
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirhodium(II)-catalysed cycloisomerization of azaenyne: rapid assembly of centrally and axially chiral isoindazole frameworks†
Shaotong
Qiu
a,
Xiang
Gao
a and
Shifa
Zhu
 *ab
*ab
aKey Lab of Functional Molecular Engineering of Guangdong Province, School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, P. R. China. E-mail: zhusf@scut.edu.cn
bGuangdong Youmei Institute of Intelligent Bio-manufacturing Co., Ltd, China
First published on 29th September 2021
Abstract
Described herein is a dirhodium(II)-catalyzed asymmetric cycloisomerization reaction of azaenyne through a cap-tether synergistic modulation strategy, which represents the first catalytic asymmetric cycloisomerization of azaenyne. This reaction is highly challenging because of its inherent strong background reaction leading to racemate formation and the high capability of coordination of the nitrogen atom resulting in catalyst deactivation. Varieties of centrally chiral isoindazole derivatives could be prepared in up to 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 d.r., 99:1 er and 99% yield and diverse enantiomerically enriched atropisomers bearing two five-membered heteroaryls have been accessed by using an oxidative central-to-axial chirality transfer strategy. The tethered nitrogen atom incorporated into the starting materials enabled easy late-modifications of the centrally and axially chiral products via C–H functionalizations, which further demonstrated the appealing synthetic utilities of this powerful asymmetric cyclization.
:1 d.r., 99:1 er and 99% yield and diverse enantiomerically enriched atropisomers bearing two five-membered heteroaryls have been accessed by using an oxidative central-to-axial chirality transfer strategy. The tethered nitrogen atom incorporated into the starting materials enabled easy late-modifications of the centrally and axially chiral products via C–H functionalizations, which further demonstrated the appealing synthetic utilities of this powerful asymmetric cyclization.
Known as one of the most significant and reliable access methods to chiral heterocycles, asymmetric cycloisomerization of conjugated enyne has caught extensive attention and interest for its wide applications in synthetic route design and mechanistic investigation.1 Specifically, asymmetric cyclization of conjugated enynone (X = C, Z = O) has been successfully developed and applied to the rapid construction of various chiral furan-containing skeletons with high efficiency in an extremely operationally simple manner (Scheme 1a).2 However, compared to the fruitful research with enynone, it is surprising that the analogous asymmetric version of azaenyne (Z = N–R) still remains underdeveloped.3 In fact, no successful example of catalytic asymmetric cyclization of azaenyne has been reported in the literature despite the apparent significance of nitrogen-containing five-membered heterocycles in the synthetic and pharmaceutical community.4 In 2004, Haley and Herges reported a detailed experimental and theoretical study of the cyclization reaction of (2-ethynylphenyl)-phenyldiazene, which is a unique azaenyne.5 According to the DFT calculations, very close and low activation barriers for 5-exo-dig and 6-endo-dig cyclization pathways under catalyst-free conditions were found, which shed light on the inherent challenges of the asymmetric reaction of azaenyne (Scheme 1b). For instance, there was usually a regioselectivity issue (5-exo and 6-endo) in the cyclization reaction of azaenyne because of their close reaction barriers where the competitive 6-endo-dig cyclization3a,6 may lead to troublesome side-product formation. In addition, the low activation barrier deriving from the strong N-nucleophilicity of azaenyne may easily lead to self-cyclization which will cause severe background reactions to interfere with the asymmetric process. More troublingly, this transformation might suffer from catalyst deactivation arising from the high coordinating capability of the nitrogen atom in both starting materials and products, which might give more opportunities to the propagation of detrimental background reactions. In some cases, even a super-stoichiometric amount of transition metal has to be used to ensure effective conversion.3a,7 Therefore, although many nonchiral approaches have been reported,3,5 catalytic asymmetric cyclization of azaenyne still remains elusive due to the inherent obstacles aforementioned. With our continuous interest in alkyne chemistry,2a,8 herein we designed a cap-tether synergistic modulation strategy to tackle these challenges, envisioning that modulation of the tethered atom and protecting cap of nitrogen in the azaenyne would intrinsically perturb and alter the reactivity of the starting material, and therefore the azaenyne motif could be effectively harnessed as a promising synthon for asymmetric transformations (Scheme 1c). It should be noted that the obtained centrally chiral product produced from intramolecular C–H insertion of donor-type metal carbene9 might be potentially converted into the axially chiral molecule via a central-to-axial chirality conversion strategy.
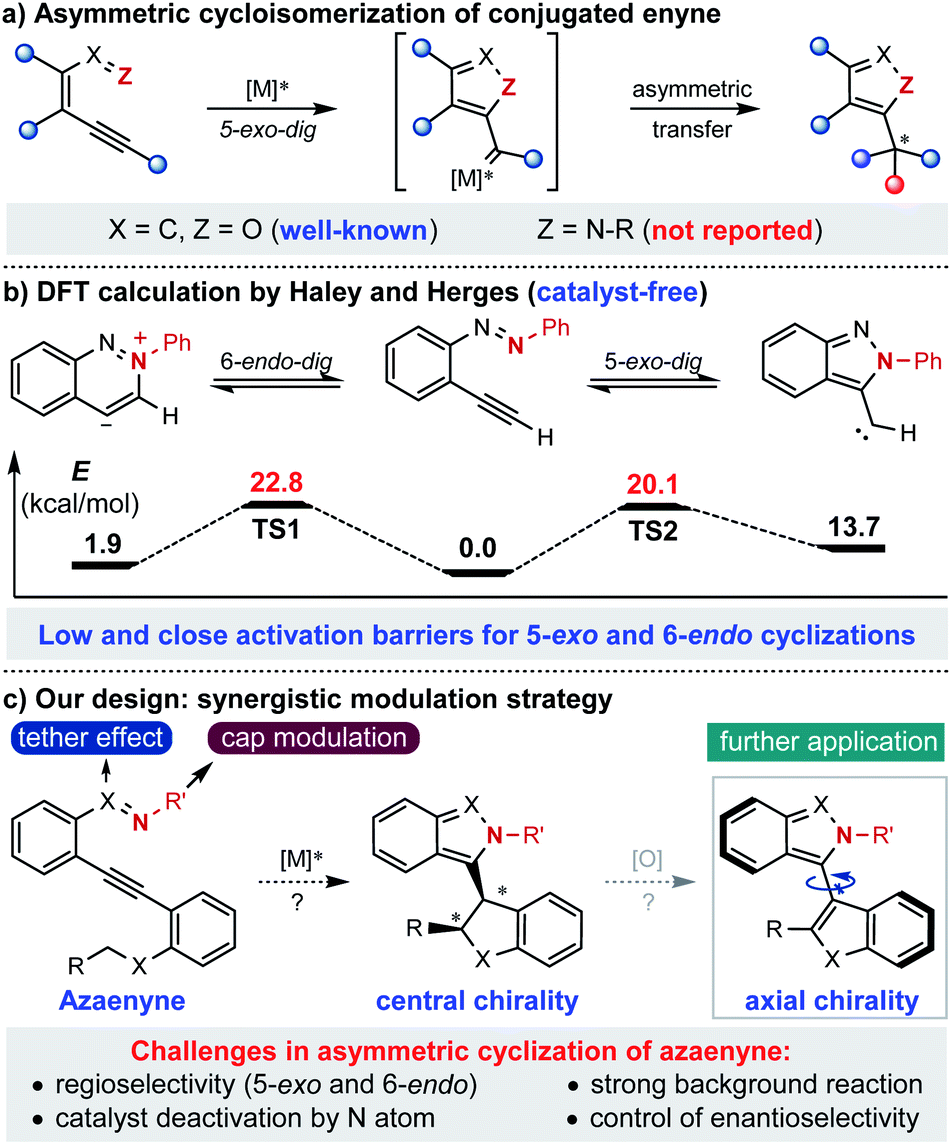 | ||
| Scheme 1 Development of the asymmetric cyclization reaction of conjugated azaenyne. | ||
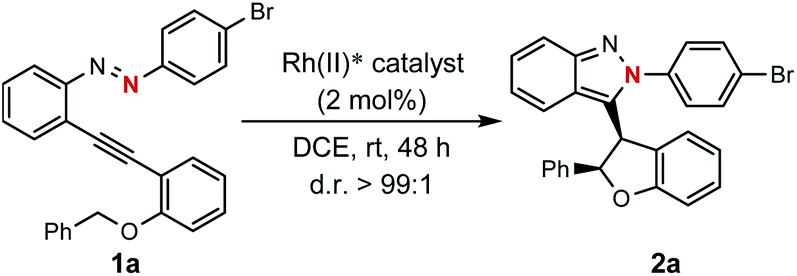
With this design in mind, different types of azaenynes bearing typical tethering atoms and capping groups were chosen to test our hypothesis and representative results are shown in Scheme 2. First, tBu-capping imine (X = C, R = tBu) was selected as a substrate to test our hypothesis.6a It was found that the imine exhibited low reactivity and the reaction temperature has to be elevated to 100 °C to initiate the transformation with or without catalyst. Unfortunately, the desired 5-exo-dig cyclization product was not detected, but isoquinoline from 6-endo-dig cyclization was obtained instead (Scheme 2a). To further regulate and control the regioselectivity and reactivity, triazene (X = N, R = N-piperidyl) was then investigated. Similarly, this substrate also showed low reactivity and it is still required to be heated at 100 °C for conversion. In the absence of a metal catalyst, an unexpected alkyne, deriving from the fragmentation of the triazene moiety, was produced in 41% yield. When 2 mol% Rh2(OPiv)4 was added as a catalyst, the side reaction could be efficiently suppressed and the reaction selectivity was apparently reversed. In this case, the target C–H insertion dihydrofuran was furnished as the major product in 30% yield but still accompanied by concomitant formation of 12% yield of undesired alkyne (Scheme 2b). The above investigations showed neither the imine nor triazene was an ideal substrate for the asymmetric reaction. Thus, we moved our attention to the diazene substrate (X = N, R = aryl). As demonstrated by Haley's and Herges' pioneering work, ortho-alkynyl diazene, compared with imine and triazene, was more unstable and tended to self-cyclization even at room temperature.5a As shown in Scheme 2c, the ortho-alkynyl diazene degrades and 5-exo-dig cyclization products could be observed even in DCE solvent without any catalyst at room temperature. When the phenyl capping group was installed in the substrate, the reaction furnished 10% yield of isoindazole derivative. The uncatalyzed self-cyclization reaction was obviously accelerated when an electron-rich capping group (4-MeO–C6H4–) was introduced, affording the corresponding product in 20% yield. Inspired by these findings, we assumed that installation of an electron deficient group on the capping phenyl would reduce the nucleophilicity of the nitrogen atom and thus the troublesome self-cyclization reaction might be effectively inhibited. To our delight, when a bromo-substituent was introduced onto the phenyl cap, the undesired self-cyclization was almost suppressed. When Rh2(OPiv)4 was added as a catalyst, the desired carbene-involved C–H insertion product was furnished in 90% yield at room temperature. Worthy of note was the total absence of any cinnoline formation from 6-endo-dig cyclization.3a,6b In short, the synthetic challenges associated with regioselectivity (5-exo-dig and 6-endo-dig), strong background reaction and catalyst deactivation could be successfully regulated and controlled via a tether-cap synergistic modulation strategy.
 | ||
| Scheme 2 Typical substrate investigation. | ||
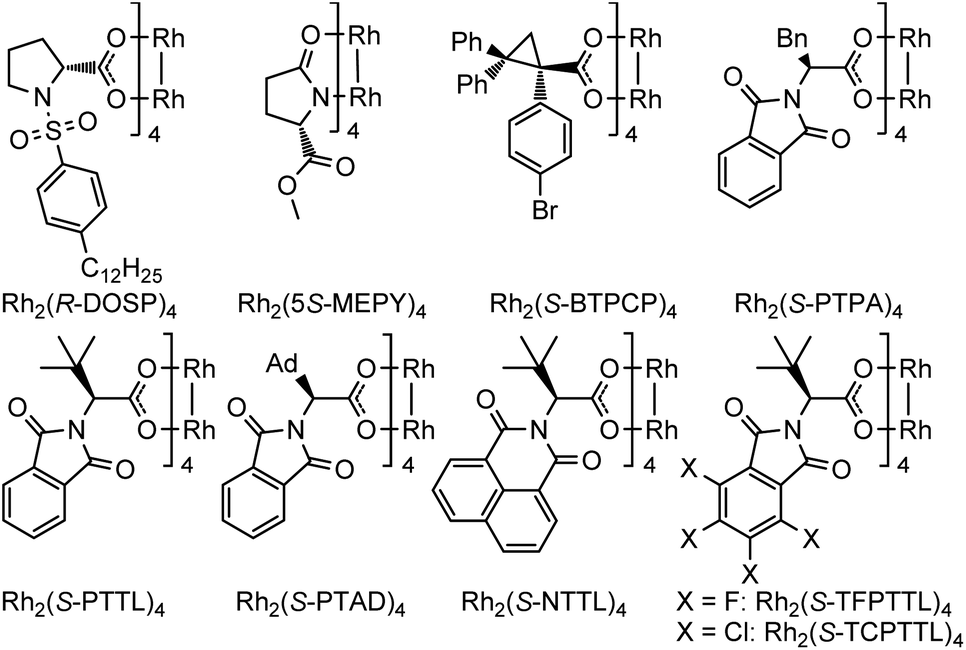
Encouraged by the above findings, ortho-alkynyl bromodiazene 1a was chosen as a model substrate and different types of chiral dirhodium catalysts10 were screened in DCE at room temperature for 48 h. As shown in Table 1, excellent diastereoselectivities (>99:1 d.r.) were observed in all these attempts with varied yields and enantioselectivities (16–98%, 50:50–98:2 er). First, the desired product 2a could be obtained in 56% yield and 29:71 er when dirhodium N-sulfonylprolinate Rh2(S-DOSP)4 was used as the chiral catalyst (entry 1). Dirhodium carboxamide Rh2(5S-MEPY)4 exhibited poorer catalytic activity with no enantioselectivity (entry 2). Dirhodium triarylcyclopropane carboxylate Rh2(S-BTPCP)4 led to an improved enantioselectivity (8:92 er), but only gave moderate yield (entry 3). To our delight, phthalimide-based dirhodium complexes proved to be better choices for this reaction, affording the target product 2a in obviously higher yields (86–98%) with effective control of enantioselectivities (91:9–98:2 er) (entries 4–9). For example, when Rh2(S-PTPA)4 was utilized as the catalyst, the desired product 2a could be obtained in 91% yield with 91:9 er (entry 4). The enantioselectivity could be further improved to 97:3 er by using a bulkier dirhodium catalyst Rh2(S-PTTL)4 (entry 5). When sterically more crowded Rh2(S-PTAD)4 and Rh2(S-NTTL)4 were tested, marginal improvement of yields (93% and 92%) was observed but with slight reduction in enantioselectivity (94:6 er and 96:4) (entries 6–7). Furthermore, electron-deficient dirhodium catalysts Rh2(S-TCPTTL)4 and Rh2(S-TFPTTL)4 were also examined (entries 8–9), where Rh2(S-TFPTTL)4 gave the best yield of 98% with the best enantioselectivity of 98:2 er (entry 9). In the tested solvents, DCE proved to be the optimal one (entries 10–13). Reduced catalyst loading provided less satisfactory results with 25% starting material recovered (entry 14).
|
|
||||
|---|---|---|---|---|
| Entry | Rh(II)* | Solvent | Yieldb [%] | erc |
| a Unless otherwise noted, reactions were performed at 0.1 M in DCE using 0.20 mmol substrate and catalyst (2 mol%) under a N2 atmosphere. b Determined by 1H NMR spectroscopy. c The er value of 2a was determined by HPLC using a chiral stationary phase. d Isolated yields. e 1 mol% catalyst was used. f 25% starting material was recovered. | ||||
| 1 | Rh2(R-DOSP)4 | DCE | 56 | 29:71 |
| 2 | Rh2(5S-MEPY)4 | DCE | 17 | 50:50 |
| 3 | Rh2(S-BTPCP)4 | DCE | 61 | 8:92 |
| 4 | Rh2(S-PTPA)4 | DCE | 91 | 91:9 |
| 5 | Rh2(S-PTTL)4 | DCE | 86 | 97:3 |
| 6 | Rh2(S-PTAD)4 | DCE | 93 | 94:6 |
| 7 | Rh2(S-NTTL)4 | DCE | 92 | 96:4 |
| 8 | Rh2(S-TCPTTL)4 | DCE | 95 | 98:2 |
| 9 | Rh 2 (S-TFPTTL) 4 | DCE | 98 |
98:2 |
| 10 | Rh2(S-TFPTTL)4 | DCM | 88 | 98:2 |
| 11 | Rh2(S-TFPTTL)4 | Toluene | 92 | 98:2 |
| 12 | Rh2(S-TFPTTL)4 | MeCN | 16 | 92:8 |
| 13 | Rh2(S-TFPTTL)4 | n-Hexane | 96 | 98:2 |
| 14e | Rh2(S-TFPTTL)4 | DCE | 65f | 96:4 |
|
||||
With the optimized reaction conditions in hand (Table 1, entry 9), the substrate scope of this asymmetric cyclization was then examined. As shown in Scheme 3, the catalytic process could be successfully applied to azaenynes 1 bearing different ether side chains. For example, in addition to 1a, various azaenyne derivatives containing benzylic ethers could be efficiently converted into the desired products 2b–i with excellent diastereoselectivities and enantioselectivities (>99:1 d.r., 97:3–99:1 er). The yields were typically higher than 90% for most substrates. Satisfyingly, the substrates with bulkier aryl groups were well-tolerated to afford the isoindazole products 2j–m in good yields with excellent diastereo- and enantiocontrol (>97:3 d.r., > 95:5 er). In addition to azaenynes with arylmethyl ether, this protocol was also successfully applied to substrates with allylic ether, propargyl ether and even aliphatic ether to furnish the cyclization products 2n–u in good yields with decent diastereo- and enantioselectivities (>93:7 d.r., > 90:10 er). In the cases of allylic and propargyl ether, only C–H insertion products (2n–p) were observed though cyclopropanation or cyclopropenation often took place competitively when using the allylic or propargyl substrate to trap the carbene intermediate.11 It was noted that the azaenynes with aliphatic ether, which represent challenging substrates2a in the asymmetric carbene transfer reactions, also showed good reactivities to afford the corresponding chiral dihydrobenzofurans (2q–u) with excellent diastereoselectivities (>93:7 d.r.) and enantioselectivities (>98:2 er). Interestingly, when phenyl and methoxyphenyl capping azaenynes, which potentially suffered from the undesired background reactions, were subjected to the standard conditions, chiral products (2v–w) could be obtained with high optical purity (>99:1 d.r., > 96:4 er) as well. These results might be attributed to the high catalytic activity of Rh2(S-TFPTTL)4 in the asymmetric cyclization process, which eventually led to complete suppression of the uncatalyzed self-cyclization. The scopes with respect to the group R1 on the fused phenyl ring were further investigated. Both electron-rich and -deficient substituents R1 were well accommodated, with the product yields ranging from 80% to 99%, enantiomeric ratios ranging from 95:5 to 97:3 and diastereomeric ratios higher than 99:1 (2x–z). In addition, azaenyne substituted with an alkyl side chain at the alkynyl carbon atom was also tested, giving tetrahydrofuran (2aa) with excellent diastereoselectivity (>99:1 d.r.), good enantioselectivity (90:10 er) and moderate yield (43%). In addition to the side chain of ether, this asymmetric protocol could even be extended to the more challenging nitrogen- and thio-tethered analogues, albeit with somewhat lower reactivities (46–65% yields) but good stereoselectivities (93:7 er and 84:16 d.r. for 2ab; 81:19 er and >99:1 d.r. for 2ac). Structures of the resulting products were confirmed by X-ray crystallographic analysis of their analogue 2h.
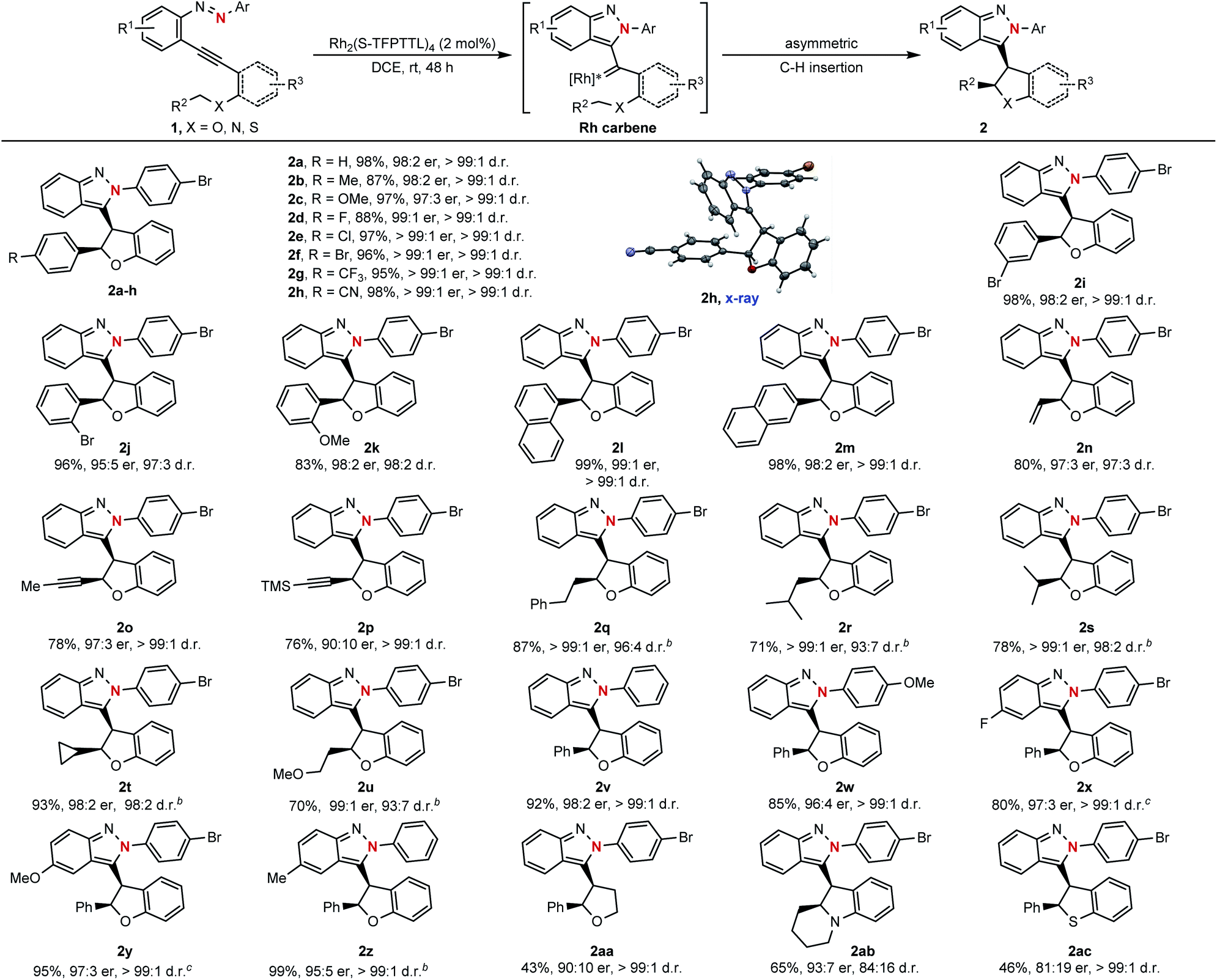 | ||
| Scheme 3 a Unless otherwise noted, the reactions were performed under standard conditions for 48 h or monitored by TLC until the starting material disappeared. b5 mol% catalyst was used. cReactions were performed in n-hexane, using 2 mol% Rh2(S-TCPTTL)4 as the catalyst. | ||
The successful preparation of centrally chiral isoindazole through the asymmetric cyclization reaction prompted us to explore the further applications of this protocol. Axially chiral biaryl skeletons are undoubtedly regarded as one of the most prominent structural motifs for their ubiquity in natural products, pharmaceuticals and useful chiral ligands in asymmetric catalysis.12 Due to the lower rotational barrier, there are only limited examples of the enantioselective synthesis of axially chiral atropisomers featuring a five-membered ring, especially those bearing two pentatomic aromatics.13 Compared with the furan analogue, the extending cap in the isoindazole scaffold provides additional ortho steric hindrance making these molecules possible candidates for the preparation of five-five-membered biaryl atropisomers. Considering the unique chiral skeleton of dihydrofuranyl isoindazole 2, we began to explore their potential application in chiral atropisomer synthesis via a central-to-axial chirality transfer strategy. As shown in Scheme 4, oxidative aromatization of representative dihydrofuran candidate 2m furnished two configurationally unstable atropisomers, which might be attributed to their relatively low rotational barriers as five-membered atropisomers especially when the furan ring was incorporated (see ESI† for details). Therefore, it was hypothesized that extending the fused phenyl to naphthyl might afford stable atropisomers by enhancing the ortho steric hindrance (Scheme 4b).
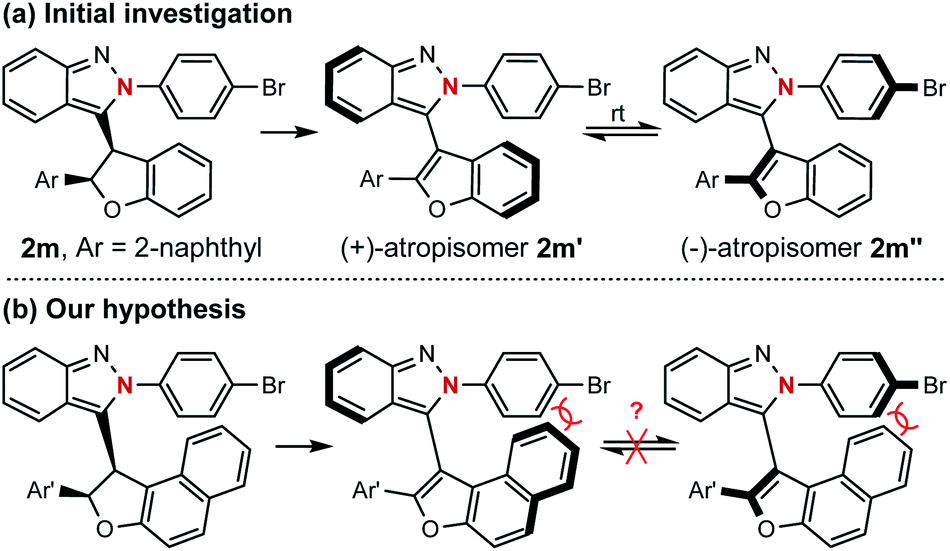 | ||
| Scheme 4 Investigation of central-to-axial chirality transfer. | ||
To our delight, as shown in Scheme 5, naphthyl-fused dihydrofurans 4 could be easily accessed through the above established dirhodium-catalyzed cyclization process and configurationally stable atropisomers 5 could be generated via further oxidative dehydrogenation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) as the oxidant (see ESI† for the proposed mechanism). For example, asymmetric cyclization reactions proceeded smoothly to give the centrally chiral compounds 4 in good yields (54–99%) with excellent diastereoselectivities (92:8–99:1 d.r.) and enantioselectivities (95:5–99:1 er) under slightly modified reaction conditions. This reaction was compatible with a variety of arylmethyl side chains in azaenynes and well-accommodated with various functional groups (F, Cl, Br, OMe, and –CO2Me). Additionally, oxidative dehydrogenation of chiral candidates 4 with DDQ smoothly resulted in the formation of axially chiral atropisomers 5 in 90–99% yields with only slight loss of chiral integrity (90:10–97:3 er). An enantiomerically pure atropisomer could be obtained through a simple recrystallization procedure as exemplified by compound 5g. The structure and absolute configuration of isoindazole 4g and atropisomer 5g were confirmed by their single-crystal X-ray diffraction analysis.
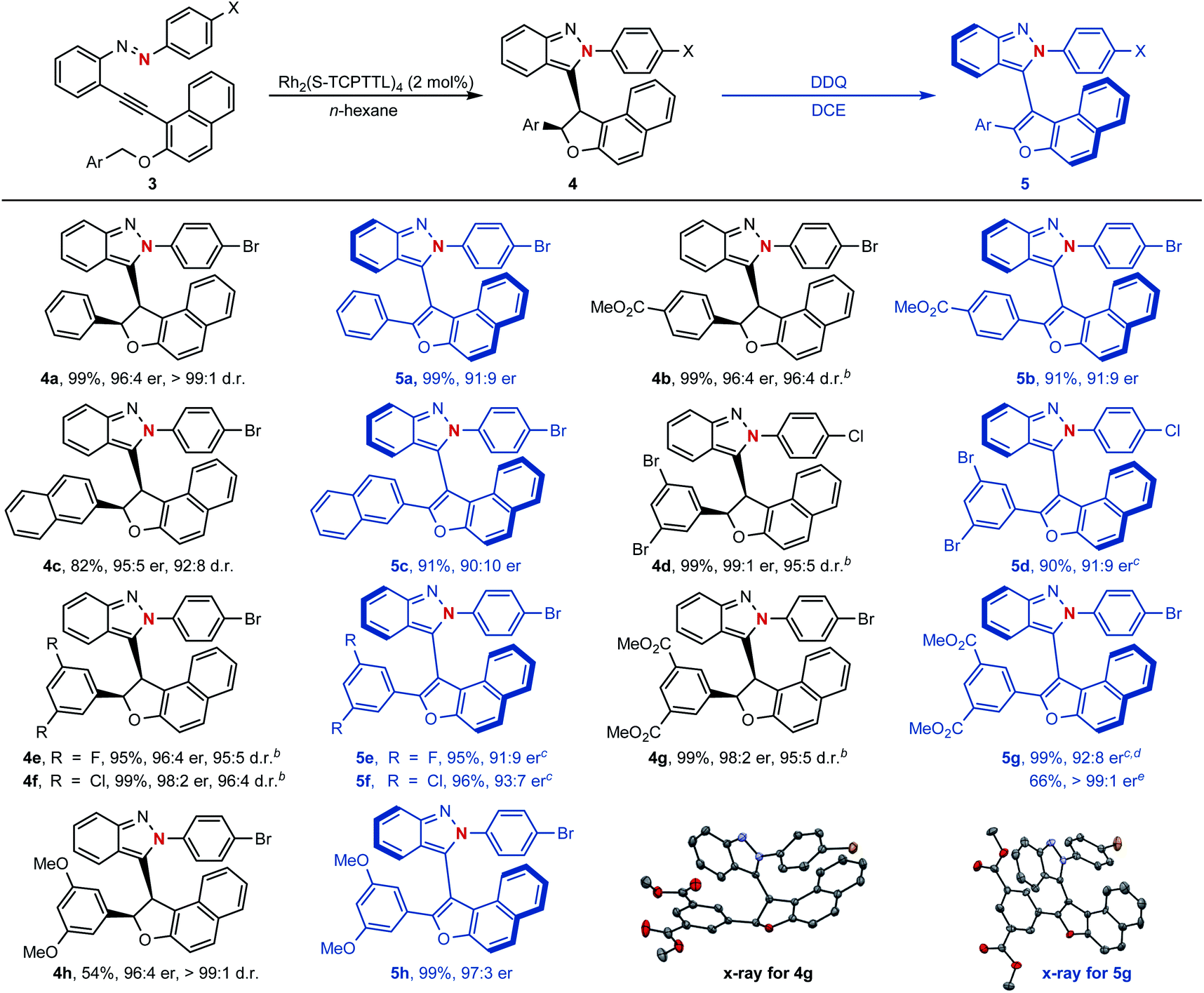 | ||
| Scheme 5 a Conditions for cyclization of azaenyne: Rh2(S-TCPTTL)4 (2 mol%), n-hexane, rt for 48 h or monitored by TLC until the starting material disappeared; conditions for oxidative chirality transfer: DDQ (2 equiv.), DCE, −20 °C for 48 h or monitored by TLC until the starting material disappeared. b45 °C. cDDQ (5 equiv.). dRoom temperature. eAfter one recrystallization. | ||
With centrally and axial chiral molecules in hand, further transformations of these compounds were also explored. The tethered nitrogen atom in azaenynes not only showed a synergetic effect with the capping group on promoting asymmetric cyclization but also served as an innate directing group for late-stage modifications via C–H functionalization. As shown in Scheme 6, a variety of functional groups could be directly introduced onto the capping aromatic rings, allowing for rapid build-up of molecular complexity. For example, synthetically valuable alkenyl,14 allyl15 and alkynyl16 groups could be easily incorporated into the final structures, which had wide potential applications in organic synthesis (6a–c). Furthermore, C–H alkylation,17 amidation18 and selenylation19 were performed smoothly to afford the desired products 6d–g. It is noteworthy that unique chiral chelation backbones were constructed by amidation and selenylation of the isoindazole moiety (6e–g). In addition to centrally chiral compounds, axial chiral atropisomers 5 themselves could be efficiently converted to their functionalized scaffolds as well (6h–i) through a similar directed C–H functionalization process.
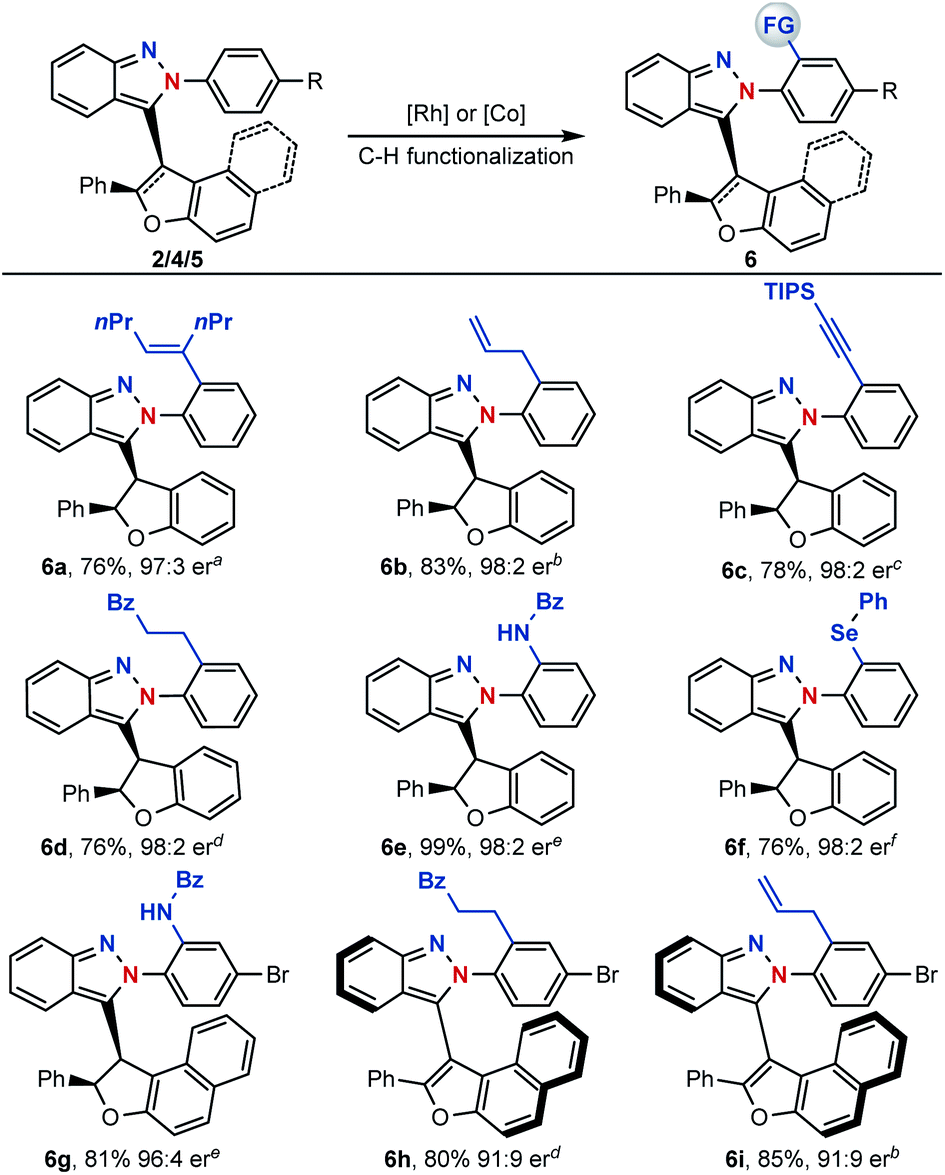 | ||
| Scheme 6 Late-stage modification of chiral isoindazoles. Reaction conditions: a4-octyne, [Rh(Cp*Cl2)]2, AgSbF6, Cu(OAc)2, DCE, 80 °C. bAllyl carbonate, [Rh(Cp*Cl2)]2, AgSbF6, PivOH, PhCl, 40 °C. cHypervalent iodine-alkyne, [Rh(Cp*Cl2)]2, Zn(OTf)2, DCE, 80 °C. dAlkene, [Rh(Cp*Cl2)]2, AgSbF6, AcOH, 1,4-dioxane, 50 °C. e3-Phenyl-1,4,2-dioxazol-5-one, [Cp*Co(MeCN)3](SbF6)2, DCE, 80 °C. fPhSeCl, [Rh(Cp*Cl2)]2, AgSbF6, THF, 60 °C. | ||
Conclusions
In summary, a dirhodium-catalyzed asymmetric cyclization reaction of azaenyne has been developed through a cap-tether synergistic modulation strategy. Benefiting from this strategy, a diverse array of centrally chiral isoindazole derivatives could be prepared in up to 99:1 d.r., 99:1 er and 99% yield in an efficient and practical manner. This reaction is highly challenging due to its inherent strong background reaction leading to racemate formation and the high capability of coordination of the nitrogen atom resulting in catalyst deactivation. Compared with the well-established enynone system, this work represents a significant advance in the asymmetric cyclization of conjugated enyne motifs where the introduction of the nitrogen tether atom and capping group provided strategic advantages in both reactivity regulation of reactants and further applications of the products. On one hand, this cycloisomerization of azaenyne features an additional cap in the formed heterocycle, which enables access to varieties of enantiomerically enriched atropisomers bearing five–five-membered heteroaryls via an oxidative central-to-axial chirality transfer strategy. On the other hand, the tethered nitrogen atom incorporated in the azaenyne served as an innate directing group of the product for late-stage modifications via transition metal catalyzed C–H functionalization. Various important functional groups were introduced into the centrally and axially chiral frameworks, demonstrating the appealing synthetic utilities of this powerful asymmetric cyclization.
Data availability
All the data have been included in the ESI.†Author contributions
S. Qiu and X. Gao performed all the experiments. S. Qiu and S. Zhu contributed to the conception of the experiments, discussion of the results and preparation of manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Nature Science Foundation of China (22071062, 21871096) and Guangdong Science and Technology Department (2018B030308007). We also thank Dr Tongxiang Cao and Rui Wu for helpful discussions and suggestions.Notes and references
- For selected reviews on conjugated enynes, see: (a) H. Wang, Y. Kuang and J. Wu, Asian J. Org. Chem., 2012, 1, 302–312 CrossRef CAS; (b) A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084–3213 CrossRef CAS PubMed; (c) A. L. S. Kumari, A. S. Reddy and K. C. K. Swamy, Org. Biomol. Chem., 2016, 14, 6651–6671 RSC; (d) J. Ma, L. Zhang and S. Zhu, Curr. Org. Chem., 2016, 20, 102–118 CrossRef CAS; (e) L. Li, D. Huang, C. Shi and G. Yan, Adv. Synth. Catal., 2019, 361, 1958–1984 CrossRef CAS.
- For selected reports on asymmetric cyclization reactions of enynone, see: (a) D. Zhu, J. Ma, K. Luo, H. Fu, L. Zhang and S. Zhu, Angew. Chem., Int. Ed., 2016, 55, 8452–8456 CrossRef CAS PubMed; (b) J. Yang, Z. Li, M. Li, Q. He, S. Zhu and Q. Zhou, J. Am. Chem. Soc., 2017, 139, 3784–3789 CrossRef CAS PubMed; (c) M. Huang, J. Yang, Y. Zhao and S. Zhu, ACS Catal., 2019, 9, 5353–5357 CrossRef CAS; (d) K. Wang, Z. Liu, G. Xu, Y. Shao, S. Tang, P. Chen, X. Zhang and J. Sun, Angew. Chem., Int. Ed., 2021, 60, 16942–16946 CrossRef CAS PubMed.
- For selected nonchiral reports on cyclization reactions of azaeyne see: (a) D. B. Kimball, R. Herges and M. M. Haley, J. Am. Chem. Soc., 2002, 124, 1572–1573 CrossRef CAS PubMed; (b) F. Nishino, K. Miki, Y. Kato, K. Ohe and S. Uemura, Org. Lett., 2003, 5, 2615–2617 CrossRef CAS PubMed; (c) H. Shen, C. Li and R. Liu, Tetrahedron Lett., 2004, 45, 9245–9247 CrossRef CAS; (d) C. Zhu, C. Feng and M. Yamane, Chem. Commun., 2017, 53, 2606–2609 RSC; (e) W. Wei, X. Li, M. Gu, H. Yao and A. Lin, Org. Biomol. Chem., 2017, 15, 8458–8462 RSC.
- (a) H. Cerecetto, A. Gerpe, M. González, V. J. Arán and C. O. de Ocáriz, Mini-Rev. Med. Chem., 2005, 5, 869–878 CrossRef CAS PubMed; (b) M. D. Angelis, F. Stossi, K. A. Carlson, B. S. Katzenellenbogen and J. A. Katzenellenbogen, J. Med. Chem., 2005, 48, 1132–1144 CrossRef PubMed.
- For selected reviews on this type of cyclization reaction, see: (a) L. D. Shirtcliff, T. J. R. Weakley and M. M. Haley, J. Org. Chem., 2004, 69, 6979–6985 CrossRef CAS PubMed; (b) L. D. Shirtcliff, S. P. McClintock and M. M. Haley, Chem. Soc. Rev., 2008, 37, 343–364 RSC; (c) B. S. Young, R. Herges and M. M. Haley, Chem. Commun., 2012, 48, 9441–9455 RSC.
- For selected reports on 6-endo-dig cyclization of azaenyne, see: (a) Q. Huang, J. A. Hunter and R. C. Larock, Org. Lett., 2001, 3, 2973–2976 CrossRef CAS PubMed; (b) X. Fang, J. Cao, W. Ding, H. Jin, X. Yu and S. Wang, Org. Lett., 2021, 23, 1228–1233 CrossRef CAS PubMed.
- Y. Fang, C. Wang, S. Su, H. Yu and Y. Huang, Org. Biomol. Chem., 2014, 12, 1061–1071 RSC.
- (a) L. Chen, K. Chen and S. Zhu, Chem, 2018, 4, 1208–1262 CrossRef CAS; (b) R. Wu, J. Lu, T. Cao, J. Ma, K. Chen and S. Zhu, J. Am. Chem. Soc., 2021, 143, 14916–14925 CrossRef CAS PubMed.
- For selected reviews on donor-donor carbenes, see: (a) D. Zhu, L. Chen, H. Fan, Q. Yao and S. Zhu, Chem. Soc. Rev., 2020, 49, 908–950 RSC; (b) B. D. Bergstrom, L. A. Nickerson, J. T. Shaw and L. W. Souza, Angew. Chem., Int. Ed., 2021, 60, 6864–6878 CrossRef CAS PubMed , for selected reports, see references 2 and:; (c) J. Barluenga, L. Riesgo, R. Vicente, L. A. Llpez and M. Tomás, J. Am. Chem. Soc., 2008, 130, 13528–13529 CrossRef CAS PubMed; (d) Y. Xia, S. Qu, Q. Xiao, Z. Wang, P. Qu, L. Chen, Z. Liu, L. Tian, Z. Huang, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2013, 135, 13502–13511 CrossRef CAS PubMed; (e) J. González, J. González, C. Pérez-Calleja, L. A. López and R. Vicente, Angew. Chem., Int. Ed., 2013, 52, 5853–5857 CrossRef PubMed; (f) C. Soldi, K. N. Lamb, R. A. Squitieri, M. González-López, M. J. D. Maso and J. T. Shaw, J. Am. Chem. Soc., 2014, 136, 15142–15145 CrossRef CAS PubMed; (g) H. Jin, B. Tian, X. Song, J. Xie, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 12688–12692 CrossRef CAS PubMed; (h) R. L. Sahani and R. Liu, Angew. Chem., Int. Ed., 2017, 56, 1026–1030 CrossRef CAS PubMed; (i) F. Hong, Z. Wang, D. Wei, T. Zhai, G. Deng, X. Lu, R. Liu and L. Ye, J. Am. Chem. Soc., 2019, 141, 16961–16970 CrossRef CAS PubMed; (j) L. Yang, D. Evans, B. Xu, W. Li, M. Li, S. Zhu, K. N. Houk and Q. Zhou, J. Am. Chem. Soc., 2020, 142, 12394–12399 CrossRef CAS PubMed; (k) J. R. Jagannathan, J. C. Fettinger, J. T. Shaw and A. K. Franz, J. Am. Chem. Soc., 2020, 142, 11674–11679 CrossRef CAS PubMed; (l) K. Dong, X. Fan, C. Pei, Y. Zheng, S. Chang, J. Cai, L. Qiu, Z. Yu and X. Xu, Nat. Commun., 2020, 11, 2363 CrossRef CAS PubMed.
- For selected reviews on the dirhodium (II) catalysed asymmetric C–H insertion reaction of metal carbenes, see: (a) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861–2904 CrossRef CAS PubMed; (b) M. P. Doyle, J. Org. Chem., 2006, 71, 9253–9260 CrossRef CAS PubMed; (c) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS PubMed; (d) H. M. L. Davies and K. Liao, Nat. Rev. Chem., 2019, 3, 347–360 CrossRef CAS PubMed.
- (a) S. Ishii, S. Zhao, G. Mehta, C. J. Knors and P. Helquist, J. Org. Chem., 2001, 66, 3449–3458 CrossRef CAS PubMed; (b) M. Honma and M. Nakada, Tetrahedron Lett., 2003, 44, 9007–9011 CrossRef CAS.
- For selected reviews, see: (a) R. Noyori, Asymmetric Catalysis in Organic Synthesis; Wiley, New York, 1994 Search PubMed; (b) M. C. Kozlowski, B. J. Morgan and E. C. Linton, Chem. Soc. Rev., 2009, 38, 3193–3207 RSC; (c) G. Bring-mann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed; (d) Privileged Chiral Ligands and Catalysts, ed. Q. Zhou, Wiley-VCH, Weinheim, Germany, 2011 Search PubMed; (e) J. E. Smyth, N. M. Butler and P. A. Keller, Nat. Prod. Rep., 2015, 32, 1562–1583 RSC.
- (a) D. Bonne and J. Rodriguez, Chem. Commun., 2017, 53, 12385–12393 RSC; (b) D. Bonne and J. Rodriguez, Eur. J. Org. Chem., 2018, 2417–2431 CrossRef CAS; (c) S. Zhang, Q. Yao, G. Liao, X. Li, H. Li, H. Chen, X. Hong and B. Shi, ACS Catal., 2019, 9, 1956–1961 CrossRef CAS.
- D. L. Davies, C. E. Ellul, S. A. Macgregor, C. L. McMullin and K. Singh, J. Am. Chem. Soc., 2015, 137, 9659–9669 CrossRef CAS PubMed.
- H. Wang, N. Schrçder and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 5386–5389 CrossRef CAS PubMed.
- F. Xie, Z. Qi, S. Yu and X. Li, J. Am. Chem. Soc., 2014, 136, 4780–4787 CrossRef CAS PubMed.
- J. A. Boerth, J. R. Hummel and J. A. Ellman, Angew. Chem., Int. Ed., 2016, 55, 12650–12654 CrossRef CAS PubMed.
- Y. Liang, Y. Liang, C. Tang, Y. Yuan and N. Jiao, Chem.–Eur. J., 2015, 21, 16395–16399 CrossRef CAS PubMed.
- S. Yu, B. Wan and X. Li, Org. Lett., 2015, 17, 58–61 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization of all compounds, copies of 1H and 13C NMR for selected compounds. CCDC 2105966 (2h), 2105967 (4g), 2105968 (5g). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04961e |
| This journal is © The Royal Society of Chemistry 2021 |