
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActive site engineering of single-atom carbonaceous electrocatalysts for the oxygen reduction reaction
Guangbo
Chen
 a,
Haixia
Zhong
a and
Xinliang
Feng
*ab
a,
Haixia
Zhong
a and
Xinliang
Feng
*ab
aCenter for Advancing Electronics Dresden (cfaed) and Faculty of Chemistry and Food Chemistry, Technische Universität Dresden, Mommsenstr. 4, 01062 Dresden, Germany. E-mail: xinliang.feng@tu-dresden.de
bDepartment of Synthetic Materials and Functional Devices, Max Planck Institute of Microstructure Physics, Weinberg 2, Halle (Saale), D-06120, Germany
First published on 10th November 2021
Abstract
The electrocatalytic oxygen reduction reaction (ORR) is the vital process at the cathode of next-generation electrochemical storage and conversion technologies, such as metal–air batteries and fuel cells. Single-metal-atom and nitrogen co-doped carbonaceous electrocatalysts (M–N–C) have emerged as attractive alternatives to noble-metal platinum for catalyzing the kinetically sluggish ORR due to their high electrical conductivity, large surface area, and structural tunability at the atomic level, however, their application is limited by the low intrinsic activity of the metal–nitrogen coordination sites (M–Nx) and inferior site density. In this Perspective, we summarize the recent progress and milestones relating to the active site engineering of single atom carbonous electrocatalysts for enhancing the ORR activity. Particular emphasis is placed on the emerging strategies for regulating the electronic structure of the single metal site and populating the site density. In addition, challenges and perspectives are provided regarding the future development of single atom carbonous electrocatalysts for the ORR and their utilization in practical use.
 Guangbo Chen | Guangbo Chen received his PhD degree in Chemistry from Technische Universität Dresden (TU Dresden) in 2021 under the supervision of Prof. Xinliang Feng. Thereafter, he continued his academic research as a postdoctoral fellow in the Chair of Molecular Functional Materials at TU Dresden. His research topic mainly focuses on the design and synthesis of carbonaceous and two-dimensional based nanostructures for energy storage and conversion applications. |
 Xinliang Feng | Xinliang Feng is a full professor and has been the head of the Chair for Molecular Functional Materials at TU Dresden since 2014. Starting from 2021, he has been the director of Max Planck Institute of Microstructure Physics, Germany. His current scientific interests include organic synthetic methodology, organic synthesis, and supramolecular chemistry of π-conjugated systems, bottom-up synthesis of graphene and graphene nanoribbons, electrochemical exfoliation of 2D crystals, 2D polymers, and supramolecular polymers as well as 2D carbon-rich conjugated polymers for optoelectronic applications, energy storage and conversion, and new energy devices and technologies. |
1. Introduction
With the rapid depletion of fossil fuels and growing environmental concerns, electrochemical energy storage and conversion technologies have emerged as promising pathways for developing clean and sustainable energy devices.1,2 The electrochemical oxygen reduction reaction (ORR) plays a significant role in determining the efficiency of next-generation energy storage and conversion systems, e.g., metal–air batteries and proton exchange membrane fuel cells (PEMFCs).3–5 At the air cathode of metal–air batteries and PEMFCs, the O2 molecules are reduced into OH− (in bases) and water (in acids) via a four proton-coupled electron transfer (PCET) pathway, respectively.6–10 However, the four electrons, multi-O-containing intermediates (i.e., OH*, O* and OOH*), and three phases (i.e., solid catalyst, liquid electrolyte and gaseous O2) involved in the process make the ORR kinetically sluggish with a large overpotential loss, calling for highly efficient ORR electrocatalysts.11–13 Until now, platinum-group-metal (PGM)-based materials have been the benchmark electrocatalysts for catalyzing the ORR.14–17 Unfortunately, the high cost, scarcity, and poor stability of PGMs seriously hamper their widespread utilization in practical energy systems.18–20Compared to PGM-based catalysts, single-metal-atom and nitrogen co-doped carbonaceous (M–N–C) PGM-free materials have been regarded as emerging alternatives for catalyzing the ORR, attributed to their high electrical conductivity, large surface areas, and structural tunability at the atomic level.21–31 In 1964, macrocyclic compounds, such as metal phthalocyanines, were initially found active toward the ORR.32 Afterward, it was discovered that high-temperature pyrolysis could effectively enhance the ORR activity and durability of these macrocycle-derived catalysts.33,34 These pioneering studies opened the window and inspired the widespread investigation of single atom carbonous materials as efficient ORR electrocatalysts. In the early 2000s, instead of expensive metal-macrocycles, common nitrogen-containing precursors, inorganic metal sources, and high-surface-area carbon supports were used to prepare M–N–C catalysts, which showed significantly promoted catalytic performance.35 Since then, numerous synthetic strategies have been devoted to the preparation of M–N–C electrocatalysts, through combining various N- and metal-containing precursors and careful optimization of the combinations and thermal treatments, resulting in highly variable coordination chemistry and densities of active sites and consequently drastically different ORR activities.36–39 Time-of-flight secondary ion mass spectrometry (MS)-based investigations indicated that the FeN2C4+ ion was detected in all synthesized Fe–N–C electrocatalysts regardless of the Fe precursor or synthesis procedures used.40 Thus, the catalytic site for the ORR in these Fe–N–C electrocatalysts was first proposed to be similar to the molecular structure of Fe–Nx in an iron macrocyclic complex.41
With the prosperous development of M–N–C electrocatalysts, advanced characterization techniques have been adopted to identify the ORR active site.42–44 Atomic-resolution aberration-corrected scanning transmission electron microscopy (AC-STEM) is a powerful technique to directly observe the distribution of single atoms and even obtain the atomic structure of active moieties.45 In 2016, Zelenay et al. combined AC-STEM and electron energy-loss spectroscopy (EELS) to investigate the structure and composition of the active moieties in a (CM + PANI)–Fe–C electrocatalyst.46 The coordination structure of individual Fe atoms could be clearly observed on the graphene sheet. The presence of N around the Fe was then confirmed by EELS, giving an average composition of Fe–N4 by quantifying the ratio of Fe-to-N.
X-ray absorption spectroscopy (XAS) is another powerful tool to analyze the coordination environment around the metal center of M–N–C electrocatalysts.47 Thereinto, X-ray absorption near edge structure (XANES) analysis reveals the metal valence state and uncovers the local atomic arrangement.48 Further, extended X-ray absorption fine structure (EXAFS) analysis offers precise structural information including the coordination number, and the bond distance between the central atom and neighboring atoms, which is widely used to identify the single atom characteristic of M–N–C electrocatalysts.49 In 2015, Jaouen et al. utilized the XAS technique to investigate the active sites of Fe–N–C electrocatalysts on the basis of the successful synthesis of atomic Fe doped carbons.50 The Fourier transforms of the EXAFS spectra indicated the absence of iron based crystalline structures. To further identify the active sites, the simulated XANES spectra were obtained and compared with the experimental results. By screening numerous possible structures, they found that FeN4C12 was the most possible active moiety for the ORR. Similarly, single atomic cobalt (Co),25,51–54 manganese (Mn),55–57 zinc (Zn),58 and copper (Cu)59,60 were detected by XAS and visualized by AC-STEM in Co–N–C, Mn–N–C, Zn–N–C and Cu–N–C electrocatalysts, respectively, indicating that the active sites were M–Nx (Fig. 1a).
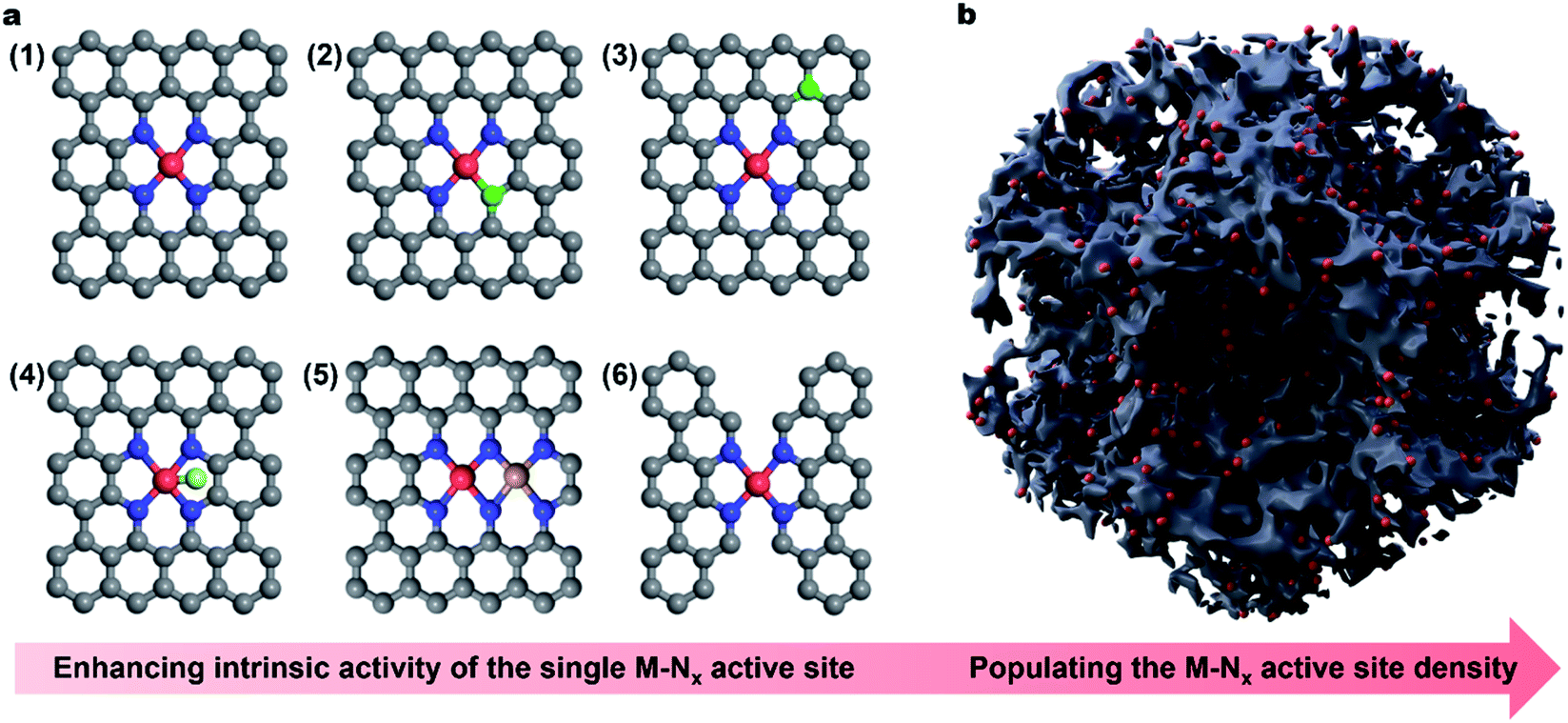 | ||
| Fig. 1 Schematic illustration of active engineering of single-atom carbonaceous M–N–C electrocatalysts for promoting the ORR activity. (a) Enhancing the intrinsic activity of the (1) single M–Nx site via (2) regulation of the coordination geometry, (3) heteroatom incorporation, (4) axial ligand modification, (5) construction of dual-metal sites and (6) regulation of the carbon skeleton geometry. The brown, blue and red spheres represent C, N, and central M atoms, respectively. The green sphere stands for the heteroatom. The light green sphere represents the ligand on the M–Nx site. The dark yellow sphere is the second metal site near the M–Nx. (b) Populating the site density by engineering porous carbon structures. | ||
The fruitful identification and insightful understanding of the M–Nx active sites enabled the rational design and preparation of various efficient M–N–C electrocatalysts for the ORR via different synthetic approaches.61,62 Currently, the representative synthetic strategies towards M–N–C electrocatalysts can be summarized as: (1) carbonization of metal-containing complexes or mixtures of specific precursors,63–66 (2) carbonization of metal–organic frameworks (MOFs),67–72 and (3) the template-assisted pyrolysis approach,73,74 which can be seen in the recently published excellent review articles, examining both the design of M–N–C materials and their ORR activities.22,38,42,75–79 Until now, M–N–C electrocatalysts have achieved outstanding ORR activity in alkaline solutions, exceeding that of the benchmark Pt/C catalyst.80 The distinguished ORR activity of such M–N–C materials empowered the as-assembled metal–air batteries with superior performance.81 Unfortunately, in an acidic electrolyte, the electrocatalytic activities of such M–N–C materials are still much lower than those of PGM-based components,82 motivating the further rational engineering of the active site at the atomic level.
In this Perspective, we do not intend to be exhaustive but aim to highlight the latest achievements on the active site engineering of single atom carbonous electrocatalysts for the ORR. Especially, we summarize the representative approaches to enhancing the intrinsic activity of the (1) M–Nx active site by modulating the electronic structure of the single metal sites, including (2) regulation of coordination geometry, (3) heteroatom incorporation (e.g., sulphur (S), phosphorus (P) or boron (B) near the M atom), (4) axial ligand modification, (5) construction of dual-metal sites and (6) regulation of the carbon skeleton geometry (Fig. 1a). Besides, we present the recent progress in improving the mass activity via populating the site density by porosity engineering (Fig. 1b). Finally, challenges and perspectives are provided regarding the future development of single atom carbonaceous M–N–C electrocatalysts for the ORR and their applications in practical energy technology. We hope that this Perspective article will shed light on the development of highly efficient carbonaceous nanomaterials via rational active site engineering as alternative electrocatalysts to PGM-based systems, eventually realizing widespread applications in metal–air batteries and PEMFCs.
2. Engineering of the M–Nx active site for enhancing the intrinsic activity
The central atom of various M–N–C electrocatalysts with different electronic structures will undoubtedly determine the ORR catalytic activity.83–85 In the past decade, much progress has been made to reveal the intrinsic ORR activity of M–N–C electrocatalysts, following the order of Fe–N–C > Co–N–C > Cu–N–C > Mn–N–C > Ni–N–C.86–91 Accordingly, the sections below will mainly focus on Fe–N–C systems, examining the strategies for enhancing their intrinsic activity. According to the Sabatier principle, the adsorption of ORR intermediates (i.e., O2*, OOH*, O* and OH*) on the active site should be neither too strong nor too weak.92 Unfortunately, like benchmark Pt, the adsorption of ORR intermediates on the Fe–Nx sites is still too strong, which seriously limits their desorption and then the ORR kinetics,93 while on the Co–Nx site, such adsorption energy is too weak, restricting the formation of corresponding intermediates.94 Engineering the local geometry of the single metal site (e.g., Fe or Co) can effectively modulate its electronic structure, hence optimizing the adsorption/desorption strength of O2 and ORR intermediates and thus promoting the intrinsic activity.2.1 Regulation of the coordination geometry of the metal center
In most of the M–N–C electrocatalysts, the central metal atom is coordinated by four N atoms, forming the M–N4 coordination site.49 For the Fe–N4 coordination geometry, the N atoms can be typically classified into pyridinic- or pyrrolic-N.95 Cao et al. proved that Fe@pyrrolic-N4 presented higher ORR activity than Fe@pyrindinic-N4 by combining both experiments and density functional theory (DFT) calculations.96 The same conclusion was also reported by Wu et al.97 They constructed high-purity pyrrole-type Fe–N4 (HP-Fe–N4) by a simple ammonia-assisted strategy and compared its performance with pyridine-type Fe–N4 in acidic media (Fig. 2a). The as-prepared HP-Fe–N4 showed a higher onset potential (Eonset, 0.95 V vs. the reversible hydrogen electrode, RHE) and a more positive E1/2 (0.80 V) than that of pyridine-type Fe–N4 (0.86 V and 0.71 V, respectively). DFT calculations demonstrate that the pyrrole-type Fe–N4 shows stronger electron depletion around the Fe atom than pyridine-type Fe–N4, enabling preferred O2 adsorption energy and complete four-electron reaction selectivity for the ORR. Recently, Jaouen and co-workers systematically investigated the durability and degradation of the Fe–N4 site by combining operando57Fe Mössbauer spectroscopy and XAS.98 They firstly identified two types of Fe–N4 sites in the Fe–N–C electrocatalyst, including a high-spin FeN4C12 moiety (denoted as S1, pyrrole-type Fe–N4) and a low- or intermediate-spin FeN4C10 moiety (denoted as S2, pyridine-type Fe–N4). Both FeN4C12 and FeN4C10 sites initially contributed to the ORR activity of Fe–N–C in an acidic medium and the FeN4C12 site was found to be more intrinsically active in the beginning. However, FeN4C12 was not durable in operating PEMFCs, quickly transforming to ferric oxides (Fig. 2b). In contrast, the number of less active FeN4C10 sites remained unchanged after 50 h operation at 0.5 V, maintaining the PEMFC performance after initial degradation. This work presented a profound comprehension of the local coordination effect on the ORR activity and a fundamental understanding of the degradation of Fe–N4 sites. Importantly, it provided a new direction for the rational design of durable Fe–N–C catalysts.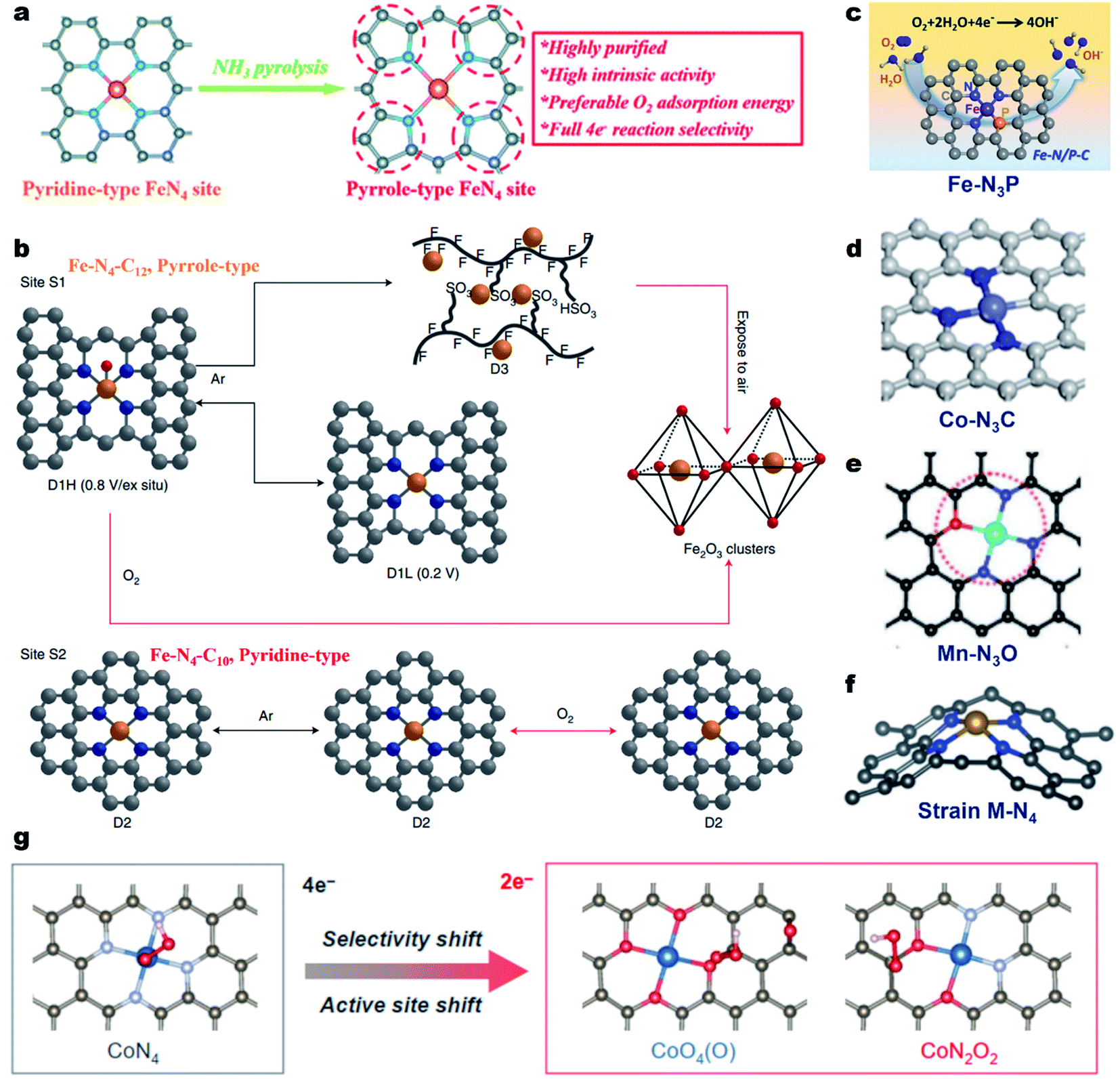 | ||
| Fig. 2 Regulation of the coordination geometry of the metal center for enhancing the ORR activity. (a) Preparation process of the high-purity pyrrole-type Fe–N4 structure. Reproduced with permission from ref. 97. Copyright (2020) Royal Society of Chemistry. (b) Coordination and structural changes of the sites S1 and S2 under in situ or operando conditions. Reproduced with permission from ref. 98. Copyright (2020) Springer Nature. (c) Schematic showing the N and P co-coordinated single Fe site (Fe–N3P). Reproduced with permission from ref. 104. Copyright (2020) American Chemical Society. (d) Schematic showing the N and C co-coordinated single Co site (Co–N3C). Reproduced with permission from ref. 106. Copyright (2020) American Chemical Society. (e) Schematic showing the N and O co-coordinated single Mn site (Mn–N3O). Reproduced with permission from ref. 109. Copyright (2018) Wiley-VCH. (f) Schematic showing the strained M–N4 site. Reproduced with permission from ref. 112. Copyright (2019) WILEY-VCH. (g) OOH* adsorption on CoN4 and CoO4(O), and CoN2O2 sites. Reproduced with permission from ref. 117. Copyright (2021) American Chemical Society. | ||
On the other hand, the N atoms in Fe–N4 could be replaced with other atoms for the formation of the Fe–N4−xAx geometry (A = C, S, P, B or O), resulting in a modulated electronic structure and tailored ORR properties.99–103 For example, Yuan et al. prepared N/P dual-coordinated Fe sites by pyrolysis of poly-pyrrole (PPy)/phytic acid/FeCl3. The aberration-corrected HAADF-STEM image, XANES and EXAFS investigations proved that the Fe was coordinated with three N atoms and one P atom, forming the Fe–N3P1. Electrochemical tests and DFT calculations demonstrated that the Fe–N3P1 sites were more favorable for oxygen intermediate adsorption/desorption, leading to accelerated reaction kinetics and promising ORR activity (Fig. 2c).104 The local coordination geometry could also affect the catalytic properties of Cu–N–C,105 Co–N–C,106–108 Mn–N–C,109 Mg–N–C,110 Ni–N–C,111 and Zn–N–C58 electrocatalysts. For instance, the ORR tests and DFT calculation results indicated that Co–N3C presented the best catalytic activity among Co–N3C1, Co–N2C2, and Co–N4 (Fig. 2d).106 The N/O dual coordinated Mn–NxOy active sites exhibited higher ORR activity than that of Mn–N4 and the Mn–N3O1 configuration displayed the best intrinsic ORR activity among all reported Mn–NxOy sites (Fig. 2e).109 Besides, the introduction of the local strain effect around M–Nx sites could also tailor the ORR performance of M–N–C materials (Fig. 2f).112–115 Tailoring the local coordination geometry can also tune the catalytic pathway and selectivity. For instance, Qiao et al. synthesized a Mo single atom catalyst with a unique O, S coordination (Mo1/OSG-H) that could catalyze the ORR via the 2e− pathway with a high H2O2 selectivity of >95% in 0.1 M KOH.116 Apart from first-shell coordination, the second coordination sphere also largely alters the electronic structures of active sites through structural effects and manipulates intermediate adsorption through the space effect, thus changing the catalytic ORR pathway and selectivity. Very recently, the same group constructed a CoNOC catalyst with N, O-dual coordination and C–O–C functional groups that demonstrated outstanding activity and a selectivity of >95% for acidic H2O2 electrosynthesis relative to the CoN4 site.117 DFT computations, poisoning experiments, and in situ attenuated total reflectance surface-enhanced infrared absorption spectroscopy suggested that an active site (especially for OOH* adsorption) shifted from the center Co atom to the O-adjacent C atom, enabling a superior 2e− ORR pathway (Fig. 2g).
These studies clearly demonstrated that the local coordination geometry will largely affect the catalytic ORR behavior. Precisely engineering the coordination configurations of the single atom site and molecular-level understanding of actual active sites are essential to correlate the structure–performance relationship and pursue advanced M–N–C electrocatalysts, but they remain a huge challenge.
2.2 Heteroatom incorporation
Aside from direct coordination with the central atom, a heteroatom (i.e., S, P, B or O) can also be incorporated into the carbon skeleton.83,118 Such an incorporated heteroatom with a different atomic size and electronegativity (compared to N and C atoms) can affect the electronic structure of the single M–Nx sites via long-range delocalization.119–122 In 2015, Sun et al. for the first time synthesized a S doped Fe–N–C catalyst and validated the promotional role of S dopant in the ORR.123 Later on, Li et al. demonstrated a volcano relationship between the ORR activity and the S incorporation content.124 In a 0.1 M KOH solution, the Fe-ISA/SNC catalyst with a N/S atomic ratio of 1/1 achieved the highest ORR activity (Fig. 3a and b) with an E1/2 of 0.896 V (vs. RHE). XAS investigation and DFT calculations revealed that the relatively low electronegativity of S could enrich the charge on a N atom, which facilitated the rate-limiting reductive release of OH* and accelerated the overall ORR process. The influence of S-containing functional groups (e.g., thiophene-S and oxidized-S species) on the ORR activity was further investigated by Lee et al.125 They found that the oxidized S functionality decreased the d-band center of iron by withdrawing electrons, thereby facilitating the ORR at the Fe–N4 site by lowering the intermediate adsorption energy. In contrast, the thiophene-S structure with electron-donating properties reduced the intrinsic ORR activity of the Fe–N4 site (Fig. 3c).126 Beside S, P,127 B,128 or co-doping S and P129 could also induce uneven charge density distribution, hence altering the intrinsic activity of Fe–Nx sites. In 2018, Wang and co-workers developed a novel MOF@polymer strategy for the construction of single Fe atomic sites supported on a N, P and S co-doped hollow carbon polyhedron (Fe–SAs/NPS-HC).129 The single Fe atom was coordinated with four N atoms for the formation of Fe–N4 and the S and P atoms were doped into the carbon skeleton (Fig. 3d). DFT calculations indicate that the surrounding S and P atoms can donate electrons to the single-atom Fe center, making the charge of Fe (Feδ+) less positive and weakening the binding of adsorbed OH species (Fig. 3e). As a result, the as-prepared Fe–SAs/NPS-HC demonstrated superior ORR activity in both alkaline and acidic solutions, achieving a high E1/2 of 0.912 V and 0.791 V (vs. RHE), respectively (Fig. 3f). Moreover, the above dopants could as well regulate the electronic structure and improve the intrinsic activity of single Co–Nx,130 Cu–Nx![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 59 and Mn–Nx131,132 sites for Co–N–C, Cu–N–C and Mn–N–C electrocatalysts, respectively.
59 and Mn–Nx131,132 sites for Co–N–C, Cu–N–C and Mn–N–C electrocatalysts, respectively.
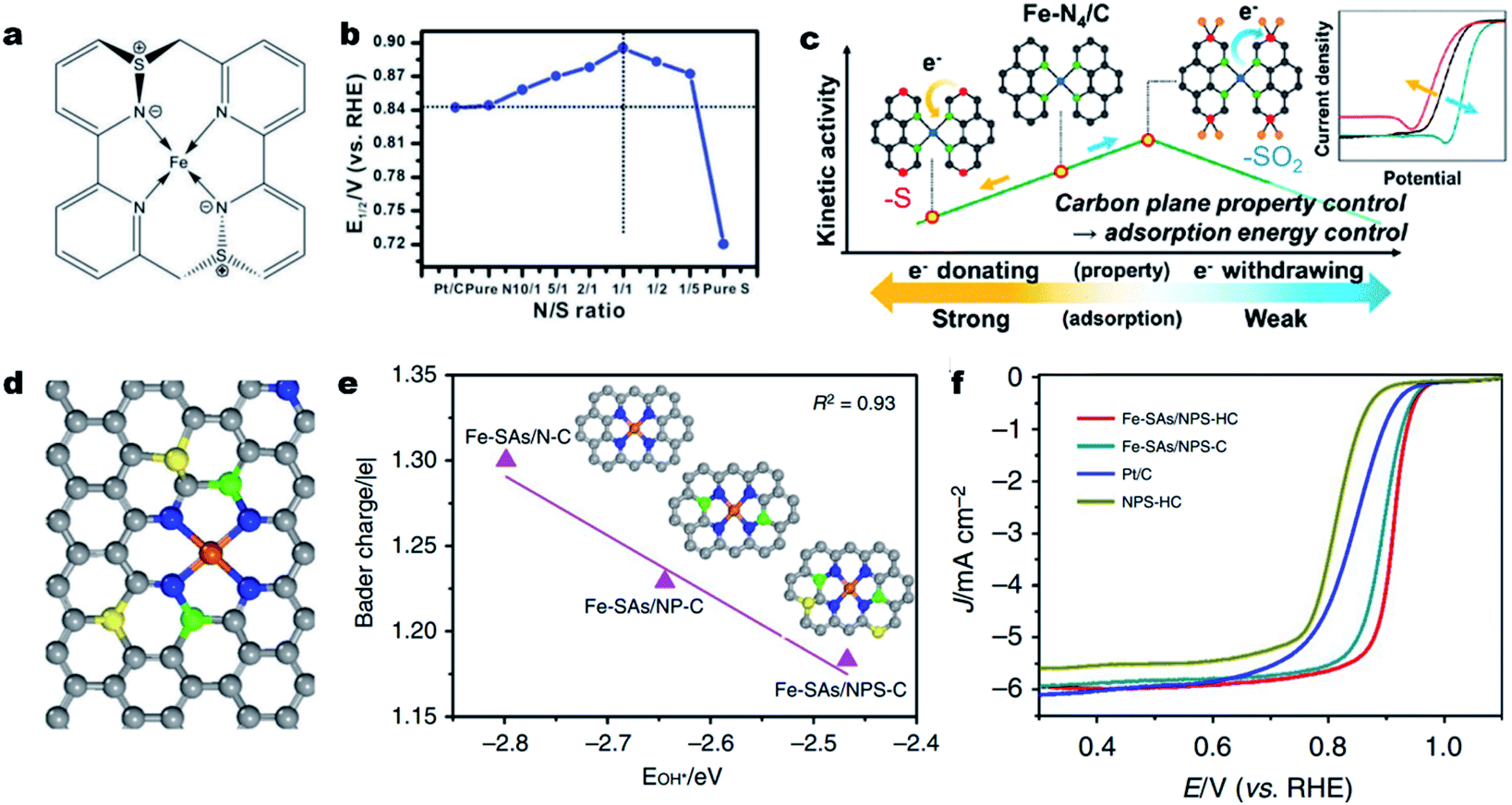 | ||
| Fig. 3 Heteroatom incorporation for promoting the ORR activity. (a) Structure of the FeN4S2 active site in Fe-ISA/SNC. (b) Line graph of the relationship between the N/S ratio and corresponding E1/2 of different Fe-ISA/SNC samples, showing that the Fe-ISA/SNC with a N/S ratio of 1/1 presents the highest E1/2. Reproduced with permission from ref. 124. Copyright (2018) WILEY-VCH. (c) Plot for the kinetic activity of a Fe–N–C catalyst as a function of the electron-donating/withdrawing properties of S dopants. Reproduced with permission from ref. 125. Copyright (2019) American Chemical Society. (d) Schematic model of Fe-SAs/NPS-HC, Fe (orange), N (blue), P (green), S (yellow) and C (gray). (e) Linear relationship between the OH* binding energy and Bader charge of single-atom iron in Fe-SAs/N–C, Fe-SAs/NP-C and Fe-SAs/NPS-C, respectively. Insets: the corresponding schematic models of samples, Fe (orange), N (blue), P (green), S (yellow) and C (gray). (f) ORR polarization curves for Fe-SAs/NPS-HC, Fe-SAs/NPS-C, NPS-HC and 20% Pt/C. Reproduced with permission from ref. 129. Copyright (2021) Springer Nature. | ||
2.3 Axial ligand modification
It is noted that the d orbitals of the single metal sites perpendicular to the M–N–C plane are not fully occupied and can potentially interact with small ligands (e.g., small molecules, or anions). Such ligands can coordinate with the single metal atoms and modulate the electronic structure of the M–Nx.133–136 Cho et al. first designed a penta-coordinated FePc–Py–CNT catalyst by immobilizing FePc molecules on pyridine-functionalized carbon nanotubes (CNTs) (Fig. 4a).137 Thanks to the synergy of the penta-coordinated structure and CNTs for efficient electron transfer, the synthesized FePc–Py–CNTs exhibited good ORR activity in alkaline solutions, better than that of Pt/C. Such a promotional role of axial ligands could also be found in Fe porphyrins. The creation of strong axial coordination can further enhance the ORR activity and stability of FePc. As shown in Fig. 4b, FePc could coordinate with oxygen-containing groups on an O2 plasma-treated acetylene black (AB-O) matrix, forming an FeAB-O catalyst. For comparison, FePc/AB was prepared by physically mixing FePc and AB. DFT calculations imply that the axial O coordination (O–FeN4) sites can greatly break the electronic distribution symmetry of Fe and lead to electron localization on O. This electron localization strengthened O2 adsorption and accelerated charge transfer from the Fe to the O2 molecule. As a result, the FeAB-O catalyst exhibited fast ORR kinetics with a remarkable E1/2 of 0.90 V (vs. RHE), which was much higher than those of FePc/AB (0.87 V) and Pt/C (0.85 V) (Fig. 4c).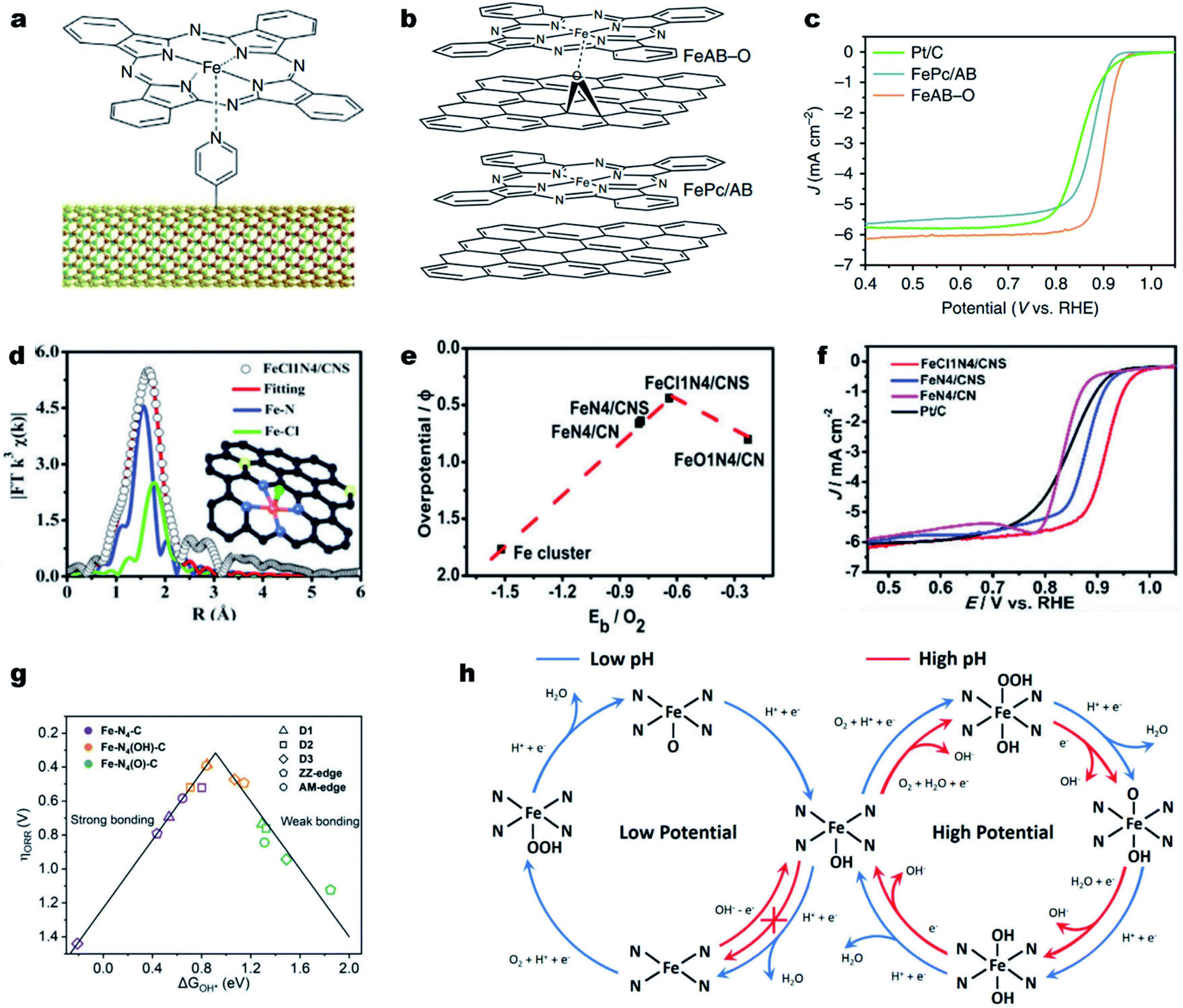 | ||
| Fig. 4 Engineering axial ligands on Fe–Nx for improving the ORR activity. (a) Schematic diagram of the structure of the FePc–Py–CNT composite. Reproduced with permission from ref. 137. Copyright (2013) Springer Nature. (b) Molecular structure models of FeAB-O and FePc/AB. (c) ORR polarization curves of FeAB-O, FePc/AB, and Pt/C in O2-saturated 0.1 M KOH. Reproduced with permission from ref. 143. Copyright (2020) Springer Nature. (d) EXAFS fitting curves of the FeCl1N4/CNS in the R space. Inset: Schematic model of FeCl1N4/CNS: Fe (red), Cl (green), S (yellow), N (blue), and C (gray). (e) The correlation of the ORR overpotentials with the O2 binding energies for the Fe catalysts. (f) ORR polarization curves of different catalysts in O2-saturated 0.1 M KOH. Reproduced with permission from ref. 138. Copyright (2018) Royal Society of Chemistry. (g) Volcano plot between ΔG*OH and the ORR overpotential for five Fe–N4–C structures with different axial ligands. (h) Schematic illusion of reaction pathways for the ORR on Fe–N4 moieties in acidic and alkaline solutions under ORR working potentials. Reproduced with permission from ref. 142. Copyright (2020) WILEY-VCH. | ||
Halogen ions, such as chloride ions (Cl−), displayed strong coordination ability with transition metal atoms. For example, Han et al. modified pristine Fe–N4 active sites with axial Cl coordination for the formation of Fe–Cl1N4 sites decorated on the N, S co-doped carbon (FeCl1N4/CNS) (Fig. 4d).138 Due to the synergistic effect of the Cl ligand and S dopant, FeCl1N4/CNS exhibited a much lower overpotential (0.44 V) than that of FeN4/CN (0.66 V) and possessed a moderate binding energy of O2 (Eb = 0.64 eV) (Fig. 4e), indicating a favorable ORR activity. In 0.1 M KOH, FeCl1N4/CNS displayed an unexpectedly excellent ORR activity with an E1/2 of 0.921 V (vs. RHE) (Fig. 4f).
Recent investigations also revealed a self-adjusting mechanism induced by its intrinsic intermediate (such as *OH) during the ORR process for enhancing the intrinsic activity of M–N–C materials.46,139,140 Using DFT simulations, Zhou and co-workers found that the single Fe site of Fe–N–C was preferentially covered with an intrinsic intermediate OH* from 0.28 to 1.00 V (vs. RHE). Such OH* became part of the Fe center for the formation of Fe(OH)N4 and optimized the intermediate binding, promoting the ORR kinetics.141 Li et al. also investigated the self-adjusting mechanism on different Fe–N4 configurations, including Fe–N4–C10 (D1), Fe–N4–C12 (D2), Fe–N4–C8 (D3), zigzag edge Fe–N4–C (ZZ-edge) and armchair edge Fe–N4–C (AM-edge) and obtained the same conclusions (Fig. 4g).142 The axial OH ligand could be formed under certain electrode potentials as an intermediate during the ORR or directly from the alkaline solution (Fig. 4h). Remarkably, the OH ligand decreased the bonding strength of ORR intermediates on a single Fe site and therefore accelerated the catalytic activity of Fe–N4 moieties. The profound understanding of the promotional effect of the axial ligand on the ORR activity on Fe–N–C electrocatalysts motivates the design of highly active and stable Fe–Nx sites, such as higher coordination configurations, Fe–N5 and Fe–N6.
2.4 Dual metal sites
The adjacent M–Nx sites can hardly influence each other's electronic structure and intrinsic activity if they are far apart in the carbon substrate.144 When the distance goes to several angstroms, the electronic structures of M–Nx and M′–Nx may affect each other.145,146 Yu et al. demonstrated strong electronic interactions between the adjacent Fe–N4 sites over the Fe–N–C material when the inter-site distance (dsite) was less than about 1.6 nm (Fig. 5a).147 When the dsite was about 0.7 nm, the Fe–N4 showed much enhanced ORR intrinsic activity (Fig. 5b). Fu et al. constructed a single-atom dispersed Zn/Cu–NC catalyst with Zn–N4 together with Cu–N4 and proposed that the adjacent Zn sites could modify the d-orbital electron configuration of Cu–N4 sites.148 The modified Cu–N4 catalytically active site could facilitate the O–O bond breakage and reduce the energy barrier for OOH* conversion, thus promoting the ORR activity. The synergistic effect could also be observed on (Fe,Co)-SA/CS, where the Fe modulated single atom Co–N4 acted as the strengthened active site.149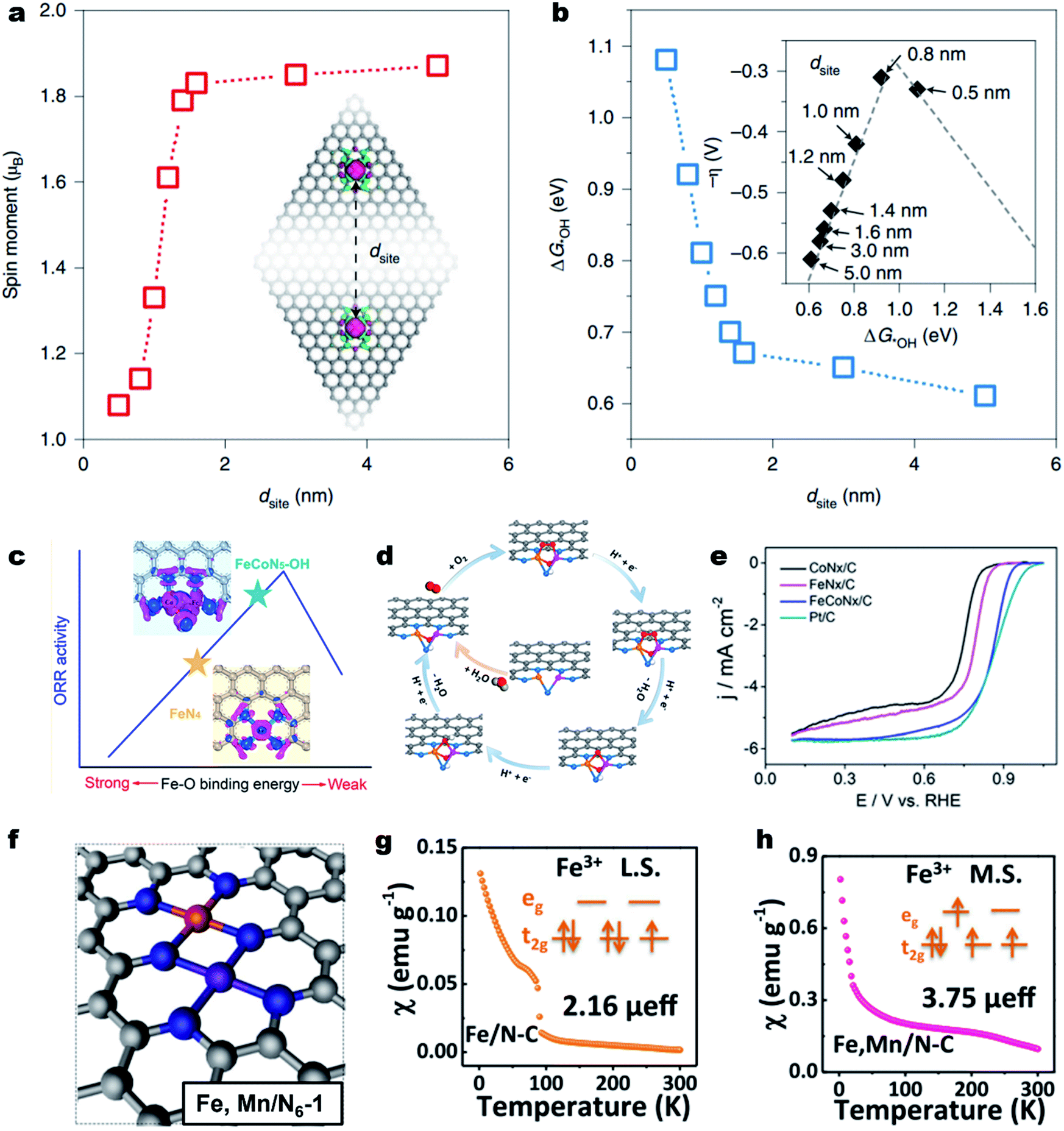 | ||
| Fig. 5 Survey of the dual metal sites for enhancing the ORR activity. (a) DFT calculated on-site magnetic moments of Fe atoms interacting with neighboring sites with different dsite values. (b) The dsite-dependent ΔG*OH obtained by DFT calculations. Inset: volcano plot of calculated overpotentials for the ORR against ΔG*OH. Reproduced with permission from ref. 147. Copyright (2021) Springer Nature. (c) Improving the intrinsic activity of Fe–N4 site by Co–Fe dual site construction. (d) Proposed ORR mechanism on the FeCoN5–OH site. (e) ORR polarization curves of CoNx/C, FeNx/C, FeCoNx–C and Pt/C in a 0.1 M HClO4 solution. Reproduced with permission from ref. 151. Copyright (2019) American Chemical Society. (f) Optimized structure of Fe,Mn/N–C. Magnetic susceptibility of (g) Fe/N–C and (h) Fe,Mn/N–C (M.S. represents medium-spin and L.S. represents low-spin). Reproduced with permission from ref. 155. Copyright (2021) Springer Nature. | ||
Further decreasing the distance, the M and M′ could coordinate with each other and form the “real” closest dual metal sites, i.e., M–M′–Nx (Fig. 5c).150,151 The M and M′ can largely alter each other's electronic structure through metal–metal interactions and charge polarization,152,153 therefore changing the O2 adsorption behavior, and the subsequent ORR pathway. For example, Wang et al. used a double-solvent method for the construction of an Fe–Co dual-site (Fe,Co)/N–C catalyst and achieved superior ORR activity in acidic media to those of single Co or Fe site.154 The HAADF-STEM image and Mössbauer spectroscopy indicated the presence of Co–Fe dual sites. The DFT calculations suggest that the Fe–Co dual-site could reduce the energy barrier of OOH* dissociation into O* and OH*, enabling a faster ORR kinetics. Further, an OH-ligand self-binding approach was discovered to enhance the Fe–Co dual-site.151 The pristine binuclear FeCoN5 site promoted a bridging-cis O2 adsorption configuration, making the O–O bond easier to cleave (Fig. 5d). In the ORR process, the in situ generated FeCoN5–OH site decreased the localized electron density around single Fe atoms, leading to a weakened Fe–O bonding and dramatically improved intrinsic ORR activity. As a result, the as-constructed FeCoN5–OH site delivered an ORR Eonset and E1/2 of up to 1.02 and 0.86 V (vs. RHE), respectively, with an intrinsic activity over 20 times higher than that of the single-atom Fe–N4 site in a 0.1 M HClO4 electrolyte solution (Fig. 5e). Besides, the Mn–N4 site can also change the spin state of the neighboring Fe–N4 site from the low spin state (t52ge0g) to the intermediate spin state (t42ge1g) (Fig. 5f). The magnetic susceptibility results indicated that the effective magnetic moment of Fe,Mn/N–C and Fe/N–C was 3.75 μeff and 2.16 μeff, respectively (Fig. 5g and h).155 DFT calculations reveal that the tailored Fe,Mn/N–C catalyst can interact with oxygen moderately, with appropriate bond length and adsorption energy, beneficial to promote the ORR kinetics. As a result, the Fe,Mn/N–C catalyst displayed excellent ORR activity in both 0.1 M HClO4 and 0.1 M KOH solutions with a high E1/2 of 0.804 and 0.928 V (vs. RHE), respectively.
Besides, a dual metal sites can be constructed using an identical metal species for the formation of M2–N–C as well.156 Xiong et al. prepared single site Fe1–N–C, dual site Fe2–N–C, and triple site Fe3–N–C by pyrolysis of specific Fe atoms as precursors encapsulated in a zeolitic imidazolate framework (ZIF-8).157 The dual site Fe2–N–C prepared from the binuclear Fe2(CO)9@ZIF-8 hybrid structure displayed excellent ORR activity and durability in an acidic solution, which was superior to those of Fe1–N–C and Fe3–N–C. They found that the dual site Fe2 cluster in Fe2–N–C and the triple site Fe3 cluster in Fe3–N–C adsorbed the O2 in a side-on configuration, enabling better O2 activation than Fe–N–C. However, Fe2–N–C had more catalytic sites and more enhanced bonding hybridization between Fe 3d and O 2p orbitals than Fe3–N–C, making Fe2–N–C more active. A similar phenomenon was reported by Xie et al., where the Fe2N6 site was more active than Fe–N4.158 These results discussed above provide new insights into the design of dual metal sites and the fundamental understanding of the ORR mechanism at the atomic level.
2.5 Regulation of the carbon skeleton geometry
The geometric configuration of the carbon skeleton can also largely alter the electronic structure of M–Nx sites, thus regulating the intrinsic ORR activity.159 For example, the Fe–N4 active moiety can exist in different forms, such as micropore-hosted Fe–N4–C8, Fe–N4–C10 and Fe–N4–C12, depending on the surrounding carbon geometry (Fig. 6a).160 Their ORR properties were theoretically investigated. As illustrated in Fig. 6b, the O2 was adsorbed on Fe–N4–C8 in a side-on configuration, while it was adsorbed on Fe–N4–C10 and Fe–N4–C12 in an end-on configuration (Fig. 6b). As a result, Fe–N4–C8 has the lowest activation energy for O2 and OOH dissociation and favors a direct four-electron pathway (Fig. 6c). Using advanced 57Fe Mössbauer spectroscopy, Wu et al. identified Fe–N4–C8 and Fe–N4–C10 in an atomically dispersed Fe–N–C catalyst and experimentally found that Fe–N4–C8 was more active for the ORR.161 Due to the rich Fe–N4–C8 site, the as-developed Fe–N–C demonstrated excellent ORR activity with a high E1/2 of 0.88 V (vs. RHE) in 0.5 M H2SO4, which was comparable to that of Pt/C.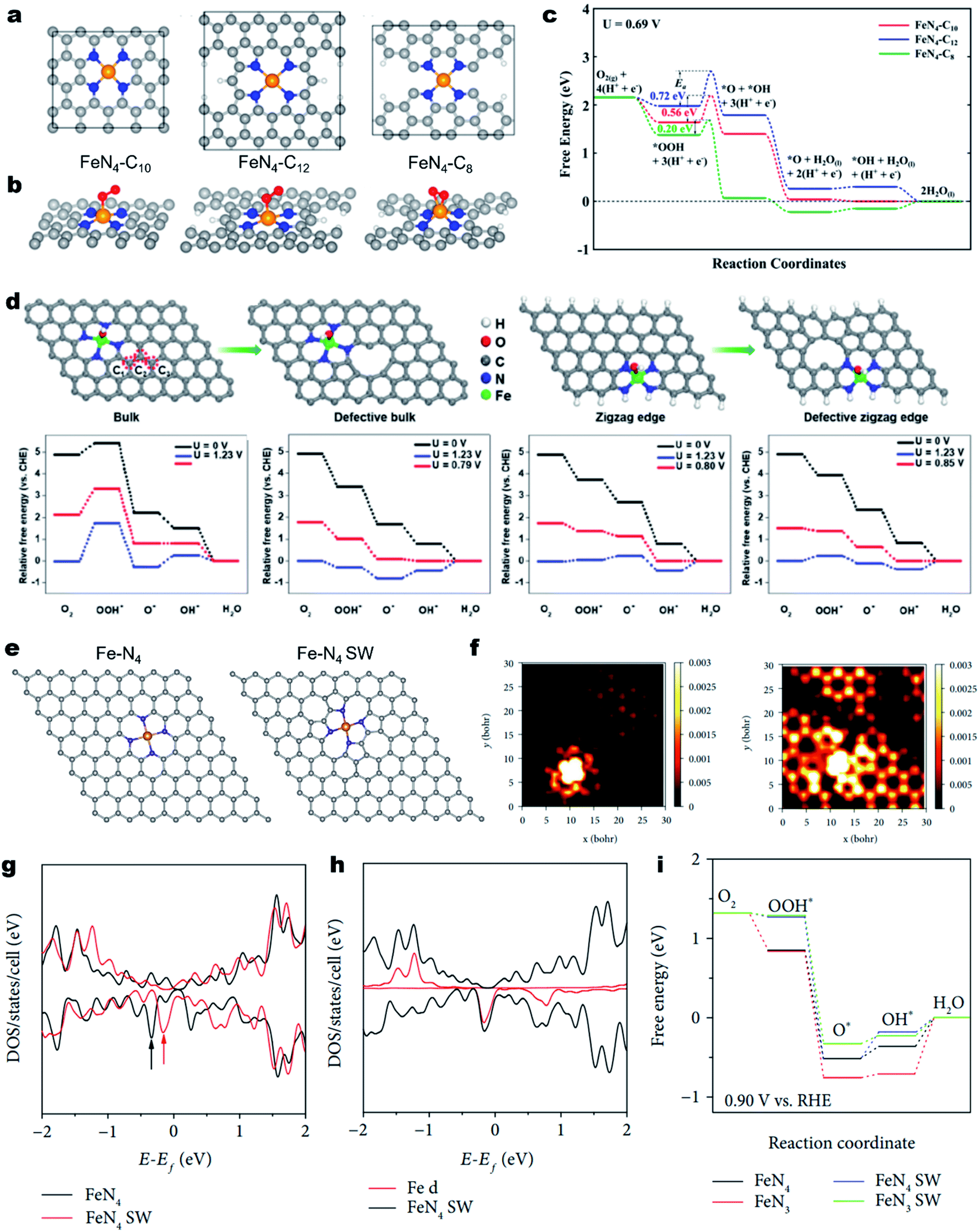 | ||
| Fig. 6 Regulating the carbon skeleton geometry for enhancing the ORR activity. (a) Atomistic configuration of the Fe–N4 moiety with different local carbon structures of Fe–N4–C10, Fe–N4–C12, and Fe–N4–C8 and (b) corresponding O2 adsorption configuration. (c) Calculated free energy diagrams for the ORR through an OOH dissociation pathway on the above three different active sites under a potential of 0.69 V in acidic medium. Reproduced with permission from ref. 160. Copyright (2017) American Chemical Society. (d) Schematic illustration of a bulk Fe–N4 site, defective bulk Fe–N4 site, zigzag Fe–N4 site, and defective zigzag Fe–N4 site and corresponding free energy diagram for the ORR. Reproduced with permission from ref. 163. Copyright (2019) Wiley-VCH. (e) Atomic models of Fe–N4 and Fe–N4 SW. (f) Simulated STM image (at a bias of −1.0 V) of normal Fe–N4 doped graphene sheets (left) and Fe–N4 SW doped graphene sheets (right). Density of states (DOS) of normal Fe–N4 and Fe–N4 SW-doped graphene sheets. (g) DOS of Fe–N4 SW and (h) Fe 3d. (i) Free energy diagrams of ORR processes on normal Fe–N4, normal Fe–N3, Fe–N4 SW, and Fe–N3 SW at the applied potential of +0.9 V vs. RHE. Reproduced with permission from ref. 165. Copyright (2019) the American Association for the Advancement of Science. | ||
Besides, carbon defects can also induce redistribution of the charge density of the single metal atoms and the neighboring N atoms. In 2017, Zelenay et al. firstly proposed that Fe–N4 moieties at the edges of graphitic domains might have higher ORR activity.46 Later on, Yao et al. demonstrated the local electronic redistribution and bandgap shrinkage of the edge-Fe–N4 site, enabling a lower free-energy barrier toward the direct four-electron ORR.162 The utilization of NH4Cl salt was found to be a promising approach for the creation of abundant edge-Fe–N4 sites.163 NH4Cl enabled the preferential formation of edge-Fe–N4 sites and favored the formation of numerous pores and N-doped edges. DFT calculations confirm that the introduced in-plane holes could lower the adsorption energy of intermediates (e.g., O2* and OOH*), thereby promoting the intrinsic ORR activity (Fig. 6d). Subsequently, the edge-Fe–N4 site anchored FeNx/GM catalysts demonstrated remarkable ORR activity in acid media. Moreover, the PEMFC using FeNx/GM as the cathode catalyst exhibited an impressive maximum power density (Pmax) value of 0.43 W cm−2 under air conditions.
In addition, the topological defect is another regulatory factor of M–Nx site's intrinsic activity of the M–Nx site. Using DFT calculations, Yang et al. revealed that the defective graphene skeleton with pentagon–octagon–pentagon (585) defects could efficiently mediate charge redistribution of the attached exfoliated monolayer iron phthalocyanine (FePc), providing the FePc with enhanced ORR activity.164 Moreover, the Fe–N4 in the Stone–Wales (SW) defect (pentagon–heptagon–heptagon–pentagon, 5775 defects) configurations also displayed enhanced catalytic performance relative to the pristine Fe–N4 (Fig. 6e).165 As depicted in Fig. 6f, the simulated scanning tunneling microscopy (STM) images demonstrated that the SW defects caused significant redistribution of electron densities of Fe–N4 and adjacent carbon atoms. Fig. 6g and h show the total density of states (DOS) of normal and SW Fe–N centers. Notably, the marked state of Fe–N4 SW is much closer to the Fermi level than that of normal Fe–N4, indicating higher ability for donating electrons and reducing oxygen. The ORR free energy diagrams further add the evidence that the Fe–N4 SW has a lower reaction overpotential than the normal counterpart (Fig. 6i).165 In contrast, the neighboring SW defect or 585 defects would lower the intrinsic ORR activity of the M–N4 sites.166
These studies highlight the essential role of carbon defects and inspire the rational design of more advanced Fe–N–C catalysts for the ORR via such a pore/edge/topological-engineering strategy. Similar to Fe–N–C materials, engineering of the local carbon structure of Co–Nx and Mn–Nx sites can also enhance their corresponding intrinsic ORR activities.167,168 Although several investigations by theoretical studies have demonstrated the promotional role of the defects in the ORR, further efforts regarding the precise identification of the edge or topological defects are indispensable, requiring accurate atomic TEM scrutinization.
3. Engineering dense active sites
Aside from improving the intrinsic ORR activity of the M–Nx active site as discussed above, it is essential to maximize the density of active sites over single atom carbonous M–N–C electrocatalysts for enhancing the ORR activity.169 However, currently reported strategies for synthesizing M–N–C materials unavoidably involve high-temperature pyrolysis of metal- and N-containing precursors. Unfortunately, during the thermal pyrolysis, metal species tend to agglomerate and eventually form low-active metal-based clusters and nanoparticles (NPs) encapsulated in carbon shells (e.g., M@C NPs). The M@C NPs are typically difficult to remove and thus seriously block the formation of active Fe–Nx moieties, leading to a low density of M–Nx active sites (less than 3.0 at%).In order to achieve highly active M–N–C electrocatalysts, it is important to prevent the migration and agglomeration of metal atoms during the high-temperature pyrolysis process.170 Thus, confining the metal atoms within a certain spatial region to prevent their migration becomes one of the effective approaches. The organic/inorganic hybrid frameworks, especially MOFs, with metal–N coordination can adequately prevent the migration of metal atoms.171 Among the various MOF materials, zeolitic imidazolate frameworks (ZIFs) (e.g., ZIF-8 and ZIF-67) are the most common precursors used due to their easy preparation, and well-defined metal–N4 configurations.167,172–177 For the synthesis of Fe–N–C electrocatalysts, Fe-doped ZIF-8 can be transformed into Fe single atoms with Zn species being removed due to their lower boiling point (∼907 °C), resulting in atomically dispersed Fe atoms that are confined in ZIF-derived N-doped carbons.178 Moreover, the Zn vaporization could generate rich micropores, which could host more Fe–N4 active sites.179–181 For example, Wu et al. synthesized an Fe-doped ZIF-8 in an oxygen-free environment and directly converted it into an atomically dispersed Fe–N–C electrocatalyst (Fig. 7a).182 In the Fe-doped ZIF-8, the Fe–N4 coordination was atomically dispersed and separated by abundant zinc nodes. The special space isolation effect enabled the pyrolyzed Fe–N–C electrocatalyst without Fe aggregation. The particle size of atomic Fe–N–C could be controlled from 20 to 1000 nm by simply adjusting the solution concentration during the synthesis of Fe-doped ZIF-8. Remarkably, the Fe–N–C electrocatalyst with an average particle size of ∼50 nm displayed the best ORR activity in 0.5 M H2SO4 electrolyte with an E1/2 of 0.85 V (vs. RHE), which was only 30 mV lower than that of the Pt/C catalyst. Apart from chemical doping of Fe ions into ZIF-8 precursors, a spatial confinement strategy was also found effective for the synthesis of Fe–N–C electrocatalysts. Li et al. developed a novel cage encapsulation strategy to synthesize an atomically dispersed Fe–N–C electrocatalyst (Fig. 7b).183 ZIF-8 with a cavity diameter of 11.6 Å as molecular cages was employed to encapsulate and separate the iron precursor Fe(acac)3 (diameter, ca. 9.7 nm). During the pyrolysis in an inert atmosphere, isolated single Fe atoms with a high content of 2.16 wt% were stabilized on N doped carbons. In a 0.1 M KOH electrolyte solution, the obtained FeISAs/CN electrocatalyst displayed an excellent ORR performance with an E1/2 of 0.90 V (vs. RHE). Similarly, ferrocene184 or FePc185 molecules could also be encapsulated in ZIF-8 for the preparation of atomically dispersed Fe–N–C electrocatalysts. Also, the short-range order nitrogen doped carbon can provide plentiful anchoring sites for the stabilization of single metal sites with strong interaction, enabling the synthesis of high-metal-loading M–N–C electrocatalysts.186–188
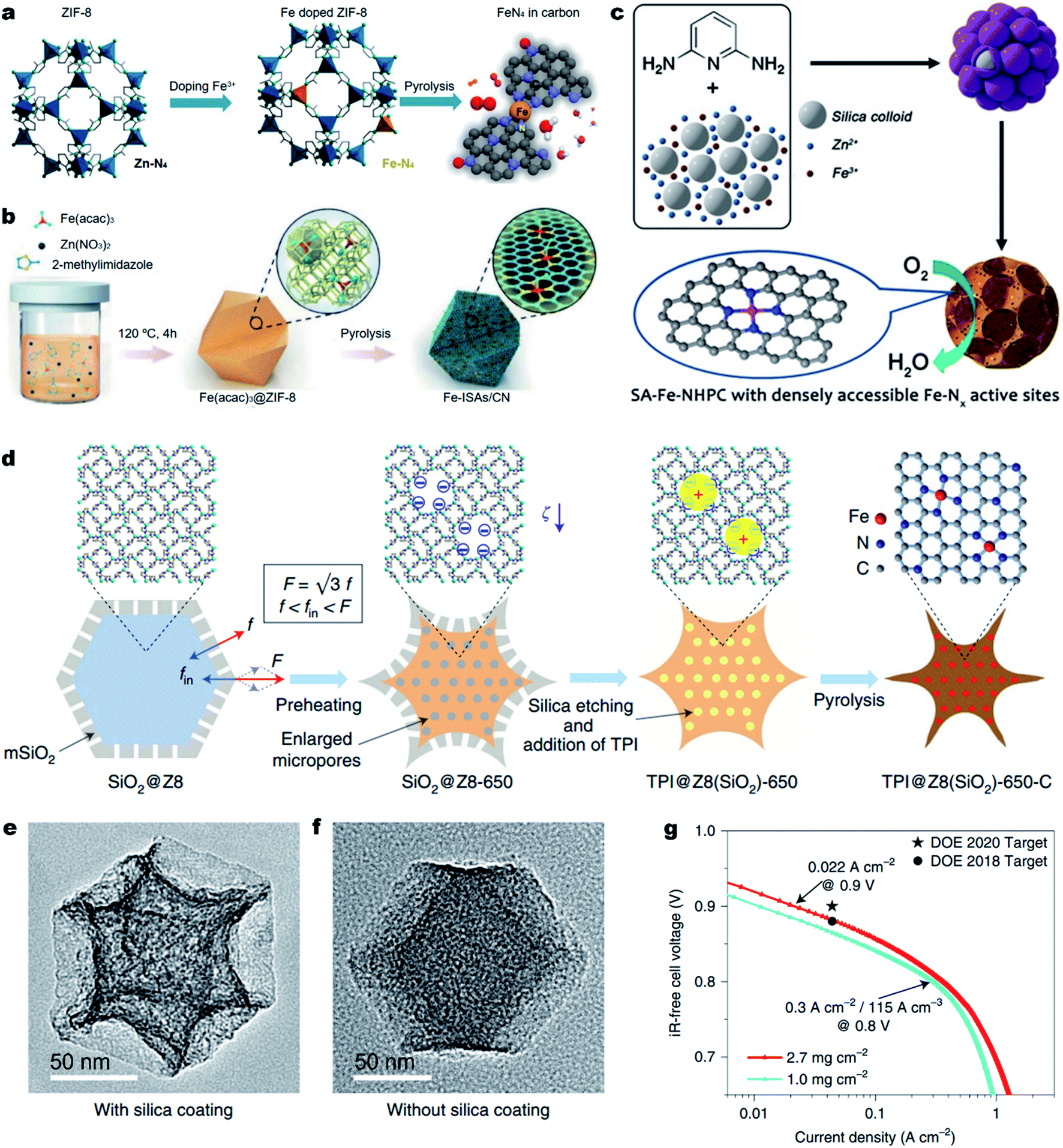 | ||
| Fig. 7 Engineering dense Fe–Nx active sites on hierarchically porous carbons. (a and b) One step synthesis of the Fe–N–C electrocatalyst. (a) Space isolation strategy by chemical doping of Fe into ZIF-8 nanocrystals. Reproduced with the permission from ref. 182. Copyright (2017) American Chemical Society. (b) Space confinement by encapsulation of Fe species into the ZIF-8 cavities. Reproduced with the permission from ref. 183. Copyright (2017) Wiley-VCH. (c–g) Enhancing the Fe–Nx site density and ORR activity by constructing hierarchically porous carbons. (c) Schematic showing the zinc mediated templated synthesis of Fe–N–C with densely accessible Fe–Nx sites. Reproduced with the permission from ref. 204. Copyright (2020) Wiley-VCH. (d) A schematic synthesis process of TPI@Z8(SiO2)-650-C by a mesoporous silica coating strategy. TEM images (e) TPI@Z8(SiO2)-650-C and (f) TPI@Z8-650-C. (g) Tafel plot for the determination of the PEMFC performance for TPI@Z8(SiO2)-650-C at 0.9 ViR-free measured under 1.0 bar H2–O2. Reproduced with the permission from ref. 205. Copyright (2019) Springer Nature. | ||
Previous studies have shown that the surface area and the porosity of M–N–C electrocatalysts play an important role in ORR performance. A high Brunauer–Emmett–Teller (BET) surface area is beneficial for the ORR catalytic process. The micropores can accommodate abundant M–Nx active sites and the meso-/macro-pores can facilitate the mass transport. Thus, construction of hierarchically porous carbon nanostructures with a high surface area is an effective strategy to improve the utilization of M–Nx sites, thus promoting the ORR activity.189–198 Template sacrificial strategies including a hard template and soft template are common methods to create hierarchically porous M–N–C electrocatalysts. The template utilizes SiO2,74,199–201 SBA-15,202 molten salts,169 or Te nanowires203 as sacrificial templates and small organic molecules, metal complexes or polymers as carbon/nitrogen precursors. In 2020, our group reported the synthesis of densely accessible single atom Fe–Nx active sites on nitrogen doped hierarchically porous carbon (SA-Fe–NHPC) by a zinc mediated template synthesis strategy.204 As depicted in Fig. 7c, during the thermal treatment of a N rich 2,6-diaminopyridine/ZnFe/SiO2 complex, the zinc prevented the formation of Fe3C@C nanoparticles and promoted the generation of micropores. In addition, the SiO2 spheres (∼12 nm) served as hard templates for the generation of a mesoporous carbon nanostructure, achieving densely accessible Fe–Nx active sites on SA-Fe–NHPC. As a result, SA-Fe–NHPC showed a very large specific surface area of 1327 m2 g−1 and a high Fe content of 1.25 wt%. Benefiting from the high density and superior accessibility of the Fe–Nx active sites, the SA-Fe–NHPC electrocatalyst demonstrated high ORR activity with an E1/2 of 0.93 V (vs. RHE) in a 0.1 M KOH aqueous solution. The assembled Zn–air battery using the SA-Fe–NHPC electrocatalyst as an air electrode showed a very high maximum power density (Pmax) of 266.4 mW cm−2 and excellent stability for 10 days. Shui et al. fabricated a cave-shaped Fe–N–C electrocatalyst (TPI@Z8(SiO2)-650-C) with rich meso-porosity, external surface area and dense Fe–N4 moieties by pyrolyzing mesoporous silica-coated ZIF-8 (Fig. 7d).205 TPI@Z8-650-C was also synthesized for comparison without the coating of mesoporous SiO2. The resultant TPI@Z8(SiO2)-650-C exhibited a large number of mesopores across its surface which presented a three-fold higher external surface area than that of TPI@Z8-650-C (Fig. 7e and f). As a result, the porous TPI@Z8(SiO2)-650-C showed much improved ORR activity compared to that of TPI@Z8-650-C in acids. Under the department of energy (DOE) of USA testing protocol, the TPI@Z8-650-C based PEMFC (loading amount: 2.7 mg cm−2) achieved a current density of 0.022 A cm−2 at 0.9 ViR-free under 1.0 bar H2–O2 conditions (Fig. 7g). Meanwhile, this TPI@Z8-650-C based PEMFC delivered a Pmax of 1.18 W cm−2 under 2.5 bar H2–O2, which approached that of the Pt/C based PEMFC (Pmax ≈ 1.30 W cm−2, loading amount: 0.2 mgPt cm−2).
Besides, engineering M–Nx sites on the surface of carbons will undoubtedly largely improve site utilization. Very recently, Jia et al. implemented a chemical vapor deposition (CVD) synthesis, flowing FeCl3 vapor over a Zn–N–C substrate at 750 °C to prepare accessible Fe–N4 sites on the surface of carbons (denoted as FeNC-CVD-750) (Fig. 8a).20657Fe Mössbauer spectroscopy, atomic-resolution annular dark-field STEM (ADF-STEM) and XAS showed the atomically dispersed Fe–N4 site in the FeNC-CVD-750 catalyst. The number of accessible Fe–N4 sites (site density, SDmass) was determined by the electrochemical stripping of NO formed by exposure to aqueous nitrite (Fig. 8 and c), and by exposure to gaseous NO (Fig. 3c), giving a SDmass value as high as 1.92 × 1020 sites g−1. According to the inductively coupled plasma-mass spectrometry (ICP-MS) and 57Fe Mössbauer spectroscopy, all the Fe–N4 sites were gas-phase and electrochemically accessible, delivering an Fe site utilization around 100%. As a result, in a 0.5 M H2SO4 electrolyte, the FeNC-CVD-750 displayed excellent high ORR activity with an E1/2 of 0.85 V (vs. RHE). When used as a cathode catalyst in a 1.0 bar H2–O2 PEMFC, FeNC-CVD-750 delivered a high current density of 33 mA cm−2 at 0.90 ViR-free, which approached the DOE 2025 target of 44 mA cm−2 (Fig. 8d). In 1.0 bar H2–air, the FeNC-CVD-750 delivered a Pmax of 0.37 W cm−2 (Fig. 8e), among the highest values reported for PGM-free cathodes to date. Other similar studies also reported the synthesis of Fe–N–C materials with a surface-hosted Fe–N4 site, achieving high site densities, high site utilization and eventually excellent ORR performance.207,208
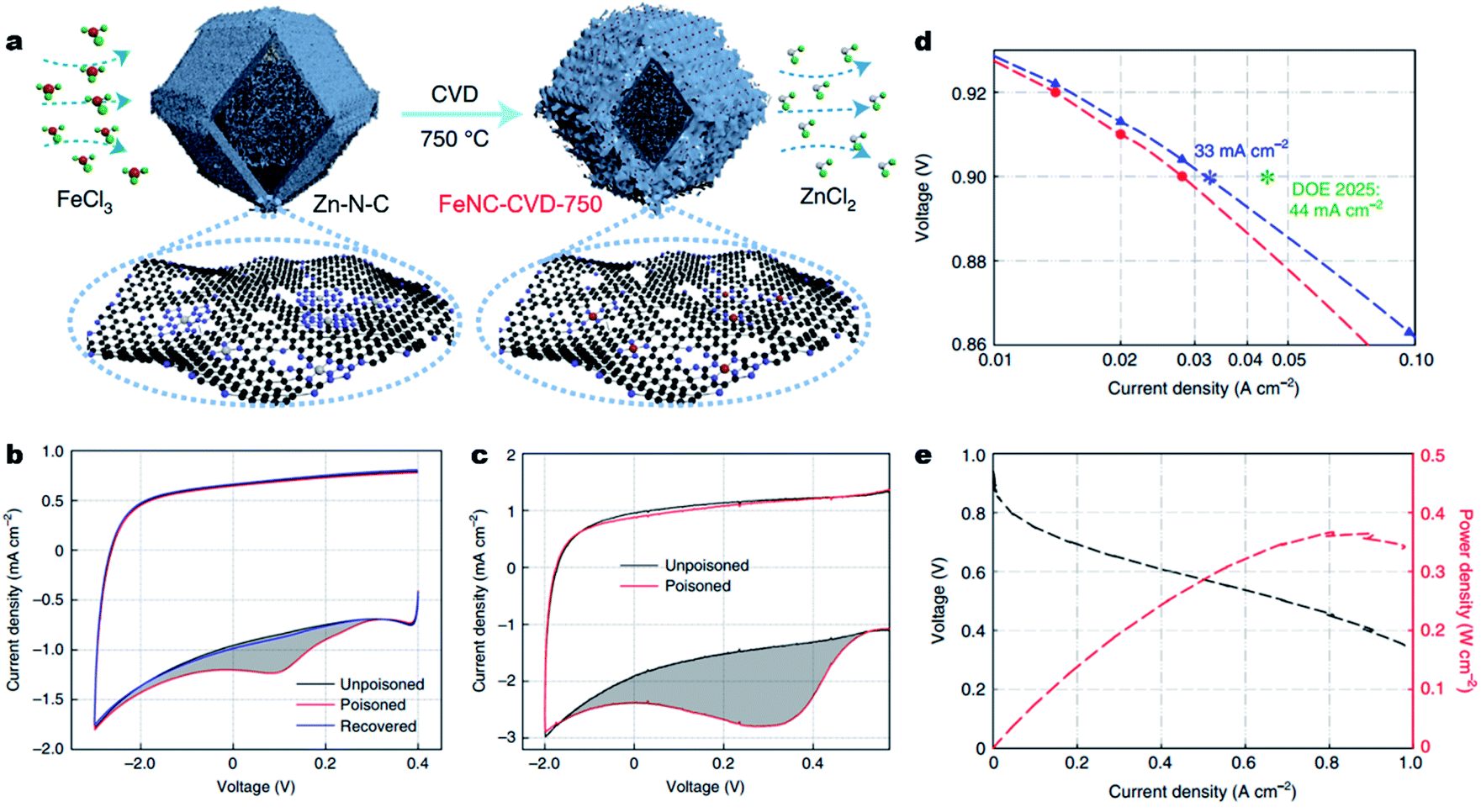 | ||
| Fig. 8 Engineering fully accessible Fe–Nx sites on the surface of carbons. (a) Synthesis of FeNC-CVD-750 via an FeCl3 CVD method. Red, green, grey, purple, and black balls represent Fe, Cl, Zn, N and C atoms, respectively. (b) Nitrite stripping voltammogram in N2-saturated 0.5 M acetate electrolyte buffer (pH 5.2) with a catalyst loading of 270 μg cm−2 and a scan rate of 10 mV s−1. (c) NO stripping voltammogram in N2-saturated 0.5 M H2SO4 with a catalyst loading of 600 μg cm−2 and a scan rate of 10 mV s−1. The grey shading represents the excess current associated with the reductive stripping of nitrite. (d) Tafel plot for the determination of FeNC-CVD-750 activity at 0.9 ViR-free. (e) The H2–air PEMFC polarization curve and power density without iR-correction for the membrane electrode assembly. Conditions: 500 ml min−1 H2 and 2000 ml min−1 air, 100% RH, 1.0 bar H2 and air partial pressure, and 80 °C. Reproduced with the permission from ref. 206. Copyright (2021) Springer Nature. | ||
In short, engineering hierarchically porous carbon nanostructures and manipulating the location of M–Nx sites can improve the site density, expose each active site accessible to reactants, and facilitate mass transfer, therefore promoting the ORR performance of M–N–C electrocatalysts.
4. Conclusions and perspectives
Single-atom carbonaceous M–N–C electrocatalysts have demonstrated promising catalytic ORR properties for applications in Zn–air batteries and PEMFCs due to their abundant resources, low cost, large surface area, structural tunability, etc. In this Perspective, we summarized two strategies for the preparation of high-performance M–N–C electrocatalysts toward the ORR, including increasing the intrinsic activity and promoting the accessibility of the M–Nx active sites. Manipulating the coordination geometry, doping heteroatoms, constructing dual-metal sites, and regulating the geometry of the carbon skeleton can effectively engineer the electronic structure of the single metal site, thus altering the adsorption behavior of O-containing intermediates and enhancing the intrinsic activity. Engineering carbon nanostructures can largely facilitate the exposure of the M–Nx site, hence increasing the site utilization and the overall catalytic activity. In alkaline media, the as-developed M–N–C materials show superb ORR performance, outperforming that of commercial Pt/C (E1/2 ≈ 0.85 V) (Table 1). These M–N–C electrocatalysts enabled the as-assembled metal–air batteries with high energy density and efficiency. However, in acidic media, M–N–C materials are still inferior to the benchmark Pt/C (E1/2 ≈ 0.85 V) (Table 1), restricting their utilization in practical PEMFCs. Therefore, rational design of highly active M–Nx sites, and fruitful identification of the active site and development of new synthetic methods for single-atom carbonaceous M–N–C electrocatalysts are still highly desirable for future practical applications. Below we provide our perspectives regarding the future directions of M–N–C electrocatalysts for the ORR.| Method | M–N–C electrocatalyst | Active site | Electrolyte | E onset (V vs. RHE) | E 1/2 (V vs. RHE) | Ref. |
|---|---|---|---|---|---|---|
| Local coordination | HP-Fe–N4 | Pyrrole-type Fe–N4 | 0.5 M H2SO4 | 0.95 | 0.80 | 97 |
| Fe–N/P–C-700 | Fe–N3P1 | 0.1 M KOH | 0.941 | 0.867 | 104 | |
| S–Cu-ISA/SNC | Cu–N3S1 | 0.1 M KOH | ∼0.98 | 0.918 | 105 | |
| Mn/C–NO | Mn–N3O | 0.1 M KOH | ∼0.98 | 0.86 | 109 | |
| Co1-SAC | Co–N3C1 | 0.1 M KOH | 0.904 | 0.824 | 106 | |
| Heteroatom incorporation | Fe/N/C–SCN | S-modified Fe–N4 | 0.1 M H2SO4 | — | 0.836 | 123 |
| Fe-ISA/SNC | S-modified Fe–N4 | 0.1 M KOH | ∼0.98 | 0.896 | 124 | |
| Fe-SAs/NPS-HC | S, P-co-modified Fe–N4 | 0.1 M KOH | ∼0.98 | 0.912 | 129 | |
| 0.5 M H2SO4 | ∼0.89 | 0.791 | ||||
| Mn–N–C–S | S-modified Mn–N4 | 0.5 M H2SO4 | ∼0.98 | 0.81 | 131 | |
| Axial ligand modification | FeCl1N4/CNS | Fe–Cl1N4 | 0.1 M KOH | ∼0.97 | 0.921 | 138 |
| Fe–N–C | Self-adjusted FeN4–OH | 0.1 M KOH | ∼0.97 | ∼0.90 | 142 | |
| Dual metal sites | (Fe,Co)/N–C | FeN3–CoN4 | 0.1 M HClO4 | 1.060 | 0.863 | 154 |
| FeCoNx/C | FeCoN5–OH | 0.1 M HClO4 | 1.02 | 0.86 | 151 | |
| Fe,Mn/N–C | Fe–Mn–N6 | 0.1 M HClO4 | ∼0.92 | 0.804 | 155 | |
| 0.1 M KOH | ∼0.99 | 0.928 | ||||
| Fe2–N–C | Fe2–N6 | 0.5 M H2SO4 | ∼0.92 | 0.78 | 157 | |
| Regulation of the carbon skeleton geometry | Fe–N–C (1.5Fe-ZIF) | Fe–N4–C8 dominated | 0.5 M H2SO4 | ∼0.98 | 0.88 | 161 |
| FeNx/GM | Edge FeN4 | 0.5 M H2SO4 | ∼0.90 | 0.80 | 163 | |
| Fe–N4–C-60 | Edge FeN4 | 0.1 M HClO4 | ∼0.95 | 0.80 | 162 | |
| ZIF-NC-0.5Fe-700 | Contracted FeN4 | 0.5 M H2SO4 | ∼0.98 | 0.84 | 112 |
(1) Precise active structures are the prerequisite for exploring the structure–activity relationship. However, until now, most single-atom carbonaceous M–N–C materials have been synthesized via high-temperature pyrolysis, which makes the synthesis process uncontrollable and thus the active structures elusive. These uncontrolled structures in M–N–C electrocatalysts further make it challenging to identify active sites and correlate the relationship between the catalyst structure and the ORR performance. Thus, the exploration of well-structure-controlled chemical methodologies and precise control of the active sites at the atomic level are essential for the development of M–N–C electrocatalysts. Furthermore, a profound understanding of the structural transformation and M–Nx site formation is essential to explore more advanced catalysts. Apart from the high temperature pyrolysis process, bottom-up synthesis is a promising strategy to construct well defined active sites. Conductive MOFs, covalent organic frameworks (COFs) and other supramolecular structures with well-defined M–Nx sites might provide a promising platform for the investigation, while significant efforts are required to improve their intrinsic activity and stability.
(2) In recent years, various advanced atomic-resolution techniques including HRTEM, XAS, 57Fe Mössbauer spectroscopy and DFT calculations have been used to identify the active sites of M–N–C electrocatalysts. However, they could only give the average structure information and the local structure around the single metal sites remains unclear. Thus, deeper insights into the electronic structure and geometric configuration of M–N–C electrocatalysts are still required, such as probing the local coordination environments of centrally active metal sites (e.g., coordination numbers, distances, and adjacent atomic species). Further, the development of in situ characterization techniques, such as in situ electron microscopy, in situ XAS and in situ57Fe Mössbauer spectroscopy is essential for providing insight into the active site formation mechanism. In addition, detailed in situ or operando measurements are necessary to deeply reveal the real interactions between the metal centers and O-containing species (i.e., O2, OOH*, O* and OH*) under working potential conditions. The insightful identification of the active sites and understanding the structure–performance relationship thus can guide the further rational design of highly active M–N–C electrocatalysts.
(3) The highly active M–Nx sites for the ORR are always essential. The rational design of advanced catalytically active sites and regulating their intrinsic activity are two fundamental approaches for promoting the sluggish ORR kinetics. The theoretical prediction can be used to guide the precise active site design, relying on the exploration of new synthetic approaches, such as supramolecular assembly or a new templating/confinement strategy.
(4) It has been reported that adsorption energies of OH* and OOH* on the active site follow the universally established scaling relationship, that is, ΔGOOH* = ΔGOH* + 3.2 ± 0.2.209,210 Overcoming or breaking the adsorption-energy scaling relationship is vital to enhance the ORR activity, requiring novel exemplification for catalyst design by the combination of computational screening, machine learning and experimental accumulation.
(5) To date, the developed M–N–C catalysts demonstrated excellent ORR activity in alkaline electrolytes, which enabled their superior performances in primary Zn–air batteries. However, the oxygen evolution reaction (OER) performances of such M–N–C were extremely poor, which seriously limited them as bifunctional electrocatalysts to drive the operation of a rechargeable Zn–air battery. Hence, the development of highly active and stable bifunctional electrocatalysts that alternately promote the OER and ORR kinetics is necessary. In this regard, the integration of OER-active atomic transition metals (e.g., Ru, Rh or Ni) into ORR active M–N–C electrocatalysts may provide a potential approach for the exploration of bifunctional single-atom carbonaceous electrocatalysts.
(6) The reactant (i.e., O2, OOH*, O* and OH*) accessible number of active sites plays a key role in the ORR process. Smart design of M–N–C materials with high single metal loading is highly necessary. Exploring new synthetic approaches via precursor design, supramolecular assembly and a new templating/confinement strategy is effective for achieving this target. Further, constructing porous carbon with a high specific surface area, a large porous structure and a tailored reaction interface can be flexible for facilitating the accessibility of the active site.
(7) For single-atom carbonaceous M–N–C electrocatalysts, translating their activity and stability observed in rotating disc electrode (RDE) tests into high performance devices remains a great challenge due to the lack of accurate nanoscale control of three-phase interfaces within electrodes. For the most promising PEMFC applications, developing new approaches for implementing the most promising Fe–N–C or Co–N–C electrocatalysts into membrane electrode assembly is required for achieving high PEMFC performance. Construction of hierarchically porous nanostructures with micro-porosity, meso-porosity and micro-porosity is an effective approach for achieving high MEA performance. Thereinto, micropores are capable of a high density of single metal active sites. Mesopores and macropores can promote the ionomer distribution and facilitate the mass transport.
(8) Aside from intrinsic activity, durability is another important parameter for M–N–C electrocatalysts in practical PEMFC applications. Acidic electrolytes might cause metal centers to be exchanged by protons, degrade the carbon support, and result in significant activity loss, especially for single atom Fe–N–C electrocatalysts due to the undesired Fenton reaction. Experimental and theoretical studies should focus on single metal site dissolution and carbon corrosion mechanisms. Furthermore, developing advanced supporting materials with high conductivity and corrosion resistance of M–N–C electrocatalysts are highly desirable for practical implementation.
Overall, active site engineering of single atom M–N–C electrocatalysts for the ORR is imperative to promote metal–air batteries and PEMFC technologies. It is expected that active site engineering via further advanced site design, modulation, and population will realize M–N–C materials for practical commercial use in the future.
Author contributions
G. C. and X. F. formulated the project and wrote the manuscript. G. C. and H. Z. conducted a literature review and all authors contributed to revising the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 819698 and GrapheneCore3: 881603), Deutsche Forschungsgemeinschaft (COORNETs, SPP 1928 and CRC 1415: 417590517).References
- Y. Huang, M. Zhu, Y. Huang, Z. Pei, H. Li, Z. Wang, Q. Xue and C. Zhi, Adv. Mater., 2016, 28, 8344–8364 CrossRef CAS PubMed.
- Z. P. Cano, D. Banham, S. Ye, A. Hintennach, J. Lu, M. Fowler and Z. Chen, Nat. Energy, 2018, 3, 279–289 CrossRef.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS PubMed.
- B. Wang, J. Power Sources, 2005, 152, 1–15 CrossRef CAS.
- X. Tian, X. F. Lu, B. Y. Xia and X. W. Lou, Joule, 2020, 4, 45–68 CrossRef CAS.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- K. Jiao, J. Xuan, Q. Du, Z. Bao, B. Xie, B. Wang, Y. Zhao, L. Fan, H. Wang, Z. Hou, S. Huo, N. P. Brandon, Y. Yin and M. D. Guiver, Nature, 2021, 595, 361–369 CrossRef CAS PubMed.
- M. M. Tellez-Cruz, J. Escorihuela, O. Solorza-Feria and V. Compañ, Polymers, 2021, 13, 3064 CrossRef CAS PubMed.
- G. Chen, J. Zhang, F. Wang, L. Wang, Z. Liao, E. Zschech, K. Müllen and X. Feng, Chem.–Eur. J., 2018, 24, 18413–18418 CrossRef CAS PubMed.
- Q. Wang, L. Shang, D. Sun-Waterhouse, T. Zhang and G. Waterhouse, SmartMat, 2021, 2, 154–175 CrossRef.
- Y.-J. Wang, N. Zhao, B. Fang, H. Li, X. T. Bi and H. Wang, Chem. Rev., 2015, 115, 3433–3467 CrossRef CAS PubMed.
- C. Zhu, H. Li, S. Fu, D. Du and Y. Lin, Chem. Soc. Rev., 2016, 45, 517–531 RSC.
- Y. Zhou, X. Tao, G. Chen, R. Lu, D. Wang, M.-X. Chen, E. Jin, J. Yang, H.-W. Liang, Y. Zhao, X. Feng, A. Narita and K. Müllen, Nat. Commun., 2020, 11, 5892 CrossRef CAS PubMed.
- S. Guo, S. Zhang and S. Sun, Angew. Chem., Int. Ed., 2013, 52, 8526–8544 CrossRef CAS.
- L. Dai, Y. Xue, L. Qu, H.-J. Choi and J.-B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- Y. Nie, L. Li and Z. Wei, Chem. Soc. Rev., 2015, 44, 2168–2201 RSC.
- A. A. Gewirth, J. A. Varnell and A. M. DiAscro, Chem. Rev., 2018, 118, 2313–2339 CrossRef CAS PubMed.
- G. Chen, T. Wang, P. Liu, Z. Liao, H. Zhong, G. Wang, P. Zhang, M. Yu, E. Zschech, M. Chen, J. Zhang and X. Feng, Energy Environ. Sci., 2020, 13, 2849–2855 RSC.
- G. Chen, T. Wang, J. Zhang, P. Liu, H. Sun, X. Zhuang, M. Chen and X. Feng, Adv. Mater., 2018, 30, 1706279 CrossRef.
- Z. Ma, Z. P. Cano, A. Yu, Z. Chen, G. Jiang, X. Fu, L. Yang, T. Wu, Z. Bai and J. Lu, Angew. Chem., Int. Ed., 2020, 59, 18334–18348 CrossRef CAS PubMed.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- Z. Li, S. Ji, Y. Liu, X. Cao, S. Tian, Y. Chen, Z. Niu and Y. Li, Chem. Rev., 2020, 120, 623–682 CrossRef CAS PubMed.
- F. Jaouen, E. Proietti, M. Lefèvre, R. Chenitz, J.-P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114–130 RSC.
- F. D. Speck, J. H. Kim, G. Bae, S. H. Joo, K. J. J. Mayrhofer, C. H. Choi and S. Cherevko, JACS Au, 2021, 1, 1086–1100 CrossRef CAS PubMed.
- Y. Han, Y.-G. Wang, W. Chen, R. Xu, L. Zheng, J. Zhang, J. Luo, R.-A. Shen, Y. Zhu, W.-C. Cheong, C. Chen, Q. Peng, D. Wang and Y. Li, J. Am. Chem. Soc., 2017, 139, 17269–17272 CrossRef CAS PubMed.
- T. Asset and P. Atanassov, Joule, 2020, 4, 33–44 CrossRef CAS.
- Z. Chen, D. Higgins, A. Yu, L. Zhang and J. Zhang, Energy Environ. Sci., 2011, 4, 3167–3192 RSC.
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang, H. Sun, P. An, W. Chen, Z. Guo, C. Lee, D. Chen, I. Shakir, M. Liu, T. Hu, Y. Li, A. I. Kirkland, X. Duan and Y. Huang, Nat. Catal., 2018, 1, 63–72 CrossRef CAS.
- B. Peng, H. Liu, Z. Liu, X. Duan and Y. Huang, J. Phys. Chem. Lett., 2021, 12, 2837–2847 CrossRef CAS PubMed.
- B. Bayatsarmadi, Y. Zheng, A. Vasileff and S.-Z. Qiao, Small, 2017, 13, 1700191 CrossRef PubMed.
- U. Martinez, S. Komini Babu, E. F. Holby, H. T. Chung, X. Yin and P. Zelenay, Adv. Mater., 2019, 31, 1806545 CrossRef.
- R. Jasinski, Nature, 1964, 201, 1212–1213 CrossRef CAS.
- V. S. Bagotzky, M. R. Tarasevich, K. A. Radyushkina, O. A. Levina and S. I. Andrusyova, J. Power Sources, 1978, 2, 233–240 CrossRef.
- J. A. R. van Veen, J. F. van Baar and K. J. Kroese, J. Chem. Soc., Faraday Trans., 1981, 77, 2827–2843 RSC.
- T. Sun, B. Tian, J. Lu and C. Su, J. Mater. Chem. A, 2017, 5, 18933–18950 RSC.
- G. Wu and P. Zelenay, Acc. Chem. Res., 2013, 46, 1878–1889 CrossRef CAS PubMed.
- Y. He, Q. Tan, L. Lu, J. Sokolowski and G. Wu, Electrochem. Energy Rev., 2019, 2, 231–251 CrossRef CAS.
- D. Zhao, Z. Zhuang, X. Cao, C. Zhang, Q. Peng, C. Chen and Y. Li, Chem. Soc. Rev., 2020, 49, 2215–2264 RSC.
- H. Zhang, H. Osgood, X. Xie, Y. Shao and G. Wu, Nano Energy, 2017, 31, 331–350 CrossRef CAS.
- M. Lefèvre, J. P. Dodelet and P. Bertrand, J. Phys. Chem. B, 2000, 104, 11238–11247 CrossRef.
- M. Lefèvre, J. P. Dodelet and P. Bertrand, J. Phys. Chem. B, 2002, 106, 8705–8713 CrossRef.
- X. Wan, W. Chen, J. Yang, M. Liu, X. Liu and J. Shui, ChemElectroChem, 2019, 6, 304–315 CrossRef CAS.
- V. A. Saveleva, K. Ebner, L. Ni, G. Smolentsev, D. Klose, A. Zitolo, E. Marelli, J. Li, M. Medarde, O. V. Safonova, M. Nachtegaal, F. Jaouen, U. I. Kramm, T. J. Schmidt and J. Herranz, Angew. Chem., Int. Ed., 2021, 60, 11707–11712 CrossRef CAS PubMed.
- U. I. Kramm, L. Ni and S. Wagner, Adv. Mater., 2019, 31, 1805623 CrossRef.
- J. M. Thomas, Proc.: Math., Phys. Eng. Sci., 2017, 473, 20160714 Search PubMed.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–484 CrossRef CAS PubMed.
- Y. Zhao, G. I. N. Waterhouse, G. Chen, X. Xiong, L. Z. Wu, C. H. Tung and T. Zhang, Chem. Soc. Rev., 2019, 48, 1972–2010 RSC.
- S. Zhao, C. Tan, C.-T. He, P. An, F. Xie, S. Jiang, Y. Zhu, K.-H. Wu, B. Zhang, H. Li, J. Zhang, Y. Chen, S. Liu, J. Dong and Z. Tang, Nat. Energy, 2020, 5, 881–890 CrossRef CAS.
- Q. Jia, E. Liu, L. Jiao, S. Pann and S. Mukerjee, Adv. Mater., 2019, 31, 1805157 CrossRef PubMed.
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937 CrossRef CAS PubMed.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- A. Zitolo, N. Ranjbar-Sahraie, T. Mineva, J. Li, Q. Jia, S. Stamatin, G. F. Harrington, S. M. Lyth, P. Krtil, S. Mukerjee, E. Fonda and F. Jaouen, Nat. Commun., 2017, 8, 957 CrossRef PubMed.
- Z. Miao, Y. Xia, J. Liang, L. Xie, S. Chen, S. Li, H.-L. Wang, S. Hu, J. Han and Q. Li, Small, 2021, 17, 2100735 CrossRef CAS PubMed.
- H. Zhang, W. Zhou, T. Chen, B. Y. Guan, Z. Li and X. W. Lou, Energy Environ. Sci., 2018, 11, 1980–1984 RSC.
- J. Li, M. Chen, D. A. Cullen, S. Hwang, M. Wang, B. Li, K. Liu, S. Karakalos, M. Lucero, H. Zhang, C. Lei, H. Xu, G. E. Sterbinsky, Z. Feng, D. Su, K. L. More, G. Wang, Z. Wang and G. Wu, Nat. Catal., 2018, 1, 935–945 CrossRef CAS.
- X. Xiong, Y. Li, Y. Jia, Y. Meng, K. Sun, L. Zheng, G. Zhang, Y. Li and X. Sun, Nanoscale, 2019, 11, 15900–15906 RSC.
- Q. Zhou, J. Cai, Z. Zhang, R. Gao, B. Chen, G. Wen, L. Zhao, Y. Deng, H. Dou, X. Gong, Y. Zhang, Y. Hu, A. Yu, X. Sui, Z. Wang and Z. Chen, Small Methods, 2021, 5, 2100024 CrossRef CAS.
- J. Wang, H. Li, S. Liu, Y. Hu, J. Zhang, M. Xia, Y. Hou, J. Tse, J. Zhang and Y. Zhao, Angew. Chem., Int. Ed., 2021, 60, 181–185 CrossRef CAS PubMed.
- Z. Jiang, W. Sun, H. Shang, W. Chen, T. Sun, H. Li, J. Dong, J. Zhou, Z. Li, Y. Wang, R. Cao, R. Sarangi, Z. Yang, D. Wang, J. Zhang and Y. Li, Energy Environ. Sci., 2019, 12, 3508–3514 RSC.
- F. Li, G.-F. Han, H.-J. Noh, S.-J. Kim, Y. Lu, H. Y. Jeong, Z. Fu and J.-B. Baek, Energy Environ. Sci., 2018, 11, 2263–2269 RSC.
- S. Ji, Y. Chen, X. Wang, Z. Zhang, D. Wang and Y. Li, Chem. Rev., 2020, 120, 11900–11955 CrossRef CAS PubMed.
- M.-X. Chen, M. Zhu, M. Zuo, S.-Q. Chu, J. Zhang, Y. Wu, H.-W. Liang and X. Feng, Angew. Chem., Int. Ed., 2020, 59, 1627–1633 CrossRef CAS PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- J. Guo, J. Huo, Y. Liu, W. Wu, Y. Wang, M. Wu, H. Liu and G. Wang, Small Methods, 2019, 3, 1900159 CrossRef.
- L. Zhao, Y. Zhang, L.-B. Huang, X.-Z. Liu, Q.-H. Zhang, C. He, Z.-Y. Wu, L.-J. Zhang, J. Wu, W. Yang, L. Gu, J.-S. Hu and L.-J. Wan, Nat. Commun., 2019, 10, 1278 CrossRef PubMed.
- G. Zhang, Y. Jia, C. Zhang, X. Xiong, K. Sun, R. Chen, W. Chen, Y. Kuang, L. Zheng, H. Tang, W. Liu, J. Liu, X. Sun, W.-F. Lin and H. Dai, Energy Environ. Sci., 2019, 12, 1317–1325 RSC.
- Z. Song, L. Zhang, K. Doyle-Davis, X. Fu, J.-L. Luo and X. Sun, Adv. Energy Mater., 2020, 10, 2001561 CrossRef CAS.
- A. Han, B. Wang, A. Kumar, Y. Qin, J. Jin, X. Wang, C. Yang, B. Dong, Y. Jia, J. Liu and X. Sun, Small Methods, 2019, 3, 1800471 CrossRef.
- L. Li, J. He, Y. Wang, X. Lv, X. Gu, P. Dai, D. Liu and X. Zhao, J. Mater. Chem. A, 2019, 7, 1964–1988 RSC.
- L. Jiao, G. Wan, R. Zhang, H. Zhou, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 8525–8529 CrossRef CAS PubMed.
- H.-F. Wang, L. Chen, H. Pang, S. Kaskel and Q. Xu, Chem. Soc. Rev., 2020, 49, 1414–1448 RSC.
- Y.-S. Wei, M. Zhang, R. Zou and Q. Xu, Chem. Rev., 2020, 120, 12089–12174 CrossRef CAS PubMed.
- C. Zhu, Q. Shi, B. Z. Xu, S. Fu, G. Wan, C. Yang, S. Yao, J. Song, H. Zhou, D. Du, S. P. Beckman, D. Su and Y. Lin, Adv. Energy Mater., 2018, 8, 1801956 CrossRef.
- Y. Chen, Z. Li, Y. Zhu, D. Sun, X. Liu, L. Xu and Y. Tang, Adv. Mater., 2019, 31, 1806312 CrossRef PubMed.
- M. Chen, Y. He, J. S. Spendelow and G. Wu, ACS Energy Lett., 2019, 4, 1619–1633 CrossRef CAS.
- M. Liu, L. Wang, K. Zhao, S. Shi, Q. Shao, L. Zhang, X. Sun, Y. Zhao and J. Zhang, Energy Environ. Sci., 2019, 12, 2890–2923 RSC.
- Y. Wang, H. Su, Y. He, L. Li, S. Zhu, H. Shen, P. Xie, X. Fu, G. Zhou, C. Feng, D. Zhao, F. Xiao, X. Zhu, Y. Zeng, M. Shao, S. Chen, G. Wu, J. Zeng and C. Wang, Chem. Rev., 2020, 120, 12217–12314 CrossRef CAS PubMed.
- E. Luo, Y. Chu, J. Liu, Z. Shi, S. Zhu, L. Gong, J. Ge, C. H. Choi, C. Liu and W. Xing, Energy Environ. Sci., 2021, 14, 2158–2185 RSC.
- M.-X. Chen, L. Tong and H.-W. Liang, Chem.–Eur. J., 2021, 27, 145–157 CrossRef CAS PubMed.
- J. Zhang, G. Chen, K. Müllen and X. Feng, Adv. Mater., 2018, 30, 1800528 CrossRef PubMed.
- Y. Wang, F. Chu, J. Zeng, Q. Wang, T. Naren, Y. Li, Y. Cheng, Y. Lei and F. Wu, ACS Nano, 2021, 15, 210–239 CrossRef CAS PubMed.
- X. X. Wang, M. T. Swihart and G. Wu, Nat. Catal., 2019, 2, 578–589 CrossRef CAS.
- C.-X. Zhao, B.-Q. Li, J.-N. Liu and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 4448–4463 CrossRef CAS PubMed.
- W.-H. Lai, Z. Miao, Y.-X. Wang, J.-Z. Wang and S.-L. Chou, Adv. Energy Mater., 2019, 9, 1900722 CrossRef CAS.
- T. Sun, S. Mitchell, J. Li, P. Lyu, X. Wu, J. Pérez-Ramírez and J. Lu, Adv. Mater., 2021, 33, 2003075 CrossRef CAS PubMed.
- H. Peng, F. Liu, X. Liu, S. Liao, C. You, X. Tian, H. Nan, F. Luo, H. Song, Z. Fu and P. Huang, ACS Catal., 2014, 4, 3797–3805 CrossRef CAS.
- Y. Zheng, D.-S. Yang, J. M. Kweun, C. Li, K. Tan, F. Kong, C. Liang, Y. J. Chabal, Y. Y. Kim, M. Cho, J.-S. Yu and K. Cho, Nano Energy, 2016, 30, 443–449 CrossRef CAS.
- F. Calle-Vallejo, J. I. Martínez and J. Rossmeisl, Phys. Chem. Chem. Phys., 2011, 13, 15639–15643 RSC.
- J. Masa, A. Zhao, W. Xia, M. Muhler and W. Schuhmann, Electrochim. Acta, 2014, 128, 271–278 CrossRef CAS.
- L. Osmieri, A. H. A. Monteverde Videla, P. Ocón and S. Specchia, J. Phys. Chem. C, 2017, 121, 17796–17817 CrossRef CAS.
- J. Kim, J. M. Yoo, H. S. Lee, Y.-E. Sung and T. Hyeon, Trends Chem., 2021, 3, 779–794 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- Y. He, S. Liu, C. Priest, Q. Shi and G. Wu, Chem. Soc. Rev., 2020, 49, 3484–3524 RSC.
- J. Qin, Z. Liu, D. Wu and J. Yang, Appl. Catal., B, 2020, 278, 119300 CrossRef CAS.
- U. I. Kramm, J. Herranz, N. Larouche, T. M. Arruda, M. Lefèvre, F. Jaouen, P. Bogdanoff, S. Fiechter, I. Abs-Wurmbach, S. Mukerjee and J.-P. Dodelet, Phys. Chem. Chem. Phys., 2012, 14, 11673–11688 RSC.
- L. Yang, D. Cheng, H. Xu, X. Zeng, X. Wan, J. Shui, Z. Xiang and D. Cao, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6626–6631 CrossRef CAS PubMed.
- N. Zhang, T. Zhou, M. Chen, H. Feng, R. Yuan, C. a. Zhong, W. Yan, Y. Tian, X. Wu, W. Chu, C. Wu and Y. Xie, Energy Environ. Sci., 2020, 13, 111–118 RSC.
- J. Li, M. T. Sougrati, A. Zitolo, J. M. Ablett, I. C. Oğuz, T. Mineva, I. Matanovic, P. Atanassov, Y. Huang, I. Zenyuk, A. Di Cicco, K. Kumar, L. Dubau, F. Maillard, G. Dražić and F. Jaouen, Nat. Catal., 2021, 4, 10–19 CrossRef CAS.
- J. Zhang, H. Yang and B. Liu, Adv. Energy Mater., 2021, 11, 2002473 CrossRef CAS.
- M. Wang, W. Yang, X. Li, Y. Xu, L. Zheng, C. Su and B. Liu, ACS Energy Lett., 2021, 6, 379–386 CrossRef CAS.
- X. Zhu, X. Tan, K.-H. Wu, C.-L. Chiang, Y.-C. Lin, Y.-G. Lin, D.-W. Wang, S. Smith, X. Lu and R. Amal, J. Mater. Chem. A, 2019, 7, 14732–14742 RSC.
- X. Li, L. Liu, X. Ren, J. Gao, Y. Huang and B. Liu, Sci. Adv., 2020, 6, eabb6833 CrossRef CAS PubMed.
- X. Li, H. Rong, J. Zhang, D. Wang and Y. Li, Nano Res., 2020, 13, 1842–1855 CrossRef CAS.
- K. Yuan, D. Lützenkirchen-Hecht, L. Li, L. Shuai, Y. Li, R. Cao, M. Qiu, X. Zhuang, M. K. H. Leung, Y. Chen and U. Scherf, J. Am. Chem. Soc., 2020, 142, 2404–2412 CrossRef CAS PubMed.
- H. Shang, X. Zhou, J. Dong, A. Li, X. Zhao, Q. Liu, Y. Lin, J. Pei, Z. Li, Z. Jiang, D. Zhou, L. Zheng, Y. Wang, J. Zhou, Z. Yang, R. Cao, R. Sarangi, T. Sun, X. Yang, X. Zheng, W. Yan, Z. Zhuang, J. Li, W. Chen, D. Wang, J. Zhang and Y. Li, Nat. Commun., 2020, 11, 3049 CrossRef CAS PubMed.
- X. Hai, X. Zhao, N. Guo, C. Yao, C. Chen, W. Liu, Y. Du, H. Yan, J. Li, Z. Chen, X. Li, Z. Li, H. Xu, P. Lyu, J. Zhang, M. Lin, C. Su, S. J. Pennycook, C. Zhang, S. Xi and J. Lu, ACS Catal., 2020, 10, 5862–5870 CrossRef CAS.
- Y. Ha, B. Fei, X. Yan, H. Xu, Z. Chen, L. Shi, M. Fu, W. Xu and R. Wu, Adv. Energy Mater., 2020, 10, 2002592 CrossRef CAS.
- F. Li, G.-F. Han, Y. Bu, H.-J. Noh, J.-P. Jeon, T. J. Shin, S.-J. Kim, Y. Wu, H. Y. Jeong, Z. Fu, Y. Lu and J.-B. Baek, Angew. Chem., Int. Ed., 2020, 59, 23678–23683 CrossRef CAS PubMed.
- Y. Yang, K. Mao, S. Gao, H. Huang, G. Xia, Z. Lin, P. Jiang, C. Wang, H. Wang and Q. Chen, Adv. Mater., 2018, 30, 1801732 CrossRef PubMed.
- S. Liu, Z. Li, C. Wang, W. Tao, M. Huang, M. Zuo, Y. Yang, K. Yang, L. Zhang, S. Chen, P. Xu and Q. Chen, Nat. Commun., 2020, 11, 938 CrossRef CAS PubMed.
- J. Zhang, Y. Zhao, C. Chen, Y.-C. Huang, C.-L. Dong, C.-J. Chen, R.-S. Liu, C. Wang, K. Yan, Y. Li and G. Wang, J. Am. Chem. Soc., 2019, 141, 20118–20126 CrossRef CAS PubMed.
- J. Li, H. Zhang, W. Samarakoon, W. Shan, D. A. Cullen, S. Karakalos, M. Chen, D. Gu, K. L. More, G. Wang, Z. Feng, Z. Wang and G. Wu, Angew. Chem., Int. Ed., 2019, 58, 18971–18980 CrossRef CAS PubMed.
- Y. He, Q. Shi, W. Shan, X. Li, A. J. Kropf, E. C. Wegener, J. Wright, S. Karakalos, D. Su, D. A. Cullen, G. Wang, D. J. Myers and G. Wu, Angew. Chem., Int. Ed., 2021, 60, 9516–9526 CrossRef CAS PubMed.
- Y. Wang, K. Liu, J. Li, X. Yang, J. Hu, T.-S. Chan, X. Qiu, W. Li and M. Liu, Chem. Eng. J., 2022, 429, 132119 CrossRef CAS.
- Z. Li, Z. Zhuang, F. Lv, H. Zhu, L. Zhou, M. Luo, J. Zhu, Z. Lang, S. Feng, W. Chen, L. Mai and S. Guo, Adv. Mater., 2018, 30, 1803220 CrossRef PubMed.
- C. Tang, Y. Jiao, B. Shi, J.-N. Liu, Z. Xie, X. Chen, Q. Zhang and S.-Z. Qiao, Angew. Chem., Int. Ed., 2020, 59, 9171–9176 CrossRef CAS PubMed.
- C. Tang, L. Chen, H. Li, L. Li, Y. Jiao, Y. Zheng, H. Xu, K. Davey and S.-Z. Qiao, J. Am. Chem. Soc., 2021, 143, 7819–7827 CrossRef CAS PubMed.
- G. Daniel, M. Mazzucato, R. Brandiele, L. De Lazzari, D. Badocco, P. Pastore, T. Kosmala, G. Granozzi and C. Durante, ACS Appl. Mater. Interfaces, 2021, 13, 42693–42705 CrossRef CAS PubMed.
- X. Wei, X. Luo, N. Wu, W. Gu, Y. Lin and C. Zhu, Nano Energy, 2021, 84, 105817 CrossRef CAS.
- J. Zhang, M. Zhang, Y. Zeng, J. Chen, L. Qiu, H. Zhou, C. Sun, Y. Yu, C. Zhu and Z. Zhu, Small, 2019, 15, 1900307 CrossRef PubMed.
- C. Shao, L. Wu, H. Zhang, Q. Jiang, X. Xu, Y. Wang, S. Zhuang, H. Chu, L. Sun, J. Ye, B. Li and X. Wang, Adv. Funct. Mater., 2021, 31, 2100833 CrossRef CAS.
- J.-C. Li, H. Zhong, M. Xu, T. Li, L. Wang, Q. Shi, S. Feng, Z. Lyu, D. Liu, D. Du, S. P. Beckman, X. Pan, Y. Lin and M. Shao, Sci. China Mater., 2020, 63, 965–971 CrossRef CAS.
- Y.-C. Wang, Y.-J. Lai, L. Song, Z.-Y. Zhou, J.-G. Liu, Q. Wang, X.-D. Yang, C. Chen, W. Shi, Y.-P. Zheng, M. Rauf and S.-G. Sun, Angew. Chem., Int. Ed., 2015, 54, 9907–9910 CrossRef CAS PubMed.
- Q. Li, W. Chen, H. Xiao, Y. Gong, Z. Li, L. Zheng, X. Zheng, W. Yan, W.-C. Cheong, R. Shen, N. Fu, L. Gu, Z. Zhuang, C. Chen, D. Wang, Q. Peng, J. Li and Y. Li, Adv. Mater., 2018, 30, 1800588 CrossRef PubMed.
- Y. Mun, S. Lee, K. Kim, S. Kim, S. Lee, J. W. Han and J. Lee, J. Am. Chem. Soc., 2019, 141, 6254–6262 CrossRef CAS PubMed.
- H. Sun, S. Liu, M. Wang, T. Qian, J. Xiong and C. Yan, ACS Appl. Mater. Interfaces, 2019, 11, 33054–33061 CrossRef CAS PubMed.
- H. Sun, M. Wang, X. Du, Y. Jiao, S. Liu, T. Qian, Y. Yan, C. Liu, M. Liao, Q. Zhang, L. Meng, L. Gu, J. Xiong and C. Yan, J. Mater. Chem. A, 2019, 7, 20952–20957 RSC.
- K. Yuan, S. Sfaelou, M. Qiu, D. Lützenkirchen-Hecht, X. Zhuang, Y. Chen, C. Yuan, X. Feng and U. Scherf, ACS Energy Lett., 2018, 3, 252–260 CrossRef CAS.
- Y. Chen, S. Ji, S. Zhao, W. Chen, J. Dong, W.-C. Cheong, R. Shen, X. Wen, L. Zheng, A. I. Rykov, S. Cai, H. Tang, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 5422 CrossRef CAS PubMed.
- S. Guo, P. Yuan, J. Zhang, P. Jin, H. Sun, K. Lei, X. Pang, Q. Xu and F. Cheng, Chem. Commun., 2017, 53, 9862–9865 RSC.
- L. Guo, S. Hwang, B. Li, F. Yang, M. Wang, M. Chen, X. Yang, S. G. Karakalos, D. A. Cullen, Z. Feng, G. Wang, G. Wu and H. Xu, ACS Nano, 2021, 15, 6886–6899 CrossRef CAS PubMed.
- H. Shang, Z. Jiang, D. Zhou, J. Pei, Y. Wang, J. Dong, X. Zheng, J. Zhang and W. Chen, Chem. Sci., 2020, 11, 5994–5999 RSC.
- P.-J. Wei, G.-Q. Yu, Y. Naruta and J.-G. Liu, Angew. Chem., Int. Ed., 2014, 53, 6659–6663 CrossRef CAS PubMed.
- L. Xie, X.-P. Zhang, B. Zhao, P. Li, J. Qi, X. Guo, B. Wang, H. Lei, W. Zhang, U.-P. Apfel and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 7576–7581 CrossRef CAS PubMed.
- A. Friedman, L. Landau, S. Gonen, Z. Gross and L. Elbaz, ACS Catal., 2018, 8, 5024–5031 CrossRef CAS.
- L. Gong, H. Zhang, Y. Wang, E. Luo, K. Li, L. Gao, Y. Wang, Z. Wu, Z. Jin, J. Ge, Z. Jiang, C. Liu and W. Xing, Angew. Chem., Int. Ed., 2020, 59, 13923–13928 CrossRef CAS PubMed.
- R. Cao, R. Thapa, H. Kim, X. Xu, M. Gyu Kim, Q. Li, N. Park, M. Liu and J. Cho, Nat. Commun., 2013, 4, 2076 CrossRef PubMed.
- Y. Han, Y. Wang, R. Xu, W. Chen, L. Zheng, A. Han, Y. Zhu, J. Zhang, H. Zhang, J. Luo, C. Chen, Q. Peng, D. Wang and Y. Li, Energy Environ. Sci., 2018, 11, 2348–2352 RSC.
- F. Wang, Y. Zhou, S. Lin, L. Yang, Z. Hu and D. Xie, Nano Energy, 2020, 78, 105128 CrossRef CAS.
- X. Li, C.-S. Cao, S.-F. Hung, Y.-R. Lu, W. Cai, A. I. Rykov, S. Miao, S. Xi, H. Yang, Z. Hu, J. Wang, J. Zhao, E. E. Alp, W. Xu, T.-S. Chan, H. Chen, Q. Xiong, H. Xiao, Y. Huang, J. Li, T. Zhang and B. Liu, Chem, 2020, 6, 3440–3454 CAS.
- Y. Wang, Y.-J. Tang and K. Zhou, J. Am. Chem. Soc., 2019, 141, 14115–14119 CrossRef CAS PubMed.
- X. Yang, D. Xia, Y. Kang, H. Du, F. Kang, L. Gan and J. Li, Adv. Sci., 2020, 7, 2000176 CrossRef CAS PubMed.
- K. Chen, K. Liu, P. An, H. Li, Y. Lin, J. Hu, C. Jia, J. Fu, H. Li, H. Liu, Z. Lin, W. Li, J. Li, Y.-R. Lu, T.-S. Chan, N. Zhang and M. Liu, Nat. Commun., 2020, 11, 4173 CrossRef CAS PubMed.
- J. Zhu and S. Mu, Chem. Commun., 2021, 57, 7869–7881 RSC.
- Z. Zhu, H. Yin, Y. Wang, C.-H. Chuang, L. Xing, M. Dong, Y.-R. Lu, G. Casillas-Garcia, Y. Zheng, S. Chen, Y. Dou, P. Liu, Q. Cheng and H. Zhao, Adv. Mater., 2020, 32, 2004670 CrossRef CAS PubMed.
- X. Han, X. Ling, D. Yu, D. Xie, L. Li, S. Peng, C. Zhong, N. Zhao, Y. Deng and W. Hu, Adv. Mater., 2019, 31, 1905622 CrossRef CAS PubMed.
- Z. Jin, P. Li, Y. Meng, Z. Fang, D. Xiao and G. Yu, Nat. Catal., 2021, 4, 615–622 CrossRef CAS.
- M. Tong, F. Sun, Y. Xie, Y. Wang, Y. Yang, C. Tian, L. Wang and H. Fu, Angew. Chem., Int. Ed., 2021, 60, 14005–14012 CrossRef CAS PubMed.
- V. Jose, H. Hu, E. Edison, W. Manalastas Jr, H. Ren, P. Kidkhunthod, S. Sreejith, A. Jayakumar, J. M. V. Nsanzimana, M. Srinivasan, J. Choi and J.-M. Lee, Small Methods, 2021, 5, 2000751 CrossRef CAS.
- J. Wang, W. Liu, G. Luo, Z. Li, C. Zhao, H. Zhang, M. Zhu, Q. Xu, X. Wang, C. Zhao, Y. Qu, Z. Yang, T. Yao, Y. Li, Y. Lin, Y. Wu and Y. Li, Energy Environ. Sci., 2018, 11, 3375–3379 RSC.
- M. Xiao, Y. Chen, J. Zhu, H. Zhang, X. Zhao, L. Gao, X. Wang, J. Zhao, J. Ge, Z. Jiang, S. Chen, C. Liu and W. Xing, J. Am. Chem. Soc., 2019, 141, 17763–17770 CrossRef CAS PubMed.
- D. Liu, B. Wang, H. Li, S. Huang, M. Liu, J. Wang, Q. Wang, J. Zhang and Y. Zhao, Nano Energy, 2019, 58, 277–283 CrossRef CAS.
- H. Li, J. Wang, R. Qi, Y. Hu, J. Zhang, H. Zhao, J. Zhang and Y. Zhao, Appl. Catal., B, 2021, 285, 119778 CrossRef CAS.
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS PubMed.
- G. Yang, J. Zhu, P. Yuan, Y. Hu, G. Qu, B.-A. Lu, X. Xue, H. Yin, W. Cheng, J. Cheng, W. Xu, J. Li, J. Hu, S. Mu and J.-N. Zhang, Nat. Commun., 2021, 12, 1734 CrossRef CAS PubMed.
- M. Xiao, H. Zhang, Y. Chen, J. Zhu, L. Gao, Z. Jin, J. Ge, Z. Jiang, S. Chen, C. Liu and W. Xing, Nano Energy, 2018, 46, 396–403 CrossRef CAS.
- W. Ye, S. Chen, Y. Lin, L. Yang, S. Chen, X. Zheng, Z. Qi, C. Wang, R. Long, M. Chen, J. Zhu, P. Gao, L. Song, J. Jiang and Y. Xiong, Chem, 2019, 5, 2865–2878 CAS.
- N. Zhang, T. Zhou, J. Ge, Y. Lin, Z. Du, C. a. Zhong, W. Wang, Q. Jiao, R. Yuan, Y. Tian, W. Chu, C. Wu and Y. Xie, Matter, 2020, 3, 509–521 CrossRef.
- C. E. Szakacs, M. Lefèvre, U. I. Kramm, J.-P. Dodelet and F. Vidal, Phys. Chem. Chem. Phys., 2014, 16, 13654–13661 RSC.
- K. Liu, G. Wu and G. Wang, J. Phys. Chem. C, 2017, 121, 11319–11324 CrossRef CAS.
- H. Zhang, H. T. Chung, D. A. Cullen, S. Wagner, U. I. Kramm, K. L. More, P. Zelenay and G. Wu, Energy Environ. Sci., 2019, 12, 2548–2558 RSC.
- X. Wang, Y. Jia, X. Mao, D. Liu, W. He, J. Li, J. Liu, X. Yan, J. Chen, L. Song, A. Du and X. Yao, Adv. Mater., 2020, 32, 2000966 CrossRef CAS PubMed.
- X. Fu, N. Li, B. Ren, G. Jiang, Y. Liu, F. M. Hassan, D. Su, J. Zhu, L. Yang, Z. Bai, Z. P. Cano, A. Yu and Z. Chen, Adv. Energy Mater., 2019, 9, 1803737 CrossRef.
- X. Yu, S. Lai, S. Xin, S. Chen, X. Zhang, X. She, T. Zhan, X. Zhao and D. Yang, Appl. Catal., B, 2021, 280, 119437 CrossRef CAS.
- T. He, B. Lu, Y. Chen, Y. Wang, Y. Zhang, J. L. Davenport, A. P. Chen, C.-W. Pao, M. Liu and Z. Sun, Research, 2019, 2019, 6813585 CAS.
- K. Khan, X. Yan, Q. Yu, S.-H. Bae, J. J. White, J. Liu, T. Liu, C. Sun, J. Kim, H.-M. Cheng, Y. Wang, B. Liu, K. Amine, X. Pan and Z. Luo, Nano Energy, 2021, 90, 106488 CrossRef CAS.
- Y. He, S. Hwang, D. A. Cullen, M. A. Uddin, L. Langhorst, B. Li, S. Karakalos, A. J. Kropf, E. C. Wegener, J. Sokolowski, M. Chen, D. Myers, D. Su, K. L. More, G. Wang, S. Litster and G. Wu, Energy Environ. Sci., 2019, 12, 250–260 RSC.
- S. Yuan, J. Zhang, L. Hu, J. Li, S. Li, Y. Gao, Q. Zhang, L. Gu, W. Yang, X. Feng and B. Wang, Angew. Chem., Int. Ed., 2021, 60, 21685–21690 CrossRef CAS PubMed.
- J. Wu, H. Zhou, Q. Li, M. Chen, J. Wan, N. Zhang, L. Xiong, S. Li, B. Y. Xia, G. Feng, M. Liu and L. Huang, Adv. Energy Mater., 2019, 9, 1900149 CrossRef.
- C. Xia, Y. Qiu, Y. Xia, P. Zhu, G. King, X. Zhang, Z. Wu, J. Y. Kim, D. A. Cullen, D. Zheng, P. Li, M. Shakouri, E. Heredia, P. Cui, H. N. Alshareef, Y. Hu and H. Wang, Nat. Chem., 2021, 13, 887–894 CrossRef CAS PubMed.
- H. Yin, H. Xia, S. Zhao, K. Li, J. Zhang and S. Mu, Energy Environ. Mater., 2021, 4, 5–18 CrossRef CAS.
- X. Wang, H. Zhang, H. Lin, S. Gupta, C. Wang, Z. Tao, H. Fu, T. Wang, J. Zheng, G. Wu and X. Li, Nano Energy, 2016, 25, 110–119 CrossRef CAS.
- X. Xu, Z. Xia, X. Zhang, R. Sun, X. Sun, H. Li, C. Wu, J. Wang, S. Wang and G. Sun, Appl. Catal., B, 2019, 259, 118042 CrossRef CAS.
- F. Xiao, G.-L. Xu, C.-J. Sun, M. Xu, W. Wen, Q. Wang, M. Gu, S. Zhu, Y. Li, Z. Wei, X. Pan, J. Wang, K. Amine and M. Shao, Nano Energy, 2019, 61, 60–68 CrossRef CAS.
- X. Han, X. Ling, Y. Wang, T. Ma, C. Zhong, W. Hu and Y. Deng, Angew. Chem., Int. Ed., 2019, 58, 5359–5364 CrossRef CAS PubMed.
- Y. Zheng and S.-Z. Qiao, Natl. Sci. Rev., 2018, 5, 626–627 CrossRef CAS.
- V. Armel, S. Hindocha, F. Salles, S. Bennett, D. Jones and F. Jaouen, J. Am. Chem. Soc., 2017, 139, 453–464 CrossRef CAS PubMed.
- T. Al-Zoubi, Y. Zhou, X. Yin, B. Janicek, C. Sun, C. E. Schulz, X. Zhang, A. A. Gewirth, P. Huang, P. Zelenay and H. Yang, J. Am. Chem. Soc., 2020, 142, 5477–5481 CrossRef CAS PubMed.
- Y. Zhou, G. Chen, Q. Wang, D. Wang, X. Tao, T. Zhang, X. Feng and K. Müllen, Adv. Funct. Mater., 2021, 31, 2102420 CrossRef CAS.
- Q. Liu, X. Liu, L. Zheng and J. Shui, Angew. Chem., Int. Ed., 2018, 57, 1204–1208 CrossRef CAS PubMed.
- C.-C. Hou, H.-F. Wang, C. Li and Q. Xu, Energy Environ. Sci., 2020, 13, 1658–1693 RSC.
- H. Zhang, S. Hwang, M. Wang, Z. Feng, S. Karakalos, L. Luo, Z. Qiao, X. Xie, C. Wang, D. Su, Y. Shao and G. Wu, J. Am. Chem. Soc., 2017, 139, 14143–14149 CrossRef CAS PubMed.
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 6937–6941 CrossRef CAS PubMed.
- J. Wang, G. Han, L. Wang, L. Du, G. Chen, Y. Gao, Y. Ma, C. Du, X. Cheng, P. Zuo and G. Yin, Small, 2018, 14, 1704282 CrossRef PubMed.
- R. Jiang, L. Li, T. Sheng, G. Hu, Y. Chen and L. Wang, J. Am. Chem. Soc., 2018, 140, 11594–11598 CrossRef CAS PubMed.
- J. Shan, C. Ye, S. Chen, T. Sun, Y. Jiao, L. Liu, C. Zhu, L. Song, Y. Han, M. Jaroniec, Y. Zhu, Y. Zheng and S.-Z. Qiao, J. Am. Chem. Soc., 2021, 143, 5201–5211 CrossRef CAS PubMed.
- S. Chen, N. Zhang, C. W. Narváez Villarrubia, X. Huang, L. Xie, X. Wang, X. Kong, H. Xu, G. Wu, J. Zeng and H.-L. Wang, Nano Energy, 2019, 66, 104164 CrossRef CAS.
- X. Zhang, S. Zhang, Y. Yang, L. Wang, Z. Mu, H. Zhu, X. Zhu, H. Xing, H. Xia, B. Huang, J. Li, S. Guo and E. Wang, Adv. Mater., 2020, 32, 1906905 CrossRef CAS PubMed.
- F. Jaouen, M. Lefèvre, J.-P. Dodelet and M. Cai, J. Phys. Chem. B, 2006, 110, 5553–5558 CrossRef CAS PubMed.
- F. Jaouen, J. Herranz, M. Lefèvre, J.-P. Dodelet, U. I. Kramm, I. Herrmann, P. Bogdanoff, J. Maruyama, T. Nagaoka, A. Garsuch, J. R. Dahn, T. Olson, S. Pylypenko, P. Atanassov and E. A. Ustinov, ACS Appl. Mater. Interfaces, 2009, 1, 1623–1639 CrossRef CAS PubMed.
- H.-W. Liang, W. Wei, Z.-S. Wu, X. Feng and K. Müllen, J. Am. Chem. Soc., 2013, 135, 16002–16005 CrossRef CAS PubMed.
- Z. Yang, Y. Wang, M. Zhu, Z. Li, W. Chen, W. Wei, T. Yuan, Y. Qu, Q. Xu, C. Zhao, X. Wang, P. Li, Y. Li, Y. Wu and Y. Li, ACS Catal., 2019, 9, 2158–2163 CrossRef CAS.
- M. Qiao, Y. Wang, Q. Wang, G. Hu, X. Mamat, S. Zhang and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 2688–2694 CrossRef CAS PubMed.
- S. Fu, C. Zhu, D. Su, J. Song, S. Yao, S. Feng, M. H. Engelhard, D. Du and Y. Lin, Small, 2018, 14, 1703118 CrossRef PubMed.
- T. Sun, S. Zhao, W. Chen, D. Zhai, J. Dong, Y. Wang, S. Zhang, A. Han, L. Gu, R. Yu, X. Wen, H. Ren, L. Xu, C. Chen, Q. Peng, D. Wang and Y. Li, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 12692–12697 CrossRef CAS PubMed.
- F. Luo, S. Wagner, I. Onishi, S. Selve, S. Li, W. Ju, H. Wang, J. Steinberg, A. Thomas, U. I. Kramm and P. Strasser, Chem. Sci., 2021, 12, 384–396 RSC.
- M. Primbs, Y. Sun, A. Roy, D. Malko, A. Mehmood, M.-T. Sougrati, P.-Y. Blanchard, G. Granozzi, T. Kosmala, G. Daniel, P. Atanassov, J. Sharman, C. Durante, A. Kucernak, D. Jones, F. Jaouen and P. Strasser, Energy Environ. Sci., 2020, 13, 2480–2500 RSC.
- X. Xie, L. Peng, H. Yang, G. I. N. Waterhouse, L. Shang and T. Zhang, Adv. Mater., 2021, 33, 2101038 CrossRef CAS PubMed.
- L. Shang, H. Yu, X. Huang, T. Bian, R. Shi, Y. Zhao, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2016, 28, 1668–1674 CrossRef CAS PubMed.
- L. Jiao, R. Zhang, G. Wan, W. Yang, X. Wan, H. Zhou, J. Shui, S.-H. Yu and H.-L. Jiang, Nat. Commun., 2020, 11, 2831 CrossRef CAS PubMed.
- H. Adabi, A. Shakouri, N. Ul Hassan, J. R. Varcoe, B. Zulevi, A. Serov, J. R. Regalbuto and W. E. Mustain, Nat. Energy, 2021, 6, 834–843 CrossRef CAS.
- H. Shen, E. Gracia-Espino, J. Ma, H. Tang, X. Mamat, T. Wagberg, G. Hu and S. Guo, Nano Energy, 2017, 35, 9–16 CrossRef CAS.
- S. Fu, C. Zhu, J. Song, M. H. Engelhard, X. Li, P. Zhang, H. Xia, D. Du and Y. Lin, Nano Res., 2017, 10, 1888–1895 CrossRef CAS.
- G. Chen, P. Liu, Z. Liao, F. Sun, Y. He, H. Zhong, T. Zhang, E. Zschech, M. Chen, G. Wu, J. Zhang and X. Feng, Adv. Mater., 2020, 32, 1907399 CrossRef CAS PubMed.
- X. Wan, X. Liu, Y. Li, R. Yu, L. Zheng, W. Yan, H. Wang, M. Xu and J. Shui, Nat. Catal., 2019, 2, 259–268 CrossRef CAS.
- L. Jiao, J. Li, L. L. Richard, Q. Sun, T. Stracensky, E. Liu, M. T. Sougrati, Z. Zhao, F. Yang, S. Zhong, H. Xu, S. Mukerjee, Y. Huang, D. A. Cullen, J. H. Park, M. Ferrandon, D. J. Myers, F. Jaouen and Q. Jia, Nat. Mater., 2021, 20, 1385–1391 CrossRef CAS PubMed.
- S. Liu, M. Wang, X. Yang, Q. Shi, Z. Qiao, M. Lucero, Q. Ma, K. L. More, D. A. Cullen, Z. Feng and G. Wu, Angew. Chem., Int. Ed., 2020, 59, 21698–21705 CrossRef CAS PubMed.
- Q. Wang, Y. Yang, F. Sun, G. Chen, J. Wang, L. Peng, W.-T. Chen, L. Shang, J. Zhao, D. Sun-Waterhouse, T. Zhang and G. I. N. Waterhouse, Adv. Energy Mater., 2021, 11, 2100219 CrossRef CAS.
- Z.-F. Huang, J. Song, S. Dou, X. Li, J. Wang and X. Wang, Matter, 2019, 1, 1494–1518 CrossRef.
- D. J. Martin, B. Q. Mercado and J. M. Mayer, Sci. Adv., 2020, 6, eaaz3318 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |