
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxime as a general photocage for the design of visible light photo-activatable fluorophores†
Lushun
Wang‡
a,
Shichao
Wang‡
a,
Juan
Tang
a,
Vanessa B.
Espinoza
a,
Axel
Loredo
 a,
Zeru
Tian
a,
R. Bruce
Weisman
a and
Han
Xiao
*abc
a,
Zeru
Tian
a,
R. Bruce
Weisman
a and
Han
Xiao
*abc
aDepartment of Chemistry, Rice University, 6100 Main Street, Houston, Texas 77005, USA. E-mail: han.xiao@rice.edu
bDepartment of Biosciences, Rice University, 6100 Main Street, Houston, Texas 77005, USA
cDepartment of Bioengineering, Rice University, 6100 Main Street, Houston, Texas 77005, USA
First published on 22nd November 2021
Abstract
Photoactivatable fluorophores have been widely used for tracking molecular and cellular dynamics with subdiffraction resolution. In this work, we have prepared a series of photoactivatable probes using the oxime moiety as a new class of photolabile caging group in which the photoactivation process is mediated by a highly efficient photodeoximation reaction. Incorporation of the oxime caging group into fluorophores results in loss of fluorescence. Upon light irradiation in the presence of air, the oxime-caged fluorophores are oxidized to their carbonyl derivatives, restoring strong fluorophore fluorescence. To demonstrate the utility of these oxime-caged fluorophores, we have created probes that target different organelles for live-cell confocal imaging. We also carried out photoactivated localization microscopy (PALM) imaging under physiological conditions using low-power light activation in the absence of cytotoxic additives. Our studies show that oximes represent a new class of visible-light photocages that can be widely used for cellular imaging, sensing, and photo-controlled molecular release.
Introduction
Photoactivatable fluorophores, also known as photocaged fluorophores, are powerful chemical probes for single-particle tracking and localization imaging.1–15 Photoactivatable fluorophores are maintained in weakly fluorescent or non-fluorescent dark states via masking by built-in photocaged groups. The photocages can be removed by irradiation with light of appropriate wavelength to restore fluorophore fluorescence. As they can be activated with high spatiotemporal resolution in complex biological environments, photoactivatable fluorophores have enjoyed a variety of important applications in biological research. These include monitoring of biological processes, detection of biomolecules, and super-resolution biological imaging beyond the diffraction limit.16–18The center for photoactivatable fluorophore design is the development of photocages or photolabile protecting groups that can alter the emission of conventional fluorescent dyes and can be cleaved upon light irradiation. In exploring the applicability of photocages, researchers have designed photocages based on a variety of different photoactivation mechanisms.19 Examples of efficient photoactivatable fluorophores include structures based on o-nitrobenzyl or its derivatives, phenacyl, 2-diazoketone, acridinyl, azidophenyl, coumarinyl and o-hydroxynaphthyl moieties.3,4,6,16,20–38 However, these photocaged dyes are generally characterized by relatively large sizes, poor water solubility, and poor biocompatibility, properties that greatly limit their biological applications.4 To minimize side effects resulting from the use of UV light, investigators have developed sensitizer-assisted light-induced cleavage20,39 and multiphoton activation strategies,40,41 resulting in efficient photocage cleavage with visible or near-infrared light. The downside of these technologies is the need for a light-capturing sensitizer or an expensive multiphoton light source. In recent years, elegant strategies based on quenching BODIPY dyes have been applied to releasing carboxylic acid and regenerating fluorescence with green light excitation >500 nm. These tactics are promising alternatives for o-nitrobenzyl and other photocaged groups.13,42–46 Quite recently, a novel light-induced protonation strategy has been employed to prepare a photoactivatable silicon rhodamine derivative.47 The utility of this fluorophore was successfully demonstrated in live-cell single molecule localization microscopy (SMLM) imaging. However, these strategies are limited to certain fluorophore scaffolds.
As a consequence of these difficulties, there is an urgent need for small photocaged moieties that are biocompatible and can be activated with visible light. In 2019, we developed a general strategy to obtain photoactivatable fluorophores across a broad emission range via a single sulfur-for-oxygen atom substitution.11 We found that a single sulfur-for-oxygen atom substitution within the fluorophore can yield heavy-atom-free photosensitizers.48 Upon irradiation with visible light, fluorophores with the thiocarbonyl group can be efficiently desulfurized to its oxo derivative, thus restoring the strong emission from the fluorophores. Because of the photosensitizer properties of these thio-caged dyes, the stability of these dyes and the potential toxicity of singlet oxygen during the photoactivity may limit their potential long-term imaging applications.
The oxime group has been recently employed as a fluorescence quencher that functions by weakening the internal charge transfer (ICT) process of fluorophores, due to its small size and weak electron-withdrawing characteristics. An ICT-based fluorophore usually contains an electron donor and an electron acceptor, which form a “push–pull” system in the excited state.49 The introduction of oxime group at the electron acceptor site deactivates the “push–pull” system, blocks the ICT process, and quenches fluorophore fluorescence. Based on this mechanism, carbonyl groups of several fluorophores have been substituted with the oxime group, creating feeble “push–pull” based photocaged fluorophores with weak fluorescence. The resulting oxime-caged fluorophores have been used for the detection of diverse chemicals, including hypochlorous acid/hypochlorite,50–55 phosgene,56,57 ions,58–60 NO2,61 trichloroisocyanuric acid,62 and organophosphorous nerve agents.63 The basis for most of these detections is a deoximation reaction that occurs in the presence of oxidizing agents. As a result, the technique has rarely been applied to cell imaging.
In this work, we have used the oxime group to prepare photoactivatable fluorophores that undergo a new kind of photoactivation process triggered by visible light (Fig. 1). We found that oxime substitution of the carbonyl group within a variety of fluorophore scaffolds weakens the ICT process, resulting in weak fluorescence. Significantly, excitation with visible light is likely to cause the resulting oxime-caged fluorophores to undergo a [2 + 2] cycloaddition with molecular oxygen, followed by spontaneous dissociation into fluorescent fluorophores with the classic “push–pull” ICT system. This process represents a new photoactivation mechanism in which fluorophore fluorescence is modulated by the photodeoximation reaction. To explore the utility of these oxime-caged photoactivatable fluorophores, we have used them for live-cell imaging of different organelles and for super-resolution imaging of proteins of interest in combination with genetically encoded tagging technology.
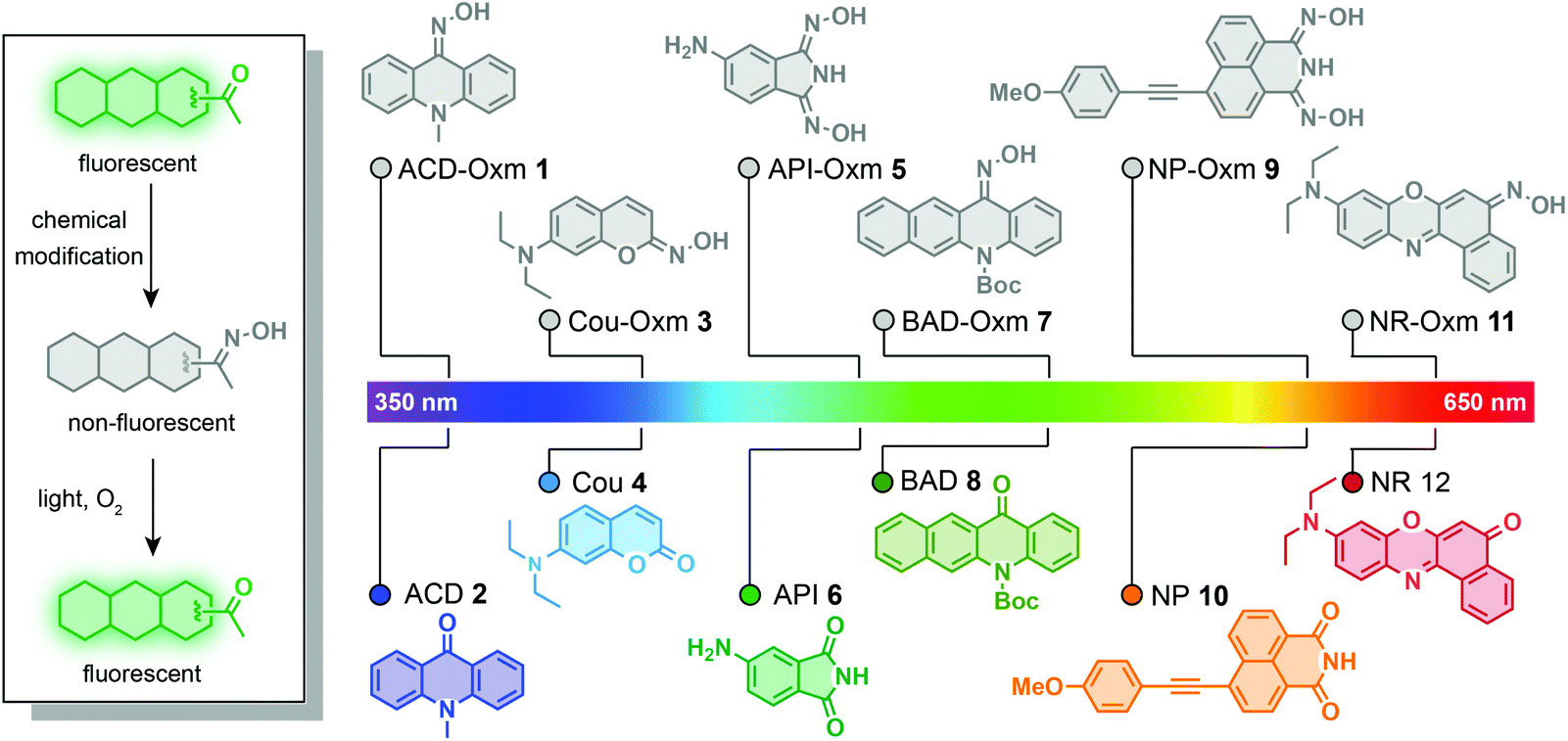 | ||
| Fig. 1 Design of oxime-caged fluorogenic dyes. Oxime substitution at the carbonyl group of fluorophores results in very weak fluorescence via an ICT off mechanism. Upon irradiation with light, the oxime group can be oxidized to its carbonyl derivatives, thus restoring the strong fluorescence of fluorophores. | ||
Results and discussion
Synthesis and characterization of oxime-caged 10-methylacridin-9(10H)-one
To assess the compatibility of the oxime caging strategy with different fluorophores, we initially synthesized oxime-caged 10-methylacridin-9(10H)-one (ACD-Oxm 1, Fig. 2) based on a method from the literature.64 The solution of ACD-Oxm in DMSO exhibited three maxima at 262 nm, 300 nm, and 383 nm accompanied by very weak fluorescence (Fig. 2A and B). The shorter blue shift of the absorption of ACD-Oxm compared to its carbonyl counterpart, 10-methylacridin-9(10H)-one (ACD 2), is likely attributable to the weaker electron-withdrawing ability of oxime relative to the ketone group. The fluorescence quantum yield of ACD-Oxm (Φf = 0.001) in DMSO is much lower than that of ACD (Φf = 0.67), indicating that the oxime should be a suitable caging group (Table 1). This also suggests the possibility of oxime modification of the fluorophore for fluorescence quenching, followed by reactivation of the fluorescence by photodeoximation. The shoulder of the absorption spectrum of ACD-Oxm extends from 400 nm up to 450 nm, suggesting that efficient photoactivation should be achieved using 430 nm light. Gratifyingly, activation of ACD-Oxm with blue light (430 nm, 62.2 μW cm−2) results in a 400-fold enhancement of fluorescence (Fig. 2D). The absorption spectrum of the resulting compound exhibits three maxima at 265 nm, 383 nm, and 401 nm, in good agreement with the absorption peaks of authentic ACD (Fig. 2B and C).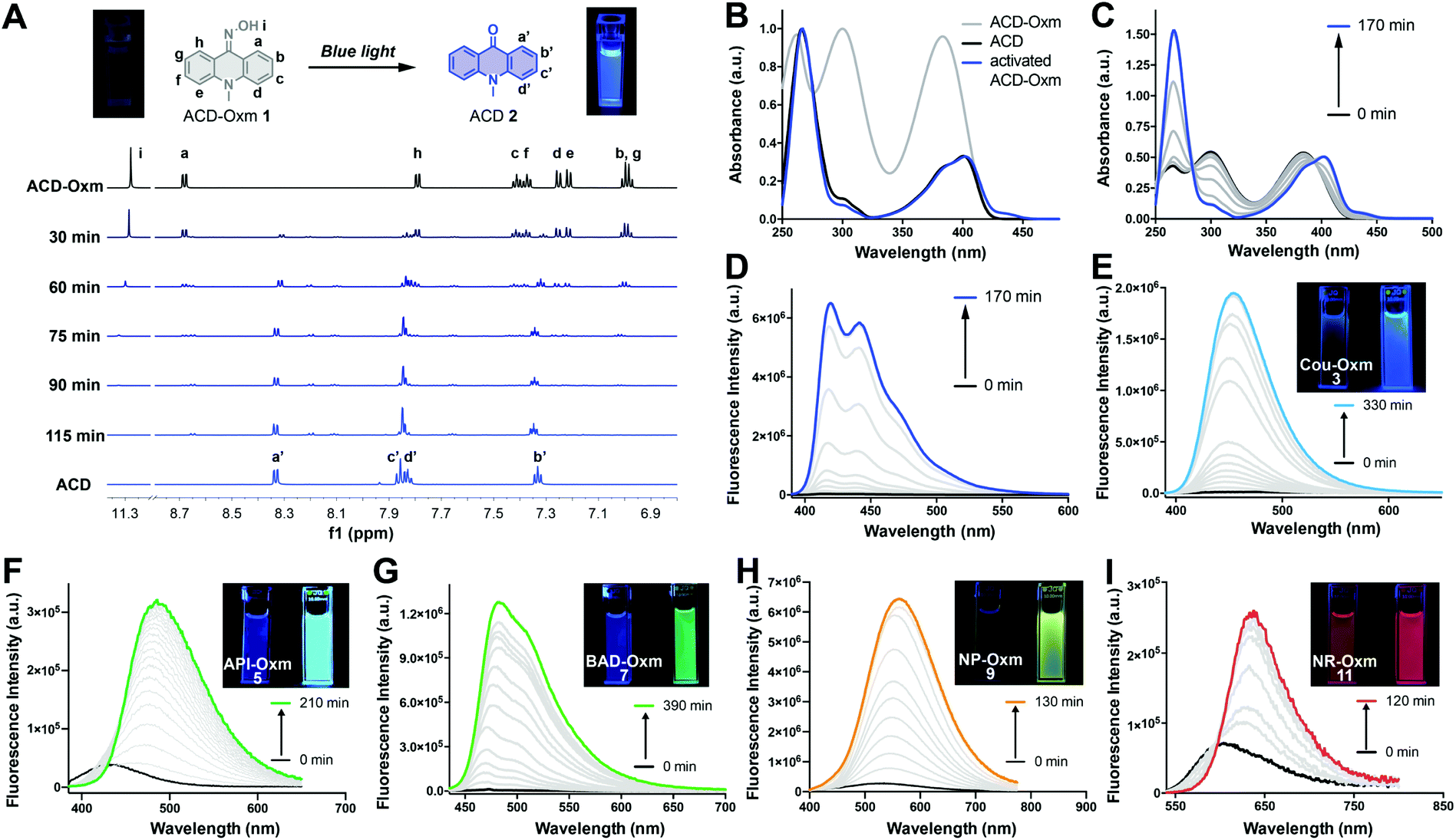 | ||
| Fig. 2 Photoactivation of oxime-caged fluorophores. (A) Overlay of 1H NMR spectra (6.8–11.5 ppm) of ACD-Oxm taken at the indicated light irradiation times (430 nm, 62.2 μW cm−2). (B) Normalized absorbance spectra of ACD, ACD-Oxm, and ACD-Oxm after photoactivation. (C) Absorbance changes in ACD-Oxm after light irradiation for different times (50 μM in DMSO, 430 nm, 62.2 μW cm−2). (D) Fluorescence spectra of ACD-Oxm (50 μM in DMSO) after light irradiation for different times (430 nm, 62.2 μW cm−2). (E–I) Fluorescence spectra of Cou-Oxm, API-Oxm, BAD-Oxm, NP-Oxm, and NR-Oxm (50 μM in DMSO) after light irradiation for different times (Cou-Oxm, BAD-Oxm, NP-Oxm and NR-Oxm by 430 nm, 62.2 μW cm−2, API-Oxm by 405 nm, 70.0 μW cm−2). | ||
| Compoundsa | λ abs (nm) | ε (×104 M−1 cm−1) | λ em (nm) | Φ f | Turn-on fold | Φ o |
|---|---|---|---|---|---|---|
| a Compounds were dissolved in DMSO (50 μM). b ε: molar extinction coefficients. c Fluorescence quantum yields were measured using rhodamine B in ethanol or quinine sulfate in 0.5 M H2SO4 as the reference. d 430 nm, 62.2 μW cm−2. e 405 nm, 70.0 μW cm−2. f 520 nm, 25.0 μW cm−2. g Photochemical quantum yields were assessed by LC-MS. | ||||||
| ACD-Oxm (1) | 300, 384 | 1.09, 1.07 | 414, 437, 471 | 0.001 | 400d | 6.1%d |
| ACD (2) | 383, 401 | 0.57, 0.70 | 420, 441, 473 | 0.67 | — | |
| Cou-Oxm (3) | 359 | 1.01 | — | 0.003 | 148d | 1.7%d |
| Cou (4) | 378 | 2.82 | 445 | 0.71 | — | |
| API-Oxm (5) | 342 | 0.79 | 417 | 0.11 | 22e | 1.9%e |
| API (6) | 311, 373 | 0.43, 0.36 | 479 | 0.60 | — | |
| BAD-Oxm (7) | 310, 352 | 0.44, 0.13 | 444 | 0.006 | 225d | 4.1%e |
| BAD (8) | 318, 408 | 0.70, 0.27 | 466, 494 | 0.44 | — | |
| NP-Oxm (9) | 387 | 0.65 | 526 | 0.003 | 26d | 0.9%d |
| NP (10) | 398 | 3.52 | 551 | 0.32 | — | |
| NR-Oxm (11) | 507 | 0.40 | 600 | 0.10 | 6f | 1.4%d |
| NR (12) | 556 | 4.17 | 634 | 0.46 | — | |
We used 1H NMR and ESI-MS analyses to characterize the photoactivation process that occurs in ACD-Oxm (Fig. 2A). The 1H NMR spectrum was evaluated at regular time intervals during irradiation of ACD-Oxm solution with blue light. Comparison of the 1H NMR spectrum of ACD-Oxm with that of the resulting compound (Fig. 2A) reveals the gradual disappearance of signals corresponding to hydroxy and phenyl protons at 11.33, 8.73, 7.84, 7.46, 7.42, 7.30, 7.26, 7.04 ppm, accompanied by the emergence of new signals at 8.35, 7.88, 7.85 and 7.35 ppm. The 1H NMR spectrum of the resulting product exactly matches that of synthesized ACD. Additional analysis using ESI-MS reveals that the observed m/z 210.0 of the photoactivated compound also corresponds to that ACD (Fig. S1 and S2†). These results confirm that visible-light photoactivation of ACD-Oxm restores the ketone structure of ACD.
Extension of the oxime-caging strategy to other fluorophores
Encouraged by the outstanding photoactivation properties of ACD-Oxm, we extended our design strategy to other commonly used fluorophores. We synthesized a variety of dyes with different scaffolds, as shown in Fig. 1. Oxime-caged 4-aminophthalimide (API-Oxm) 5 was resynthesized as previously reported.65 For oxime-caged coumarin (Cou-Oxm) 3 and oxime-caged Nile Red (NR-Oxm) 11 syntheses, we used Lawesson's reagent to generate SCou and SNile Red,48,66 followed by treatment of the resulting thiocarbonyl compounds with hydroxylamine to yield the oxime moiety (ESI Section 2†). Tert-butyl-12-(hydroxyamino)benzo[b]acridine-5(12H)-carboxylate (BAD-Oxm) 7 was obtained via a synthetic strategy similar to that used for ACD-Oxm 1. Generation of the oxime caged 4-((4-methoxyphenyl)ethynyl)-1,8-naphthalimide (NP-Oxm) 9via Sonogashira alkynylation of aryl bromide with 1-ethynyl-4-methoxybenzene initially produced 4-methoxyphenyl acetylide in THF/NEt3 in 86% yield. Subsequent condensation of hydroxylamine with bisnitrile in refluxing ethanol/water generated the desired imidedioxime NP-Oxm in moderate yield.67 The photophysical properties and visual fluorescence of these fluorophores were then investigated in DMSO solution at room temperature. As shown in Fig. 2 and Table 1, the absorption spectra of oxime-caged fluorophores exhibit blue-shifts compared to their carbonyl analogs (Fig. S3†). This can be attributed to the fact that the oxime group has a weaker electron-withdrawing ability than carbonyl groups. As expected, the introduction of the oxime group in all selected fluorophores led to dramatic hypsochromic shifts of the emission maxima along with significant reductions in fluorescence quantum yield (Table 1). To evaluate the photoactivation efficiency of the oxime-caged fluorophores, we recorded absorption and emission spectra after different irradiation times (Fig. 2E–I and S4–S13†). Upon light irradiation, the fluorescence intensity of Cou-Oxm, API-Oxm, BAD-Oxm, NP-Oxm, and NR-Oxm exhibited 148-fold, 22-fold, 225-fold, 26-fold, and 6-fold enhancements, respectively. We speculated that the relatively low turn-on efficiency of NR-Oxm may be attributed to a minimal ICT effect change after introducing the oxime group. This hypothesis was confirmed by density functional theory calculation at the TD-DFT/B3YLP/6-31G level (Fig. S14, S15, Tables S1, S2 and S5†). Furthermore, the dramatic shifts of the emission maxima and the good fluorescence quantum yields indicate that API-Oxm and NR-Oxm can be promising ratiometric fluorescent probes for the detection of bioactive molecules.50,54,68 These results demonstrate that incorporation of the oxime group into various fluorophores provides a general method for generating photoactivatable dyes.Mechanism of oxime-caged fluorophore activation
To study the mechanism responsible for fluorescence quenching in oxime caged fluorophores, we performed time-dependent density functional theory (TD-DFT/B3LYP) calculations for the optimized structures of ACD-Oxm and ACD. Highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) of ACD-Oxm are spread throughout the entire ACD-Oxm structure, suggesting a very weak ICT effect in this molecule (Fig. S16†). This was confirmed by electrostatic potential surfaces (ESP) map and Mulliken population analysis in the ground state and excited state (Fig. S17, Tables S3 and S5†). In contrast, ACD HOMOs and LUMOs are mainly located in the carbonyl area and the benzene conjugated structure, respectively. ESP map and Mulliken population analysis exhibit marked electron transfer from donor to acceptor upon excitation, indicative of a strong ICT effect and robust fluorescence emission under visible light irradiation (Fig. S17, Tables S4 and S5†). These data are consistent with our observations on ACD-Oxm and ACD fluorescence.To explore the photoactivation mechanism in oxime-caged fluorophores, we studied the solvent effect and carried out several control experiments using ACD-Oxm (ESI Section 3†). The photoactivation of ACD-Oxm in toluene (aprotic/non-polar solvent), acetonitrile (aprotic/polar solvent), DMSO (aprotic/polar solvent), and t-BuOH (protic/polar solvent), generated product with 78%, 71%, 85%, and 80% yield, respectively, indicating no obvious solvent effect on the photoactivation reaction. Then, the light dependence of ACD-Oxm activation was evaluated. No activation product was detected in the dark, confirming that light irradiation is necessary to promote this transformation (Fig. 3A, entry 1). Next, the photo-oxidation process was characterized. As shown in Fig. 3A, entry 2, no activation product was observed in the absence of oxygen. Isotopic analysis revealed that H2O was not involved in the photoactivation (Fig. 3A, entries 3 and 4). Several groups have reported singlet oxygen (1O2)-mediated oxidation of ketoximes and aldoximes to the corresponding carbonyl compounds and nitric acid. In these cases, the singlet oxygen was generated by irradiation of catalysts (rose bengal, platinum(II) terpyridyl acetylide complex, and potassium poly(heptazine imide)).69–71 To identify the oxygen species that participate in ACD-Oxm photoactivation, we used near-IR phosphorimetry to detect the production of singlet oxygen following excitation of ACD-Oxm in air-saturated methanol. Singlet oxygen exhibits characteristic phosphorescence at 1275 nm.72–75 However, none of our oxime-caged fluorophores showed detectable phosphorescence emission peaks around 1275 nm, indicating that singlet oxygen does not participate in their photoactivation (Fig. 3B and S18†). As a positive control, Ru(bpy)32+ did show a clear emission peak at 1274 nm (Fig. 3B and S18†). An alternative mechanism may involve the regeneration of bi-radical intermediates when imine and oxime compounds are exposed to direct irradiation.76–79 We investigated this type of radical-mediated mechanism for the photoactivation of oxime-caged fluorophores. Knowing that 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) is a free radical scavenger, we found that the addition of TEMPO dramatically inhibits the photoactivation of oxime-caged fluorophores (Fig. 3A, entry 5 and Fig. 3C), suggesting the involvement of radical species in the photolysis process. Furthermore, we showed that activation of ACD-Oxm occurs in the dark at 80 °C in the presence of the free radical initiator azodiisobutyronitrile (AIBN), producing ACD with an 80% yield (Fig. 3A, entries 6 and 7). Taking these experimental data and previous literature into account,76–79 we propose a tentative mechanism for the photoactivation of oxime-caged fluorophores. When irradiated by light, oxime-caged fluorophores are activated to the intermediate bi-radical species A*. Subsequently, oxidation of the radical moiety by dissolved oxygen yields B,76 which dissociates to produce the carbonyl compound C (Fig. 3D).
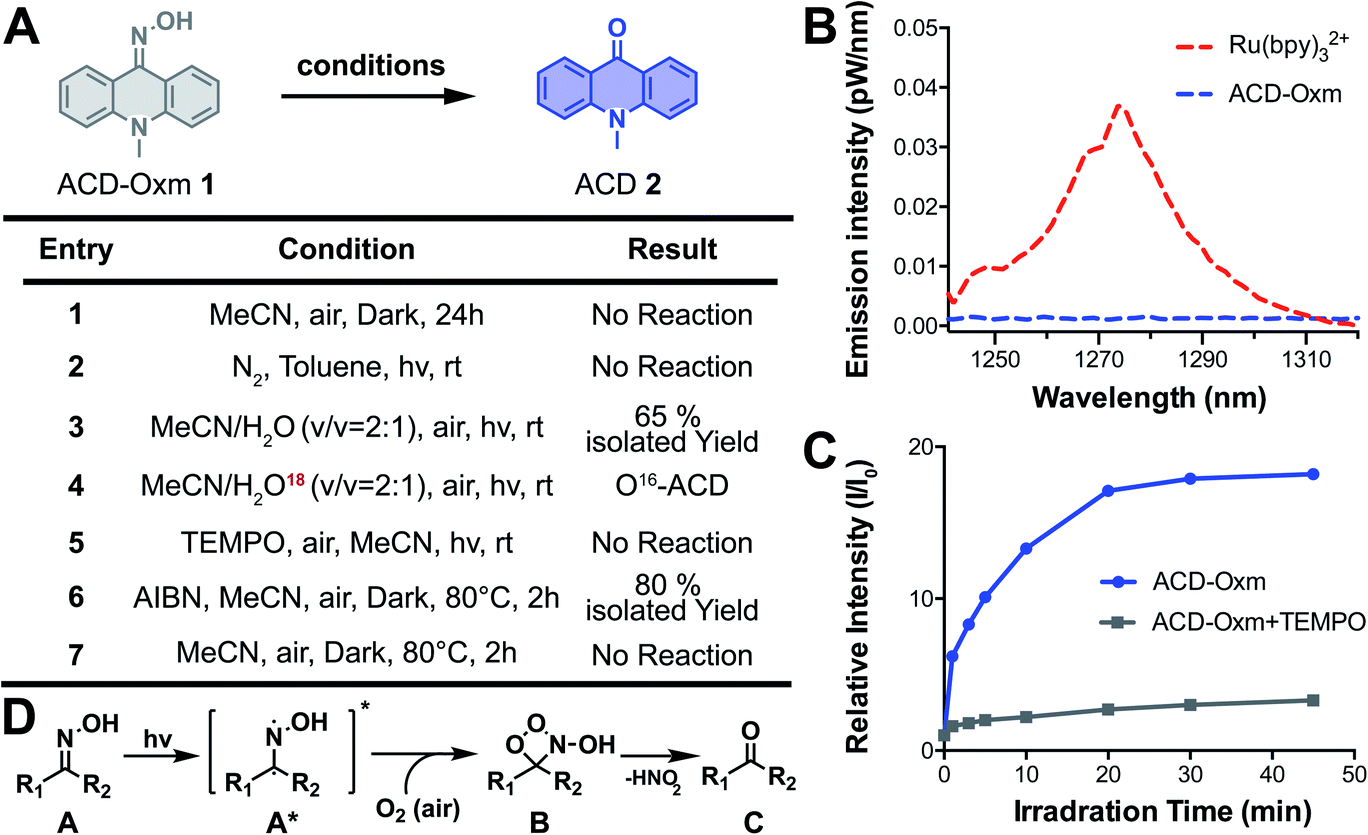 | ||
| Fig. 3 Mechanism of oxime-caged fluorophore activation. (A) Control experiments for studying the reaction mechanism. (B) Near-IR phosphorescence spectra of singlet oxygen generated by excitation of ACD-Oxm and the reference (Ru(bpy)32+) in oxygen-saturated methanol (50 μM) with 405 nm laser pulses at 25 °C. (C) Fluorescence change in ACD-Oxm (50 μM in DMSO) in the presence (1.1 equivalent TEMPO) and absence of TEMPO under light irradiation (430 nm, 62.2 μW cm−2). (D) Proposed photoactivation mechanism of oxime-caged fluorophores. | ||
Live-cell confocal imaging using oxime-caged fluorophores
Having demonstrated the applicability of the oxime-caging strategy to several different fluorophores, we turned our attention to the use of oxime-caged probes for biological imaging. Prior to live-cell imaging, oxime-caged probes were evaluated for stability, pH sensitivity, and photoactivation efficiency in aqueous buffer (Fig. S19–S26†). To our delight, oxime-caged fluorophores showed excellent stability and clean conversion. As shown in Fig. S27–S29,† BAD-Oxm exhibits superb photostability and negligible phototoxicity. Because of its large increase in fluorescence signal (Fig. S26 and Table S6†) and its low overlap with cellular autofluorescence, BAD-Oxm was employed for live-cell confocal imaging. Exposure of living HeLa cells to BAD-Oxm (10 μM) showed that the compound has good cell permeability at incubation times no longer than 1 h. In intracellular photoactivation experiments with BAD-Oxm, we observed a 22-fold enhancement of fluorescence as shown in Video S1,† along with very low cellular autofluorescence and negligible background signal (Fig. 4B and S30–S32†). These results indicate that BAD-Oxm is well suited for live-cell imaging.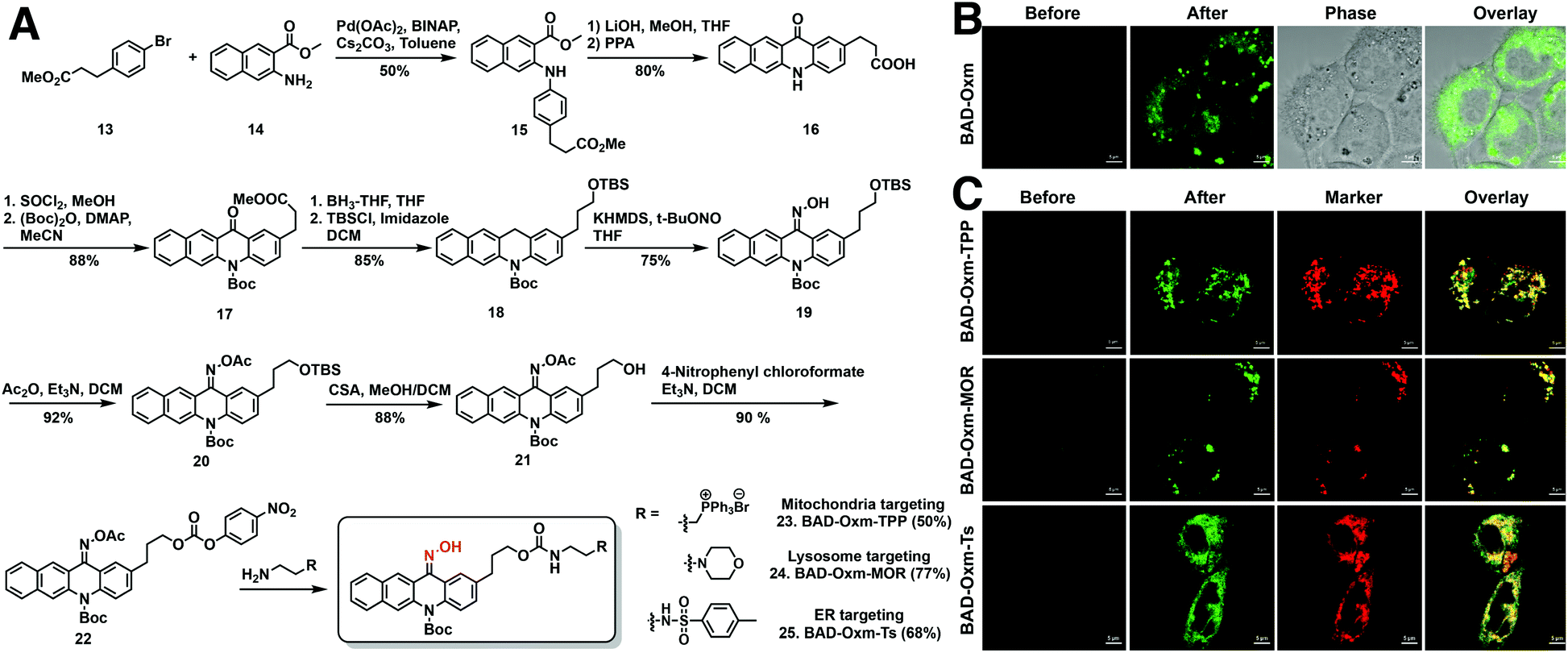 | ||
| Fig. 4 Live-cell confocal imaging using oxime-caged fluorophores. (A) Synthesis of organelle-targeting BAD-Oxm fluorogenic fluorophores. (B) Photoactivation of BAD-Oxm (5 μM) in A431 cells using 405 nm laser activation. Confocal images were captured before and after 405 nm light activation of BAD-Oxm. Scale bar = 5 μm. (C) Photoactivation of organelle-targeting BAD-Oxm probes (2 μM) in A431 cells. Confocal images were captured from cells incubated with BAD-Oxm-TPP, BAD-Oxm-MOR, and BAD-Oxm-Ts along with corresponding commercial markers for mitochondria, lysosomes, and endoplasmic reticulum, respectively. Commercial MitoView™ 633, LysoView™ 633, and ERTracker™ Red (BODIPY™ TR Glibenclamide) were used as markers for mitochondria, lysosomes, and endoplasmic reticulum, respectively. Scale bar = 5 μm. | ||
Fluorescence imaging of organelles in live cells
Organelles, such as the nucleus, endoplasmic reticulum (ER), endosomes, lysosomes, mitochondria, and the Golgi apparatus play significant roles in maintaining subcellular microenvironments. To investigate the properties and functions of these subcellular compartments, investigators have described many elaborately designed probes for fluorescence imaging of organelles.80–82 Our study explores the utility of the oxime-caging strategy for organelle-targeted fluorescence imaging. Synthesis of the organelle-targeted probes commenced with Buchwald–Hartwig cross-coupling amination between commercially available methyl 3-(4-bromophenyl) propionate 12 and methyl 3-aminonaphthalene-2-carboxylate 14 to provide di-ester 15 in 50% yield (Fig. 4A). Hydrolysis of the di-ester 15 with LiOH was followed by a Friedel–Crafts ring-closing reaction to yield benzo[b]acridin-12(5H)-one 16.83 Benzo[b]acridin-12(5H)-one 16 was then transformed into the key intermediate 18 in a 4 step reaction involving (1) esterification of the carboxylic acid with thionyl chloride and methanol; (2) protection of the arylamine with (Boc)2O; (3) reduction of the ketone moiety with BH3-THF; (4) protection of the resulting primary alcohol as a TBS ether. The reaction of intermediate 18 with t-BuONO and KHMDS generated the desired oxime as a Z/E isomer mixture in 75% yield.64 With key intermediates 19 in hand, the oxime hydroxyl moiety was protected with an acetyl group, followed by removal of the TBS group under acidic conditions and activation of the resulting primary hydroxy group as a carbonate ester 22. The activated BAD-Oxm carbonate ester was then functionalized with triphenylphosphonium (BAD-Oxm-TPP 23 for mitochondrial targeting), morpholine (BAD-Oxm-MOR 24 for lysosomal targeting), and phenylsulfonamide (BAD-Oxm-Ts 25 for endoplasmic reticulum targeting) (Fig. 4A). All three organelle-targeting probes exhibited satisfactory stability in the dark, and over 90-fold fluorescence enhancements could be induced by photoactivation in DMSO/PBS (buffer) (Table S7 and Fig. S33–S37†). Confocal laser scanning microscopy was then used to determine the respective abilities of the three fluorophores to localize to the endoplasmic reticulum, mitochondria, and lysosomes in living cells. Commercial MitoView™ 633, LysoView™ 633, and ERTracker™ Red (BODIPY™ TR Glibenclamide) were used as markers to define the target organelles. Organelle-targeting probes were incubated with A431 cells for 1 h, at which time the excess probe was cleared by washing with PBS. As shown in Fig. 4C, negligible fluorescence was observed before photoactivation. Dramatic fluorescence enhancement was then observed upon irradiation with the 405 nm laser. Co-localization experiments confirmed that the fluorescence of BAD-Oxm compounds exhibited good co-localization with the respective commercial organelle-specific dyes (Fig. 4C). The high Pearson's correlation coefficients for the organelle markers and BAD-Oxm-TPP (γ = 0.78), BAD-Oxm-MOR (γ = 0.90), and BAD-Oxm-Ts (γ = 0.90) indicate that the respective double-staining patterns overlap precisely. These results verify that the photoactivation of BAD-Oxm probes provides an excellent platform for organelle imaging.Super-resolution imaging of HaloTag proteins using oxime-caged fluorophores
In recent years, super-resolution microscopy techniques have been well developed for biological imaging that overcomes the diffraction limit for light microscopy.84,85 Photoactivatable or photoswitchable fluorophores have been widely applied to the development of SMLM techniques,7,8,36,47,86 such as photoactivated localization microscopy (PALM)17 and stochastic optical reconstruction microscopy (STORM).18 The excellent photoactivation properties of BAD-Oxm in solution and living cells make it an attractive probe for use in PALM. Accordingly, we synthesized BAD-Oxm-Halo 26 (Fig. 5A) and evaluated its properties in a PBS buffer/DMSO solution. BAD-Oxm-Halo small molecule and its labeled protein displayed 103-, 80-fold increases in fluorescence, respectively, upon irradiation with blue light (Fig. 5B, Table S7, and Fig. S34, S38 and S39†). We next evaluated the suitability of BAD-Oxm-Halo for protein super-resolution imaging in live cells. HaloTag labeling technology relies on the formation of a covalent bond between HaloTag-fused proteins and a synthetic probe containing a HaloTag ligand. HaloTag methodology has been used for site-specific labeling of proteins of interest.87–90 Following this line of investigation, we transiently transfected CHO–K1 cells with histone-2B–Halo and stained with BAD-Oxm-Halo. Fig. 5C shows that irradiation with the 405 nm laser produced a 7-fold fluorescence enhancement, which was recorded by Video S2,† accompanied by a distinct pattern of nuclear labeling. This was further confirmed by the co-incubation of BAD-Oxm-Halo with the commercial nuclear dye, DRAQ5 (Fig. S40†), as imaged with the confocal laser scanning microscope. Following up on this result, we performed PALM imaging of histone-2B in live CHO–K1 cells. The reconstructed image of histone-2B proteins, obtained from 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–30000 imaging frames, exhibits significant enhancement of resolution (average localization precision of 55.7 nm) compared to the corresponding wide-field image (Fig. 5D and E). This result demonstrates the utility of oxime-caged fluorophores for PALM imaging under physiological conditions without the addition of toxic enhancers. The oxime-based fluorophores developed in our work possess an extended functionality for specific labeling of existing proteins, thus offering great potential for super-resolution imaging in live cells.
000–30000 imaging frames, exhibits significant enhancement of resolution (average localization precision of 55.7 nm) compared to the corresponding wide-field image (Fig. 5D and E). This result demonstrates the utility of oxime-caged fluorophores for PALM imaging under physiological conditions without the addition of toxic enhancers. The oxime-based fluorophores developed in our work possess an extended functionality for specific labeling of existing proteins, thus offering great potential for super-resolution imaging in live cells.
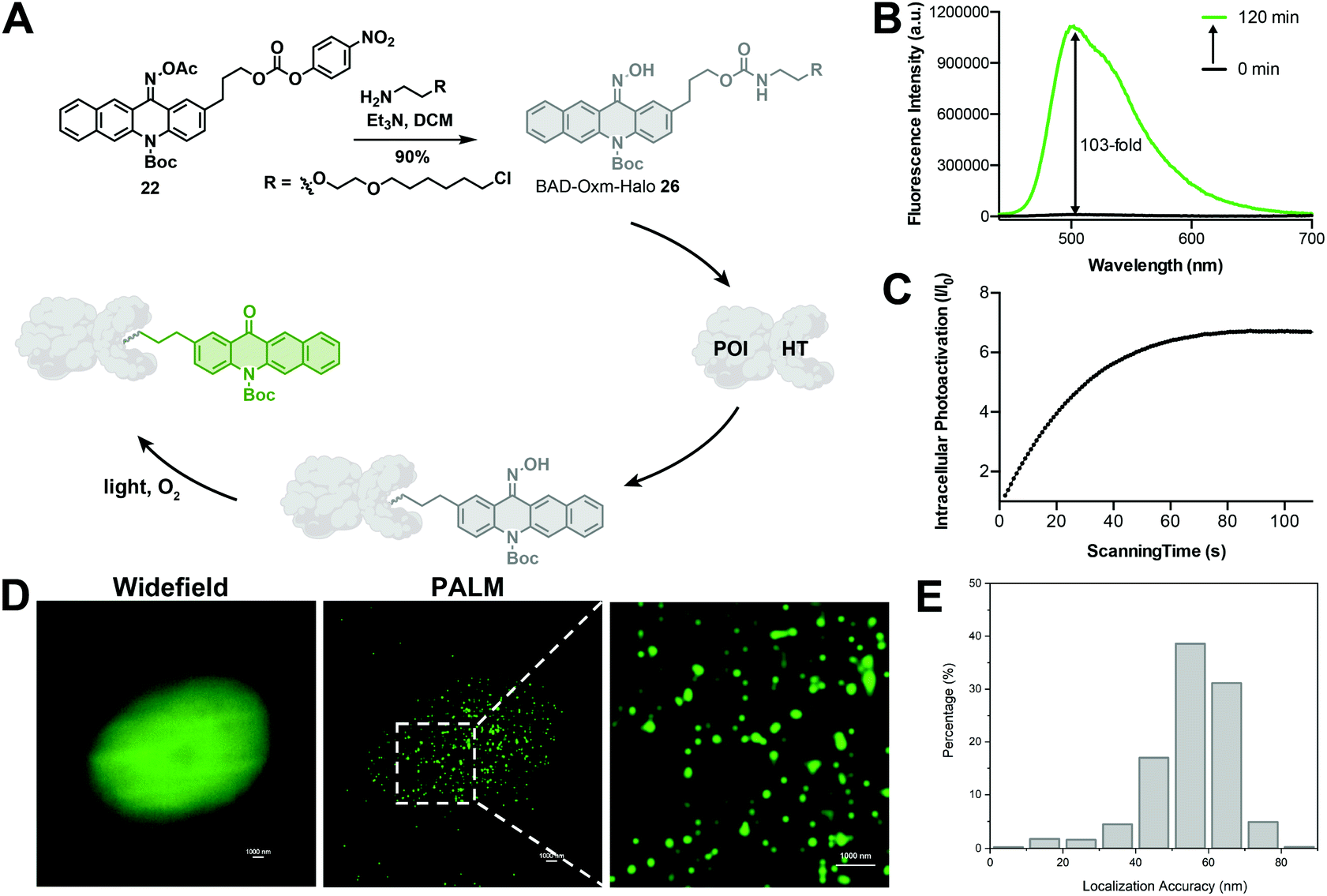 | ||
| Fig. 5 Super-resolution imaging of HaloTag proteins using oxime-caged fluorophores. (A) Scheme for labeling the protein of interest (POI) with BAD-Oxm-Halo ligand for fluorescence imaging. (B) Fluorescence change of BAD-Oxm-Halo in the presence of 430 nm light (62.2 μW cm−2). (C) Intracellular fluorescence change in BAD-Oxm-Halo (5 μM) during photoactivation at 405 nm in CHO–K1 cells. (D) Widefield image and corresponding PALM images of H2B labeled with H2B-HaloTag in CHO–K1 cells. (E) Histogram plot of the localization accuracy of PALM images in (D). Scale bar = 1 μm. | ||
Conclusions
In summary, we have developed a series of novel photoactivatable probes by incorporating the oxime group into a large group of fluorophores. The introduction of the oxime into fluorophores produces a reduced ICT effect and significantly weakened fluorescence. This caging effect can be reversed via a novel photodeoximation reaction triggered by exposure to air and visible light. We demonstrate the application of these photoactivatable oxime-probes for confocal imaging of different organelles. Going further, we apply oxime-caged probes to super-resolution imaging of proteins of interest. We expect that oxime-caged photoactivatable probes and the photodeoximation strategy will significantly expand strategies for living cell imaging and sensing.Data availability
Experimental data associated with this work have been provided in the ESI.†Author contributions
L. W. and H. X. designed all the experiments. L. W., S. W., J. T., V. E., A. L., and T. Z. performed research; L. W., S. W., R. B. W., and H. X. analyzed data; and L. W. and H. X. wrote the manuscript. H. X. conceived and supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Cancer Prevention Research Institute of Texas (CPRIT, RR170014 to H. X.), NIH (R35-GM133706 to H. X., R21-CA255894 to H. X.), U.S. Department of Defense (W81XWH-21-1-0789 to H. X.), the Robert A. Welch Foundation (C-1970 to H. X. and C-0807 to R. B. W.), the National Science Foundation (CHE-1803066 to R. B. W.), the Hamill Innovation Award (Hamill Foundation), the John S. Dunn Foundation Collaborative Research Award (Gulf Coast Consortia). H. X. is a Cancer Prevention & Research Institute of Texas (CPRIT) Scholar in Cancer Research.Notes and references
- L. D. Lavis, T.-Y. Chao and R. T. Raines, ACS Chem. Biol., 2006, 1, 252–260 CrossRef CAS PubMed.
- D. Puliti, D. Warther, C. Orange, A. Specht and M. Goeldner, Bioorg. Med. Chem., 2011, 19, 1023–1029 CrossRef CAS PubMed.
- C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2012, 51, 8446–8476 CrossRef CAS PubMed.
- W. Li and G. Zheng, Photochem. Photobiol. Sci., 2012, 11, 460 CrossRef CAS PubMed.
- T. J. Chozinski, L. A. Gagnon and J. C. Vaughan, FEBS Lett., 2014, 588, 3603–3612 CrossRef CAS PubMed.
- J. B. Grimm, B. P. English, H. Choi, A. K. Muthusamy, B. P. Mehl, P. Dong, T. A. Brown, J. Lippincott-Schwartz, Z. Liu, T. Lionnet and L. D. Lavis, Nat. Methods, 2016, 13, 985–988 CrossRef CAS PubMed.
- M. Minoshima and K. Kikuchi, JBIC, J. Biol. Inorg. Chem., 2017, 22, 639–652 CrossRef CAS PubMed.
- Z. Ye, H. Yu, W. Yang, Y. Zheng, N. Li, H. Bian, Z. Wang, Q. Liu, Y. Song, M. Zhang and Y. Xiao, J. Am. Chem. Soc., 2019, 141, 6527–6536 CrossRef CAS PubMed.
- S. Hauke, A. von Appen, T. Quidwai, J. Ries and R. Wombacher, Chem. Sci., 2017, 8, 559–566 RSC.
- F. M. Raymo, J. Phys. Chem. Lett., 2012, 3, 2379–2385 CrossRef CAS PubMed.
- J. Tang, M. A. Robichaux, K.-L. Wu, J. Pei, N. T. Nguyen, Y. Zhou, T. G. Wensel and H. Xiao, J. Am. Chem. Soc., 2019, 141, 14699–14706 CrossRef CAS PubMed.
- S.-Y. Dai and D. Yang, J. Am. Chem. Soc., 2020, 142, 17156–17166 CrossRef CAS PubMed.
- Y. Zhang, K.-H. Song, S. Tang, L. Ravelo, J. Cusido, C. Sun, H. F. Zhang and F. M. Raymo, J. Am. Chem. Soc., 2018, 140, 12741–12745 CrossRef CAS PubMed.
- Y. Zhang and F. M. Raymo, Bioconjugate Chem., 2020, 31, 1052–1062 CrossRef CAS PubMed.
- A. Loredo, J. Tang, L. Wang, K.-L. Wu, Z. Peng and H. Xiao, Chem. Sci., 2020, 11, 4410–4415 RSC.
- P. Sengupta, S. B. van Engelenburg and J. Lippincott-Schwartz, Chem. Rev., 2014, 114, 3189–3202 CrossRef CAS PubMed.
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS PubMed.
- M. J. Rust, M. Bates and X. Zhuang, Nat. Methods, 2006, 3, 793–796 CrossRef CAS PubMed.
- E. Abou Nakad, J. Chaud, C. Morville, F. Bolze and A. Specht, Photochem. Photobiol. Sci., 2020, 19, 1122–1133 CrossRef CAS PubMed.
- R. R. Nani, A. P. Gorka, T. Nagaya, H. Kobayashi and M. J. Schnermann, Angew. Chem., 2015, 127, 13839–13842 CrossRef.
- V. N. Belov, C. A. Wurm, V. P. Boyarskiy, S. Jakobs and S. W. Hell, Angew. Chem., Int. Ed., 2010, 49, 3520–3523 CrossRef CAS PubMed.
- G. A. Krafft, W. R. Sutton and R. T. Cummings, J. Am. Chem. Soc., 1988, 110, 301–303 CrossRef CAS.
- R. Misra and S. P. Bhattacharyya, Intramolecular Charge Transfer: Theory and Applications, Wiley-VCH, Weinheim, 2018 Search PubMed.
- J. P. Olson, M. R. Banghart, B. L. Sabatini and G. C. R. Ellis-Davies, J. Am. Chem. Soc., 2013, 135, 15948–15954 CrossRef CAS PubMed.
- L. A. P. Antony, T. Slanina, P. Šebej, T. Šolomek and P. Klán, Org. Lett., 2013, 15, 4552–4555 CrossRef CAS PubMed.
- R. Sharma, J. D. Knoll, P. D. Martin, I. Podgorski, C. Turro and J. J. Kodanko, Inorg. Chem., 2014, 53, 3272–3274 CrossRef CAS PubMed.
- S. Arumugam and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 11892–11899 CrossRef CAS PubMed.
- Y. Zhang, K.-H. Song, S. Tang, L. Ravelo, J. Cusido, C. Sun, H. F. Zhang and F. M. Raymo, J. Am. Chem. Soc., 2018, 140, 12741–12745 CrossRef CAS PubMed.
- Z. Zou, Z. Luo, X. Xu, S. Yang, Z. Qing, J. Liu and R. Yang, TrAC, Trends Anal. Chem., 2020, 125, 115811 CrossRef CAS.
- E. A. Halabi, Z. Thiel, N. Trapp, D. Pinotsi and P. Rivera-Fuentes, J. Am. Chem. Soc., 2017, 139, 13200–13207 CrossRef CAS PubMed.
- D.-P. Klötzner, K. Klehs, M. Heilemann and A. Heckel, Chem. Commun., 2017, 53, 9874–9877 RSC.
- S. J. Lord, N. R. Conley, H. D. Lee, R. Samuel, N. Liu, R. J. Twieg and W. E. Moerner, J. Am. Chem. Soc., 2008, 130, 9204–9205 CrossRef CAS PubMed.
- H. D. Lee, S. J. Lord, S. Iwanaga, K. Zhan, H. Xie, J. C. Williams, H. Wang, G. R. Bowman, E. D. Goley, L. Shapiro, R. J. Twieg, J. Rao and W. E. Moerner, J. Am. Chem. Soc., 2010, 132, 15099–15101 CrossRef CAS PubMed.
- J. B. Grimm, T. Klein, B. G. Kopek, G. Shtengel, H. F. Hess, M. Sauer and L. D. Lavis, Angew. Chem., Int. Ed., 2016, 55, 1723–1727 CrossRef CAS PubMed.
- L. M. Wysocki, J. B. Grimm, A. N. Tkachuk, T. A. Brown, E. Betzig and L. D. Lavis, Angew. Chem., Int. Ed., 2011, 50, 11206–11209 CrossRef CAS PubMed.
- S. Hauke, Chem. Sci., 2017, 8, 559–566 RSC.
- Y. Zhang, S. Swaminathan, S. Tang, J. Garcia-Amoros, M. Boulina, B. Captain, J. D. Baker and F. M. Raymo, J. Am. Chem. Soc., 2015, 137, 4709–4719 CrossRef CAS PubMed.
- A. V. Anzalone, Z. Chen and V. W. Cornish, Chem. Commun., 2016, 52, 9442–9445 RSC.
- A. Atilgan, E. TanriverdiEçik, R. Guliyev, T. B. Uyar, S. Erbas-Cakmak and E. U. Akkaya, Angew. Chem., Int. Ed., 2014, 53, 10678–10681 CrossRef CAS PubMed.
- E. B. Brown, J. B. Shear, S. R. Adams, R. Y. Tsien and W. W. Webb, Biophys. J., 1999, 76, 489–499 CrossRef CAS PubMed.
- C. Tran, T. Gallavardin, M. Petit, R. Slimi, H. Dhimane, M. Blanchard-Desce, F. C. Acher, D. Ogden and P. I. Dalko, Org. Lett., 2015, 17, 402–405 CrossRef CAS PubMed.
- P. P. Goswami, A. Syed, C. L. Beck, T. R. Albright, K. M. Mahoney, R. Unash, E. A. Smith and A. H. Winter, J. Am. Chem. Soc., 2015, 137, 3783–3786 CrossRef CAS PubMed.
- P. Shrestha, K. C. Dissanayake, E. J. Gehrmann, C. S. Wijesooriya, A. Mukhopadhyay, E. A. Smith and A. H. Winter, J. Am. Chem. Soc., 2020, 142, 15505–15512 CrossRef CAS PubMed.
- N. Rubinstein, P. Liu, E. W. Miller and R. Weinstain, Chem. Commun., 2015, 51, 6369–6372 RSC.
- T. Slanina, P. Shrestha, E. Palao, D. Kand, J. A. Peterson, A. S. Dutton, N. Rubinstein, R. Weinstain, A. H. Winter and P. Klán, J. Am. Chem. Soc., 2017, 139, 15168–15175 CrossRef CAS PubMed.
- J. A. Peterson, C. Wijesooriya, E. J. Gehrmann, K. M. Mahoney, P. P. Goswami, T. R. Albright, A. Syed, A. S. Dutton, E. A. Smith and A. H. Winter, J. Am. Chem. Soc., 2018, 140, 7343–7346 CrossRef CAS PubMed.
- M. S. Frei, P. Hoess, M. Lampe, B. Nijmeijer, M. Kueblbeck, J. Ellenberg, H. Wadepohl, J. Ries, S. Pitsch, L. Reymond and K. Johnsson, Nat. Commun., 2019, 10, 4580 CrossRef PubMed.
- J. Tang, L. Wang, A. Loredo, C. Cole and H. Xiao, Chem. Sci., 2020, 11, 6701–6708 RSC.
- R. Misra and S. P. Bhattacharyya, Intramolecular Charge Transfer, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2018, pp. 29–69 Search PubMed.
- W. Lin, L. Long, B. Chen and W. Tan, Chem.–Eur. J., 2009, 15, 2305–2309 CrossRef CAS PubMed.
- X. Cheng, H. Jia, T. Long, J. Feng, J. Qin and Z. Li, Chem. Commun., 2011, 47, 11978–11980 RSC.
- M. Emrullahoğlu, M. Üçüncü and E. Karakuş, Chem. Commun., 2013, 49, 7836–7838 RSC.
- C. Ma, G. Zhong, Y. Zhao, P. Zhang, Y. Fu and B. Shen, Spectrochim. Acta, Part A, 2020, 240, 118545 CrossRef CAS PubMed.
- J. Kang, F. Huo, Y. Yue, Y. Wen, J. Chao, Y. Zhang and C. Yin, Dyes Pigm., 2017, 136, 852–858 CrossRef CAS.
- L. Wang, J. Liu, H. Zhang and W. Guo, Sens. Actuators, B, 2021, 334, 129602 CrossRef CAS.
- B. Ma, X. Wang, S. Gao, L. Qi, Y. Xu, J. Yang and G. Zuo, Dyes Pigm., 2020, 177, 108279 CrossRef CAS.
- T.-I. Kim, B. Hwang, J. Bouffard and Y. Kim, Anal. Chem., 2017, 89, 12837–12842 CrossRef CAS PubMed.
- K. Chen, J. W. Bats and M. Schmittel, Inorg. Chem., 2013, 52, 12863–12865 CrossRef CAS PubMed.
- A. Balamurugan and H. Lee, Sens. Actuators, B, 2015, 216, 80–85 CrossRef CAS.
- J. Sivamani, V. Sadhasivam and A. Siva, Sens. Actuators, B, 2017, 246, 108–117 CrossRef CAS.
- L. A. Juárez, A. M. Costero, M. Parra, S. Gil, J. Ródenas, F. Sancenón and R. Martínez-Máñez, RSC Adv., 2016, 6, 43719–43723 RSC.
- S. K. Lee, M. G. Choi and S.-K. Chang, Tetrahedron Lett., 2014, 55, 7047–7050 CrossRef CAS.
- H. Lee and H.-J. Kim, Tetrahedron, 2014, 70, 2966–2970 CrossRef CAS.
- H. Tokuyama, H. Cho, Y. Iwama, T. Noro and K. Okano, Heterocycles, 2014, 88, 1433 CrossRef.
- M. Carboni, C. W. Abney, K. M. L. Taylor-Pashow, J. L. Vivero-Escoto and W. Lin, Ind. Eng. Chem. Res., 2013, 52, 15187–15197 CrossRef CAS.
- A. Loredo, L. Wang, S. Wang and H. Xiao, Dyes Pigm., 2021, 186, 109014 CrossRef CAS PubMed.
- C. D. Grant, S. O. Kang and B. P. Hay, J. Org. Chem., 2013, 78, 7735–7740 CrossRef CAS PubMed.
- Q. Jiang, Z. Wang, M. Li, J. Song, Y. Yang, X. Xu, H. Xu and S. Wang, Analyst, 2020, 145, 1033–1040 RSC.
- C. C. Wamser and J. W. Herring, J. Org. Chem., 1976, 41, 1476–1477 CrossRef CAS.
- Y. Yang, D. Zhang, L.-Z. Wu, B. Chen, L.-P. Zhang and C.-H. Tung, J. Org. Chem., 2004, 69, 4788–4791 CrossRef CAS PubMed.
- A. Savateev, N. V. Tarakina, V. Strauss, T. Hussain, K. ten Brummelhuis, J. M. S. Vadillo, Y. Markushyna, S. Mazzanti, A. P. Tyutyunnik, R. Walczak, M. Oschatz, D. M. Guldi, A. Karton and M. Antonietti, Angew. Chem., Int. Ed., 2020, 59, 15061–15068 CrossRef CAS PubMed.
- T. Tsuchiya, A. Kikuchi, N. Oguchi-Fujiyama, K. Miyazawa and M. Yagi, Photochem. Photobiol. Sci., 2015, 14, 807–814 CrossRef CAS PubMed.
- S. Fukuchi, M. Yagi, N. Oguchi-Fujiyama, J. Kang and A. Kikuchi, Photochem. Photobiol. Sci., 2019, 18, 1556–1564 CrossRef CAS PubMed.
- S. Kitasaka, M. Yagi and A. Kikuchi, Photochem. Photobiol. Sci., 2020, 19, 913–919 CrossRef CAS PubMed.
- C.-W. Lin, S. M. Bachilo and R. B. Weisman, J. Am. Chem. Soc., 2020, 142, 21189–21196 CrossRef CAS PubMed.
- N. Toshima and H. Hirai, Tetrahedron Lett., 1970, 11, 433–436 CrossRef.
- C. Castro, M. Dixon, I. Erden, P. Ergonenc, J. R. Keeffe and A. Sukhovitsky, J. Org. Chem., 1989, 54, 3732–3738 CrossRef CAS.
- H. Li, X. Jing, Y. Shi and L. Yu, React. Chem. Eng., 2021, 6, 119–124 RSC.
- M. R. Becker, A. D. Richardson and C. S. Schindler, Nat. Commun., 2019, 10, 5095 CrossRef PubMed.
- P. Gao, W. Pan, N. Li and B. Tang, Chem. Sci., 2019, 10, 6035–6071 RSC.
- G. Lukinavičius, L. Reymond, K. Umezawa, O. Sallin, E. D'Este, F. Göttfert, H. Ta, S. W. Hell, Y. Urano and K. Johnsson, J. Am. Chem. Soc., 2016, 138, 9365–9368 CrossRef PubMed.
- S. Ye, H. Zhang, J. Fei, C. H. Wolstenholme and X. Zhang, Angew. Chem., Int. Ed., 2021, 60, 1339–1346 CrossRef CAS PubMed.
- I. Sungwienwong, J. J. Ferrie, J. V. Jun, C. Liu, T. M. Barrett, Z. M. Hostetler, N. Ieda, A. Hendricks, A. K. Muthusamy, R. M. Kohli, D. M. Chenoweth, G. A. Petersson and E. J. Petersson, J. Phys. Org. Chem., 2018, 31, e3813 CrossRef PubMed.
- A. M. Sydor, K. J. Czymmek, E. M. Puchner and V. Mennella, Trends Cell Biol., 2015, 25, 730–748 CrossRef CAS PubMed.
- S. W. Hell, Angew. Chem., Int. Ed., 2015, 54, 8054–8066 CrossRef CAS PubMed.
- J. Tang, M. Zhang, H.-Y. Yin, J. Jing, D. Xie, P. Xu and J.-L. Zhang, Chem. Commun., 2016, 52, 11583–11586 RSC.
- C. G. England, H. Luo and W. Cai, Bioconjugate Chem., 2015, 26, 975–986 CrossRef CAS PubMed.
- G. V. Los, L. P. Encell, M. G. McDougall, D. D. Hartzell, N. Karassina, C. Zimprich, M. G. Wood, R. Learish, R. F. Ohana, M. Urh, D. Simpson, J. Mendez, K. Zimmerman, P. Otto, G. Vidugiris, J. Zhu, A. Darzins, D. H. Klaubert, R. F. Bulleit and K. V. Wood, ACS Chem. Biol., 2008, 3, 373–382 CrossRef CAS PubMed.
- Y. Liu, K. Miao, N. P. Dunham, H. Liu, M. Fares, A. K. Boal, X. Li and X. Zhang, Biochemistry, 2017, 56, 1585–1595 CrossRef CAS PubMed.
- Y. Liu, K. Miao, Y. Li, M. Fares, S. Chen and X. Zhang, Biochemistry, 2018, 57, 4663–4674 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc05351e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |