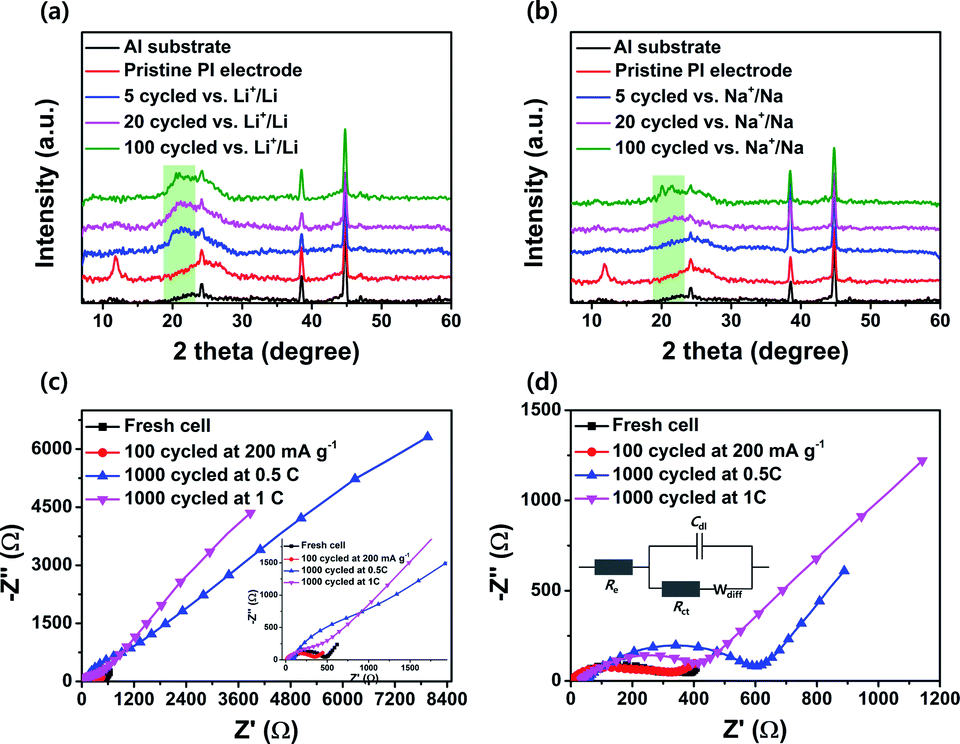
To gain more insight into the internal structural stability of the cycled PI electrodes, we disassembled both batteries after the charge–discharge cycles in the different states, and their cathodes were recovered after rinsing with dimethyl carbonate electrolyte. We characterized the crystal structure of the PI using ex situ XRD patterns for the pristine PI electrodes and different states of cycled electrodes. The main diffraction pattern peaks at the 2θ = 11.79°, 13.21° and very broad peaks in the range of 2θ = 21–28° corresponded to the pristine PI electrode owing to the crystallinity of the parent PTCDA unit, indicating that the PI retained its crystallinity (Fig. 7a and b).46,47,49 However, the PI electrodes in both battery cells completely lost their crystal properties after continuous charge–discharge cycles over 100 times. This was because the continuous metal-ion insertion and de-insertion processes in the aromatic cores of the PI changed its crystallinity into an amorphous structure. These results corresponded with the results of Jung et al.46 Moreover, the broad peak intensity located at 2θ = 19–23.5° were clearly enhanced as cycles increased from 5 to 100 in both battery cells; this can be attributed to the metal-ion kinetics of the (de)-insertion. These results confirmed that the broadening of the diffraction peaks in the range of 2θ = 19–23.5° in cycled electrodes can be correlated with the microcrystallization of the PI particles or smaller aggregated PI nanoparticles during the metal-ion (de)-insertion process.49,79 This characteristic factor leads to the shorter diffusion pathways for Li+/Na+ ions, facilitating a shorter travel distance for both electrons and ions to reach the redox entirety of the PI. The closer packing and aggregates size of PI increased the electron hopping rate between the redox units of the PI, which in turn increased the stable cycling performance of the PI electrodes, particularly in SIBs. This result was further well-supported by the SEM images of the cycled electrodes, as described above (Fig. S8e and f†). Moreover, the PI-based SIB cells exhibited excellent long-term stable cycling performance compared with that of the PI-based LIBs. This was attributed to many factors including the side reactions that chemically degraded the electrodes after many cycles at the high rates and the changes in the geometry of the PI during the charge–discharge process that caused the PI to isolate itself from the Super P network or an overall lack of crystallinity of the PI in the electrodes for Li+/Li.79,80 This phenomenon is further well-supported with the reversible capacity of PI in the rate performance when the current density goes back to 50 mA g−1 from high charging rates, especially for Li+/Li (Fig. 5a).
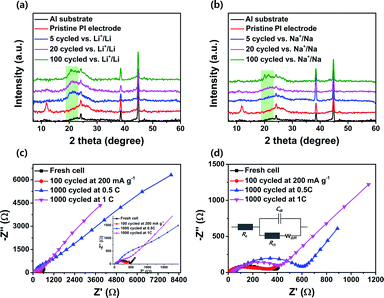 |
| | Fig. 7
Ex situ XRD patterns for (a) the pristine PI electrode and different cycled PI electrodes vs. Li+/Li; (b) pristine PI electrode and different cycled PI electrodes vs. Na+/Na. Nyquist plots for (c) PI-based fresh and different cycled electrodes vs. Li+/Li and (d) PI-based fresh and different cycled electrodes vs. Na+/Na. The insets of (c) show the expanded spectrum. The insets of (d) depict the corresponding equivalent circuits for battery cells. | |
Impedance evaluation of the Li/PI and Na/PI battery cells
To investigate the electrolyte resistance (Re) and charge transfer resistances (Rct) of the PI in both battery cells, we performed EIS measurements on fresh and tested cells after cycling under the different conditions (Fig. 7c and d; the equivalent circuits for both battery cells are shown in the inset of Fig. 7d). The intercept on the Z′-axis represents a small Re and the diameter of the semicircle in the Nyquist plot indicates the Rct. These interfacial resistance values (Re and Rct) for the PI in both battery cells are collated in Table S1.† We observed that the Rct value of the PI electrode in LIBs decreased from 392 to 337 Ω after 100 cycles at a current density of 200 mA g−1. In SIBs, the PI electrode exhibits a decrease in the Rct value from 272 to 243 Ω under the same cycled condition. These results implied that a better electrolyte/electrode interface was built gradually, which resulted in higher conductivity and fast charge transfer kinetics. In contrast, the PI electrodes exhibited significantly higher Re and Rct values after 1000 cycles at 0.5C in both battery cells. However, these values decreased noticeably in both battery cells after being cycled at a high current rate of 1C over 1000 times. In SIBs, the PI electrodes exhibited a small decrease in the charge transfer resistance compared with that of LIBs. These results indicated that the electrolyte might have not infiltrated the PI electrode completely after over 1000 cycles owing to the PI aggregated morphology in the electrode; therefore, high charge transfer resistances were obtained. This observation was further confirmed from both the ex situ XRD results (Fig. 7a and b) and ex situ SEM images of cycled PI electrodes (Fig. S8†). These results demonstrated that the PI exhibited high-rate performance and better long-life cycling stability, which are directly correlated with the unique perylene-based aromatic PI demonstrated by its multiple fast redox kinetics owing to the high rate of carbonyl groups (three or multi-carbonyl groups) acting as the multiple redox-active sites for the metal-ion uptake; its high crystallinity resulted in a short range-order of interchain hopping of electrons between the PI aggregates, and the small aggregate in the PI electrode that enabled short metal-ion diffusion pathways.
4. Conclusions
We synthesized a perylene-based aromatic multi-carbonyl PI as the cathode material for both LIBs and SIBs; the PI incorporated two distinct types of redox-active units through polymerization of the PTCDA unit with the 2,6-DAAQ moiety. The PI exhibited a three-electron transfer redox process, resulting in a practical initial discharge capacity of 209 mA h g−1 (for Li+/Li) and 207 mA h g−1 (for Na+/Na) at a high current rate of 200 mA g−1 with high discharge voltage plateaus (1.9–2.4 V for Li+/Li and Na+/Na), representing ∼65% of its theoretical capacity (135 mA h g−1). The PI exhibited a better rate performance with average reversible capacities of 58 mA h g−1 (for Li+/Li) and 77 mA h g−1 (for Na+/Na) over 20 cycles at 50 mA g−1 and attained approximately 57% (for Li+/Li) and 42% (for Na+/Na) of 50 mA g−1 at 500 mA g−1 over 20 cycles. The PI exhibit better long-life cycling stability with a discharge ability at a current density of 1C (∼15 mA h g−1 for Li+/Li; and ∼78 mA h g−1 for Na+/Na over 1000 cycles) compared with the rate of 0.5C (∼18 mA h g−1 for Li+/Li; and ∼120 mA h g−1 for Na+/Na). Furthermore, the PI exhibited a high-rate performance with a better long-term cycling stability and significantly decreased the intrinsic stability problems in both LIBs and SIBs owing to its high crystallinity and unique π-conjugated aromatic structures consisting of multiple carbonyl groups. We believe that our results can provide new insight for constructing multiple redox units in the design of perylene-based aromatic multi-carbonyl conjugated PIs/polymers for next-generation sustainable organic batteries.
Conflicts of interest
The authors declare no conflicting financial interest.
Acknowledgements
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (No. NRF-2019R1I1A3A01046928) and the “Human Resources Program in Energy Technology” of the Korea Institute of Energy Technology Evaluation and Planning (KETEP), and granted financial resources from the Ministry of Trade, Industry & Energy, Republic of Korea (No. 20204010600100).
References
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS.
- N. Nitta and G. Yushin, Part. Part. Syst. Charact., 2014, 31, 317–336 CrossRef CAS.
- J. Lu, Z. Chen, F. Pan, Y. Cui and K. Amine, Electrochem. Energy Rev., 2018, 1, 35–53 CrossRef CAS.
- Q. Wang, J. Xu, W. Zhang, M. Mao, Z. Wei, L. Wang, C. Cui, Y. Zhu and J. Ma, J. Mater. Chem. A, 2018, 6, 8815–8838 RSC.
- M. V. Reddy, G. V. S. Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113(7), 5364–5457 CAS.
- T. B. Schon, B. T. McAllister, P.-F. Li and D. S. Seferos, Chem. Soc. Rev., 2016, 45, 6345–6404 RSC.
- H. Banda, D. Damien, K. Nagarajan, A. Raj, M. Hariharan and M. M. Shaijumon, Adv. Energy Mater., 2017, 7, 1701316 CrossRef.
- Y. Fang, L. Xiao, Z. Chen, X. Ai, Y. Cao and H. Yang, Electrochem. Energy Rev., 2018, 1, 294–323 CrossRef CAS.
- J. Tian, D. Cao, X. Zhou, J. Hu, M. Huang and C. Li, ACS Nano, 2018, 12, 3424–3435 CrossRef CAS.
- H. J. Kim, Y. Kim, J. Shim, K. H. Jung, M. S. Jung, H. Kim, J. C. Lee and K. T. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 3479–3486 CrossRef CAS.
- Y. Lu, Q. Zhang, L. Li, Z. Niu and J. Chen, Chem, 2018, 4, 2786–2813 CAS.
- Z. Guo, Y. Ma, X. Dong, J. Huang, Y. Wang and Y. Xia, Angew. Chem., Int. Ed., 2018, 57, 11737–11741 CrossRef CAS.
- B. Häupler, A. Wild and U. S. Schubert, Adv. Energy Mater., 2015, 5, 1402034 CrossRef.
- J. Xie, P. Gu and Q. Zhang, ACS Energy Lett., 2017, 2, 1985–1996 CrossRef CAS.
- C. Wang, W. Tang, Z. Yao, B. Cao and C. Fan, Chem. Commun., 2019, 55, 1801–1804 RSC.
- Y. Wang, Z. Liu, C. Wang, Y. Hu, H. Lin, W. Kong, J. Ma and Z. Jin, Energy Storage Materials, 2020, 26, 494–502 CrossRef.
- C. Wang, Z. Yang, Y. Wang, P. Zhao, W. Yan, G. Zhu, L. Ma, B. Yu, L. Wang, G. Li, J. Liu and Z. Jin, ACS Energy Lett., 2018, 3, 2404–2409 CrossRef CAS.
- F. A. Obrezkov, A. F. Shestakov, V. F. Traven, K. J. Stevenson and P. A. Troshin, J. Mater. Chem. A, 2019, 7, 11430–11437 RSC.
- K. Sarang, A. Miranda, H. An, E.-S. Oh, R. Verduzco and J. L. Lutkenhaus, ACS Appl. Polym. Mater., 2019, 1, 1155–1164 CrossRef CAS.
- S. Wang, F. Li, A. D. Easley and J. L. Lutkenhaus, Nat. Mater., 2019, 18, 69–75 CrossRef CAS.
- D.-Y. Wang, W. Guo and Y. Fu, Acc. Chem. Res., 2019, 52, 2290–2300 CrossRef CAS.
- Q. Zhang, H. Wei, L. Wang, J. Wang, L. Fan, H. Ding, J. Lei, X. Yu and B. Lu, ACS Appl. Mater. Interfaces, 2019, 11, 44352–44359 CrossRef CAS.
- E. Vitaku, C. N. Gannett, K. L. Carpenter, L. Shen, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2020, 142, 16–20 CrossRef CAS.
- R. Shi, L. Liu, Y. Lu, C. Wang, Y. Li, L. Li, Z. Yan and J. Chen, Nat. Commun., 2020, 11, 178 CrossRef CAS.
- M. Wu, Y. Zhao, B. Sun, Z. Sun, C. Li, Y. Han, L. Xu, Z. Ge, Y. Ren, M. Zhang, Q. Zhang, Y. Lu, W. Wang, Y. Ma and Y. Chen, Nano Energy, 2020, 70, 104498 CrossRef CAS.
- D. J. Kim, K. R. Hermann, A. Prokofjevs, M. T. Otley, C. Pezzato, M. Owczarek and J. F. Stoddart, J. Am. Chem. Soc., 2017, 139, 6635–6643 CrossRef CAS.
- K. S. Weeraratne, A. A. Alzharani and H. M. El-Kaderi, ACS Appl. Mater. Interfaces, 2019, 11, 23520–23526 CrossRef CAS.
- T. B. Schon, S. Y. An, A. J. Tilley and D. S. Seferos, ACS Appl. Mater. Interfaces, 2019, 11, 1739–1747 CrossRef CAS.
- L. Zhu, G. Ding, L. Xie, X. Cao, J. Liu, X. Lei and J. Ma, Chem. Mater., 2019, 31, 8582–8612 CrossRef CAS.
- Q. Li, D. Li, H. Wang, H.-g. Wang, Y. Li, Z. Si and Q. Duan, ACS Appl. Mater. Interfaces, 2019, 11, 28801–28808 CrossRef CAS.
- Y. Hu, H. Ding, Y. Bai, Z. Liu, S. Chen, Y. Wu, X. Yu, L. Fan and B. Lu, ACS Appl. Mater. Interfaces, 2019, 11, 42078–42085 CrossRef CAS.
- W. Yan, C. Wang, J. Tian, G. Zhu, L. Ma, Y. Wang, R. Chen, Y. Hu, L. Wang, T. Chen, J. Ma and Z. Jin, Nat. Commun., 2019, 10, 2513 CrossRef.
- M. E. Bhosale, S. Chae, J. M. Kim and J.-Y. Choi, J. Mater. Chem. A, 2018, 6, 19885–19911 RSC.
- C. Yuan, Q. Wu, Q. Li, Q. Duan, Y. Li and H.-g. Wang, ACS Sustainable Chem. Eng., 2018, 6, 8392–8399 CrossRef CAS.
- H. G. Wang, S. Yuan, D. I. Ma, X. L. Huang, F. I. Meng and X. B. Zhang, Adv. Energy Mater., 2014, 4, 1301651–1301657 CrossRef.
- K. Amin, L. Mao and Z. Wei, Macromol. Rapid Commun., 2019, 40, 1800565 CrossRef.
- A. Mauger, C. Julien, A. Paolella, M. Armand and K. Zaghib, Materials, 2019, 12, 1770 CrossRef CAS.
- H. Oubaha, J.-F. Gohy and S. Melinte, ChemPlusChem, 2019, 84, 1179–1214 CrossRef CAS.
- Y. Wang, Z. Liu, H. Liu, H. Liu, B. Li and S. Guan, Small, 2018, 1704094 CrossRef.
- Q. Li, D. Li, H. Wang, H.-g. Wang, Y. Li, Z. Si and Q. Duan, ACS Appl. Mater. Interfaces, 2019, 11, 28801–28808 CrossRef CAS.
- B. Tian, J. Zheng, C. Zhao, C. Liu, C. Su, W. Tang, X. Lie and G.-H. Ning, J. Mater. Chem. A, 2019, 7, 9997–10003 RSC.
- X. Cao, J. Liu, L. Zhu and L. Xie, Energy Technol., 2019, 7, 1800759 CrossRef.
- H. Banda, D. Damien, K. Nagarajan, M. Hariharan and M. M. Shaijumon, J. Mater. Chem. A, 2015, 3, 10453–10458 RSC.
- G. Hernández, N. Casado, A. M. Zamarayeva, J. K. Duey, M. Armand, A. C. Arias and D. Mecerreyes, ACS Appl. Energy Mater., 2018, 1, 7199–7205 CrossRef.
- J. Manuel, X. Zhao, K.-K. Cho, J.-K. Kim and J.-H. Ahn, ACS Sustainable Chem. Eng., 2018, 6, 8159–8166 CrossRef CAS.
- M.-H. Jung and R. V. Ghorpade, J. Electrochem. Soc., 2018, 165, A2476–A2482 CrossRef CAS.
- M. R. Raj, R. V. Mangalaraja, D. Contreras, K. Varaprasad, M. V. Reddy and S. Adams, ACS Appl. Energy Mater., 2020, 3, 240–252 CrossRef.
- H.-g. Wang, S. Yuan, Z. Si and X.-b. Zhang, Energy Environ. Sci., 2015, 8, 3160–3165 RSC.
- Y. Liu, X. Zhao, C. Fang, Z. Ye, Y.-B. He, D. Lei, J. Yang, Y. Zhang, Y. Li, Q. Liu, Y. Huang, R. Zeng, L. Kang, J. Liu and Y.-H. Huang, Chem, 2018, 4, 2463–2478 CAS.
- F. Xu, J. Xia and W. Shi, Electrochem. Commun., 2015, 60, 117–120 CrossRef CAS.
- H. P. Wu, Q. Yang, Q. H. Meng, A. Ahmad, M. Zhang, L. Y. Zhu, Y. G. Liu and Z. X. Wei, J. Mater. Chem. A, 2016, 4, 2115–2121 RSC.
- F. Xu, H. Wang, J. Lin, X. Luo, S.-a. Cao and H. Yang, J. Mater. Chem. A, 2016, 4, 11491–11497 RSC.
- Z. Luo, L. Liu, J. Ning, K. Lei, Y. Lu, F. Li and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 9443–9446 CrossRef CAS.
- J. He, Y. Liao, Q. Hu, Z. Zeng, L. Yi, Y. Wang, H. Lu and M. Pan, J. Power Sources, 2020, 451, 227792 CrossRef CAS.
- Z. Ba, Z. Wang, M. Luo, H.-b. Li, Y. Li, T. Huang, J. Dong, Q. Zhang and X. Zhao, ACS Appl. Mater. Interfaces, 2020, 12, 807–817 CrossRef CAS.
- Y. Zu, Y. Xu, L. Ma, Q. Kang, H. Yao and J. Hou, ACS Appl. Mater. Interfaces, 2020, 12, 18457–18464 CrossRef CAS.
- Q. Zhao, R. R. Gaddam, D. Yang, E. Strounina, A. K. Whittaker and X. S. Zhao, Electrochim. Acta, 2018, 265, 702–708 CrossRef CAS.
- H. Peng, Q. Yu, S. Wang, J. Kim, A. E. Rowan, A. K. Nanjundan, Y. Yamauchi and J. Yu, Adv. Sci., 2019, 1900431 CrossRef.
- P. Sharma, D. Damien, K. Nagarajan, M. M. Shaijumon and M. Hariharan, J. Phys. Chem. Lett., 2013, 4, 3192–3197 CrossRef CAS.
- M. R. Raj, R. V. Mangalaraja, G. Lee, D. Contreras, K. Zaghib and M. V. Reddy, ACS Appl. Energy Mater., 2020, 3, 6511–6524 CrossRef.
- M. R. Raj, R. Margabandu, R. V. Mangalaraja and S. Anandan, Soft Matter, 2017, 13, 9179–9191 RSC.
- X. Han, C. Chang, L. Yuan, T. Sun and J. Sun, Adv. Mater., 2007, 19, 1616–1621 CrossRef CAS.
- H. Wu, K. Wang, Y. Meng, K. Lu and Z. Wei, J. Mater. Chem. A, 2013, 1, 6366–6372 RSC.
- Z. Song, H. Zhan and Y. Zhou, Angew. Chem., Int. Ed., 2010, 49, 8444–8448 CrossRef CAS.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CrossRef CAS.
- T. Brezesinski, J. Wang, S. H. Tolbert and B. Dunn, Nat. Mater., 2010, 9, 146–151 CrossRef CAS.
- N. Kim, M. R. Raj and G. Lee, Nanotechnology, 2020, 31, 415401 CrossRef CAS.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597–1614 RSC.
- K. Amin, L. Mao and Z. Wei, Macromol. Rapid Commun., 2019, 40, 1800565 CrossRef.
- Y. Wen, K. He, Y. Zhu, F. Han, Y. Xu, I. Matsuda, Y. Ishii, J. Cumings and C. Wang, Nat. Commun., 2014, 5, 4033 CrossRef CAS.
- X. Zhong, Y. Li, L. Zhang, J. Tang, X. Li, C. Liu, M. Shao, Z. Lu, H. Pan and B. Xu, ACS Appl. Mater. Interfaces, 2019, 11, 2970–2977 CrossRef CAS.
- M. J. Tauber, R. F. Kelley, J. M. Giaimo, B. Rybtchinski and M. R. Wasielewski, J. Am. Chem. Soc., 2006, 128, 1782–1783 CrossRef CAS.
- K. Gu and Y.-L. Loo, J. Polym. Sci., Part B: Polym. Phys., 2019, 57, 1559–1571 CrossRef CAS.
- B. Tian, G.-H. Ning, W. Tang, C. Peng, D. Yu, Z. Chen, Y. Xiao, C. Su and K. P. Loh, Mater. Horiz., 2016, 3, 429–433 RSC.
- Z. Man, P. Li, D. Zhou, R. Zang, S. Wang, P. Li, S. Liu, X. Li, Y. Wu, X. Liang and G. Wang, J. Mater. Chem. A, 2019, 7, 2368–2375 RSC.
-
J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-Ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data, Perkin-Elmer Corporation, Physical Electronics Division, 1992 Search PubMed.
- K. Granath, M. Bodegård and L. Stolt, Sol. Energy Mater. Sol. Cells, 2000, 60, 279–293 CrossRef CAS.
- S. Wu, W. Wang, M. Li, L. Cao, F. Lyu, M. Yang, Z. Wang, Y. Shi, B. Nan, S. Yu, Z. Sun, Y. Liu and Z. Lu, Nat. Commun., 2016, 7, 13318 CrossRef CAS.
- S. Y. An, T. B. Schon and D. S. Seferos, ACS Omega, 2020, 5, 1134–1141 CrossRef CAS.
- T. B. Schon, A. J. Tilley, E. L. Kynaston and D. S. Seferos, ACS Appl. Mater. Interfaces, 2017, 9, 15631–15637 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, characterization of materials, synthetic procedure for the synthesis of the perylene-based aromatic polyimide (PI), high-resolution XPS spectra of the PI, ex situ XPS spectra of the PI, ex situ SEM images of the PI, the shadow region of the CV profile for the charge-storage contribution and impedance performance of PI for both the battery cells. See DOI: 10.1039/d0se01246g |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Nangyeong
Kim
and
Gibaek
Lee
,
Nangyeong
Kim
and
Gibaek
Lee