
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUncommon carbene insertion reactions
Ming-Yao
Huang
and
Shou-Fei
Zhu
 *
*
Frontiers Science Center for New Organic Matter, State Key Laboratory and Institute of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: sfzhu@nankai.edu.cn
First published on 2nd November 2021
Abstract
Transition-metal-catalysed carbene insertion reaction is a straightforward and efficient protocol for the construction of carbon–carbon or carbon–heteroatom bonds. Compared to the intensively studied and well-established “common” carbene insertion reactions, including carbene insertion into C–H, Si–H, N–H, O–H, and S–H bonds, several “uncommon” carbene insertion reactions, including carbene insertion into B–H, Sn–H, Ge–H, P–H, F–H, C–C, and M–M bonds, have been neglected for a long time. However, more and more studies on uncommon carbene insertion reactions have been disclosed recently, and clearly demonstrate the great synthetic potential of these reactions. The current perspective reviews the history and the newest advances of uncommon carbene insertion reactions, discusses their potential applications and challenges, and also presents an outlook of this promising field.
Introduction
Carbene or carbenoid is an important active intermediate in organic reactions. Among them, free carbene and Fischer-type1,2 carbene species are electrophilic and can undergo insertion reactions with various σ-bonds. These carbene insertion reactions, especially those promoted by transition metal catalysts, are a straightforward and efficient method for the construction of carbon–carbon or carbon–heteroatom bonds. In the past few decades, the C–H bond and various ubiquitous X–H (X = Si, N, O, S) bonds were generally used as “common” traps for carbene insertion reactions, which marvellously promoted the development of carbene chemistry. There are quite some excellent and elegant reviews covering these areas.3–8 However, compared to those that are well established, some “uncommon” carbene insertion reactions, including carbene insertion into B–H, Sn–H, Ge–H, P–H, F–H, C–C, and M–M bonds, which also displayed great synthetic potential have been neglected for a long time. There may be two main reasons for this situation. On the one hand, because the polarity of these chemical bonds is very large (e.g., B–H, Sn–H, Ge–H, P–H, and F–H) or very small (e.g., C–C and M–M), special catalysts or conditions are required to enable the corresponding insertion reactions with carbenes. On the other hand, only a few types of carbenes have been applied in these uncommon insertion reactions, which leads to poor structural diversity of the insertion products and makes the reactions difficult to use in synthesis. Fortunately, recent increasing studies on uncommon carbene insertion reactions have clearly demonstrated their great synthetic potential. Herein, we set our sights on uncommon carbene insertion reactions, especially the inadequately investigated X–H (X = B, Ge, Sn, P, F) bond, C–C σ bond and metal–metal bond insertion reactions. It's noteworthy that the homologation reaction between organoborons and organolithium reagents with an α-leaving group9,10 or diazo compounds11–14 is a formal C–B bond insertion reaction with significant synthetic potential, but this reaction undergoes a carbene-free mechanism and will not be discussed here.Carbene insertion into X–H (X = B, Ge, Sn) bonds
Since B, Ge, and Sn are the diagonal or congener elements of Si, B–H, Ge–H, and Sn–H bonds are also suitable σ-bonds to capture carbene intermediates to form corresponding organoboron, organogermanium, and organotin compounds. Mechanistically, the above X–H bonds don't have lone pairs on their heteroatoms and exhibit a similar insertion mechanism to that of the Si–H bond.B–H bond insertion
The B–H bond insertion is a reaction that provides a straightforward approach to construct the C–B bond and bridges carbene chemistry with organoboron synthesis. Generally speaking, the electron deficient B–H bond is inert to electrophilic carbene species, but after forming an adduct with phosphine, amine or N-heterocyclic carbene (NHC), the electron density of the B–H bond is increased and thus the insertion of the B–H bond becomes possible.As early as 1983, Jones et al.15 reported that the carbethoxylcarbene generated from ethyl diazoacetate could insert into the B–H bond of carborane under irradiation, heat or in the presence of CuCl (Scheme 1a). Although the site selectivity of the reaction is poor, this reaction represents the pioneering attempt in the B–H bond insertion reaction. Later in 1990s and 2000s, classical dichlorocarbene,16,17 and methylene–carbene18 species were found to be efficient to react with borane amine or phosphine adducts, affording various methyl borane derivatives (Scheme 1b). In 2003, Sierra et al.19 reported the reaction of alkynyl alkoxy pentacarbonyl chromium carbene toward the B–H bond of NaCNBH3, which unambiguously proved that Fischer-type metal carbene is reactive in the B–H bond insertion reaction (Scheme 1c).
 | ||
| Scheme 1 Early reports on B–H bond insertion reactions. | ||
However, the lack of effective catalytic protocols in B–H bond insertion reactions resulted in a limited choice of carbene precursors, low efficiency and poor selectivity. These drawbacks dramatically restricted the synthetic utilities of the B–H bond insertion reaction. In 2013, Curran et al.20 reported a Rh-catalysed B–H bond insertion reaction of α-diazocarbonyls and NHC borane adducts (Scheme 2a). Almost at the same time, Zhu and Zhou et al.21 independently reported a Cu-catalysed B–H bond insertion reaction between α-diazoesters and phosphine borane adducts or amine borane adducts along with its enantioselective version by using a chiral spirobisoxazoline ligand (Ra,S,S)-2, providing an efficient approach for chiral organoboranes with enantioselectivity up to 94% ee (Scheme 2b). The ester moiety of the diazoesters plays crucial roles in chiral induction. A concerted mechanism was proposed for the Cu-catalysed B–H bond insertion reaction: copper complex catalyses the decomposition of the diazo group to form a copper carbene, which then undergoes a concerted B–H bond insertion through a 3-member-ring transition state to afford the insertion product (Scheme 2c). A negligible KIE was observed and suggested that the H transfer step was not involved in the rate-determining step, which indicated that the former step, the diazo-decomposition, was probably the rate-determining step. These pioneering studies opened up the door for transition-metal-catalysed B–H bond insertions with various carbene precursors.
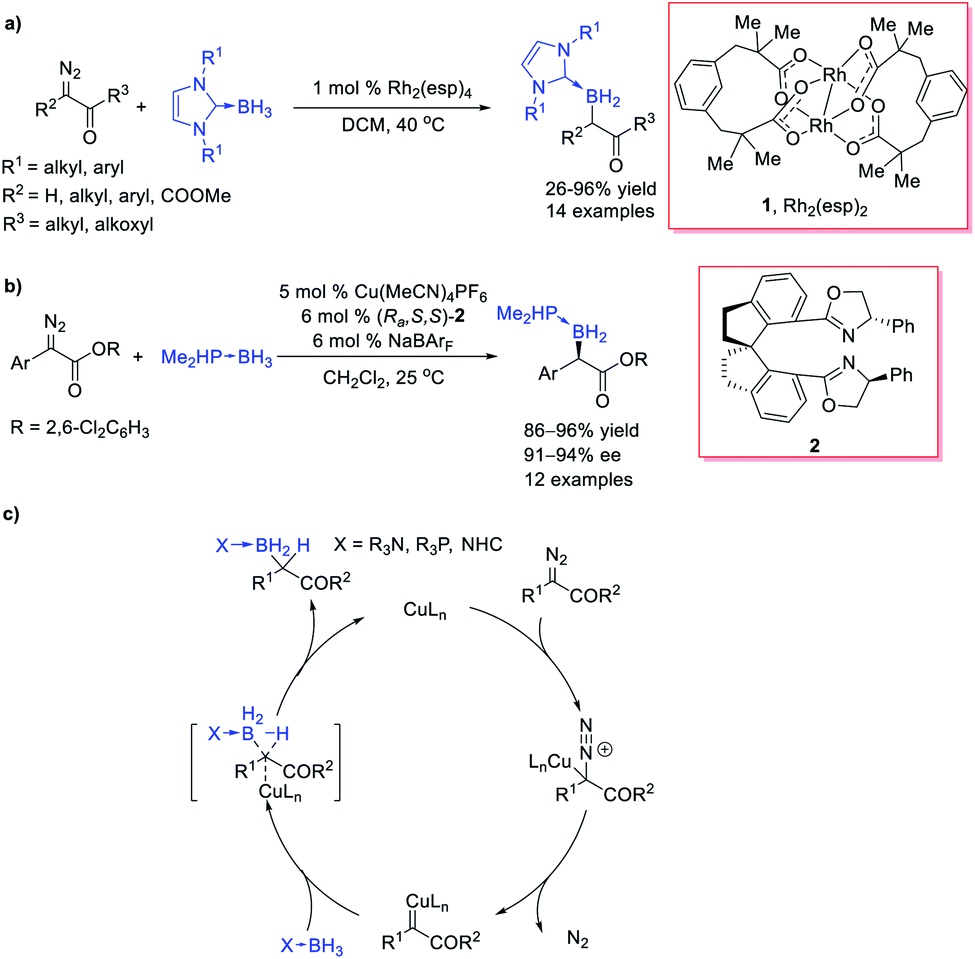 | ||
| Scheme 2 Rh or Cu-catalysed B–H bond insertion of diazocarbonyls. | ||
Xu et al.22 reported a highly enantioselective B–H bond insertion reaction of α-aryl-α-diazoesters and α-aryl-α-diazoketones under the catalysis of C1-symmetric chiral diene-ligated Rh(I) complexes, affording various α-boryl esters and ketones with satisfactory yields and enantioselectivities (up to 99% ee) (Scheme 3a). Almost at the same time, Zhu et al.23 disclosed a Cu/chiral spiroxazoline catalysed B–H bond insertion reaction of α-aryl-α-diazoketones and α-methyl-α-diazoketones with good enantioselectivities (up to 83% ee) (Scheme 3b). The ortho-substituents of α-aryl of diazoketones are helpful for getting higher enantioselectivity. Iwasa et al.24 developed a series of Ru(II)-phenyloxazoline complexes, which displayed excellent performance in the asymmetric B–H bond insertion reaction of dinaphthylenyl diazopropionate with various borane adducts (Scheme 3c). The bulky ester group is indispensable for satisfactory enantioselectivity. Vilotijevic et al.25 further expanded the scope to the α-alkenyl-diazocarbonyls, providing an efficient approach to allylboranes, which were versatile reagents in organic synthesis (Scheme 3d). Unfortunately, the preliminary attempt of the asymmetric version of the reaction of Scheme 3d only afforded very modest enantioselectivity (30% ee).
 | ||
| Scheme 3 Transition-metal-catalysed B–H bond insertion of diazocarbonyls. | ||
Gouverneur et al.26 reported an example of asymmetric B–H bond insertion of α-CF3–α-aryl diazomethanes and achieved good enantioselectivity (81% ee) (Scheme 4a). Arnold et al.27 realized a biocatalytic B–H bond insertion of α-diazo propionates and α-CF3–α-phenyl diazomethane with the NHC borane adduct. The Escherichia coli cells harbouring engineered variants of wildtype cytochrome c from Rhodothermus marinus (Rma cyt c) were found to exhibit excellent activity (providing up to 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 turnovers, a turnover frequency of 6100 h−1) as well as selectivity (a 99:1 enantiomeric ratio and 100% chemoselectivity) in this B–H bond insertion reaction (Scheme 4b). This biocatalytic system is suitable for gram-scale biosynthesis and either enantiomer of the organoborane products could be obtained through tuning the biocatalyst. Using a similar directed evolution strategy, Arnold and Houk et al.28 successfully expanded the scope to α-alkyl-α-CF3 diazomethanes, establishing a biocatalytic platform for the synthesis of α-trifluoromethylated organoborons (Scheme 4c).
300 turnovers, a turnover frequency of 6100 h−1) as well as selectivity (a 99:1 enantiomeric ratio and 100% chemoselectivity) in this B–H bond insertion reaction (Scheme 4b). This biocatalytic system is suitable for gram-scale biosynthesis and either enantiomer of the organoborane products could be obtained through tuning the biocatalyst. Using a similar directed evolution strategy, Arnold and Houk et al.28 successfully expanded the scope to α-alkyl-α-CF3 diazomethanes, establishing a biocatalytic platform for the synthesis of α-trifluoromethylated organoborons (Scheme 4c).
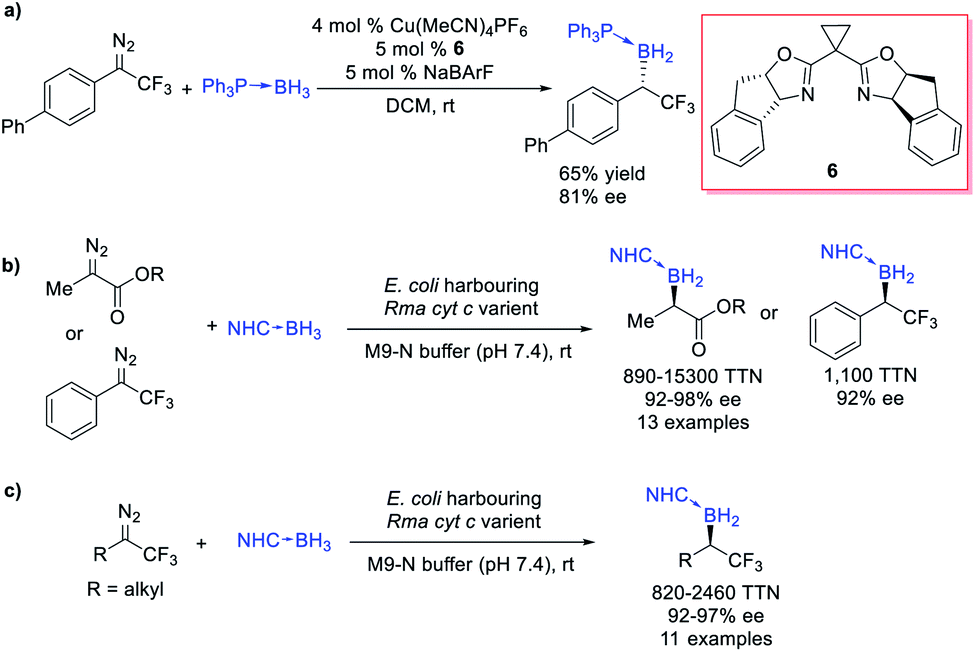 | ||
| Scheme 4 B–H bond insertion of diazocarbonyls and α-CF3 diazo compounds. | ||
Diazo compounds have long been regarded as efficient and convenient precursors in carbene transfer reactions. However, the explosive and toxic nature restricts their applications in B–H bond insertion reactions, and most importantly, the need of stabilizing the electron-withdrawing group greatly limits the structural diversity of organoborane products. To overcome these barriers, various carbene precursors were developed in the B–H bond insertion reaction.
Zhu et al.29 reported a Rh-catalysed B–H bond insertion reaction of unstabilized diazo compounds generated in situ from N-tosylhydrazones. By using chiral dirhodium catalysts 7 or 8, the reaction exhibited good yields and high enantioselectivities (up to 96% ee) (Scheme 5a). The protocol also exhibited good functional group tolerance and enabled one-pot transformation of a carbonyl group to a boryl group enantioselectively. This catalytic system is sensitive to the steric property of the diazo compounds, and the bulkier substituents of the diazo compounds resulted in lower yields and enantioselectivities. The B–H bond insertion products could be easily converted into chiral alcohols and other widely used organoboron reagents without the loss of optical purity. Quite recently, the same group developed a Rh-catalysed asymmetric B–H bond insertion reaction of borane adducts and diaryl diazomethanes as a straightforward protocol for various gem-diarylmethine borane compounds with high yield, excellent enantioselectivity, and good functional group tolerance (Scheme 5b).30 The borane compounds synthesized by this method could be efficiently transformed into synthetically important diaryl methanol, diaryl methyl amine, and triaryl methane derivatives with good stereospecificity. Hammett analysis indicated that the enantioselectivity depended strongly on electronic differences between the two aryl groups of diazomethane.
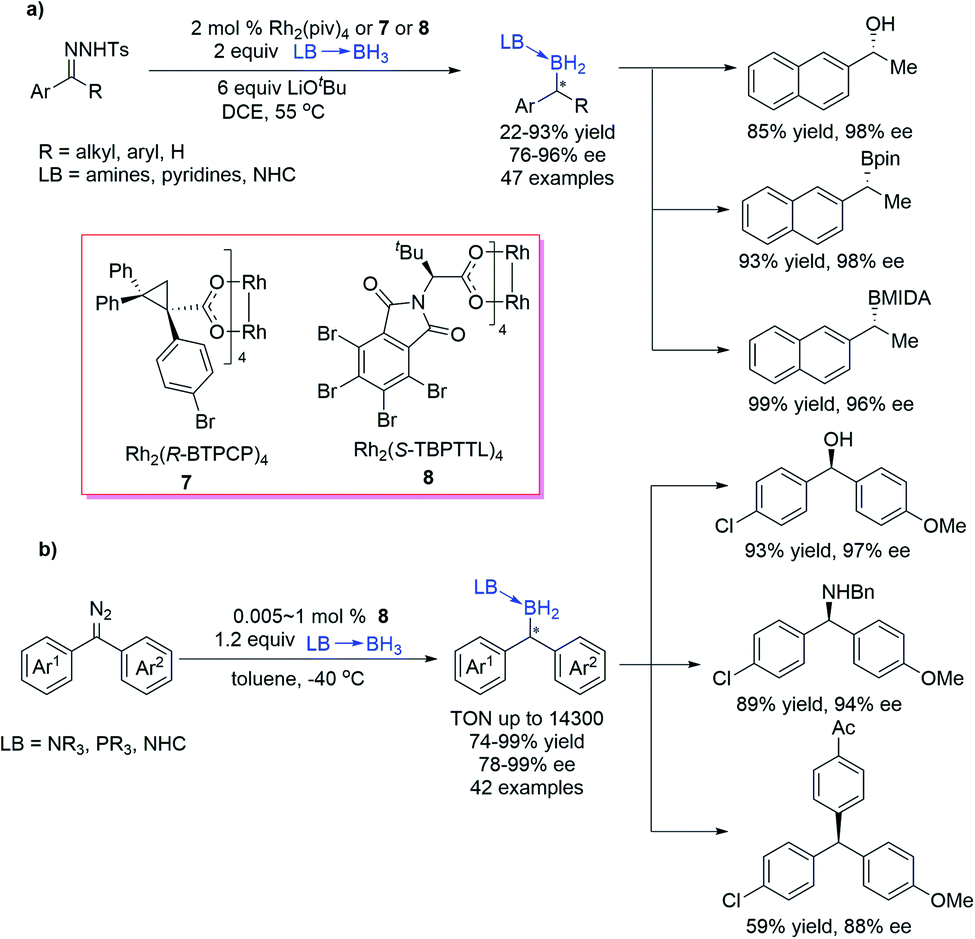 | ||
| Scheme 5 Rh-catalysed B–H bond insertion of N-tosylhydrazones. | ||
Sulfoxonium ylides can form metal carbene intermediates smoothly through releasing dimethyl sulfoxide. Wang, Wu et al.31 reported an iridium-catalysed B–H bond insertion between borane adducts and α-carbonyl activated sulfoxonium ylides (Scheme 6a). In addition, analogues of sulfoxonium ylides, such as sulfonium salts and sulfonium ylides could also be amenable to the reaction. Zhang S., Zhang L et al.32 independently reported a similar transformation and realized the B–H bond insertion of bulkier α-ester disubstituted sulfoxonium ylides (Scheme 6b). This reaction featured a broad substrate scope and high functional group compatibility and could be applied in late-stage modification of structurally complex drug compounds.
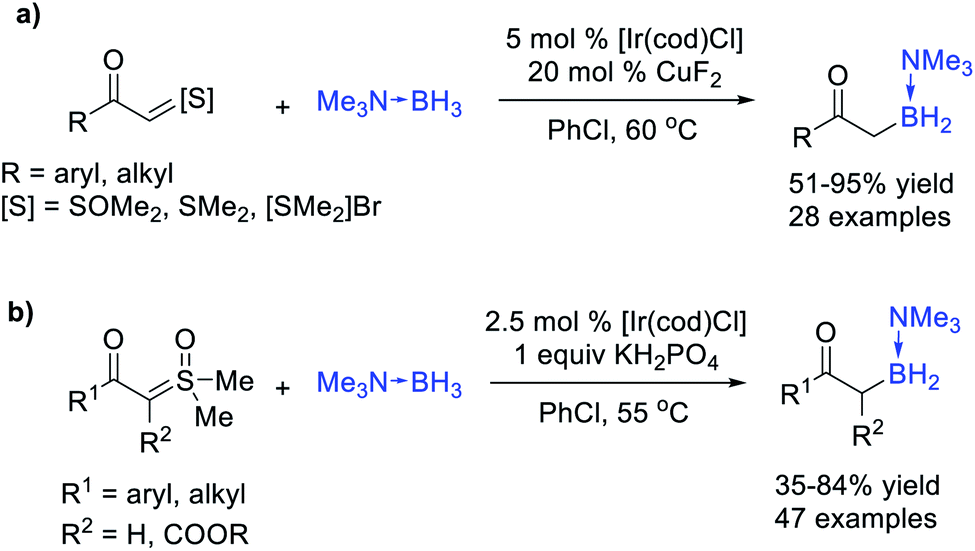 | ||
| Scheme 6 Ir-catalysed B–H bond insertion of sulfoxonium ylides. | ||
Compared with diazo compounds, alkynes are safe and easily available carbene precursors. Zhu, Zhou et al.33 developed a B–H bond insertion reaction using conjugated alkynes as carbene precursors in the presence of CuCl as a catalyst (Scheme 7a). By using chiral dirhodium catalysts, a highly enantioselective transformation was rendered feasible (with up to 96% ee). The reaction exhibits excellent atom-economy and efficiency (up to 99% yield). Chiral organoboranes could be conveniently transformed to various versatile borates and diaryl methanol compounds with well retained enantiopurity, which demonstrates its potential utility in organic synthesis. This reaction is triggered by Cu(I) catalysed 5-exo-dig cyclization to form an α-furyl carbene intermediate, which undergoes concerted B–H bond insertion to afford the corresponding product. Mechanistic studies indicated that the cyclization process was probably the rate-determining step.
 | ||
| Scheme 7 B–H bond insertion reactions using alkynes as precursors. | ||
In addition to functionalized alkynes, terminal alkynes could also serve as potential carbene precursors for B–H bond insertion reactions. Zhu, Zhou et al.34 reported an Au-catalysed oxidative coupling of terminal alkynes and borane adducts for the synthesis of an α-boryl carbonyl compound (Scheme 7b). High efficiency and specific regioselectivity were observed in this reaction, and excellent functional group tolerance made this protocol effective in late-stage modification. The reaction is initiated by Au-promoted oxidation of the alkyne by the N-oxide, which generates a highly active gold carbene intermediate. Insertion of this gold carbene intermediate into the electron-rich B–H bond of the borane adduct via a 3-member-ring transition state gives the product and regenerates the catalyst. The result of the kinetic isotopic effect experiment indicates that B–H bond insertion is a fast process, and quinoline dissociation may be involved in the rate-limiting step.
Transition-metal-catalysed ring-opening of cyclopropenes generates vinyl metal carbenes, which could be captured by borane adducts to produce allylboranes. Quit recently, Zhu et al.35 presented a Cu(I)-catalysed ring-opening/borylation reaction of cyclopropenes, delivering γ,γ-disubstituted allylboranes with moderate to good yields and regioselectivities (Scheme 8a). Interestingly, the regioselectivity arises from the steric bias between the two substituents installed at the 3,3-position of cyclopropenes in the ring-opening step, and for 3-alkyl-3-aryl cyclopropenes, the regioselectivity can be further regulated because the Z-carbene tends to undergo intramolecular C–H bond insertion, leaving the E-type products only (Scheme 8b). The allylborane product could react with an aldehyde in the presence of water, affording corresponding homoallylic alcohols with good yield and specific stereoselectivity. Mechanistic studies revealed that the ring-opening process is probably the rate-determining step (RDS).
 | ||
| Scheme 8 Cu-catalysed B–H bond insertion of cyclopropenes. | ||
In general, the electron deficient B–H bond of non-ligated boranes (i.e. HBpin) is inactive towards electrophilic carbene, but it could serve as a suitable trapper for some special nucleophilic carbene species. Glorius et al.36 reported a visible-light-induced B–H bond insertion of HBpin with acylsilanes (Scheme 9). Under the irradiation of visible light, nucleophilic α-siloxycarbenes were generated from acylsilane through 1,2-Brook rearrangement, and then directly insert into the B–H bond of electron deficient HBpin to access valuable α-alkoxyorganoboronate esters with almost quantitative yields. This metal-free and operationally simple protocol features a broad substrate scope, excellent functional group tolerance and good scalability.
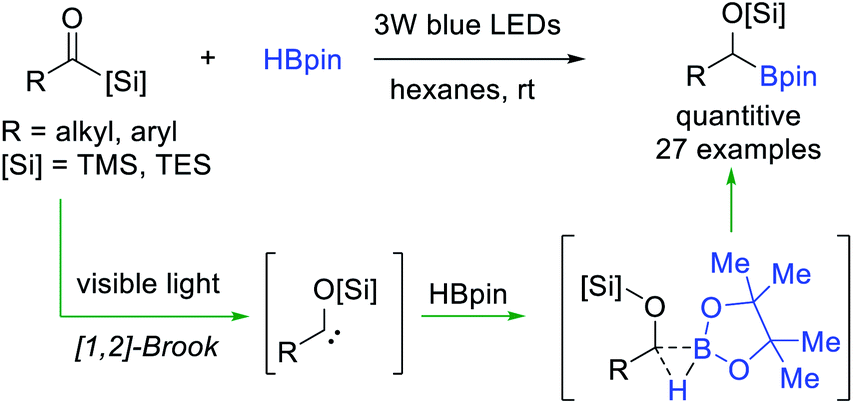 | ||
| Scheme 9 Visible-light-induced B–H bond insertion of acylsilanes with HBpin. | ||
The carbene intermediates mentioned above are all saturated carbenes (that is, disubstituted carbene), actually, unsaturated carbenes can also be utilized in the B–H bond insertion reaction to afford alkenyl boranes. Zhu, Zhou et al.37 developed a protocol of the B–H bond insertion of alkylidene carbenes, enabling construction of the C(sp2)–B bond (Scheme 10). Starting from readily available alkenyl triflates, free alkylidene carbene was formed through base triggered 1,1-elimination, which then undergoes the B–H bond insertion process, producing alkenyl boranes with acceptable yields. The intramolecular insertion provides an efficient approach for the construction of B–N heterocycles, meanwhile avoiding the potential regioselective issues. Notably, not only the transformation of alkenyl boranes to valuable alkenyl borates was convenient, but the borane products themselves served as an effective partner in the Pd-catalysed C–C or Cu-catalysed C–N coupling reactions.
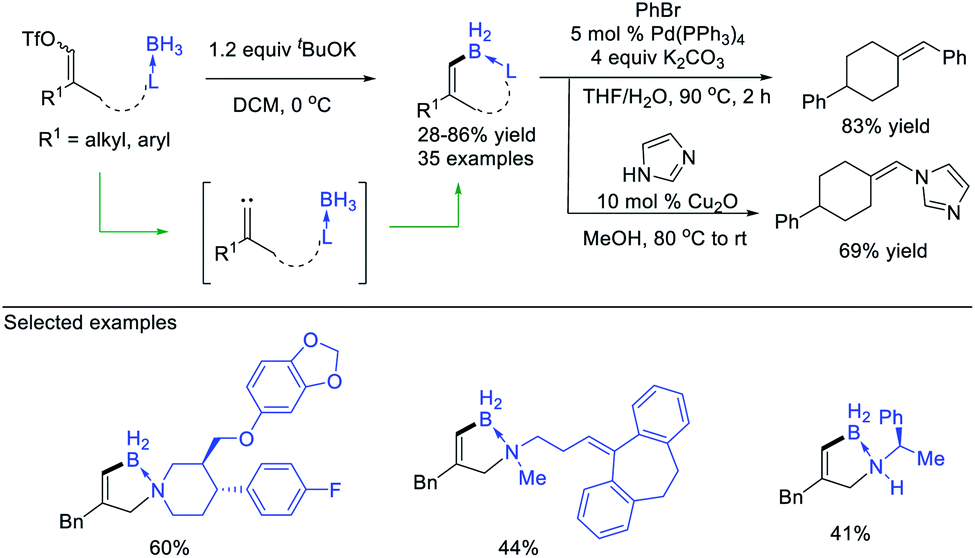 | ||
| Scheme 10 B–H bond insertion of alkylidene carbenes. | ||
Sn–H and Ge–H bond insertion
Similar to B–H bond insertion reactions, dihalocarbene,38,39 and methylcarbene40 species were first utilized in the Ge–H and Sn–H bond insertion reaction. Although transition-metal-catalysed Si–H bond insertion of diazo compounds was intensively investigated in the past few decades, the Ge–H and Sn–H bond insertion reactions received rare concern, which was probably due to the inevitable demetallization process of the resulting α-EWG (electron-withdrawing group) substituted products in work up procedures.41 However, the development of novel carbene intermediates and catalytic methodologies have attracted increasing attention for these reactions in recent years.Wang et al.42 reported a metal-free coupling reaction of N-tosylhydrazones with Bu3SnH in the presence of base and phase transfer catalysts, affording a series of functionalized benzyl- and alkyltributylstannanes in moderate to good yields (up to 85% yield) (Scheme 11a). The steric hindrance of the diazo compounds exhibited a negative effect on the yields. The benzyltributylstannanes could be subjected to Stille cross-coupling without purification and afford diarylmethane derivatives in good yields (up to 75% yield). Mechanistic experiments suggested that a free-carbene-process is likely involved in the stannylation transformation. Almost at the same time, Bi et al.43 realized an Ag-catalysed Sn–H and Ge–H bond insertion reaction of in situ generated benzylic carbenes from N-nosylhydrazones with Bu3SnH and Bu3GeH at lower temperature (Scheme 11b). Xu, Che et al.44 reported an Fe(TPP)Cl-prompted insertion reaction of carbene species generated from N-tosylhydrazones into the Ge–H or Sn–H bond (Scheme 11c). N-Tosylhydrazones derived from aldehydes afforded better results than N-tosylhydrazones derived from ketones.
 | ||
| Scheme 11 Ge–H and Sn–H bond insertion reactions. | ||
López, Vicente et al.45 reported a zinc-catalysed coupling reaction between cyclopropenes and triethylgermane. The reaction starts from zinc-catalysed ring-opening of cyclopropenes to form alkenyl zinc carbenes, which then inserts into the Ge–H bond, forming allylgermanes with moderate to good yields (up to 82% yield) (Scheme 11d).
P–H bond insertion
Organophosphorus compounds are widely used in organic synthesis, catalysis, biochemistry, and materials chemistry. Transition-metal-catalysed P–H bond insertion reaction was a promising protocol for constructing the C–P bond. Notably, pentavalent H-phosphonates or H-phosphine oxides rather than trivalent phosphines with a lone pair were generally used as P–H donors to avoid poisoning of the metal catalysts in this reaction. Arbuzov et al.46 and Polozov et al.47 verified the feasibility of P–H bond insertion of α-diazoesters, α-diazoketones and diaryldiazomethane compounds with dialkyl H-phosphonates in the presence of copper salts, enabling the facile synthesis of dialkyl phosphonates in moderate yields (Scheme 12a). Wu et al.48 and Tang et al.49 independently reported a Cu-promoted P–H bond insertion reaction of alkyl- and benzyl carbenes generated in situ from N-sulfonylhydrazones with dialkoxyl-H-phosphonates and diaryl-phosphine oxides (Scheme 12b and c). In these reactions, Cu(I) decomposes the diazo compound generated from N-tosylhydrazone under basic and thermal conditions to form a Cu carbene. This carbene intermediate is then attacked by a nucleophilic phosphoryl anion at the carbene carbon to produce a Cu carbanion, which is protonated to afford the phosphate product.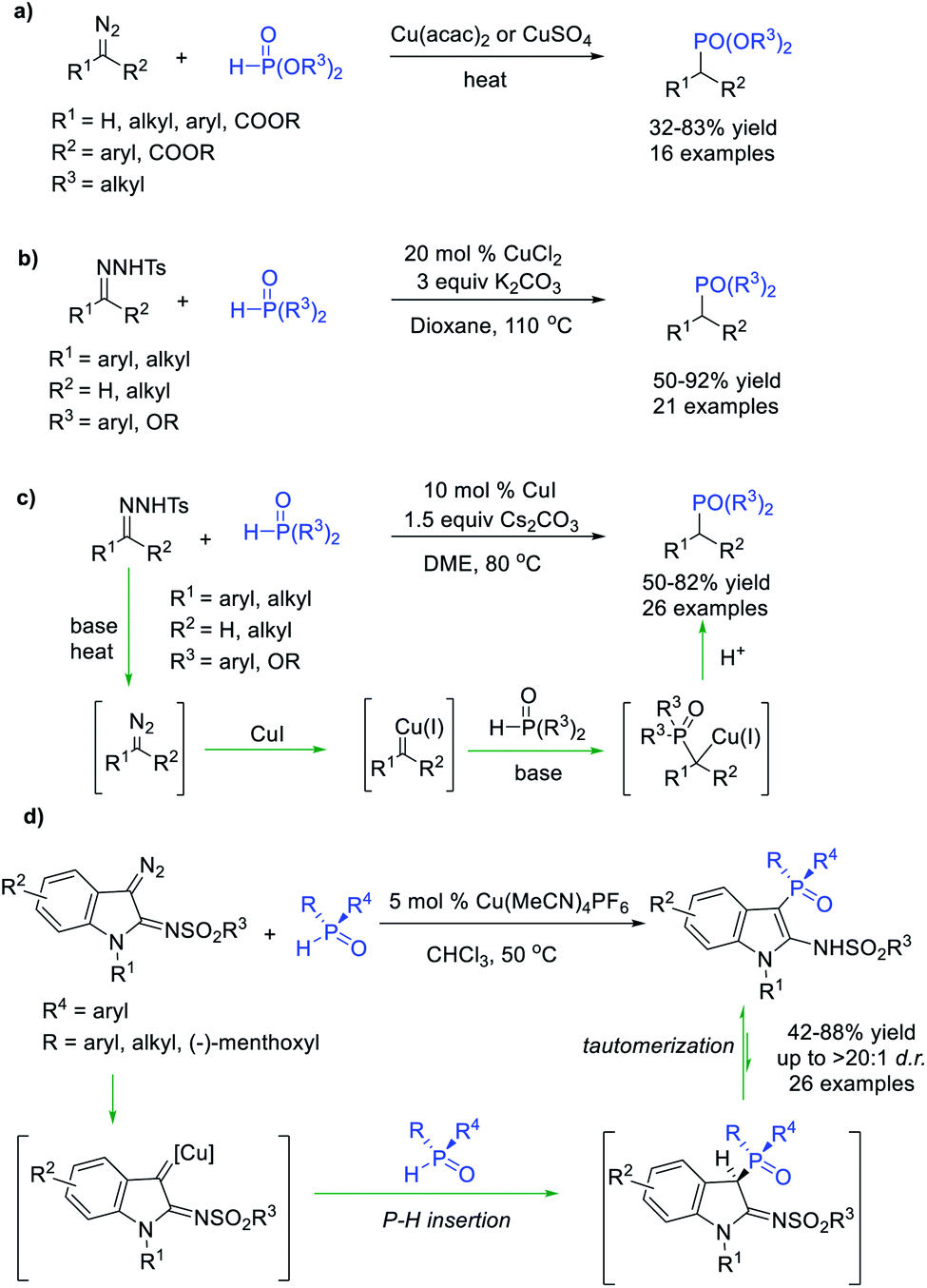 | ||
| Scheme 12 Cu-catalysed P–H bond insertion reaction of diazo compounds. | ||
Jiang et al.50 realized a highly efficient P–H bond insertion of α-imino copper carbene into H-phosphine oxides, leading to 3-phosphinoylindoles with broad substrate diversity and good chemoselectivity (Scheme 12d). Interestingly, chiral 3-phosphoindoles could be obtained with high stereoselectivities by using P-stereogenic H-phosphine oxides. This unique C(sp2)–P bond construction methodology proceeds through insertion into the P–H bond followed by tautomerization to afford the products.
By using carbonyl-ene-ynes as carbene precursors, Wu, Jiang et al.51 realized a Cu-catalysed P–H bond insertion of α-furyl carbene and H-phosphonates, affording various phosphorylated furans in good yields (Scheme 13).
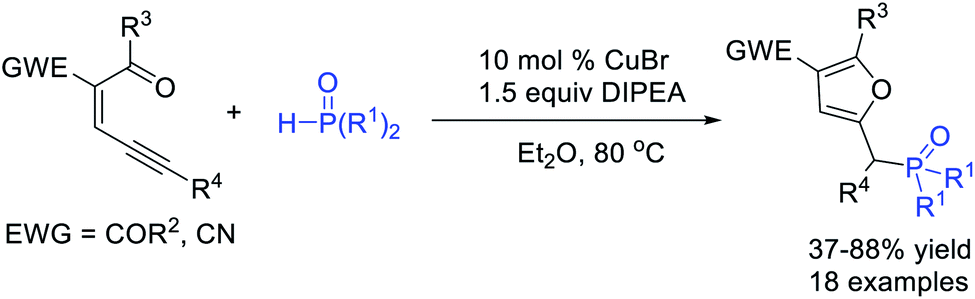 | ||
| Scheme 13 Cu-catalysed P–H bond insertion reaction of carbonyl-ene-ynes. | ||
F–H bond insertion
Organofluorine compounds display broad utility as valuable pharmaceuticals, agrochemicals, materials and tracers for positron emission tomography (PET). Treatment of diazocarbonyls with HF52 or HBF4 (ref. 53) rapidly produced α-fluoro-carbonyl compounds through an acid-promoted protonation/fluoride substitution process without involving the carbene intermediate. To avoid direct employment of intensively toxic and corrosive HF and improve the efficiency and selectivity, various “HF” surrogates and catalytic protocols were developed for F–H bond insertion reactions.Davies et al.54 reported an Ag-catalysed vinylogous fluorination of vinyl diazoacetates using Et3N–3HF as the F–H donor and synthesized a series of secondary and tertiary allylic fluorides. This reaction finished within 5 minutes and was potentially useful for 18F labelling (Scheme 14a).
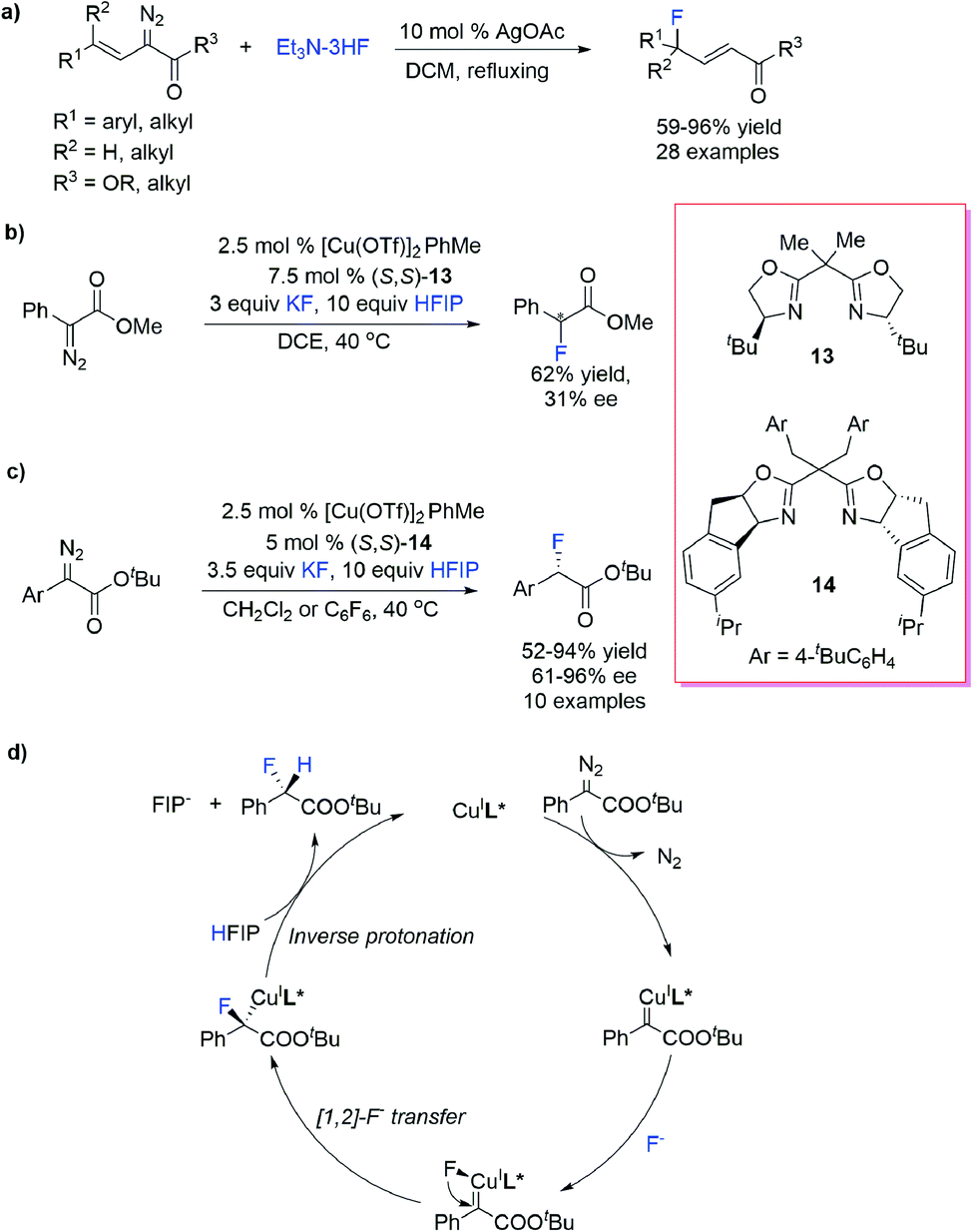 | ||
| Scheme 14 Transition-metal-catalysed F–H bond insertion reactions. | ||
Doyle et al.55 reported a Cu/bisoxazoline prompted H–F insertion reaction of α-diazocarbonyl compounds and the “H–F” source generated in situ from KF and strong protic additive hexafluoroisopropanol (HFIP) (Scheme 14b). Albeit with moderate enantioselectivities (up to 31% ee), this protocol enabled fast C–F/18F construction under mild conditions and was readily acceptable for direct, late-stage radio fluorination using available [18F]KF and Kryptofix 2.2.2 (K222) as the “18F” source. Under similar conditions, Fürstner et al.56 reported Cu/indane-based bisoxazoline catalysed asymmetric F–H bond insertion with α-aryl-diazoesters (Scheme 14c). DFT calculations revealed an F− transfer/protonation mechanism of the H–F insertion process: the F− nucleophilic attack takes place at the Cu-center rather the electrophilic carbene C-atom, forming a F− coordinated Cu carbene intermediate, and subsequent selective [1,2]-F− transfer produces a Cu carbanion, which undergoes HFIP-assisted protonation to give an insertion product with an inverse configuration (Scheme 14d).
Carbene insertion into the C–C σ bond
The selective C–C σ bond cleavage and subsequent functionalization are of pivotal significance in organic chemistry. Energetic carbene intermediates are promising species that may formally insert into the C–C σ bond to produce homologues plus one carbon. Notably, base or acid promoted homologation reaction of ketones with diazo compounds, which proceeds via a carbene-free process, has been intensively explored and comprehensively reviewed.57 In this chapter, metal-carbene or free carbene mediated formal C–C σ bond insertion reaction is emphasized.Zercher et al.58 reported a zinc-mediated homologation of β-keto esters, leading to various cyclic and acyclic γ-keto esters in good yields (Scheme 15a). This reaction starts from Simmons–Smith cyclopropanation of the enolate form of β-keto esters, followed by ring-opening of the resultant cyclopropane to produce the chain-extensive organozinc intermediate, which is quenched by an acid to afford γ-keto esters. By using a similar strategy, they further accomplished the homologation of β-keto phosphonates (Scheme 15b).59 In addition to the methylene carbene species, metal carbenes could also selectively insert into the EWG-activated C–C σ bond through an analogous cyclopropanation/ring-opening mechanism. Bi et al.60 reported an Ag-catalysed formal C–C bond insertion reaction of 1,3-diketones with α-aryl-α-diazoesters, α-aryl-α-phosphonates or diaryl diazo compounds, which provides efficient access to polycarbonyl compounds with an all-carbon quaternary center (Scheme 15c). Zhu et al.61 independently realized a similar C–C bond insertion reaction of α-aryl-α-diazoester under the catalysis of a gold complex (Scheme 15d). Compared with the Bi's Ag-catalysed system, the Zhu's Au-catalysed system exhibits higher activity and can be extended to alkyl 1,3-ketones. By using N-nosylhydrazones as diazo surrogates, Bi et al.62 developed a formal carbene insertion into the unstrained C–C σ bonds of 1,3-dicarbonyls, enabling the preparation of various synthetically useful polysubstituted γ-diketones, γ-keto esters, and γ-ketoamides in high yields (Scheme 15e).
 | ||
| Scheme 15 Formal carbene insertion into the C–C σ bonds of 1,3-carbonyls. | ||
Transition-metal-catalysed Buchner ring-expansion between aromatic rings and α-diazoesters is a classical formal C–C σ bond insertion reaction. This reaction serves as an efficient tool of preparing seven-membered rings via a “one-pot, two-step” mechanism: cyclopropanation of arenes followed by a ring-opening process. This reaction has been widely utilized in total synthesis and well summarized by several reviews.6,63 With the development of chiral catalysts, typically the chiral dirhodium carboxylates, significant advances have been made in the asymmetric intramolecular Buchner reaction, enabling the facile construction of chiral fused rings containing heptatrienes.
Iwasa et al.64 reported a highly enantioselective intramolecular Buchner reaction of diazoacetamides by using Ru(II)–Pheox as a catalyst, which affords various γ-lactam fused 5,7-bicyclic-heptatriene derivatives in excellent yields and enantioselectivities (up to 99% yield and 99% ee) (Scheme 16a). By using β-cyano α-diazoacetamide, Darses et al.65 realized an asymmetric intramolecular Buchner reaction, which allowed the highly stereoselective installation of a quaternary stereogenic center in the 5,7- or 6,7-bicyclic skeletons in the presence of chiral dirhodium catalysts (Scheme 16b). Yamaguchi et al.66 established a highly enantioselective Buchner ring expansion reaction of α-alkyl-α-diazoesters under the catalysis of chiral dirhodium carboxylates (Scheme 16c). This protocol could effectively refrain from β-hydrogen migration and constructed all-carbon fused 5,7-bicyclic systems under mild conditions. Hu, Xu et al.67 reported a Rh-catalysed intramolecular Buchner reaction of alkyne embodied diazoesters and provided a variety of functionalized 13- to 20-membered cycloalkynes with moderate to good yields (up to 86% yield) (Scheme 16d). As all the above asymmetric Buchner reactions are initiated with electron-deficient Fischer-type carbenes, substrates with electron-rich arenes are required for satisfactory results.
 | ||
| Scheme 16 Rh-catalysed intramolecular Buchner reactions. | ||
Carbene could also insert into the C–C σ bond of strained rings to form expansive rings. Wang et al.68 reported a selective ring expansion reaction of benzocyclobutenols with diazoesters under the catalysis of Rh(I) complexes, obtaining indanol derivatives bearing an all-carbon quaternary center in the ring with good yields and stereoselectivities (Scheme 17a). Different from the cyclopropanation-triggered homologation process mentioned above, this C–C bond insertion reaction proceeds through a “cut and saw” mechanism: the η2-coordination of the arene moiety to the alkoxyl-ligated rhodium center dominates the cleavage at the C(sp2)-C(sp3) bond through the β-carbon elimination process, leading to an aryl Rh(I) intermediate bearing a ketone moiety, which then reacts with diazoesters to generate the Rh(I) carbene complex; subsequent carbene migratory insertion followed by an intramolecular aldol reaction provides a ring-expansive alkoxyl Rh(I) species, which is then pronated to give the product. The diastereoselectivity arises from the sterically favourable cis-fused chairlike transition state in the aldol cyclization process. Murakami et al.69 realized a highly enantioselective C–C bond insertion reaction of cyclobutanols and benzyl carbenes generated in situ from N-tosylhydrazone (Scheme 17b). By using Rh(I) catalysts combined with chiral bisphosphine ligands, a series of cyclopentanols having two consecutive stereogenic centers were obtained in moderate yields and excellent enantioselectivities. Notably, the diastereoselectivities can be regulated to a certain extent by using ligands with different backbones.
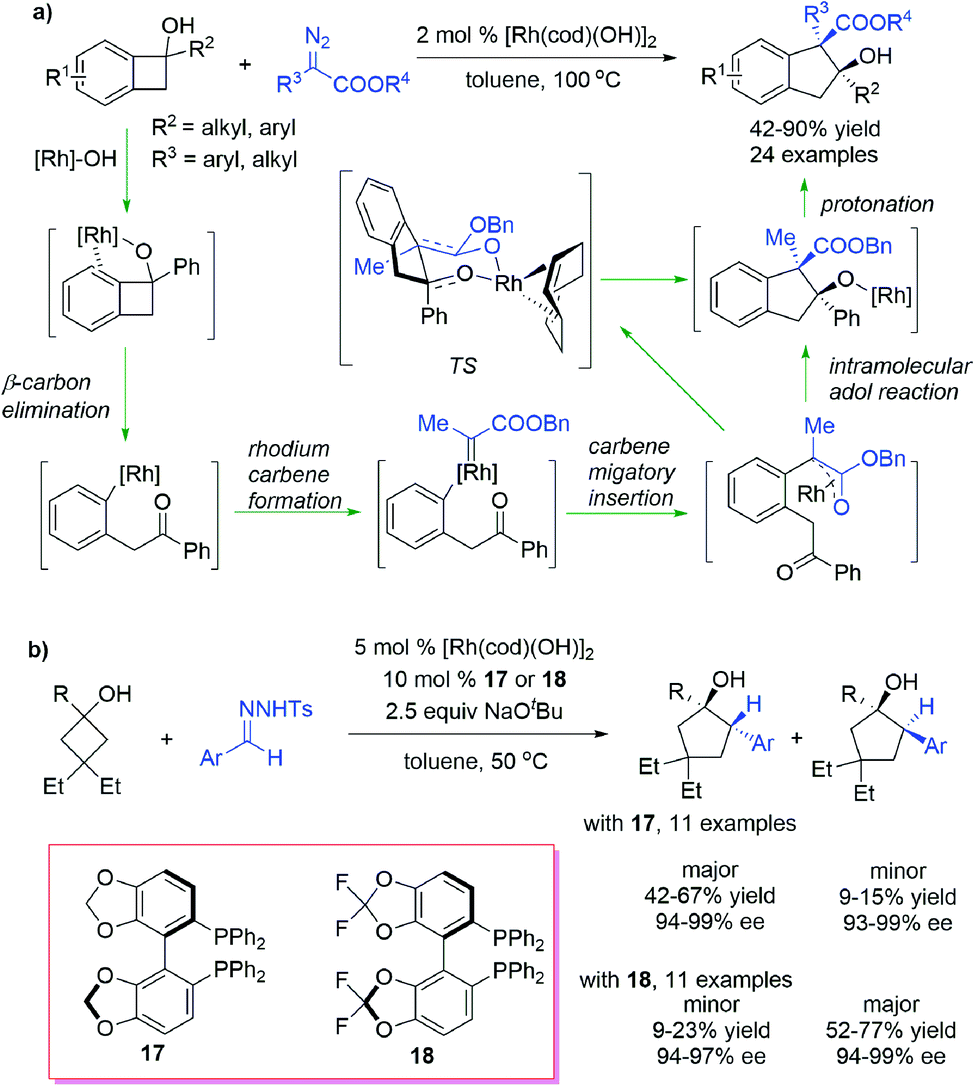 | ||
| Scheme 17 Rh-catalysed C–C σ bond insertion into strained rings. | ||
The bicyclo[1.1.1]pentane (BCP) motif has been utilized as a bioisostere in drug candidates to replace phenyl, tert-butyl, and alkynyl fragments in order to improve physicochemical properties. Hirst et al.70 developed a strategy toward the synthesis of BCP analogues which involves a dichlorocarbene insertion into bicyclo[1.1.0]butanes, and the resulting gem-dichloro-bicyclo[1.1.1]pentanes were reduced to BCPs in the subsequent steps (Scheme 18a). Later, Ma et al.71 realized the selective synthesis of 2,2-difluorobicyclo[1.1.1]pentane analogues (BCP–F2) through a formal C–C insertion into bicyclo[1.1.0]butanes of difluorocarbene (:CF2) generated in situ from FSO2CF2CO2TMS/NaF mixture (Scheme 18b). Almost at the same time, Mykhailiuk et al.72 also reported a similar protocol by using the CF3TMS/NaI mixture to release the difluorocarbene intermediate (Scheme 18c). Experimental physicochemical properties, acidity, basicity, lipophilicity and water solubility were measured for these BCP-F2 compounds, suggesting their potential to be novel saturated mimics for the benzene ring in medicinal chemistry projects.
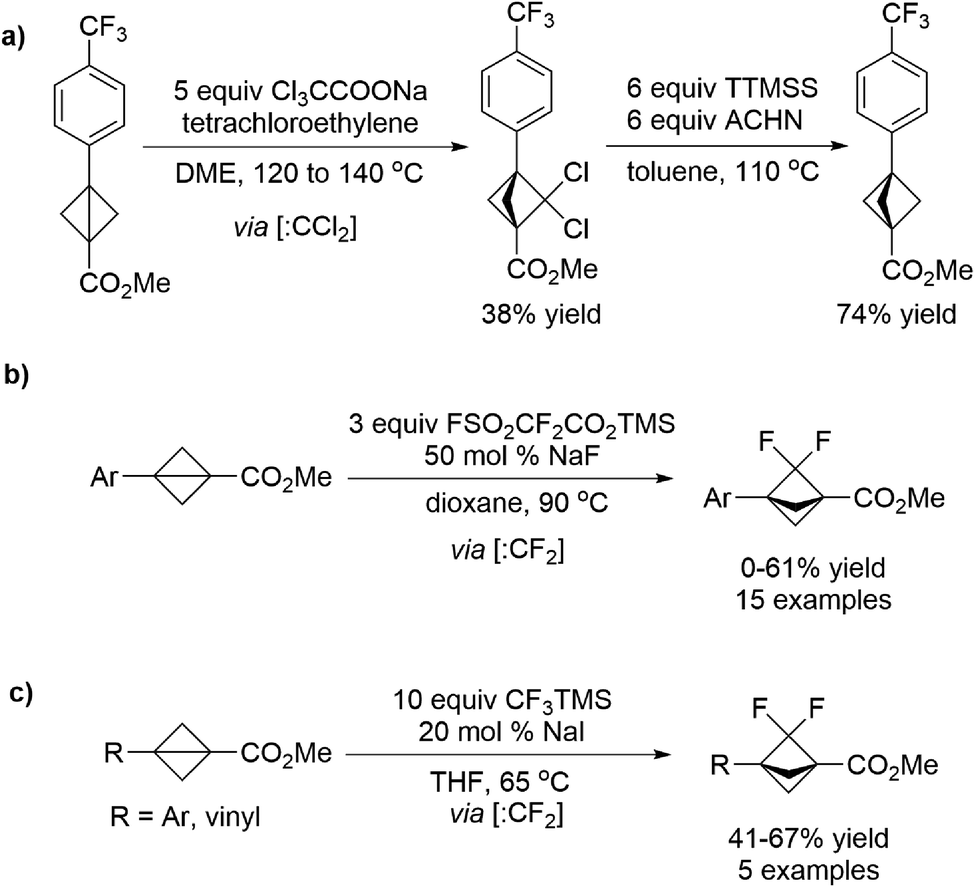 | ||
| Scheme 18 Dichloro- or difluorocarbene incorporated formal C–C bond insertion of strained rings. | ||
Carbene insertion into M–M (M = B, Si, Sn) bonds
Organic gem-dimetallics exhibit multi-functional utilities in organic synthesis. Among them, B, Si and Sn derived gem-dimetallics have attracted considerable attention due to their stability, availability, versatility and unique reactivities. Carbene insertion reaction of the M–M (M = B, Si, Sn) bond is undoubtedly a promising approach for the synthesis of these compounds.Diazo compounds or carbanions with an α-leaving group generated from dihalides or alcohol derivatives can react with diboron reagents to form “ate” complexes, which then undergo an intramolecular 1,2-boryl-migration process to afford diborated products. This carbene-free reaction is regarded as an efficient protocol of preparing diboron compounds and comprehensively reviewed.73,74 Apart from that, transition-metal-mediated carbene insertion reactions provide an alternative approach.
Srebnik et al.75 reported a Pt-catalysed B–B bond insertion reaction of B2pin2 and diazomethane or aryl-stabilized diazo compounds under thermal conditions, affording various stable gem-diborates in good yields (Scheme 19a). Kingsbury et al.76 extended the scope of this reaction to dialkyl-substituted diazo compounds (Scheme 19b). These reactions were thought to involve B–B bond activation by oxidative addition of B2pin2 to the Pt(0) complex to form a bis-(boryl) Pt(II) intermediate, which reacts with the diazo compounds to form the Pt carbene intermediates, followed by subsequent boryl-migratory insertion and reductive elimination to produce the gem-diborates (Scheme 19c).
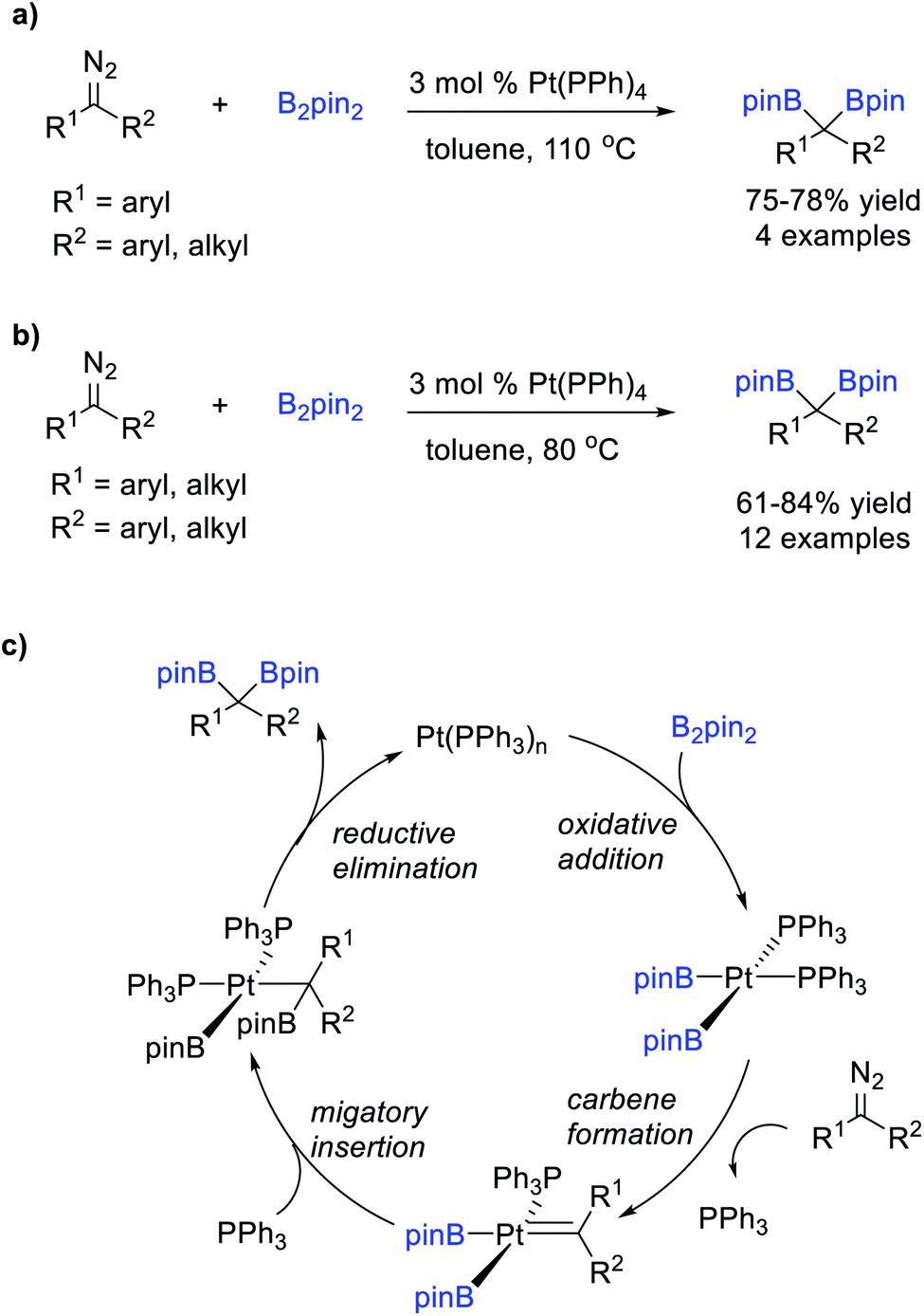 | ||
| Scheme 19 Pt-catalysed B–B bond insertion of diazo compounds. | ||
In addition to diborates, cleavage of Si–Si and Sn–Sn bonds by a transition metal could also trigger disilylation or distannylation of diazo compounds through a carbene-mediated migratory insertion/reductive elimination sequence. Wang et al.77 reported a Pd-catalysed Si–Si and Sn–Sn bond insertion reaction using N-tosylhydrazones as carbene precursors, affording a large variety of geminal bis(silane) and geminal bis(stannane) derivatives (Scheme 20a). Notably, more electrophilic FMe2SiSiMe2F rather than Me3SiSiMe3 or PhMe2SiSiMe2Ph was employed as the Si–Si source to facilitate the oxidative addition to the Pd center. This protocol also displayed a broad substrate scope and good efficiency (up to 99% yield), and the natural product (−)-perillaldehyde could be converted to corresponding dimetallic compounds in a two-step sequence in good yields (Scheme 20b).
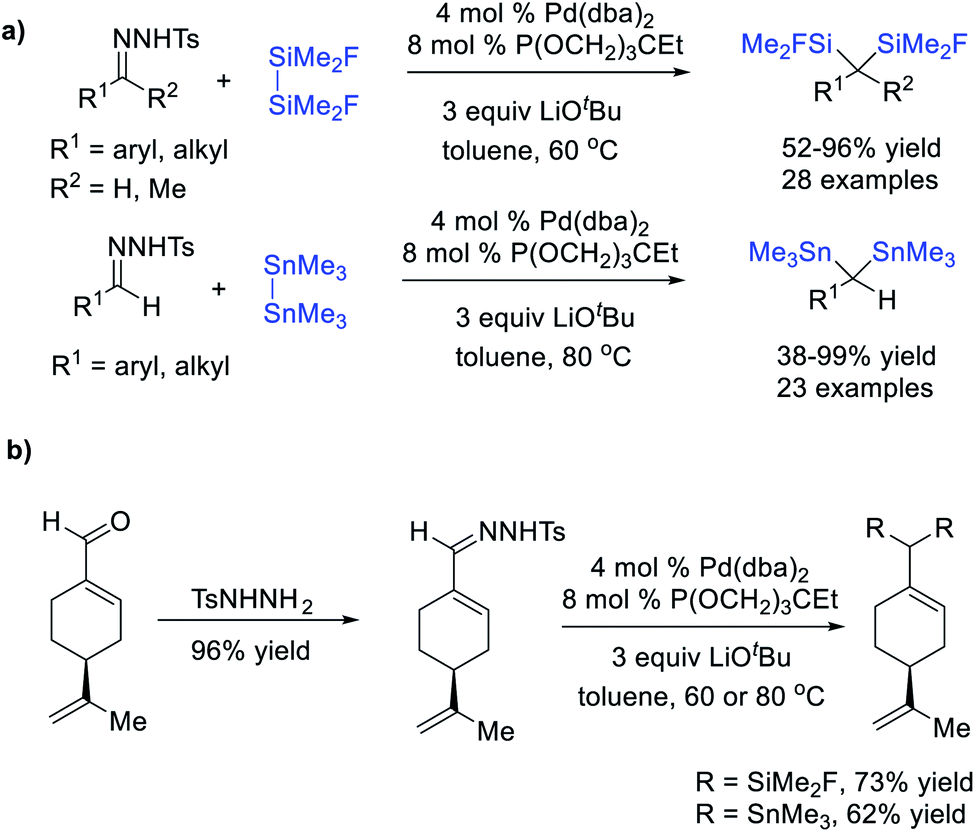 | ||
| Scheme 20 Pd-catalysed Si–Si or Sn–Sn bond insertion reactions. | ||
Summary and outlook
Carbene is a fundamental and important intermediate in organic chemistry that displays high reactivity, abundant structural diversities and good scalability with the incorporation of metal complexes.78 Transition-metal-catalysed carbene insertion reactions with the C–H bond, and several X–H bonds (e.g., Si–H, N–H, O–H, S–H) have been extensively studied in the past decades; however, recent studies on the other uncommon catalytic carbene insertion reactions described in this paper provide new growth points for carbene chemistry. Among them, B–H bond insertion reactions display broad applicability to a number of carbene precursors including α-EWG stabilized diazo compounds, N-sulfonylhydrazones, sulfoxonium ylides, alkynes, cyclopropenes, acylsilanes and alkenyl triflates under the mediation of metal complexes, radiation or bases, building up a platform for structurally diverse organoborane adducts with high optical purity. These stable products can be easily transformed into organoborates, meanwhile themselves are effective nucleophilic partners in Suzuki cross-coupling reactions. It's envisioned that more and more structurally distinctive organoboranes may spring up with the development of new catalytic B–H bond insertion reactions with different carbenes or carbenoids. The structurally diverse and relatively stable organoboranes prepared through B–H bond insertion probably could be employed in the preparation of functional polymers with ligated nitrogen–boron bonds79,80 or be used to find lead compounds. Other X–H (X = Ge, Sn, P, F) insertion reactions are sluggishly developed due to various factors, including product stability, catalyst-compatibility, usage of hazardous reagents, etc. More studies are highly desired to overcome these drawbacks and to develop efficient protocols for the enantioselective version and expand their synthetic utilities in organic synthesis. EWG-activated 1,3-dicarbonyls, arene rings and restrained rings were successfully applied in the formal C–C σ bond insertion reactions, and the employment of inactivated C–C σ bonds81 in carbene insertion reactions may lead to the development of new C–C bond forming reactions. Transition-metal-mediated carbene insertion into M–M (M = B, Si, Sn) enabled the straightforward synthesis of gem-homodimetallics, while the more synthetically useful chiral gem-heterodimetallics from asymmetric M–N bond insertion are certainly appealing. The studies on “uncommon” carbene insertion reactions not only have enriched carbene transfer reactions but also open new space for C–C and C–X bond formation. The development of new carbene transfer catalysts, new carbene precursors, and new insertion partners is the key to advance this field to a mature state.Author contributions
S.-F. Zhu and M.-Y. Huang conceived the idea of the perspective and prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (21625204, 21971119), the “111” project (B06005) of the Ministry of Education of China, National Program for Support of Top-notch Young Professionals, the Key-Area Research and Development Program of Guangdong Province (2020B010188001), and the Frontiers Science Center for New Organic Matter of Nankai University (63181206) for financial support. We appreciate the careful proofreading of the language by Y.-T. Zhao. Special gratitude to our co-workers, whose names are cited in the references, for their experimental or intellectual contributions to the field of catalytic carbene insertion reactions. This perspective was dedicated to the 100th Anniversary of Chemistry at Nankai University.References
- E. O. Fischer and A. Maasbol, Angew. Chem., Int. Ed., 1964, 3, 580–581 CrossRef.
- J. Barluenga, M. A. Fernandez-Rodrguez and E. Aguilar, J. Organomet. Chem., 2005, 690, 539–587 CrossRef CAS.
- H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861–2904 CrossRef CAS PubMed.
- M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS PubMed.
- S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365–1377 CrossRef CAS PubMed.
- A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981–10080 CrossRef CAS PubMed.
- D. Zhu, L.-F. Chen, H.-L. Fan, Q.-L. Yao and S.-F. Zhu, Chem. Soc. Rev., 2020, 49, 908–950 RSC.
- B. D. Bergstrom, L. A. Nickerson, J. T. Shaw and L. W. Souza, Angew. Chem., Int. Ed., 2021, 60, 6864–6878 CrossRef CAS PubMed.
- D. S. Matteson and D. Majumdar, J. Am. Chem. Soc., 1980, 102, 7588–7590 CrossRef CAS.
- D. Leonori and V. K. Aggarwal, Acc. Chem. Res., 2014, 47, 3174–3183 CrossRef CAS PubMed.
- O. A. Argintaru, D. Ryu, I. Aron and G. A. Molander, Angew. Chem., Int. Ed., 2013, 52, 13656–13660 CrossRef CAS PubMed.
- C. Battilocchio, F. Feist, A. Hafner, M. Simon, D. N. Tran, D. M. Allwood, D. C. Blakemore and S. V. Ley, Nat. Chem., 2016, 8, 360–367 CrossRef CAS PubMed.
- A. Greb, J.-S. Poh, S. Greed, C. Battilocchio, P. Pasau, D. C. Blakemore and S. V. Ley, Angew. Chem., Int. Ed., 2017, 56, 16602–16605 CrossRef CAS PubMed.
- Y. Yang, J. Tsien, A. B. David, J. M. E. Hughes, R. R. Merchant and T. Qin, J. Am. Chem. Soc., 2021, 143, 471–480 CrossRef CAS PubMed.
- G.-X. Zheng and M. Jones, J. Am. Chem. Soc., 1983, 105, 6487–6488 CrossRef CAS.
- C. Bedel and A. Foucaud, Tetrahedron Lett., 1993, 34, 311–314 CrossRef CAS.
- L. Monnier, J.-G. Delcrosb and B. Carboni, Tetrahedron, 2000, 56, 6039–6046 CrossRef CAS.
- T. Imamoto and Y. Yamanoi, Chem. Lett., 1996, 705–706 CrossRef CAS.
- P. Ramírez-López, M. A. Sierra, M. Gómez-Gallego, M. J. Mancheño and H. Gornitzka, Organometallics, 2003, 22, 5092–5099 CrossRef.
- X.-B. Li and D. P. Curran, J. Am. Chem. Soc., 2013, 135, 12076–12081 CrossRef CAS PubMed.
- Q.-Q. Cheng, S.-F. Zhu, Y.-Z. Zhang, X.-L. Xie and Q.-L. Zhou, J. Am. Chem. Soc., 2013, 135, 14094–14097 CrossRef CAS PubMed.
- D. Chen, X. Zhang, W.-Y. Qi, B. Xu and M.-H. Xu, J. Am. Chem. Soc., 2015, 137, 5268–5271 CrossRef CAS PubMed.
- Q.-Q. Cheng, H. Xu, S.-F. Zhu and Q.-L. Zhou, Acta Chim. Sin., 2015, 73, 326–329 CrossRef CAS.
- N. Otog, S. Chanthamath, I. Fujisawa and S. Iwasa, Eur. J. Org. Chem., 2021, 1564–1567 CrossRef CAS.
- D. Drikermann, R. S. Mößel, W. K. Al-Jammal and I. Vilotijevic, Org. Lett., 2020, 22, 1091–1095 CrossRef CAS PubMed.
- S. Hyde, J. Veliks, B. Liégault, D. Grassi, M. Taillefer and V. Gouverneur, Angew. Chem., Int. Ed., 2016, 55, 3785–3789 CrossRef CAS PubMed.
- S. B. J. Kan, X. Huang, Y. Gumulya, K. Chen and F. H. Arnold, Nature, 2017, 552, 132 CrossRef CAS PubMed.
- X.-Y. Huang, M. Garcia-Borràs, K. Miao, S. B. J. Kan, A. Zutshi, K. N. Houk and F. H. Arnold, ACS Cent. Sci., 2019, 5, 270–276 CrossRef CAS PubMed.
- Y. Pang, Q. He, Z.-Q. Li, J.-M. Yang, J.-H. Yu, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2018, 140, 10663–10668 CrossRef CAS PubMed.
- Y.-T. Zhao, Y.-X. Su, X.-Y. Li, M.-Y. Huang and S.-F. Zhu, Angew. Chem., Int. Ed., 2021, 60, 24214–24219 CrossRef CAS PubMed.
- J.-L. Li, H. He, M.-Y. Huang, Y.-C. Chen, Y. Luo, K.-C. Yan, Q.-T. Wang and Y. Wu, Org. Lett., 2019, 21, 9005–9008 CrossRef CAS PubMed.
- S.-S. Zhang, H. Xie, B. Shu, T. Che, X.-T. Wang, D.-M. Peng, F. Yang and L.-Y. Zhang, Chem. Commun., 2020, 56, 423–426 RSC.
- J.-M. Yang, Z.-Q. Li, M.-L. Li, Q. He, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2017, 139, 3784–3789 CrossRef CAS PubMed.
- J.-M. Yang, Y.-T. Zhao, Z.-Q. Li, X.-S. Gu, S.-F. Zhu and Q.-L. Zhou, ACS Catal., 2018, 8, 7351–7355 CrossRef CAS.
- M.-Y. Huang, Y.-T. Zhao, H. Chai, C.-D. Zhang and S.-F. Zhu, CCS Chem., 2021, 3, 1721–1726 CrossRef.
- J.-H. Ye, L. Quach, T. Paulisch and F. Glorius, J. Am. Chem. Soc., 2019, 141, 16227–16231 CrossRef CAS PubMed.
- J.-M. Yang, F.-K. Guo, Y.-T. Zhao, Q. Zhang, M.-Y. Huang, M.-L. Li, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2020, 142, 20924–20929 CrossRef CAS PubMed.
- D. Seyferth and J. M. Burlitch, J. Am. Chem. Soc., 1963, 85, 2667–2668 CrossRef.
- E. Abele, K. Rubina, P. Arsenyan, Y. Popelis, S. Grinberga, A. Mishnev and E. Lukevics, Main Group Met. Chem., 1999, 22, 65–70 CAS.
- H. Kondo, Y. Yamanoi and H. Nishihara, Chem. Commun., 2011, 47, 6671–6673 RSC.
- K. Kobayashi, M. Kawanisi, T. Hitomi and S. Kozima, Chem. Lett., 1984, 497–500 CrossRef CAS.
- D. Qiu, S. Wang, H. Meng, S.-B. Tang, Y. Zhang and J.-B. Wang, J. Org. Chem., 2017, 82, 624–632 CrossRef CAS PubMed.
- Z.-L. Liu, Q.-Q. Li, Y. Yang and X.-H. Bi, Chem. Commun., 2017, 53, 2503–2506 RSC.
- E.-H. Wang, Y.-J. Ping, Z.-R. Li, H.-L. Qin, Z.-J. Xu and C.-M. Che, Org. Lett., 2018, 20, 4641–4644 CrossRef CAS PubMed.
- S. Mata, L. A. López and R. Vicente, Angew. Chem., Int. Ed., 2017, 56, 7930–7934 CrossRef CAS PubMed.
- B. A. Arbuzov, N. A. Polezhaeva and A. M. Polozov, Dokl. Akad. Nauk SSSR, 1986, 287, 849 CAS.
- A. M. Polozov, N. A. Polezhaeva, A. H. Mustaphin, A. V. Khotinen and B. A. Arbuzov, Synthesis, 1990, 515–517 CrossRef CAS.
- L. Wu, X. Zhang, Q.-Q. Chen and A.-K. Zhou, Org. Biomol. Chem., 2012, 10, 7859–7862 RSC.
- W.-J. Miao, Y.-Z. Gao, X.-Q. Li, Y.-X. Gao, G. Tang and Y.-F. Zhao, Adv. Synth. Catal., 2012, 354, 2659–2664 CrossRef CAS.
- L.-S. Wang, Y. Wu, Y. Liu, H. Yang, X. Liu, J.-Y. Wang, X.-X. Li and J. Jiang, Org. Lett., 2017, 19, 782–785 CrossRef CAS PubMed.
- Y. Yu, S.-J. Yi, C.-L. Zhu, W.-G. Hu, B.-J. Gao, Y. Chen, W.-Q. Wu and H.-F. Jiang, Org. Lett., 2016, 18, 400–403 CrossRef CAS PubMed.
- E. L. Setti and O. A. Mascaretti, J. Chem. Soc., Perkin Trans. 1, 1988, 2059–2060 RSC.
- R. Pasceri, H. E. Bartrum, C. J. Hayes and C. J. Moody, Chem. Commun., 2012, 48, 12077–12079 RSC.
- C.-M. Qin and H. M. L. Davies, Org. Lett., 2013, 13, 6152–6154 CrossRef PubMed.
- E. E. Gray, M. K. Nielsen, K. A. Choquette, J. A. Kalow, T. J. A. Graham and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 10802–10805 CrossRef CAS PubMed.
- M. Buchsteiner, L. Martinez-Rodriquez, P. Jerabek, I. Pozo, M. Patzer, N. Nöthling, C. W. Lehmann and A. Fürstner, Chem.–Eur. J., 2020, 26, 2509–2525 CrossRef CAS PubMed.
- N. R. Candeias, R. Paterna and P. M. P. Gois, Chem. Rev., 2016, 116, 2937–2981 CrossRef CAS PubMed.
- J. B. Brogan and C. K. Zercher, J. Org. Chem., 1997, 62, 6444–6446 CrossRef CAS.
- C. A. Verbicky and C. K. Zercher, J. Org. Chem., 2000, 65, 5615–5622 CrossRef CAS PubMed.
- Z.-H. Liu, P. Sivaguru, G. Zanoni, E. A. Anderson and X.-H. Bi, Angew. Chem., Int. Ed., 2018, 57, 8927–8931 CrossRef CAS PubMed.
- Y.-Y. Ren, M. Chen, K. Li and S.-F. Zhu, Chem.–Asian J., 2018, 13, 2606–2610 CrossRef CAS PubMed.
- Z.-H. Liu, X.-Y. Zhang, M. Virelli, G. Zanoni, E. A. Anderson and X.-H. Bi, iScience, 2018, 8, 54–60 CrossRef CAS PubMed.
- S. E. Reisman, R. R. Nani and S. Levin, Synlett, 2011, 2437–2442 CrossRef CAS.
- N. P. T. Thanh, M. Tone, H. Inoue, I. Fujisawa and S. Iwasa, Chem. Commun., 2019, 55, 13398–13401 RSC.
- B. Darses, P. Maldivi, C. Philouze, P. Dauban and J.-F. Poisson, Org. Lett., 2021, 23, 300–304 CrossRef CAS PubMed.
- T. Hoshi, E. Ota, Y. Inokuma and J. Yamaguchi, Org. Lett., 2019, 21, 10081–10084 CrossRef CAS PubMed.
- Q. Zeng, K.-Y. Dong, C. Pei, S.-L. Dong, W.-H. Hu, L.-H. Qiu and X.-F. Xu, ACS Catal., 2019, 9, 10773–10779 CrossRef CAS.
- Y. Xia, Z.-X. Liu, Z. Liu, R. Ge, F. Ye, M. Hossain, Y. Zhang and J.-B. Wang, J. Am. Chem. Soc., 2014, 136, 3013–3015 CrossRef CAS PubMed.
- A. Yada, S. Fujita and M. Murakami, J. Am. Chem. Soc., 2014, 136, 7217–7220 CrossRef CAS PubMed.
- N. D. Measom, K. D. Down, D. J. Hirst, C. Jamieson, E. S. Manas, V. K. Patel and D. O. Somers, ACS Med. Chem. Lett., 2017, 8, 43–48 CrossRef CAS PubMed.
- X.-S. Ma, D. L. Sloman, Y.-X. Han and D. J. Bennett, Org. Lett., 2019, 21, 7199–7203 CrossRef CAS PubMed.
- R. M. Bychek, V. Hutskalova, Y. P. Bas, O. A. Zaporozhets, S. Zozulya, V. V. Levterov and P. K. Mykhailiuk, J. Org. Chem., 2019, 84, 15106–15117 CrossRef CAS PubMed.
- R. Nallagonda, K. Padala and A. Masarwa, Org. Biomol. Chem., 2018, 16, 1050–1064 RSC.
- C.-Q. Wu and J.-B. Wang, Tetrahedron Lett., 2018, 59, 2128–2140 CrossRef CAS.
- H. A. Ali, I. Goldberg, D. Kaufmann, C. Burmeister and M. Srebnik, Organometallics, 2002, 21, 1870–1876 CrossRef CAS.
- A. J. Wommack and J. S. Kingsbury, Tetrahedron Lett., 2014, 55, 3163–3166 CrossRef CAS.
- Z.-X. Liu, H.-C. Tan, T.-R. Fu, Y. Xia, D. Qiu, Y. Zhang and J.-B. Wang, J. Am. Chem. Soc., 2015, 137, 12800–12803 CrossRef CAS PubMed.
- M. Jia and S.-M. Ma, Angew. Chem., Int. Ed., 2016, 55, 9134–9166 CrossRef CAS PubMed.
- A. Staubitz, A. P. Soto and I. Manners, Angew. Chem., Int. Ed., 2008, 47, 6212–6215 CrossRef CAS PubMed.
- V. Pons and R. T. Baker, Angew. Chem., Int. Ed., 2008, 47, 9600–9602 CrossRef CAS PubMed.
- F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613–8661 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |