
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective palladium-catalyzed C(sp2)–C(sp2) σ bond activation of cyclopropenones by merging desymmetrization and (3 + 2) spiroannulation with cyclic 1,3-diketones†
Han-Qi
Zhou‡
a,
Xing-Wei
Gu‡
a,
Xiao-Hua
Zhou
a,
Li
Li
a,
Fei
Ye
a,
Guan-Wu
Yin
a,
Zheng
Xu
a and
Li-Wen
Xu
 *ab
*ab
aKey Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education, Key Laboratory of Organosilicon Material Technology of Zhejiang Province, Hangzhou Normal University, No. 2318, Yuhangtang Road, Hangzhou 311121, P. R. China. E-mail: liwenxu@hznu.edu.cn
bState Key Laboratory for Oxo Synthesis and Selective Oxidation, Suzhou Research Institute and Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, P. R. China
First published on 20th September 2021
Abstract
Catalytic asymmetric variants for functional group transformations based on carbon–carbon bond activation still remain elusive. Herein we present an unprecedented palladium-catalyzed (3 + 2) spiro-annulation merging C(sp2)–C(sp2) σ bond activation and click desymmetrization to form synthetically versatile and value-added oxaspiro products. The operationally straightforward and enantioselective palladium-catalyzed atom-economic annulation process exploits a TADDOL-derived bulky P-ligand bearing a large cavity to control enantioselective spiro-annulation that converts cyclopropenones and cyclic 1,3-diketones into chiral oxaspiro cyclopentenone–lactone scaffolds with good diastereo- and enantio-selectivity. The click-like reaction is a successful methodology with a facile construction of two vicinal carbon quaternary stereocenters and can be used to deliver additional stereocenters during late-state functionalization for the synthesis of highly functionalized or more complex molecules.
Introduction
The carbon–carbon bond exists widely and stably in nature due to its high stability (332 kJ mol−1), constructing an extremely rich and useful organic molecular library. Accordingly, the construction and breaking of carbon–carbon bonds is one of the most important research topics in modern synthetic chemistry and has attracted much attention from chemists in the past few decades.1–11 Although numerous new reaction strategies have been developed, the improvement of the adaptability of catalyst or reaction systems through carbon–carbon bond activation for the enantioselective construction of chiral molecules with complex structures is still not an easy task. In addition, carbon–carbon bond activation of small rings for the synthesis and functionalization of cyclic compounds is intrinsically challenging in a variety of synthetic chemistry and pharmaceutical applications. Among the small rings, cyclopropenones, a privileged class of small and strained ring compounds bearing two functional groups such as ketone and olefin, have drawn increasing attention because of their ability of versatile reactivity for the construction of functional polymers,12–17 fluorogenic probes,18,19 natural products or biologically useful molecules in medicinal chemistry,20–24 and as organocatalysts.25–29 Since their pioneering synthesis by Breslow in 1959,30,31 cyclopropenones have often been developed as valuable building blocks for the synthesis of complex molecules, and they are notable for their amphiphilic properties as both electrophiles and nucleophiles, and other unusual properties (Fig. 1), including high basicity, light-responsive properties, large dipole moments, aromatic characters, and significant angle strain.32–40 Although the origin of cyclopropenone rings in nature has not been fully elucidated, several cyclopropenone-containing natural products (Fig. 2A) have been isolated, synthesized, and studied on the structure–activity relationships for cytotoxicity and enzyme inhibition.41 In particular, the transition-metal-catalyzed transformations of cyclopropenones involving carbon–carbon bond activation have been an active research area in the past few years. Among various synthetic approaches based on the chemical reactivity of cyclopropenones,42–59 the enantioselective version of transition-metal-catalyzed carbon–carbon bond activation and subsequent ring opening or ring expansion reactions of cyclopropenones remains underexplored.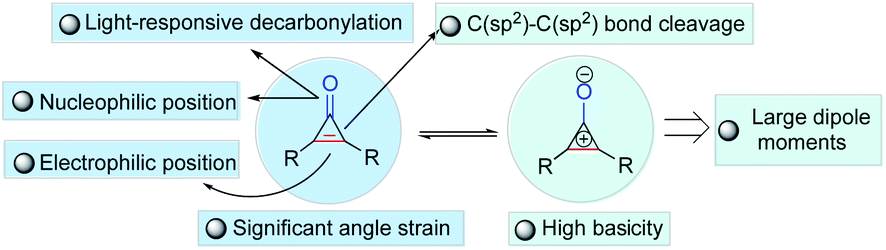 | ||
| Fig. 1 Unusual properties of cyclopropenone structures. | ||
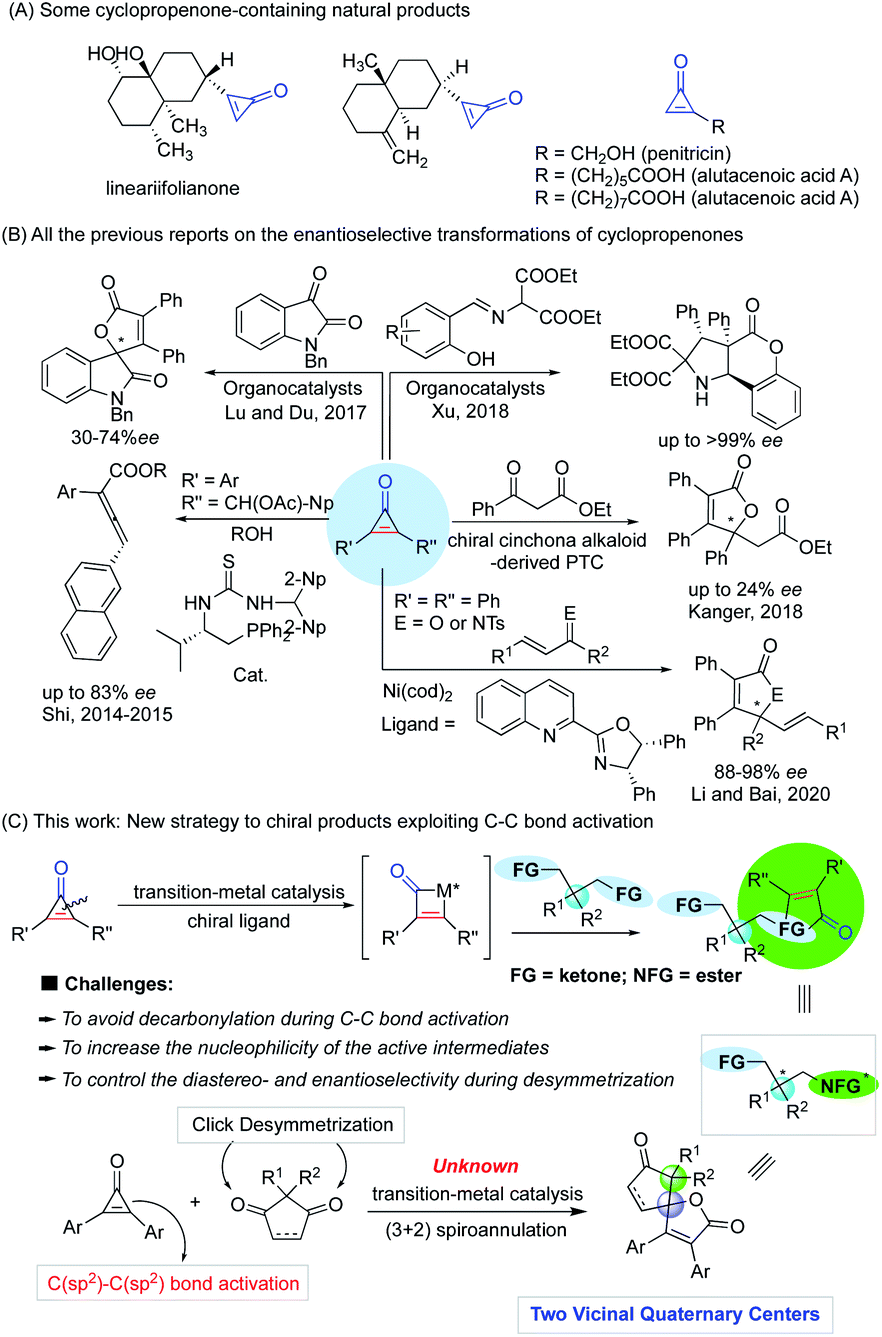 | ||
| Fig. 2 Comprehensive summary of cyclopropenone chemistry. (A) Representative structures of cyclopropane-containing natural products; (B) various reactivities of cyclopropenone compounds and their previous explorations. (C) Our new hypothesis on the enantioselective synthesis of chiral molecules based on carbon–carbon bond activation by merging the C(sp2)–C(sp2) σ bond activation of cyclopropenones and desymmetrization of cyclopentene-1,3-dione. | ||
Most of the related studies of enantioselective transformations based on the carbon–carbon bond cleavage of cyclopropenones are unsuccessful because of their low enantioselectivity (Fig. 2B), such as the organocatalytic synthesis of allenic esters (up to 83% ee),60,61 spirooxindoles from formal (3 + 2) cycloaddition with isatins (30–74% ee),62o-hydroxy aromatic aldimine-derived heterocyclic compounds,63 and other explorations with low enantioselectivity.64,65 Furthermore, the synthesis of chiral oxaspiranic compounds from cyclopropenones is especially challenging as previously reported synthetic procedures toward these species of spirocycles are based on Lewis acid66 and gold catalysis67 without suitable chiral ligands. Thus, much work is needed to develop highly stereoselective strategies with more flexible synthetic features, to expand the structural and functional pattern of heterocyclic compounds bearing at least a carbon-stereogenic center. Very recently, Li and co-workers reported a notable example that chiral nickel catalysis could complete an enantioselective (3 + 2) annulation of cyclopropenones and α,β-unsaturated ketones/imines with good ees.68 This work demonstrated for the only example that enantioselective transformations of cyclopropenones can also be achieved by a chiral ligand-controlled transition metal-catalyzed carbon–carbon bond activation process. Nevertheless, there is no precedent for the palladium-catalyzed enantioselective variant of the carbon–carbon bond functionalization of cyclopropenones involving a sequential C(sp2)–C(sp2) bond activation/(3 + 2) annulation process.
Considered the powerful potential of C–C bond activation and chiral ligands for the enantioselective construction of a single quaternary stereocenter,69 we propose a new strategy for the construction of more quaternary stereocenters that can use cyclopropenones as the active species to achieve the desymmetrization of bifunctional compounds (Fig. 2C). In this regard, one of the potential problems to control the diastereo- and enantio-selectivity lies in the precise recognition function of active intermediates of metal catalysts that are formed from the oxidative addition of the carbon–carbon bond of cyclopropenones to metal species, during the desymmetrization of the bifunctional groups before they undergo (3 + 2) annulation.
To address current limitations on the enantioselective and rarely reported transition-metal-catalyzed (3 + 2) spiro-annulation of cyclopropenones with ketones, we expect to design rigid and cyclic diketone substrates to realize a new strategy of constructing two vicinal carbon quaternary stereocenters based on C(sp2)–C(sp2) bond activation and desymmetrization that is similar to atom-economic (3 + 2) click additions.70 However, the utilization and re-organization of two small rings in transition-metal-catalyzed (3 + 2) spiro-annulation to create oxaspiro cyclopentenone–lactone scaffolds remain elusive.
Herein, we report a new strategy for the ring expansion and click cycloaddition of two small rings to access highly enantioselective transition-metal-catalyzed (3 + 2) spiro-annulation of cyclopropenones with cyclopentene-1,3-diones, which is achieved for the facile construction of complex spiro-molecules with two adjacent stereocenters. The reaction via palladium-promoted tandem carbon–carbon bond cleavage and (3 + 2) annulation featured with producing two quaternary carbon stereocenters by desymmetrization.
Results and discussion
Initially, we began our studies to develop an enantioselective palladium-catalyzed (3 + 2) spiro-annulation by the treatment of cyclopentene-1,3-dione 1a with cyclopropenone 2a as a model reaction. With Pd2(dba)3 as the Pd source, commonly employed ligands BINOL-derived phosphoramidites (L1 and L2) and TADDOL-derived phosphine ligands (L3 and L4) resulted in both low yields and low to moderate enantiomeric excess (entries 2–5 of Table 1). We found that among the above four types of chiral ligands, TADDOL-derived phosphoramidite ligands can achieve promising stereoselectivity (57% ee with 16% yield), which allows us to further optimize the structure of the TADDOL-type framework to enhance the catalytic activity and improve the enantioselective induction of the chiral palladium complex. Encouraged by the initial results, we carried out preliminary and extensive studies with the aid of chiral TADDOL-derived phosphoramidite ligands that were previously employed for asymmetric palladium catalysis in our group.69 Unfortunately, these reported P-ligands failed to provide the best level of enantioselectivities and yields (see Tables S1–S7 of the ESI†). After a judicious modification of the chiral ligands and the choice of reaction parameters, the P-ligand L15 was found to be critical for the palladium-catalyzed (3 + 2) spiro-annulation of 1a and 2a in toluene or chlorobenzene at room temperature (87% yield and 92% ee, see entry 1 of Table 1). As shown in entries 6–9, the evaluation of the palladium sources revealed that the Pd(0) source had similar activity and Pd(II) exhibited no function to promote the carbon–carbon bond activation. And subtle changes in the steric environment or electronic properties of the TADDOL core had an unexpected and negligible impact on the catalytic activity of palladium species and the enantioselective induction of the phosphine ligand (see Tables S1, S3 and S6 of the ESI†). We also checked the solvent effect with eleven different organic solvents (see Table S7†) and further confirmed that toluene or chlorobenzene was the best choice for this reaction. In addition, lower temperature was beneficial to achieving higher enantioselectivity (entries 14–16). Notably, the evaluation of nickel catalysis was not effective in this reaction (entry 17), which supported the privileged role of the Pd/L15 catalyst system in this enantioselective (3 + 2) spiro-annulation process.|
|
||||
|---|---|---|---|---|
| Entry | Variation from “standard conditions” | Yieldb (%) | ee (%) | dr |
| a Unless otherwise noted, the standard reaction conditions were as follows: 1a (0.2 mmol), 2a (0.4 mmol), and in solvent (0.4 mL). For entries 2–9, the solvent is toluene. b The yield and dr value were determined by crude 1H NMR analysis using dibromomethane as an internal standard. c The enantiomeric excess of 3a was determined using chiral UPLC. d Under the reported reaction conditions with Ni catalysis,15 no product of the annulation reaction (nr) was detected in this case. | ||||
| 1 | None | 87 | 92 | >19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
| 2 | With L1 instead of L15 | <5 | — | — |
| 3 | With L2 instead of L15 | 30 | 22 | >19:1 |
| 4 | With L3 instead of L15 | <5 | — | — |
| 5 | With L4 instead of L15 | 16 | 57 | 12:1 |
| 6 | With Pd2(dba)3 as Pd source | 61 | 90 | >19:1 |
| 7 | With Pd(dba)2 as Pd source | 59 | 89 | >19:1 |
| 8 | With Pd(MeCN)2Cl2 as the Pd source | nr | — | — |
| 9 | With PdBr2 as the Pd source | nr | — | — |
| 10 | With THF as solvent | 32 | 92 | 13:1 |
| 11 | With DMF as solvent | 69 | 94 | >19:1 |
| 12 | With NMP as solvent | 67 | 67 | >19:1 |
| 13 | With DMAc as solvent | 74 | 92 | >19:1 |
| 14 | With toluene at 0 °C | 44 | 93 | >19:1 |
| 15 | With toluene at 40 °C | 21 | 78 | 16:1 |
| 16 | With toluene at 80 °C | 34 | 70 | 16:1 |
| 17 | With Ni(cod)2 under Li's conditionsd,15 | nr | — | — |

Having optimized the reaction conditions, we then explored the substrate scope of the catalytic asymmetric (3 + 2) spiro-annulation reaction. Notably, there is no other reaction process that could be applied for the enantioselective construction of such oxaspiro cyclopentenone–lactone scaffolds. As such, catalytic access to structurally diverse products bearing different substituents on the starting materials, cyclopentene-1,3-diones or cyclopropenones, is valuable. The results in Fig. 3 show that highly efficient (3 + 2) spiro-annulation takes place for cyclopentene-1,3-diones, where the aromatic rings of benzyl groups are substituted with methyl or halide groups at the para, ortho, or meta position, with the corresponding oxaspiro molecules 3 being produced with high enantio- and diastereo-selectivity (90–94% ee, up to >19:1 dr). The yields were also high for the aryl cyclopentene-1,3-diones bearing ortho-substituted groups, such as 3b, 3e, 3i and 3n with 81–99%. Notably, cyclopentene-1,3-dione substrate 1p containing a transition-metal-sensitive nitro group was really suitable for this reaction because of its good enantioselectivity (92% ee) and moderate yield (56%), where it is generally believed that the nitro group is not conducive to the stereocontrol of transition metal complexes and likely problematic in catalytic cycles. It is also possible to use thiophene-substituted substrate 1r to finish the (3 + 2) spiro-annulation with excellent diastereo- and enantio-selectivity (92% ee and >19:1 dr). This provides access to a valuable S– heterocyclic oxaspiro molecule. In addition, alkynyl and other substrates bearing electron-withdrawing groups, such as CO2Me, CN, and CF3, were found to undergo efficient and highly diastereo- and enantio-selective cycloaddition with cyclopropenones (Fig. 3). These products bearing such functional groups provide additional access to the synthesis of more complex molecules. Interestingly, when the methyl group on the cyclopentenone is replaced with an ethyl group, the steric hindrance effect exhibits negative function, and accordingly the yields of 3v–3x are very low under the same optimized reaction conditions. Therefore, the reaction temperature needs to be increased to 50 °C to obtain an improved yield, and correspondingly, its stereoselectivity is also significantly reduced to a moderate level. As expected, substituted cyclopropenones gave the desired oxaspiro products 3y–3aa with good diastereo- and enantio-selectivity in moderate to good yields. Encouraged by these reaction results, we next sought to extend the catalytic asymmetric (3 + 2) spiro-annulation to unusual cyclopentene-1,3-diones or cyclopropenones. Using spiro-type cyclopentene-1,3-dione 1y as an acceptor allowed access to the enantioselective construction of double spiro cyclopentenone–lactone scaffold bearing two vicinal carbon quaternary stereocenters with 83% ee and 94% yield (eqn (1) of Fig. 4), whereas moderate diastereoselective control presumably occurred due to the unfavorable steric repulsion between rigid spiro substrate 1y and nucleophilic palladium-2a species.
 | ||
| Fig. 3 Scope of the catalytic asymmetric (3 + 2) spiro-annulation of cyclopentene-1,3-diones with cyclopropenones. Yields represent the isolated yield of purified products and the ee values are determined by chiral UPLC and HPLC. The absolute configuration was determined by X-ray analysis. a The reaction is performed at 50 °C. | ||
 | ||
| Fig. 4 Exploration of the catalytic asymmetric (3 + 2) spiro-annulation with other unusual substrates. | ||
Then, unsymmetrical cyclopropenone 2f bearing phenyl and i-propyl groups was evaluated to access the desired product 3ac (eqn (2) of Fig. 4), demonstrating the viability of (3 + 2) spiro-annulation with controllable chemo-, diastereo-, and enantioselectivity by palladium catalysis. In addition, an alkyl oxaspiro cyclopentenone–lactone scaffold was also accessed by using propyl cyclopropenone 2g and cyclopentene-1,3-dione 1a as coupling partners, and the desired product 3ad could be achieved with excellent diastereoselectivity and moderate yield and enantioselectivity due to the lack of aromatic interaction in comparison to that of phenyl cyclopropenone. To further demonstrate the substrate scope and applicability of the palladium-catalyzed (3 + 2) annulation reaction, we next investigated the use of 5-(tert-butyldimethylsilyloxy)naphthalene-1,4-dione 4 as an acceptor in this cycloaddition. Although the structure of 4 is largely different from that of cyclopentene-1,3-diones, the desired product 5 was obtained with excellent enantioselectivity (92% ee) and promising regioselectivity (4:1 rr) as well as good yield (86%), and we reasoned that this reaction would provide useful information on the reaction mechanism of palladium-catalyzed (3 + 2) spiro-annulation.
On the basis of experimental results and 31P NMR analysis (see Fig. S1–S3 of the ESI†), a plausible catalytic cycle for the palladium-catalyzed (3 + 2) spiro-annulation of cyclopropenones with cyclic 1,3-diketones is depicted in Fig. 5. First, the Pd0–L complex is coordinated with the three-membered cyclopropenone to obtain the complex A, and the intermediate B is obtained by oxidative addition with carbon–carbon bond activation. And then the coordination of Pd species B with the carbon–carbon double bond of cyclopentene-1,3-dione is affected by the steric repulsion of the substituted group, giving a favorable coordinated model as complex C that the Pd center located on the methyl side because of less steric hindrance, and subsequent migratory insertion of a Pd–C bond into the same-directional ketone group to give intermediate E. In this regard, the control experiment with 2-benzyl-2- methylcyclopentane-1,3-dione or other enones as a substrate instead of 1a in this reaction resulted in no reaction (Fig. S2†), which revealed the importance of both cyclic 1,3-diketone and carbon–carbon double bonds of 1a in the catalyst–substrate interaction. And finally, the product 3a is obtained by reductive elimination and the release of Pd0–L continues to participate in the next catalytic cycle. And the experimental results determined that the recognition of one of the carbonyl groups is a key step in this desymmetrization and (3 + 2) annulation reaction, indicating the crucial role of the TADDOL-derived bulky P-ligand bearing a large cavity in the alkene-directed migratory insertion of the Pd–C intermediate to the carbon–carbon double bond. The formation of Pd/substrate species D is necessary for the high level of enantioselective induction during the stereospecific migratory insertion and subsequent formation of an oxaspiro skeleton.
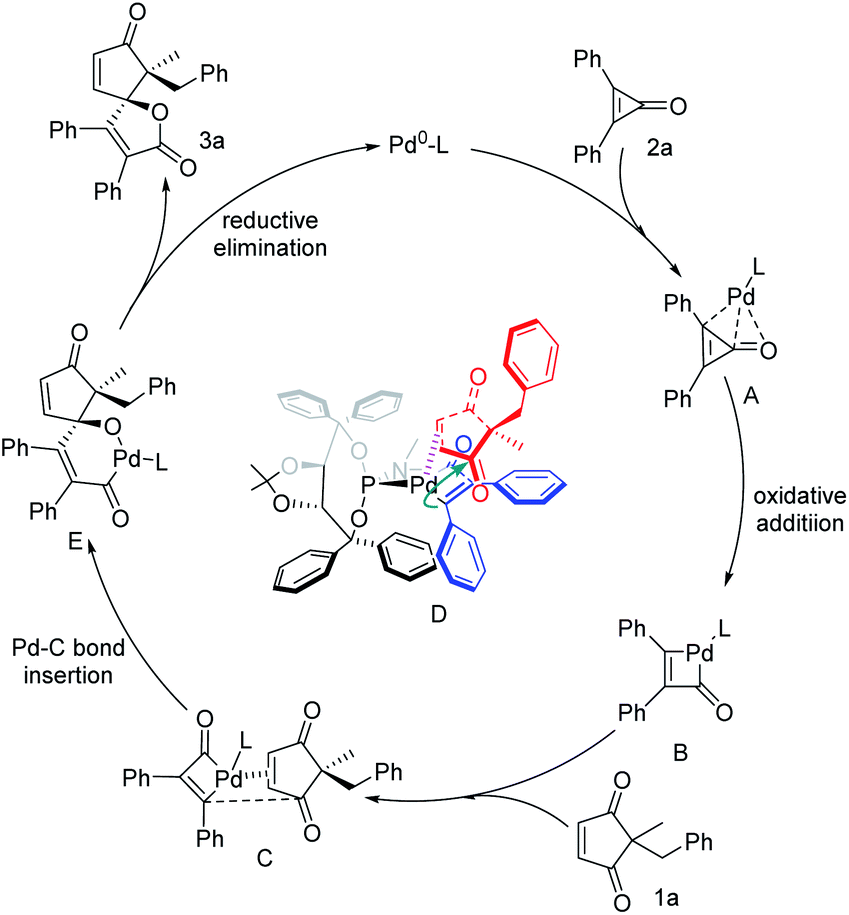 | ||
| Fig. 5 Mechanistic consideration for the palladium-catalyzed (3 + 2) spiro-annulation. | ||
To evaluate whether this (3 + 2) spiro-annulation and its natural product-like complex molecules could be applied to late-stage functionalization, we performed a gram-scale experiment to provide the starting material for the downstream transformations (Fig. 6). Next, we focused on our attention on the further functionalization of the cyclopentenone moiety. The strong base (NaOMe)-promoted transesterification reaction of 3a with methanol resulted in sequential oxa-Michael addition of alcohol to the intramolecular cyclopentenone moiety, which gave unexpected product 6 bearing an epoxide moiety with good enantioselectivity. And the treatment of 3a with t-BuOOH in the presence of DBU gave the corresponding product 7 with almost quantitative yield and good enantioselectivity (99% yield and 90% ee, Fig. 6). In addition, an enantioselective conjugate addition reaction of azide was also performed because of its importance in synthetic chemistry. The desired product 8, having an additional azide moiety on the oxaspiro cyclopentenone–lactone scaffold, was obtained with good enantioselectivity. These representative transformations supported the powerful potential of the oxaspiro cyclopentenone–lactone scaffold in synthetic chemistry.
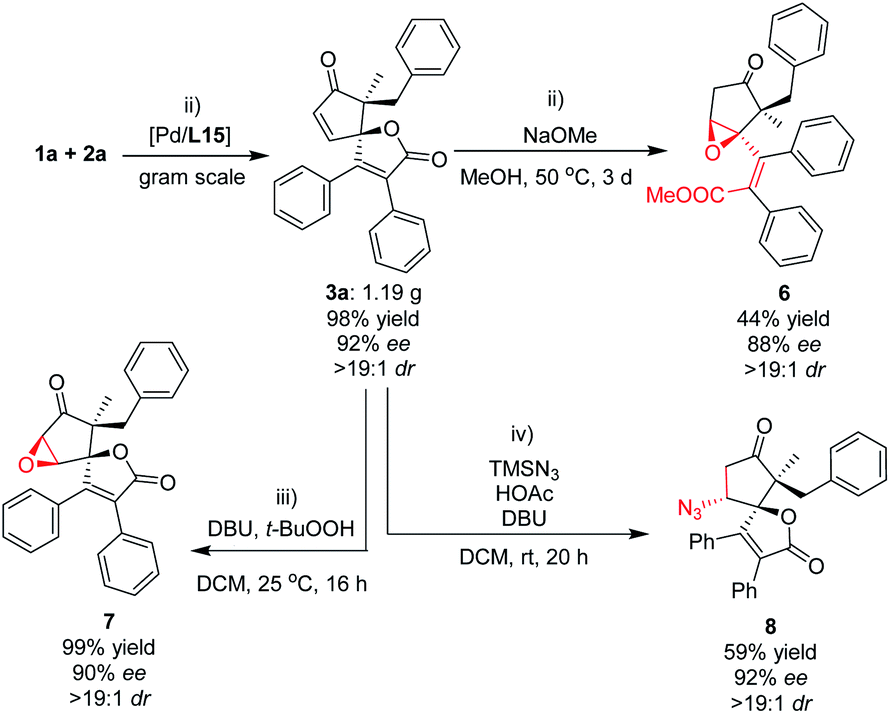 | ||
| Fig. 6 Synthetic application of the oxaspiro cyclopentenone–lactone scaffold for the downstream transformations. (i) Gram-scale synthesis of 3a; (ii) transesterification-induced intramolecular oxa-Michael addition; (iii) epoxidation reaction for the enantioselective synthesis of 7 bearing an additional epoxide; (iv) conjugate addition reaction of TMSN3/HOAc for the enantioselective synthesis of 8 bearing an additional azide. | ||
Conclusions
In summary, we developed a new strategy with C–C bond activation for atom-economic desymmetrization of cyclic 1,3-diketones by a palladium-catalyzed (3 + 2) spiro-annulation reaction of cyclopropenones with cyclopentene-1,3-diones. The reaction worked well for a series of small-ring substrates bearing electron-neutral, -withdrawing or -donating groups at the ortho-, meta- or para-positions of aromatic substituents. The corresponding highly functionalized oxaspiro molecules with a cyclopentenone–lactone scaffold were obtained in good yields, high diastereoselectivity and excellent enantioselectivity. In addition, it is the first example of an asymmetric palladium-catalyzed (3 + 2) annulation reaction of cyclopropenones with ketones that led to an unprecedented construction of oxaspiro molecules bearing two vicinal carbon quaternary stereocenters. Given the compatibility of the structurally diverse oxaspiro cyclopentenone–lactone scaffold and the fact that the palladium catalysis and corresponding chiral P-ligand are readily available, we believe that these promising findings based on palladium-catalyzed (3 + 2) annulation will open up a new avenue for further exploration of carbon–carbon bond activation and its enantioselective variants.Data availability
For all the data generated and analysed in this study, including the experimental details and spectra for all unknown compounds, see the ESI.† All data underlying the findings of this work are available from the corresponding author upon reasonable request.Author contributions
L. W. X. conceived the concept. H. Q. Z. and X. W. G. developed the Pd-catalyzed [3 + 2] spiro-annulation and its reaction mechanism. X. H. Z., F. Y. and G. W. Y. assisted with the preparation of substrates and structural analysis of the new product, and L. L. and Z. X. assisted with the theoretical studies of the reaction mechanism. All authors discussed the results and participated in writing the manuscript. L. W. X. supervised the project.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We are grateful for financial support from the grants of the National Natural Science Foundation of China (No. 22072035, 21773051, 21801056, and 21901056) and Zhejiang Provincial Natural Science Foundation of China (LZ18B020001 and LQ19B040001). The authors also thank Dr K. Z.Jiang, Dr K. F. Yang, Dr J. Cao, and X. Q. Xiao for their assistance in the spectral analysis of the new products.Notes and references
- I. Marek, Chem. Rev., 2021, 121, 1–2 CrossRef CAS PubMed , editorial..
- T. R. McDonald, L. R. Mills, M. S. West and S. A. Rousseaux, Chem. Rev., 2021, 121, 3–79 CrossRef CAS PubMed.
- O. O. Sokolova and J. F. Bower, Chem. Rev., 2021, 121, 80–109 CrossRef CAS PubMed.
- J. Wang, S. A. Blaszczyk, X. Li and W. Tang, Chem. Rev., 2021, 121, 110–139 CrossRef CAS PubMed.
- Y. Cohen, A. Cohen and I. Marek, Chem. Rev., 2021, 121, 140–161 CrossRef CAS.
- R. Vicente, Chem. Rev., 2021, 121, 162–226 CrossRef CAS.
- V. Pirenne, B. Muriel and J. Waser, Chem. Rev., 2021, 121, 227–263 CrossRef CAS PubMed.
- M. Murakami and N. Ishida, Chem. Rev., 2021, 121, 264–299 CrossRef CAS PubMed.
- M. D. R. Lutz and B. Morandi, Chem. Rev., 2021, 121, 300–326 CrossRef CAS PubMed.
- Y. Nakao, Chem. Rev., 2021, 121, 327–344 CrossRef CAS PubMed.
- K. Nogi and H. Yorimitsu, Chem. Rev., 2021, 121, 345–364 CrossRef CAS PubMed.
- Z. Li, S. Kosuri, H. Foster, J. Cohen, C. Jumeaux, M. M. Stevens, R. Chapman and A. J. Gormley, J. Am. Chem. Soc., 2019, 141, 19823–19830 CrossRef CAS PubMed.
- A. M. Laradji, C. D. McNitt, N. S. Yadavalli, V. V. Popik and S. Minko, Macromolecules, 2016, 49, 7625–7631 CrossRef CAS.
- L. Qu, Y. Wu, P. Sun, K. Zhang and Z. Liu, Polymer, 2017, 114, 36–43 CrossRef CAS.
- C. Shao, H. Duan, Y. Min and X. Zhang, Chin. Chem. Lett., 2020, 31, 299–302 CrossRef CAS.
- P. A. Peart and J. D. Tovar, J. Org. Chem., 2010, 75, 5689–5696 CrossRef CAS PubMed.
- X. Wang, F. W. Seidel and K. Nozaki, Angew. Chem., Int. Ed., 2019, 58, 12955–12959 CrossRef CAS PubMed.
- F. Friscourt, C. J. Fahrni and G.-J. Boons, J. Am. Chem. Soc., 2012, 134, 18809–18815 CrossRef CAS PubMed.
- C. Favre, L. de. Cremoux, J. Badaut and F. Friscourt, J. Org. Chem., 2018, 83, 2058–2066 CrossRef CAS PubMed.
- H.-W. Shih and J. A. Prescher, J. Am. Chem. Soc., 2015, 137, 10036–10039 CrossRef CAS PubMed.
- R. D. Row and J. A. Prescher, Org. Lett., 2018, 20, 5614–5617 CrossRef CAS PubMed.
- R. D. Row, S. S. Nguyen, A. J. Ferreira and J. A. Prescher, Chem. Commun., 2020, 56, 10883–10887 RSC.
- R. D. Row, H.-W. Shih, A. T. Alexander, R. A. Mehl and J. A. Prescher, J. Am. Chem. Soc., 2017, 139, 7370–7375 CrossRef CAS PubMed.
- S. V. Mayer, A. Murnauer, M.-K. von. Wrisberg, M.-L. Jokisch and K. Lang, Angew. Chem., Int. Ed., 2019, 58, 15876–15882 CrossRef CAS.
- C. M. Vanos and T. H. Lambert, Angew. Chem., Int. Ed., 2011, 50, 12222–12226 CrossRef CAS PubMed.
- T. Stach, J. Dräger and P. H. Huy, Org. Lett., 2018, 20, 2980–2983 CrossRef CAS PubMed.
- E. D. Nacsa and T. H. Lambert, Org. Lett., 2013, 15, 38–41 CrossRef CAS PubMed.
- B. D. Kelly and T. H. Lambert, Org. Lett., 2011, 13, 740–743 CrossRef CAS PubMed.
- A. Rai and L. D. S. Yadav, Eur. J. Org. Chem., 2013, 1889–1893 CrossRef CAS.
- R. Breslow, R. Haynie and J. Mirra, J. Am. Chem. Soc., 1959, 81, 247–248 CrossRef CAS.
- R. Breslow, T. Eicher, A. Krebs, R. A. Peterson and J. Posner, J. Am. Chem. Soc., 1965, 87, 1320–1325 CrossRef CAS.
- A. Poloukhtine and V. V. Popik, J. Org. Chem., 2003, 68, 7833–7840 CrossRef CAS PubMed.
- M. Martínek, L. Filipová, J. Galeta and L. Ludvíková, Org. Lett., 2016, 18, 4892–4895 CrossRef PubMed.
- K. Mishiro, Y. Yushima and M. Kunishima, Org. Lett., 2017, 19, 4912–4915 CrossRef CAS PubMed.
- Y. Gong, Y. Zhang, W. Xiong, K. Zhang, Y. Che and J. Zhao, J. Am. Chem. Soc., 2017, 139, 10649–10652 CrossRef CAS PubMed.
- B. J. Levandowski, T. A. Hamlin, R. C. Helgeson, F. M. Bickelhaupt and K. N. Houk, J. Org. Chem., 2018, 83, 3164–3170 CrossRef CAS PubMed.
- K. Mishiro, Y. Yushima and M. Kunishima, J. Org. Chem., 2018, 83, 13595–13603 CrossRef CAS PubMed.
- Z. M. Strater, M. Rauch, S. Jockusch and T. H. Lambert, Angew. Chem., Int. Ed., 2019, 58, 8049–8052 CrossRef CAS.
- K. Mishiro, T. Kimura, T. Furuyama and M. Kunishima, Org. Lett., 2019, 21, 4101–4105 CrossRef CAS.
- M. V. Anokhin and E. E. Nesterov, J. Phys. Chem. Lett., 2020, 11, 8745–8750 CrossRef CAS PubMed.
- K. P. Reber and I. W. Gilbert, J. Org. Chem., 2019, 84, 5524–5534 CrossRef CAS PubMed.
- T. Kondo, Y. Kaneko, Y. Taguchi, A. Nakamura, T. Okada, M. Shiotsuki, Y. Ura, K. Wada and T.-a. Mitsudo, J. Am. Chem. Soc., 2002, 124, 6824–6825 CrossRef CAS PubMed.
- P. A. Wender, T. J. Paxton and T. J. Williams, J. Am. Chem. Soc., 2006, 128, 14814–14815 CrossRef CAS PubMed.
- F. Xie, S. Yu, Z. Qi and X. Li, Angew. Chem., Int. Ed., 2016, 55, 15351–15355 CrossRef CAS PubMed.
- L. H. Li, Y. Jiang, J. Hao, Y. Wei and M. Shi, Adv. Synth. Catal., 2017, 359, 3304–3310 CrossRef CAS.
- J. L. Xu, H. Tian, J. J. Kang, W. X. Kang, W. Sun, R. Sun, Y. M. Li and M. Sun, Org. Lett., 2020, 22, 6739–6743 CrossRef CAS PubMed.
- T. K. Heiss and J. A. Prescher, J. Org. Chem., 2019, 84, 7443–7448 CrossRef CAS PubMed.
- P. Zhou, W.-T. Yang, A. U. Rahman, G. Li and B. Jiang, J. Org. Chem., 2020, 85, 360–366 CrossRef CAS PubMed.
- J. Wallbaum, P. G. Jones and D. B. Werz, J. Org. Chem., 2015, 80, 3730–3734 CrossRef CAS PubMed.
- L. Kong, X. Zhou, Y. Xu and X. Li, Org. Lett., 2017, 19, 3644–3647 CrossRef CAS.
- J.-T. Ren, J.-X. Wang, H. Tian, J.-L. Xu, H. Hu, M. Aslam and M. Sun, Org. Lett., 2018, 20, 6636–6639 CrossRef CAS PubMed.
- J. Wu, W.-X. Gao, X.-B. Huang, Y.-B. Zhou, M.-C. Liu and H.-Y. Wu, Org. Lett., 2020, 22, 5555–5560 CrossRef CAS.
- F. Jamshaid, V. V. R. Kondakal, C. D. Newman, R. Dobson, H. Joao, C. R. Rice, J. M. Mwansa, B. Thapa and K. Hemming, Tetrahedron, 2020, 76, 131570 CrossRef CAS.
- T. Nanda and P. C. Ravikumar, Org. Lett., 2020, 22, 1368–1374 CrossRef CAS PubMed.
- T. Nanda, P. Biswal, B. V. Pati, S. K. Banjare and P. C. Ravikumar, J. Org. Chem., 2021, 86, 2682 CrossRef CAS PubMed.
- Y.-F. Yang, X. .-B. Huang, W.-X. Gao, Y.-B. Zhou, M.-C. Liu and H.-Y. Wu, Org. Biomol. Chem., 2020, 18, 5822–5825 RSC.
- J. Chen, B. Tang, X. Liu, G. Lv, Y. Shi, T. Huang, H. Xing, X. Guo, L. Hai and Y. Wu, Org. Chem. Front., 2020, 7, 2944–2949 RSC.
- S. S. Nguyen, A. J. Ferreira, Z. G. Long, T. K. Heiss, R. S. Dorn, R. D. Row and J. A. Prescher, Org. Lett., 2019, 21, 8695–8699 CrossRef CAS PubMed.
- A. Haito and N. Chatani, Chem. Commun., 2019, 55, 5740–5742 RSC.
- Y. Wei, W.-T. Zhao, Y.-L. Yang, Z. Zhang and M. Shi, ChemCatChem, 2015, 7, 3340–3349 CrossRef CAS.
- Y.-L. Yang, Z. Zhang, X.-N. Zhang, D. Wang, Y. Wei and M. Shi, Chem. Commun., 2014, 50, 115–117 RSC.
- J. Xu, J. Cao, C. Fang, T. Lu and D. Du, Org. Chem. Front., 2017, 4, 560–564 RSC.
- J. Cao, R. Fang, J.-Y. Liu, H. Lu, Y.-C. Luo and P.-F. Xu, Chem.–Eur. J., 2018, 24, 18863–18867 CrossRef CAS.
- X. Li, C. Han, H. Yao and A. Lin, Org. Lett., 2017, 19, 778–781 CrossRef CAS PubMed.
- K. Reitel, K. Kriis, I. Järving and T. Kanger, Chem. Heterocycl. Compd., 2018, 54, 929–933 CrossRef CAS.
- A. R. Rivero, I. Fernández, C. R. de. Arellano and M. A. Sierra, J. Org. Chem., 2015, 80, 1207–1213 CrossRef CAS PubMed.
- T. Matsuda and Y. Sakurai, J. Org. Chem., 2014, 79, 2739–2745 CrossRef CAS PubMed.
- D. Bai, Y. Yu, H. Guo, J. Chang and X. Li, Angew. Chem., Int. Ed., 2020, 59, 2740–2744 CrossRef CAS PubMed.
- F. Ye, Z. Xu and L. W. Xu, Acc. Chem. Res., 2021, 54, 452–470 CrossRef CAS PubMed.
- W. D. G. Brittain, B. R. Buckley and S. J. Fossey, ACS Catal., 2016, 6, 3629–3636 CrossRef CAS.
Footnotes |
| † Electronic supplementary information available. CCDC 2024499. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04558j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |