
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organocatalytic cycloaddition–elimination cascade for atroposelective construction of heterobiaryls†
Wen-Lei
Xu‡
,
Wei-Ming
Zhao‡
,
Ru-Xia
Zhang
,
Jie
Chen
and
Ling
Zhou
 *
*
Key Laboratory of Synthetic and Natural Functional Molecule of the Ministry of Education, College of Chemistry & Materials Science, Northwest University, Xi'an 710102, P. R. China. E-mail: zhoul@nwu.edu.cn
First published on 30th October 2021
Abstract
The first chiral phosphoric acid (CPA) catalyzed cycloaddition–elimination cascade reaction of 2-naphthol- and phenol-derived enecarbamates with azonaphthalenes has been established, providing a highly atroposelective route to an array of axially chiral aryl-C3-benzoindoles in excellent yields with excellent enantioselectivities. The success of this strategy derives from the stepwise process involving CPA-catalyzed asymmetric formal [3 + 2] cycloaddition and subsequent central-to-axial chirality conversion by elimination of a carbamate. In addition, the practicality of this reaction had been verified by varieties of transformations towards functionalized atropisomers.
Introduction
Axially chiral biaryls serve as the core motifs of abundant natural products, clinical drugs and functional materials.1–3 They also constitute privileged skeletons in lots of organocatalysts and ligands.4 Therefore, the development of novel and efficient methods to assemble structurally diverse axially chiral frameworks has attracted considerable attention from synthetic chemists.1a,5 As is well known, the central-to-axial chirality conversion (CACC) strategy has been developed into forceful tools to construct biaryl atropisomers and natural products that are not readily accessible otherwise.6,7 Despite the progress, most of these reactions require two steps including preparation of centrally chiral intermediates and then a chirality conversion step. Except for the driving force from in situ aromatization,6f,g,8 exogenous stoichiometric reagents, such as oxidants,6e,i,m–p Lewis acids6c,j and bases6h,k are needed to trigger central-to-axial chirality conversion, which hampers the efficiency and is incongruous with the principle of step economy (Scheme 1a). In sharp contrast, only one example successively circumvents these drawbacks. Sparr and co-workers disclosed a secondary amine-catalyzed intramolecular asymmetric aldol condensation with concomitant dehydration for accessing various axially chiral skeletons (Scheme 1a).7 Given a lack of highly efficient routes in terms of the CACC strategy, the development of an alternative catalytic mode continues to be enthralling but challenging.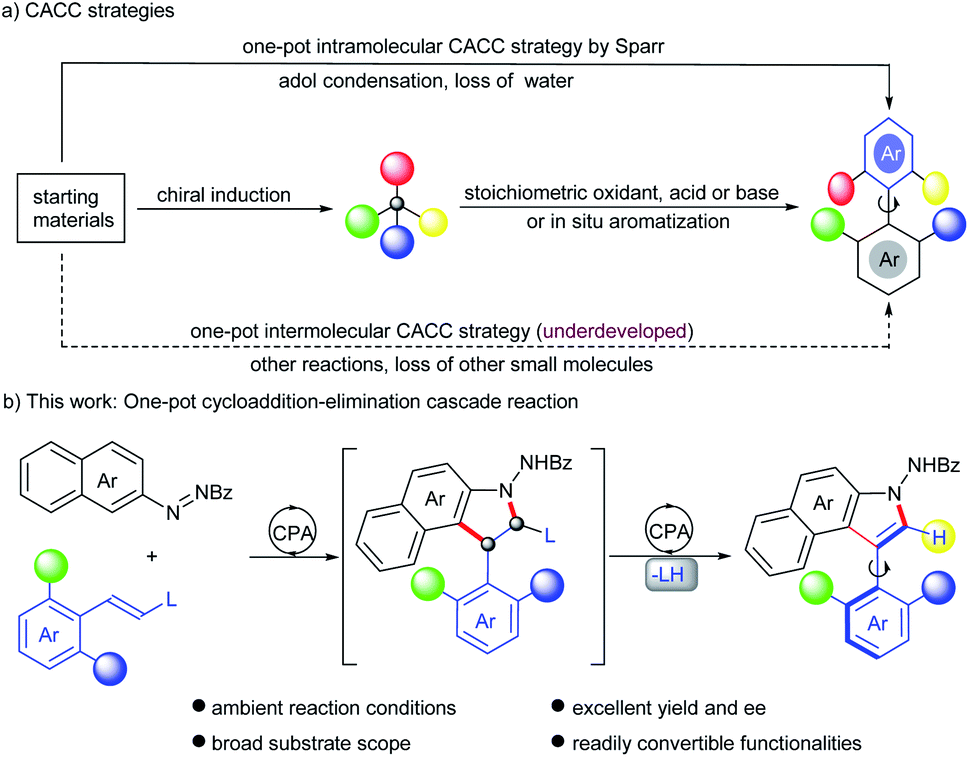 | ||
| Scheme 1 (a) CACC strategies; (b) this work: one-pot cycloaddition–elimination cascade reaction. | ||
Indoles possessing significant bioactivity are diversely found in many natural alkaloids and pharmaceuticals.9 Particularly, indole-based axially chiral skeletons hold vast potential in bioactive molecules and asymmetric catalysis.10 Therefore, a suite of synthetic attempts were devoted to procuring such optically active compounds, including dynamic kinetic resolution,11 cyclization,12 atroposelective arylation,8c,i,13de novo construction of the indole ring14 and the CACC strategy.6m,n Nevertheless, almost all generated atropisomeric indoles are installed with bulky substituents at the ortho-position around the axis, due to a lower rotation barrier and inferior conformational stability, which dramatically restricted the downstream functionalization as well as diversity-oriented synthesis of these chiral atropisomers. Exceptionally, Yan and co-workers devised an asymmetric annulation of ortho-alkynlanilines to produce axially chiral C3-unsubstituted naphthyl-indole scaffolds.14d However, the enantioselective establishment of C2-unsubstituted indole-based biaryls remains underdeveloped. Inspired by conventional CACC strategies and asymmetric cycloadditon of alkenes with azonaphthalenes,6m,n,8c,15 we envisioned that axially chiral C2-unsubstituted aryl-indole skeletons might be obtained after sequential cycloaddition and elimination by introducing a leaving group into ortho disubstituted arylethylene substrates, via a catalytic CACC mode (Scheme 1b). However, several challenges were still imbedded in this strategy, including (1) finding a feasible leaving group with catalytic sites to activate arylethylene substrates; (2) selecting powerful catalysts to increase the reactivity and promote elimination of the leaving group as well; (3) efficiently inducing stereocontrol in the sequential cycloaddition and elimination steps. To address these challenges, the alkoxycarbonylamino group was selected as the leaving group because enecarbamates have long served as a reactive dipolarophile to deliver enantioenriched compounds bearing contiguous chiral centers,16 especially upon activation by chiral phosphoric acid (CPA).16c–i In addition, the elimination of carbamate has proven to be feasible under strong acidic conditions;17 here CPA is expected to serve as an acid to realize the elimination. To the best of our knowledge, the construction of axially chiral biaryls by this designed CACC strategy has not been reported. Herein, we describe the first CPA-catalyzed cycloaddition–elimination cascade reaction of aryl enecarbamates with azonaphthalenes, providing a straightforward approach toward axially chiral aryl-C3-benzoindoles with excellent yields and enantioselectivities.
Results and discussion
To probe the feasibility of this assumption, our reaction development commenced with 2-naphthol-derived benzyl enecarbamate 1A and benzoyl azonaphthalene 2a in the presence of chiral phosphoric acid (R)-C1 (10 mol%) at room temperature. As expected, the desired product 3a was isolated smoothly in 83% yield with 81% ee without any cycloaddition intermediate remained (Table S1,† entry 1).18 This proof-of-principle result demonstrated that the proposed CPA-catalyzed cycloaddition–elimination cascade reaction seemed to be feasible. Based on this promising result, reaction parameters such as temperatures and solvents were screened, and excellent yield and high ee values were observed in DCM at 0 °C for 30 h and then 30 °C for 6 h (Table S2,† entry 1).18 To further improve the enantioselectivity, a series of BINOL-, H8-BINOL- and SPINOL-derived CPA catalysts were evaluated (Table 1, entries 2–9), which indicated that the CPA (R)-C5 could give rise to good enantiocontrol (Table 1, entry 5). In addition, the stereochemistry was also affected by different additives and O-protecting groups (Table 1, entries 10–15). An additive survey revealed Na2SO4 as the preferred choice with respect to enantioselectivity (Table 1, entry 10; Table S3†).18 Furthermore, the leaving group effect on enantioselectivity was tested in detail (Table 1, entries 16–19), and 2-naphthol-derived enecarbamate 1a bearing the 2-furanylmethoxy group afforded axially chiral benzoindole derivative 3a with 92% ee (Table 1, entry 19). Reduction of catalyst loading to 5 mol% resulted in retained ee but lower yield (Table 1, entry 20). The addition of H2O or EtOH caused significant decreases in the reaction stereoselectivity, which helps explain why hydrophilic additives produced slightly improved ee values (Table 1, entries 21–22). We finally identified the optimal conditions as follows: 1a (0.12 mmol), 2a (0.1 mmol), (R)-C5 (10 mol%), Na2SO4 (40 mg), in DCM (4.0 mL) at 0 °C for 30 h and then 30 °C for 6 h, affording product 3a in 94% yield with 92% ee. The structure of 3a and its absolute configuration was confirmed by X-ray crystallography analysis of the 2-bromobenzoindole derivative (vide infra).|
|
|||||
|---|---|---|---|---|---|
| Entry | 1 (R1/R2) | CPA | Additive | Yieldb (%) | eec (%) |
| a Reactions were carried out with 1 (0.12 mmol), 2a (0.10 mmol) and additive (40 mg) CPA (10 mol%) in DCM (4.0 mL) at 0 °C for 30 h and then 30 °C for further 6 h. b Isolated yield. c The ee value was determined by chiral HPLC analysis. d 5 mol% of CPA was used. e 10 mg H2O was added. f 10 mg EtOH was added. Pro = n-propoxy. Nmo = 2-naphthylmethoxy. Fmo = 2-furanylmethoxy. | |||||
| 1 | 1A (BnO/Me) | (R)-C1 | — | 3a, 90 | 86 |
| 2 | 1A (BnO/Me) | (R)-C2 | — | 3a, 78 | 55 |
| 3 | 1A (BnO/Me) | (R)-C3 | — | 3a, 91 | 71 |
| 4 | 1A (BnO/Me) | (R)-C4 | — | 3a, n.r. | — |
| 5 | 1A (BnO/Me) | (R)-C5 | — | 3a, 93 | 88 |
| 6 | 1A (BnO/Me) | (R)-C6 | — | 3a, 90 | 64 |
| 7 | 1A (BnO/Me) | (R)-C7 | — | 3a, 69 | 70 |
| 8 | 1A (BnO/Me) | (R)-C8 | — | 3a, 8 | 11 |
| 9 | 1A (BnO/Me) | (R)-C9 | — | 3a, trace | — |
| 10 | 1A (BnO/Me) | (R)-C5 | Na2SO4 | 3a, 95 | 90 |
| 11 | 1B (BnO/Et) | (R)-C5 | Na2SO4 | 3B, 95 | 84 |
| 12 | 1C (BnO/n-Pr) | (R)-C5 | Na2SO4 | 3C, 87 | 83 |
| 13 | 1D (BnO/i-Pr) | (R)-C5 | Na2SO4 | 3D, 93 | 89 |
| 14 | 1E (BnO/All) | (R)-C5 | Na2SO4 | 3E, 92 | 78 |
| 15 | 1F (BnO/Bn) | (R)-C5 | Na2SO4 | 3F, 90 | 67 |
| 16 | 1G (Pro/Me) | (R)-C5 | Na2SO4 | 3a, 91 | 72 |
| 17 | 1H (Nmo/Me) | (R)-C5 | Na2SO4 | 3a, 93 | 90 |
| 18 | 1I (BnS/Me) | (R)-C5 | Na2SO4 | 3a, 95 | 90 |
| 19 | 1a (Fmo/Me) | (R)-C5 | Na2SO4 | 3a, 94 | 92 |
| 20d | 1a (Fmo/Me) | (R)-C5 | Na2SO4 | 3a, 83 | 92 |
| 21e | 1a (Fmo/Me) | (R)-C5 | Na2SO4 | 3a, 87 | 81 |
| 22f | 1a (Fmo/Me) | (R)-C5 | Na2SO4 | 3a, 71 | 24 |

With the optimum conditions identified, we next explored the substrate generality of the asymmetric cycloaddition–elimination cascade reaction. Firstly, the 2-naphthol-derived enecarbamates 1a–t were examined and the results are summarized in Table 2. Regardless of the type of functional groups, such as alkyl (Me, Et, i-Pr, t-Bu, and Bn), aryl (Ph), alkoxyl (OMe) and halogen (Br), at the C5- or C6- position on the naphthyl ring, the reaction proceeded smoothly with benzoyl azonaphthalene 2a to generate the corresponding products (3a–n) in excellent yields with excellent enantioselectivities. However, the electronic properties of substituents at the C7-position on the naphthyl ring had an obvious effect on stereoselectivity. Compared with electron-withdrawing and -neutral groups, the electron-donating group at the C7-position returned product 3r with outstanding results in both reactivity and enantiocontrol. 2-Naphthol-derived enecarbamates with C3-substituents on the naphthyl ring were also tolerated, furnishing the expected products 3s and 3t with good reaction outcomes by extending the reaction time, presumably due to the steric reason. Subsequently, the substrate scope in terms of the benzoyl group of azonaphthalenes was investigated. Phenyl rings with para- and meta-substituents were compatible, delivering products 3ab–ag in remarkable results. Reduced enantiocontrol was gained by introducing substituents into the ortho-position of the phenyl ring, probably due to steric hindrance (3ah, 3ai). To investigate the configurational stability of these prepared axially chiral aryl-C3-benzoindoles, a thermal racemization experiment was conducted. The rotation barrier of compound 3a was calculated to be 31.1 kcal mol−1 at 100 °C, corresponding to a half-life of 107.0 years at 25 °C.
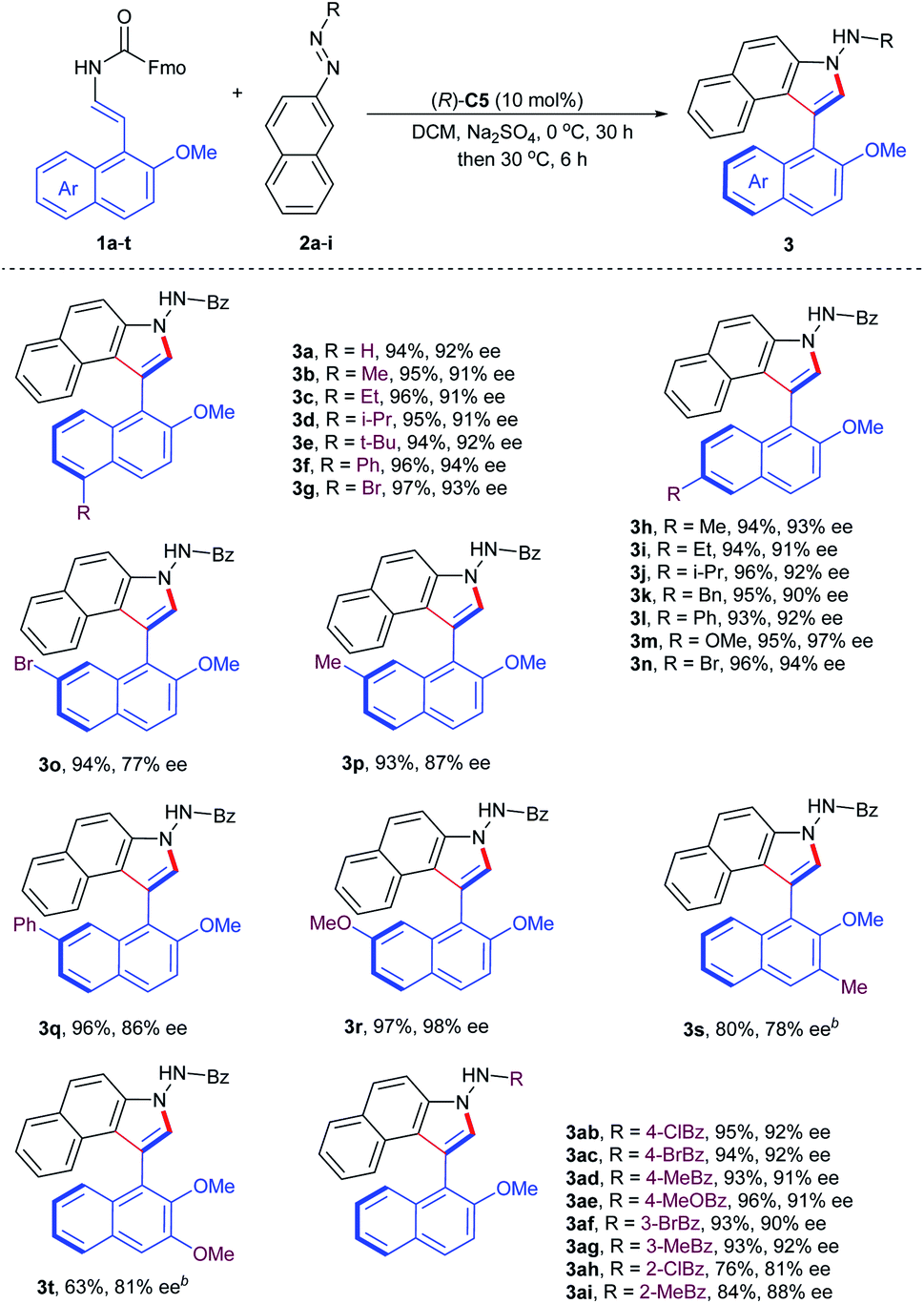
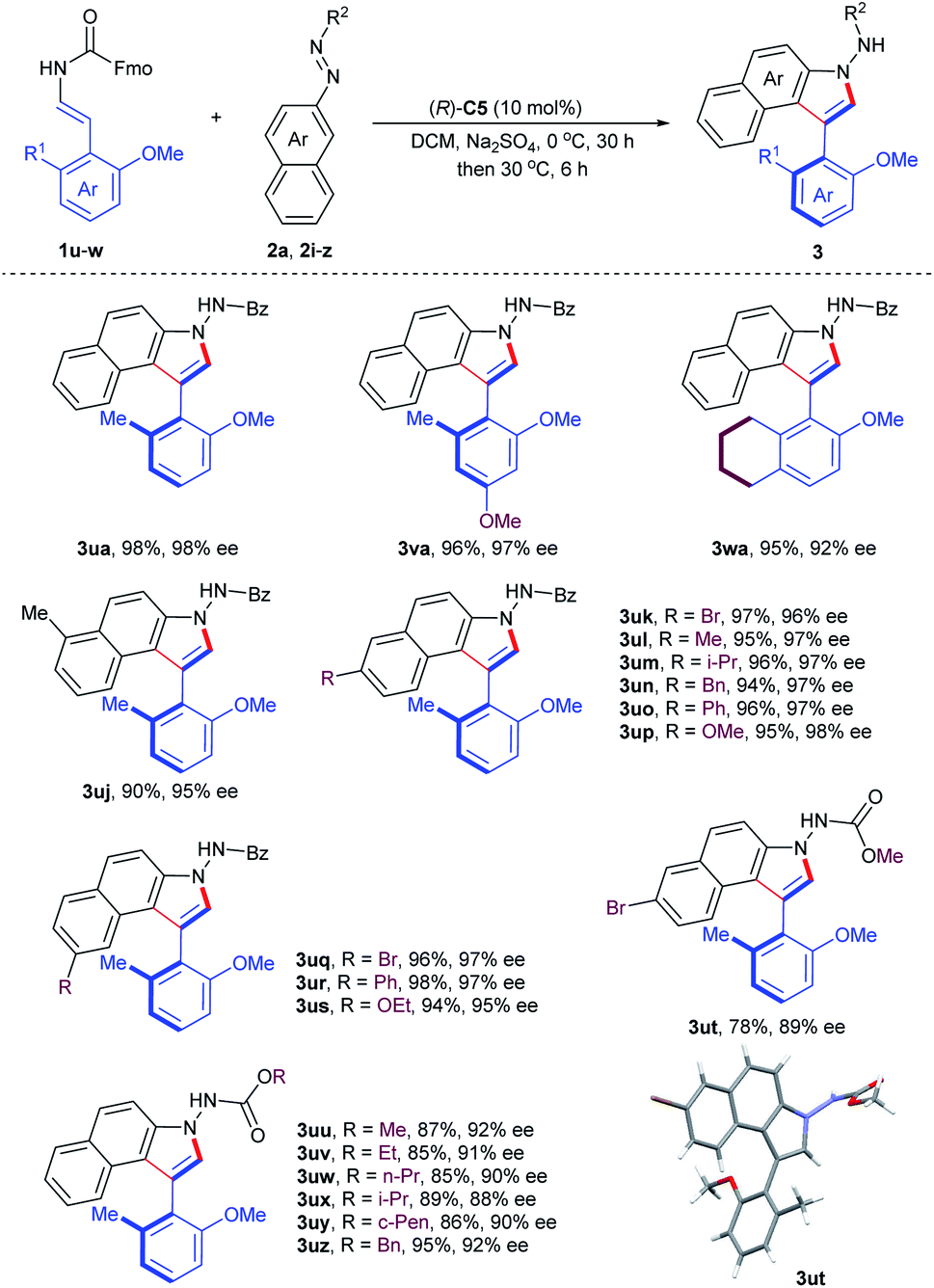
To further demonstrate the utility of this organocatalytic CACC methodology, we then turned our attention to the construction of axially chiral phenyl-benzoindoles (Table 3). The phenol-derived enecarbamates 1u–w reacted effectively with benzoyl azonaphthalene 2a under the optimal conditions, giving the desired products 3ua–wa with outstanding enantioselectivities and yields. Then, the scope of benzoyl azonaphthalenes was examined. The positions and electron properties of substituents on benzoyl azonaphthalenes did not dramatically influence the results, since the corresponding products 3uj–us were obtained in high yields with an enantiomeric excess range of 95–98%. Moreover, carbalkoxyl azonaphthalenes were further explored and generally provided the products 3ut–uz with similar enantiocontrol. The absolute configuration of 3ut was determined by an X-ray crystallographic study and other products were assigned analogously.19
| a Reactions were carried out with 1 (0.12 mmol), 2 (0.10 mmol), Na2SO4 (40 mg), and (R)-C5 (10 mol%) in DCM (4.0 mL) at 0 °C for 30 h and then 30 °C for further 6 h. Isolated yield. The ee value was determined by chiral HPLC analysis. |
|---|
|
To substantiate the practicability of this new strategy, gram-scale synthesis of 3a was performed under the optimal reaction conditions, which was obtained in comparable yield without deterioration of enantiomeric excess (95% yield, 92% ee; Scheme 2). Debenzoylation of compound 3a gave the aminobiaryl 4 in 87% yield with 91% ee. Furthermore, a range of derivatizations of 3a were conducted to expand the potential synthetic utility of this reaction. Halogenation of 3a with corresponding halogen sources, such as NIS, NBS and NCS, afforded 2-iodobenzoindole 5, 2-bromobenzoindole 6 and 2-chlorobenzoindole 7 in high yield with negligible erosion of optical purity, which serve as useful functional handles for further transformations. Atropisomeic 2-nitrobenzoindole 8 was also successfully synthesized directly from 3a in moderate yield with retention of enantiopurity. More importantly, treatment of 3a with sodium thioethylate at 70 °C led to a facile demethylation reaction, providing 9 in 91% yield with 90% ee. This finding enables the methyl moiety to play a temporary role, thus allowing downstream functionalization at the C2-position on the naphthyl ring. Further conversion of 4 with isocyanatoarene or isothiocyanatoarene proceeded smoothly to generate the axially chiral thiourea 10 and urea 11, two potential organocatalysts, in good yields with no loss of enantiomeric purity. In addition, cleavage of the N–N bond was realized to produce N-unsubstituted naphthyl-C3-benzoindole 12 in moderate yield with the same enantiopurity. To further highlight the versatile reactivities of the C2-position on the benzoindolyl ring, the axially chiral product 12 was transformed into 1-methyl-2-bromobenzoindole 13 and 2-formylbenzoindole 14, respectively. Moreover, the methoxy group of compound 12 could be readily converted to triflate, which is a crucial precursor to chiral monophosphorus ligands.13
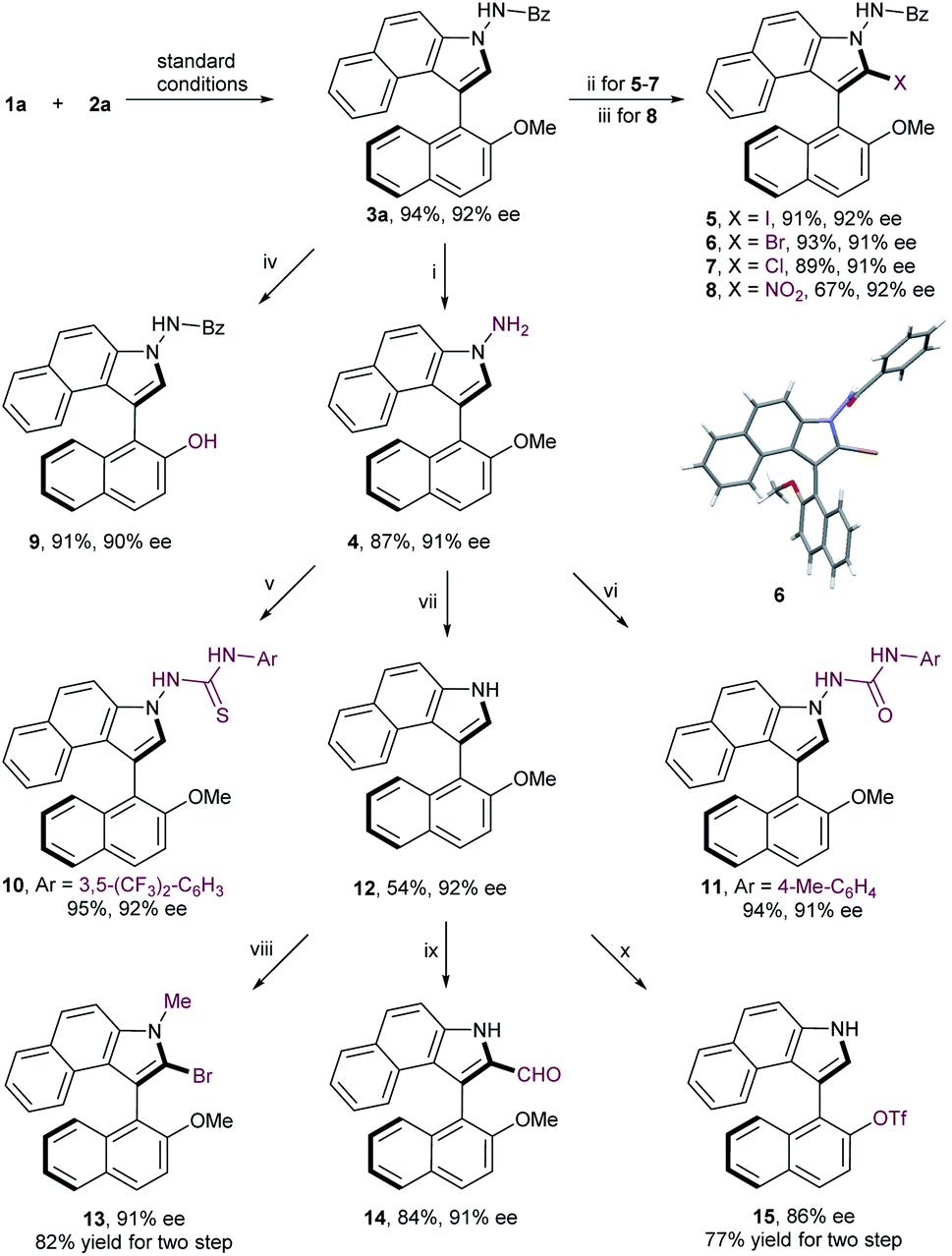 | ||
| Scheme 2 Gram-scale synthesis and further derivatizations. Reagents and conditions: (i) concentrated HCl/EtOH (1/2 v/v), 70 °C; (ii) NXS, TFA, MeCN, 0 °C; (iii) t-BuONO, MeCN, r.t.; (iv) NaSEt, DMF, 70 °C; (v) 1-isothiocyanato-3,5-bis(trifluoromethyl)benzene, MeCN, 0 °C; (vi) 1-isocyanato-4-methylbenzene, THF, r.t.; (vii) NaNO2, H2O/EtOH (1/2 v/v), 0 °C; (viii) NaH, MeI, THF, 0 °C; NBS, TFA, MeCN, 0 °C; (ix) DMF, POCl3, 0 °C; (x) BBr3, DCM, −78 °C; Tf2O, Et3N, DCM, 0 °C. | ||
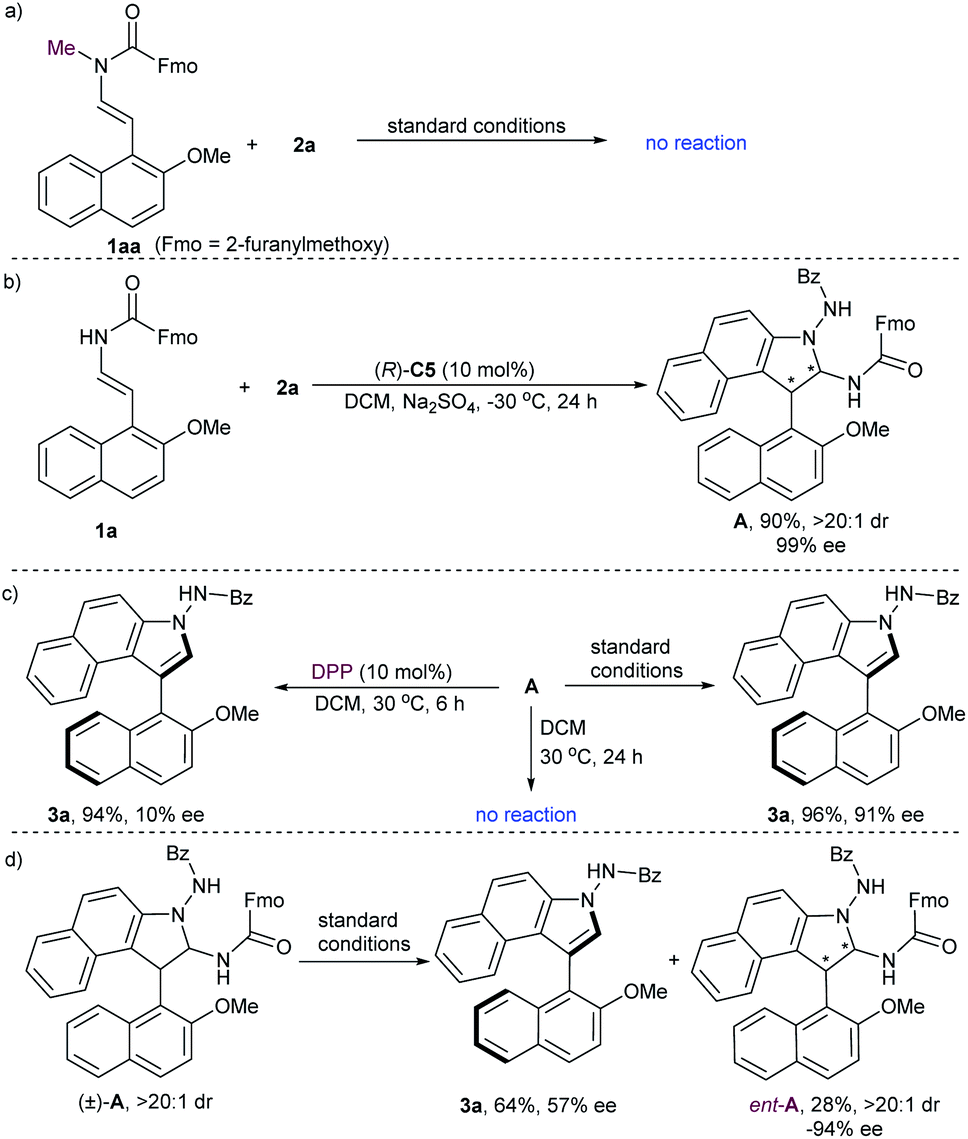
To gain some insight into the mechanism of this reaction, several control experiments were carried out (Scheme 3). No reaction proceeded when N-methyl-protected 1aa was employed in the initial cycloaddition reaction, which suggested that the hydrogen-bonding between the N–H group and the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety of the catalyst is important for the reactivity (Scheme 3a). In addition, we performed a reaction between 2-naphthol-derived enecarbamate 1a and benzoyl azonaphthalene 2a under the catalysis of CPA (R)-C5 at −30 °C for 24 h. The formal [3 + 2] cycloaddition intermediate A with diastereoisomeric conformers could be isolated in 90% yield with 99% ee and excellent diastereoselectivity (Scheme 3b).18,20 Unfortunately, these rotamers are inseparable, and the attempt to identify the absolute configuration of compound A failed. Nonetheless, some useful clues were observed from this centrally chiral intermediate. The desired axially chiral product 3a was prepared from intermediate A with comparable yield and ee under the standard conditions, while poor chirality conversion was observed under the catalysis of diphenyl phosphate (DPP, 10% ee). On the other hand, treatment of A in the absence of CPA in DCM at 30 °C for 24 h afforded only the recovered A. These results implied that the CPA catalyst plays a crucial role in controlling the reactivity and enantioselectivity during the central-to-axial chirality conversion (Scheme 3c). When the racemic cycloaddition intermediate (±)-A was treated with CPA (R)-C5 in DCM for 6 h, the product 3a was obtained in 64% yield with 57% ee, and the starting material was recovered in 28% yield with opposite enantioselectivity (ent-A, −94% ee). These results suggested that the kinetic resolution of (±)-A through CPA-catalyzed elimination of a carbamate occurred (Scheme 3d). The preferential elimination of the fast-reacting enantiomer for racemic aminal intermediate (±)-A can then be rationalized through minimization of unfavourable steric hindrance between the aminal intermediate and the (R)-configurated CPA to form a steric configuration-matched iminium-phosphate ion pair.
O moiety of the catalyst is important for the reactivity (Scheme 3a). In addition, we performed a reaction between 2-naphthol-derived enecarbamate 1a and benzoyl azonaphthalene 2a under the catalysis of CPA (R)-C5 at −30 °C for 24 h. The formal [3 + 2] cycloaddition intermediate A with diastereoisomeric conformers could be isolated in 90% yield with 99% ee and excellent diastereoselectivity (Scheme 3b).18,20 Unfortunately, these rotamers are inseparable, and the attempt to identify the absolute configuration of compound A failed. Nonetheless, some useful clues were observed from this centrally chiral intermediate. The desired axially chiral product 3a was prepared from intermediate A with comparable yield and ee under the standard conditions, while poor chirality conversion was observed under the catalysis of diphenyl phosphate (DPP, 10% ee). On the other hand, treatment of A in the absence of CPA in DCM at 30 °C for 24 h afforded only the recovered A. These results implied that the CPA catalyst plays a crucial role in controlling the reactivity and enantioselectivity during the central-to-axial chirality conversion (Scheme 3c). When the racemic cycloaddition intermediate (±)-A was treated with CPA (R)-C5 in DCM for 6 h, the product 3a was obtained in 64% yield with 57% ee, and the starting material was recovered in 28% yield with opposite enantioselectivity (ent-A, −94% ee). These results suggested that the kinetic resolution of (±)-A through CPA-catalyzed elimination of a carbamate occurred (Scheme 3d). The preferential elimination of the fast-reacting enantiomer for racemic aminal intermediate (±)-A can then be rationalized through minimization of unfavourable steric hindrance between the aminal intermediate and the (R)-configurated CPA to form a steric configuration-matched iminium-phosphate ion pair.
 | ||
| Scheme 3 Mechanistic studies. | ||
On the basis of these observations as well as previous literature,4f,8c,16i,21 a catalytic mechanism was proposed in Fig. 1. Firstly, the mechanistic studies suggest a stereochemical model for the enantioselective [3 + 2] cycloaddition, in which the activation of both 2-naphthol-derived enecarbamate 1a and benzoyl azonaphthalene 2a by a bifunctional catalyst through dual hydrogen-bonding facilitates nucleophilic attack at the α-position of 2a to give the dearomatized intermediate B. Facile aromatization of intermediate B generates the arylhydrazine intermediate C. Then, cyclization occurs via intramolecular aminalization to afford cycloaddition aminal intermediate A. Subsequently, the CPA catalyst could accelerate elimination of a carbamate from intermediate A to form the identical steric configuration-matched iminium-phosphate ion pair D or D′. Finally, further release of CPA by β-H elimination leads to aromatization to accomplish the central-to-axial chirality conversion, which ultimately affords the axially chiral product 3a.
 | ||
| Fig. 1 Proposed mechanism. | ||
In summary, we have developed the first well-designed CPA-catalyzed asymmetric cycloaddition–elimination cascade reactions of 2-naphthol- or phenol-derived enecarbamates with azonaphthalenes. A wide range of naphthyl-C3-benzoindoles and phenyl-C3-benzoindoles with functionality versatility could be obtained in high yields with good to excellent enantioselectivities. This reaction shuns the use of extra steps and exogenous stoichiometric reagents, and thus represents as a step- and atom-economical concept. Furthermore, the synthetic utility of this protocol was explored via convenient functional group transformation methods. Mechanistic studies disclosed that chiral phosphoric acid played significant roles in controlling both the reactivity as well as enantioselectivity to the cycloaddition and central-to-axial chirality conversion and showed efficient kinetic resolution performance on this type of aminal. This work will not only provide a straightforward alternative to access C2-unsubstituted axially chiral aryl-C3-benzoindoles, but will also open new avenues for conventional central-to-axial chirality conversion. Further investigation of the mechanism in detail and utilization of this strategy for the synthesis of substantial atropisomeric biaryl backbones are currently undergoing in this laboratory.
Data availability
All experimental and crystallographic data is available in the ESI.†Author contributions
W.-L. X., W.-M. Z. and R.-X. Z. performed all the experiments. W.-L. X., J. C. and L. Z. contributed to the conception of the experiments, discussion of the results and preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (NSFC 21672170), and Natural Science Basic Research Plan in Shaanxi Province of China (2021JZ-40 and 2018JC-020) for financial support.Notes and references
- (a) G. Bringmann, A. J. Price Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384–5427 CrossRef CAS PubMed; (b) M. C. Kozlowski, B. J. Morgan and E. C. Linton, Chem. Soc. Rev., 2009, 38, 3193–3207 RSC; (c) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed.
- (a) J. Clayden, W. J. Moran, P. J. Edwards and S. R. LaPlante, Angew. Chem., Int. Ed., 2009, 48, 6398–6401 CrossRef CAS PubMed; (b) S. R. LaPlante, P. J. Edwards, L. D. Fader, A. Jakalian and O. Hucke, ChemMedChem, 2011, 6, 505–513 CrossRef CAS PubMed; (c) S. R. LaPlante, L. D. Fader, K. R. Fandrick, D. R. Fandrick, O. Hucke, R. Kemper, S. P. F. Miller and P. J. Edwards, J. Med. Chem., 2011, 54, 7005–7022 CrossRef CAS PubMed; (d) A. Zask, J. Murphy and G. A. Ellestad, Chirality, 2013, 25, 265–274 CrossRef CAS PubMed; (e) P. W. Glunz, Bioorg. Med. Chem. Lett., 2018, 28, 53–60 CrossRef CAS PubMed.
- (a) L. Pu, Chem. Rev., 1998, 98, 2405–2494 CrossRef CAS PubMed; (b) L. Pu, Acc. Chem. Res., 2012, 45, 150–163 CrossRef CAS PubMed; (c) Y. Wang, A. Urbas and Q. Li, J. Am. Chem. Soc., 2012, 134, 3342–3345 CrossRef CAS PubMed; (d) S. Feuillastre, M. Pauton, L. Gao, A. Desmarchelier, A. J. Riives, D. Prim, D. Tondelier, B. Geffroy, G. Muller, G. Clavier and G. Pieters, J. Am. Chem. Soc., 2016, 138, 3990–3993 CrossRef CAS PubMed; (e) K. Takaishi, M. Yasui and T. Ema, J. Am. Chem. Soc., 2018, 140, 5334–5338 CrossRef CAS PubMed; (f) M. Li, Y.-F. Wang, D. Zhang, L. Duan and C.-F. Chen, Angew. Chem., Int. Ed., 2020, 59, 3500–3504 CrossRef CAS PubMed.
- (a) Y. Chen, S. Yekta and A. K. Yudin, Chem. Rev., 2003, 103, 3155–3212 CrossRef CAS PubMed; (b) P. Kočovský, Š. Vyskočil and M. Smrčina, Chem. Rev., 2003, 103, 3213–3246 CrossRef PubMed; (c) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029–3070 CrossRef CAS PubMed; (d) M. l. Berthod, G. r. Mignani, G. Woodward and M. Lemaire, Chem. Rev., 2005, 105, 1801–1836 CrossRef CAS PubMed; (e) M. Shibasaki and S. Matsunaga, Chem. Soc. Rev., 2006, 35, 269–279 RSC; (f) T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS PubMed; (g) J. M. Brunel, Chem. Rev., 2007, 107, PR1–PR45 CrossRef CAS; (h) M. Terada, Chem. Commun., 2008, 4097–4112 RSC; (i) J.-H. Xie and Q.-L. Zhou, Acc. Chem. Res., 2008, 41, 581–593 CrossRef CAS PubMed; (j) M. P. Carroll and P. J. Guiry, Chem. Soc. Rev., 2014, 43, 819–833 RSC; (k) Y. Xiao, Z. Sun, H. Guo and O. Kwon, Beilstein J. Org. Chem., 2014, 10, 2089–2121 CrossRef PubMed; (l) T. Akiyama and K. Mori, Chem. Rev., 2015, 115, 9277–9306 CrossRef CAS PubMed; (m) J. Wencel-Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418–3430 RSC; (n) Y.-N. Ma, S.-X. Li and S.-D. Yang, Acc. Chem. Res., 2017, 50, 1480–1492 CrossRef CAS PubMed; (o) Y.-B. Wang and B. Tan, Acc. Chem. Res., 2018, 51, 534–547 CrossRef CAS PubMed.
- (a) G. Bencivenni, Synlett, 2015, 26, 1915–1922 CrossRef CAS; (b) E. Kumarasamy, R. Raghunathan, M. P. Sibi and J. Sivaguru, Chem. Rev., 2015, 115, 11239–11300 CrossRef CAS PubMed; (c) G. Ma and M. P. Sibi, Chem.–Eur. J., 2015, 21, 11644–11657 CrossRef CAS PubMed; (d) J. Wencel-Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418–3430 RSC; (e) P. Loxq, E. Manoury, R. Poli, E. Deydier and A. Labande, Coord. Chem. Rev., 2016, 308, 131–190 CrossRef CAS; (f) S. Shirakawa, S. Liu and S. Kaneko, Chem.–Asian J., 2016, 11, 330–341 CrossRef CAS PubMed; (g) D. Bonne and J. Rodriguez, Chem. Commun., 2017, 53, 12385–12393 RSC; (h) Y.-B. Wang and B. Tan, Acc. Chem. Res., 2018, 51, 534–547 CrossRef CAS PubMed; (i) B. Zilate, A. Castrogiovanni and C. Sparr, ACS Catal., 2018, 8, 2981–2988 CrossRef CAS; (j) D.-J. Cheng and Y.-D. Shao, Adv. Synth. Catal., 2020, 362, 3081–3099 CrossRef CAS; (k) V. Corti and G. Bertuzzi, Synthesis, 2020, 52, 2450–2468 CrossRef CAS; (l) J. A. Carmona, C. Rodríguez-Franco, R. Fernández, V. Hornillos and J. M. Lassaletta, Chem. Soc. Rev., 2021, 50, 2968–2983 RSC; (m) J. K. Cheng, S.-H. Xiang, S. Li, L. Ye and B. Tan, Chem. Rev., 2021, 121, 4805–4902 CrossRef CAS PubMed.
- For reviews, see: (a) T. T. Nguyen, Org. Biomol. Chem., 2019, 17, 6952–6963 RSC; (b) H. Yang, J. Chen and L. Zhou, Chem.–Asian J., 2020, 15, 2939–2951 CrossRef CAS PubMed. For selected examples, see: (c) F. Guo, L. C. Konkol and R. J. Thomson, J. Am. Chem. Soc., 2011, 133, 18–20 CrossRef CAS PubMed; (d) O. Quinonero, C. Bressy and X. Bugaut, Angew. Chem., Int. Ed., 2014, 53, 10861–10863 CrossRef CAS PubMed; (e) O. Quinonero, M. Jean, N. Vanthuyne, C. Roussel, D. Bonne, T. Constantieux, C. Bressy, X. Bugaut and J. Rodriguez, Angew. Chem., Int. Ed., 2016, 55, 1401–1405 CrossRef CAS PubMed; (f) J.-Z. Wang, J. Zhou, C. Xu, H. Sun, L. Kürti and Q.-L. Xu, J. Am. Chem. Soc., 2016, 138, 5202–5205 CrossRef CAS PubMed; (g) Y.-H. Chen, D.-J. Cheng, J. Zhang, Y. Wang, X.-Y. Liu and B. Tan, J. Am. Chem. Soc., 2015, 137, 15062–15065 CrossRef CAS PubMed; (h) J. D. Jolliffe, R. J. Armstrong and M. D. Smith, Nat. Chem., 2017, 9, 558–562 CrossRef CAS PubMed; (i) V. S. Raut, M. Jean, N. Vanthuyne, C. Roussel, T. Constantieux, C. Bressy, X. Bugaut, D. Bonne and J. Rodriguez, J. Am. Chem. Soc., 2017, 139, 2140–2143 CrossRef CAS PubMed; (j) A. Link and C. Sparr, Angew. Chem., Int. Ed., 2018, 57, 7136–7139 CrossRef CAS PubMed; (k) X.-L. He, H.-R. Zhao, X. Song, B. Jiang, W. Du and Y.-C. Chen, ACS Catal., 2019, 9, 4374–4381 CrossRef CAS; (l) S.-C. Zheng, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2019, 58, 9215–9219 CrossRef CAS PubMed; (m) Y.-L. Hu, Z. Wang, H. Yang, J. Chen, Z.-B. Wu, Y. Lei and L. Zhou, Chem. Sci., 2019, 10, 6777–6784 RSC; (n) G. D. Bisag, D. Pecorari, A. Mazzanti, L. Bernardi, M. Fochi, G. Bencivenni, G. Bertuzzi and V. Corti, Chem.–Eur. J., 2019, 25, 15694–15701 CrossRef CAS PubMed; (o) S. Zhu, Y.-H. Chen, Y.-B. Wang, P. Yu, S.-Y. Li, S.-H. Xiang, J.-Q. Wang, J. Xiao and B. Tan, Nat. Commun., 2019, 10, 4268 CrossRef PubMed; (p) X. Bao, J. Rodriguez and D. Bonne, Chem. Sci., 2020, 11, 403–408 RSC; (q) D. Zhang, Z. Su, Q. He, Z. Wu, Y. Zhou, C. Pan, X. Liu and X. Feng, J. Am. Chem. Soc., 2020, 142, 15975–15985 CrossRef CAS PubMed.
- (a) A. Link and C. Sparr, Angew. Chem., Int. Ed., 2014, 53, 5458–5461 CrossRef CAS PubMed; (b) V. C. Fäseke and C. Sparr, Angew. Chem., Int. Ed., 2016, 55, 7261–7264 CrossRef PubMed; (c) D. Lotter, M. Neuburger, M. Rickhaus, D. Häussinger and C. Sparr, Angew. Chem., Int. Ed., 2016, 55, 2920–2923 CrossRef CAS PubMed; (d) R. M. Witzig, V. C. Fäseke, D. Häussinger and C. Sparr, Nat. Catal., 2019, 2, 925–930 CrossRef CAS.
- (a) M. Moliterno, R. Cari, A. Puglisi, A. Antenucci, C. Sperandio, E. Moretti, A. DiSabato, R. Salvio and M. Bella, Angew. Chem., Int. Ed., 2016, 55, 6525–6529 CrossRef CAS PubMed; (b) Y.-H. Chen, L.-W. Qi, F. Fang and B. Tan, Angew. Chem., Int. Ed., 2017, 56, 16308–16312 CrossRef CAS PubMed; (c) L.-W. Qi, J.-H. Mao, J. Zhang and B. Tan, Nat. Chem., 2018, 10, 58–64 CrossRef CAS PubMed; (d) J.-Y. Liu, X.-C. Yang, Z. Liu, Y.-C. Luo, H. Lu, Y.-C. Gu, R. Fang and P.-F. Xu, Org. Lett., 2019, 21, 5219–5224 CrossRef CAS PubMed; (e) D.-L. Lu, Y.-H. Chen, S.-H. Xiang, P. Yu, B. Tan and S. Li, Org. Lett., 2019, 21, 6000–6004 CrossRef CAS PubMed; (f) L.-W. Qi, S. Li, S.-H. Xiang, J. Wang and B. Tan, Nat. Catal., 2019, 2, 314–323 CrossRef CAS; (g) G. Coombs, M. H. Sak and S. J. Miller, Angew. Chem., Int. Ed., 2020, 59, 2875–2880 CrossRef CAS PubMed; (h) W.-Y. Ding, P. Yu, Q.-J. An, K. L. Bay, S.-H. Xiang, S. Li, Y. Chen, K. N. Houk and B. Tan, Chem, 2020, 6, 2046–2059 CrossRef CAS; (i) W. Xia, Q.-J. An, S.-H. Xiang, S. Li, Y.-B. Wang and B. Tan, Angew. Chem., Int. Ed., 2020, 59, 6775–6779 CrossRef CAS PubMed.
- (a) G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875–2911 CrossRef CAS PubMed; (b) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608–9644 CrossRef CAS PubMed; (c) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489–4497 CrossRef CAS PubMed.
- (a) G. N. Anilkumar, C. A. Lesburg, O. Selyutin, S. B. Rosenblum, Q. Zeng, Y. Jiang, T.-Y. Chan, H. Pu, H. Vaccaro, L. Wang, F. Bennett, K. X. Chen, J. Duca, S. Gavalas, Y. Huang, P. Pinto, M. Sannigrahi, F. Velazquez, S. Venkatraman, B. Vibulbhan, S. Agrawal, N. Butkiewicz, B. Feld, E. Ferrari, Z. He, C.-k. Jiang, R. E. Palermo, P. McMonagle, H. C. Huang, N.-Y. Shih, G. Njoroge and J. A. Kozlowski, Bioorg. Med. Chem. Lett., 2011, 21, 5336–5341 CrossRef CAS PubMed; (b) J. G. Luz, M. W. Carson, B. Condon, D. Clawson, A. Pustilnik, D. T. Kohlman, R. J. Barr, J. S. Bean, M. J. Dill, D. K. Sindelar, M. Maletic and M. J. Coghlan, J. Med. Chem., 2015, 58, 6607–6618 CrossRef CAS PubMed; (c) K. Sharma, E. R. Baral, M. S. Akhtar, Y. R. Lee, S. H. Kim and Y.-J. Wee, Res. Chem. Intermed., 2017, 43, 2387–2399 CrossRef CAS; (d) S. Mishra, J. Liu and A. Aponick, J. Am. Chem. Soc., 2017, 139, 3352–3355 CrossRef CAS PubMed; (e) T.-Z. Li, S.-J. Liu, W. Tan and F. Shi, Chem.–Eur. J., 2020, 26, 15779–15792 CrossRef CAS PubMed.
- (a) F. Jiang, K.-W. Chen, P. Wu, Y.-C. Zhang, Y. Jiao and F. Shi, Angew. Chem., Int. Ed., 2019, 58, 15104–15110 CrossRef CAS PubMed; (b) C. Ma, F. Jiang, F.-T. Sheng, Y. Jiao, G.-J. Mei and F. Shi, Angew. Chem., Int. Ed., 2019, 58, 3014–3020 CrossRef CAS PubMed.
- (a) C. He, M. Hou, Z. Zhu and Z. Gu, ACS Catal., 2017, 7, 5316–5320 CrossRef CAS; (b) A. Kim, A. Kim, S. Park, S. Kim, H. Jo, K. M. Ok, S. K. Lee, J. Song and Y. Kwon, Angew. Chem., Int. Ed., 2021, 60, 12279–12283 CrossRef CAS PubMed.
- H.-H. Zhang, C.-S. Wang, C. Li, G.-J. Mei, Y. Li and F. Shi, Angew. Chem., Int. Ed., 2017, 56, 116–121 CrossRef CAS PubMed.
- (a) N. Ototake, Y. Morimoto, A. Mokuya, H. Fukaya, Y. Shida and O. Kitagawa, Chem.–Eur. J., 2010, 16, 6752–6755 CrossRef CAS PubMed; (b) M. Tian, D. Bai, G. Zheng, J. Chang and X. Li, J. Am. Chem. Soc., 2019, 141, 9527–9532 CrossRef CAS PubMed; (c) L. Wang, J. Zhong and X. Lin, Angew. Chem., Int. Ed., 2019, 58, 15824–15828 CrossRef CAS PubMed; (d) L. Peng, K. Li, C. Xie, S. Li, D. Xu, W. Qin and H. Yan, Angew. Chem., Int. Ed., 2019, 58, 17199–17204 CrossRef CAS PubMed; (e) Y.-P. He, H. Wu, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2020, 59, 2105–2109 CrossRef CAS PubMed; (f) L. Sun, H. Chen, B. Liu, J. Chang, L. Kong, F. Wang, Y. Lan and X. Li, Angew. Chem., Int. Ed., 2021, 60, 8391–8395 CrossRef CAS PubMed; (g) Y.-P. He, J. Cao, H. Wu, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2021, 60, 7093–7097 CrossRef CAS PubMed.
- C. Li, D.-N. Xu, C. Ma, G.-J. Mei and F. Shi, J. Org. Chem., 2018, 83, 9190–9200 CrossRef CAS PubMed.
- (a) R. Matsubara, Y. Nakamura and S. Kobayashi, Angew. Chem., Int. Ed., 2004, 43, 1679–1681 CrossRef CAS PubMed; (b) R. Matsubara, Y. Nakamura and S. Kobayashi, Angew. Chem., Int. Ed., 2004, 43, 3258–3260 CrossRef CAS PubMed; (c) M. Terada and K. Sorimachi, J. Am. Chem. Soc., 2007, 129, 292–293 CrossRef CAS PubMed; (d) A. l. Alix, C. Lalli, P. Retailleau and G. r. Masson, J. Am. Chem. Soc., 2012, 134, 10389–10392 CrossRef CAS PubMed; (e) G. Dagousset, W. Erb, J. Zhu and G. Masson, Org. Lett., 2014, 16, 2554–2557 CrossRef CAS PubMed; (f) A. Dumoulin, C. Lalli, P. Retailleau and G. Masson, Chem. Commun., 2015, 51, 5383–5386 RSC; (g) C. Lebée, A. O. Kataja, F. Blanchard and G. Masson, Chem.–Eur. J., 2015, 21, 8399–8402 CrossRef PubMed; (h) J. Pous, T. Courant, G. Bernadat, B. I. Iorga, F. Blanchard and G. Masson, J. Am. Chem. Soc., 2015, 137, 11950–11953 CrossRef CAS PubMed; (i) W.-Y. Ma, C. Gelis, D. Bouchet, P. Retailleau, X. Moreau, L. Neuville and G. r. Masson, Org. Lett., 2021, 23, 442–448 CrossRef CAS PubMed.
- (a) S.-L. Cui, J. Wang and Y.-G. Wang, Org. Lett., 2008, 10, 13–16 CrossRef CAS PubMed; (b) C. Zhou and D. Ma, Chem. Commun., 2014, 50, 3085–3088 RSC.
- See ESI† for details.
- CCDC 2101872 (3ut) and 2101876 (6) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre†.
- N. Di Iorio, G. Filippini, A. Mazzanti, P. Righi and G. Bencivenni, Org. Lett., 2017, 19, 6692–6695 CrossRef CAS PubMed.
- (a) M. Terada, Synthesis, 2010, 1929–1982 CrossRef CAS; (b) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2017, 117, 10608–10620 CrossRef CAS PubMed; (c) A. Gualandi, G. Rodeghiero and P. G. Cozzi, Asian J. Org. Chem., 2018, 7, 1957–1981 CrossRef CAS; (d) X. Li and Q. Song, Chin. Chem. Lett., 2018, 29, 1181–1192 CrossRef CAS; (e) R. Maji, S. C. Mallojjala and S. E. Wheeler, Chem. Soc. Rev., 2018, 47, 1142–1158 RSC; (f) J. Merad, C. Lalli, G. Bernadat, J. Maury and G. Masson, Chem.–Eur. J., 2018, 24, 3925–3943 CrossRef CAS PubMed; (g) Y.-D. Shao and D.-J. Cheng, ChemCatChem, 2021, 13, 1271–1289 CrossRef CAS; (h) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 CrossRef CAS PubMed; (i) D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356–5357 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2101872 and 2101876. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05161j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |