
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBispidine as a β-strand nucleator: from a β-arch to self-assembled cages and vesicles†
Hanuman
Singh
 a,
Akshay
Chenna
b,
Upanshu
Gangwar
a,
Julie
Borah
b,
Gaurav
Goel
*b and
V.
Haridas
*a
a,
Akshay
Chenna
b,
Upanshu
Gangwar
a,
Julie
Borah
b,
Gaurav
Goel
*b and
V.
Haridas
*a
aDepartment of Chemistry, Indian Institute of Technology Delhi, Hauz Khas, New Delhi-110016, India. E-mail: haridasv@chemistry.iitd.ac.in
bDepartment of Chemical Engineering, Indian Institute of Technology Delhi, Hauz Khas, New Delhi-110016, India. E-mail: goelg@chemical.iitd.ac.in
First published on 25th October 2021
Abstract
The development of synthetic scaffolds that nucleate well-folded secondary structures is highly challenging. Herein, we designed and synthesized a series of core-modified peptides (F1, F2, F3, and F4) that fold into β-strand structures. These bispidine-scaffolded peptides were studied by CD, IR, NMR, single crystal XRD, and Molecular Dynamics (MD) simulations to investigate their conformational preferences. Solid-state and solution studies revealed that bispidine is a versatile scaffold that could be placed either at the terminal or at the middle of the peptide strand for nucleating the β-strand structure. Scaffolds that nucleate an isolated β-strand conformation are rare. Bispidine placed at the C-terminus of the peptide chain could nucleate a β-strand conformation, while bispidine placed at the middle resulted in a β-arch conformation. This nucleation activity stems from the ability to restrict the psi torsion angle (ψ) through intramolecular C5 hydrogen bonding between the equatorial hydrogen(s) of bispidine and the carbonyl oxygen(s) of the amino acid close to the scaffold. Furthermore, the bispidine peptidomimetic with a super secondary structure, namely β-arch, assembled into single-hole submicron cages and spherical vesicles as evident from microscopic studies. The design logic defined here will be a significant strategy for the development of β-strand mimetics and super secondary structures.
Introduction
Proteins have uniquely folded secondary structures (α-helix and β-strand) that undergo well-controlled self-assembly to form hierarchically ordered assemblies. This supramolecular assembly called a quaternary structure ranges from nanometers to micrometers and beyond.1 The functional aspect of proteins is intimately linked to their secondary and quaternary structures. However, the evolution of the quaternary structure from the secondary structure is not well understood.2 In addition, the development of biomimetic systems with the ability to form a sophisticated assembly will have far-reaching applications in areas like therapeutics, and in the development of artificial enzymes.3,4 In this perspective, the de novo design of secondary structure mimetics with the ability to self-assemble has multiple applications. The secondary structure mimetics can act as an effective agent to perturb protein–protein interactions (PPIs)5 and are expected to be useful in studying bioactive conformations. Many studies have already explored α-helix mimics while β-sheet mimics are less studied because of the lack of good chemical models.6–9 Similarly, helical PPIs have been extensively targeted, while the inhibition of the β-sheet interface is very limited.10 Peptidomimetic foldamers that can mimic β-sheet interfaces and form soluble β-sheet aggregates can be excellent candidates for therapeutic intervention against protein aggregation.11 However, in general, short peptides do not have a single dominant solution conformation. The insertion of an unnatural backbone in the peptide sequence is a better strategy to overcome these issues. The groups of Kelly,12 Nowick,13 Hamilton,14 and Gellman15 have designed various β-sheet peptidomimetics.Scaffolds that could enforce peptide conformation to specific secondary structures such as an α-helix or β-sheet are scarce. The nucleation of secondary structures is achieved by the introduction of a rigid unit that favors the initial hydrogen-bonding pattern of the α-helix or β-strand as per the nucleating preference of the scaffold.6–9,16–18 However, there are limited examples of isolated β-strands. Most of the designed scaffolds act by facilitating initial hydrogen bonding to form a sheet that further propagates the β-sheet structure. Furthermore, turns are proposed to be the nucleators of secondary structures without loss in chain entropy.19 Bispidine serves as a versatile scaffold for nucleating secondary structures such as reverse turns, helices, and sheets in peptides.20,21 The nature of the linkage between the peptide fragments and the bispidine scaffold is crucial for nucleating specific secondary structures. Here we show that bispidine could nucleate isolated β-strand and β-arch super secondary structure along with their intrinsic ability to self-assemble. The unique geometry of the bispidine scaffold facilitates intramolecular C5-hydrogen bonding, thereby nucleating the β-strand structure on the attached peptides. Bispidine could be placed either at the C-terminus of the peptide or in the middle of the peptide strand for nucleating the β-strand.
Results and discussion
We show that the introduction of an artificial rigid structure that enables chain reversal could lead to the nucleation of secondary structures. In this report, we incorporate a rigid bicyclic entity reminiscent of an artificial turn in a peptide. The bispidine unit could be considered as a folded diaminopropane due to its bicyclic architecture (Fig. 1). This bicyclic unit could be considered as a turn since it provides a kink to the peptide backbone. Bispidine could be attached to the peptide portion through amide bonds between the amino units of bispidine and the C-terminal of peptide fragments. The bispidine scaffold can be synthesized easily and allows easy incorporation of peptide units.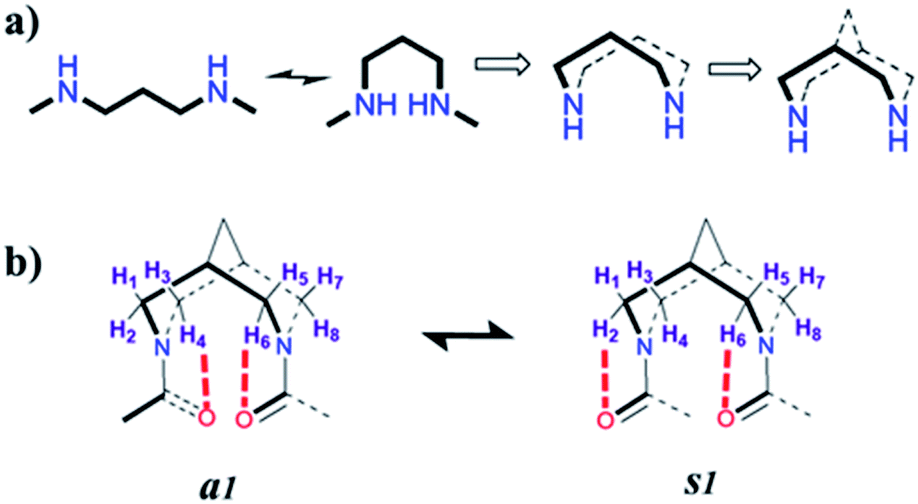 | ||
| Fig. 1 Evolution of the bispidine scaffold. (a) The most preferred conformer of the 1,3-diaminopropane linker along with the folded conformer. Cyclization of this folded conformer results in conformational restriction. This cyclic structure is further interlocked by a methylene bridge resulting in a bicyclic architecture. (b) The conformations of bispidine diamides, showing the equatorial hydrogen atoms that are proximal to the carbonyl oxygen atoms. The anti (a1) and syn (s1) forms are shown. | ||
Herein, we incorporated bispidine as a non-peptidic molecular scaffold that can act as a promising template for the nucleation of β-strand secondary structures and further assemble into a vesicular quaternary structure. Bispidine was synthesized from Boc protected piperidone, benzylamine, and formaldehyde through a double Mannich reaction, followed by Wolff–Kishner reduction. The bispidine peptide conjugate adopts a conformation based on the nature of the linkage between the peptide and scaffold.20,21 Bispidine was incorporated into the peptide by a typical peptide coupling procedure (Scheme S1†). A series of bispidine–peptide conjugates with an increasing number of amino acids were designed and synthesized (Fig. 2). A dipeptide F1, tetrapeptide F2, hexapeptide F3, and deprotected water-soluble hexapeptide F4 were chosen for conformational studies. The conformations of bispidine peptide conjugates F2–F4 were studied by CD, 1D, and 2D NMR, variable temperature NMR (VT-NMR), FT-IR, and all-atom MD simulations. The crystallographic coordinates of F1 were used to create an initial structure for MD simulations of bispidine anchored peptides (simulation details in Section S 2.1†). For each system, we generated a 200 ns trajectory and calculated averages from the last 50 ns portion. All structural properties, such as the distribution of (φ, ψ) dihedrals, converged on the simulation time scale (Fig. S11†).
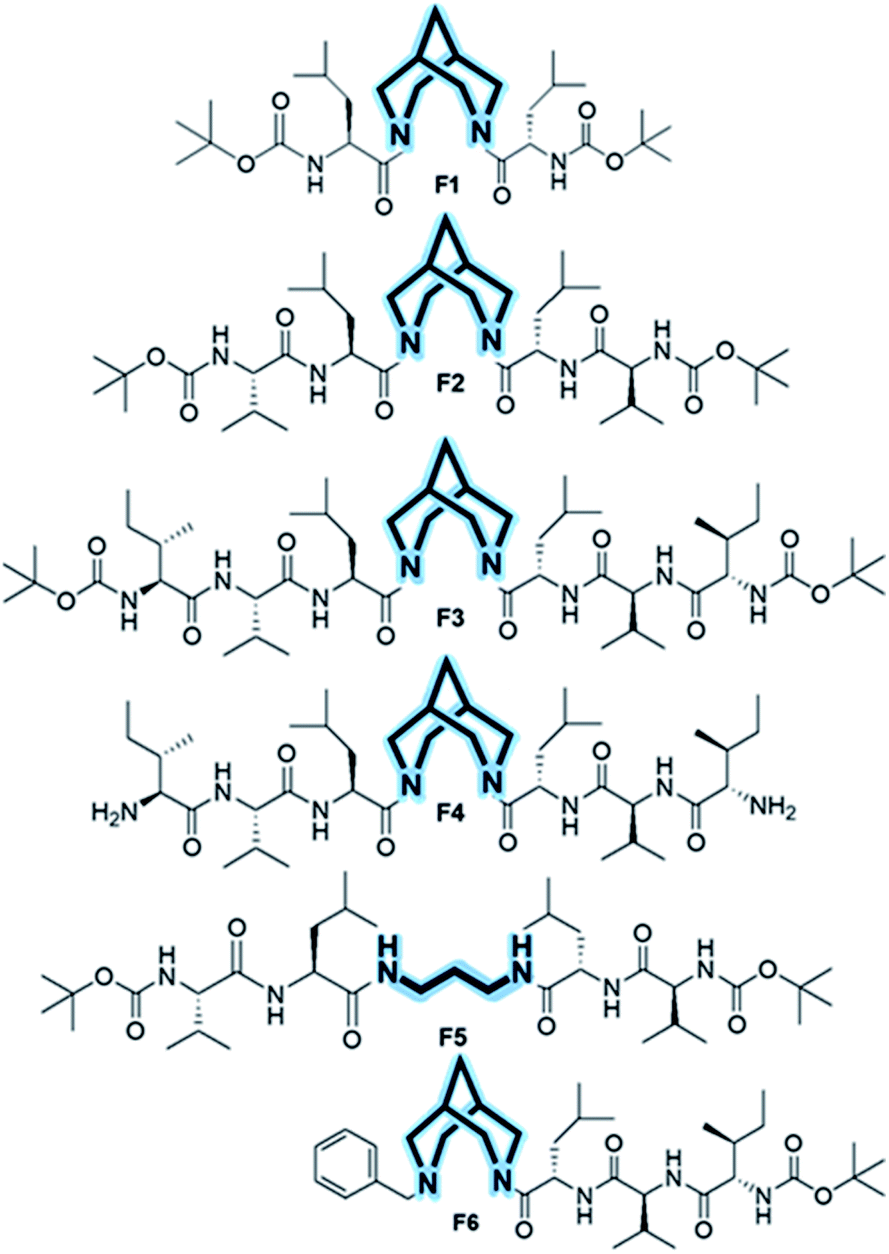 | ||
| Fig. 2 Chemical structures of foldamers (F1–F4) in which the bispidine scaffold is at the middle. Control compound F5 with a 1,3 diaminopropane spacer instead of bispidine and F6 with bispidine placed at the terminal. | ||
The 1H NMR spectra of F2 and F3 showed two interconverting conformers, syn and anti. The J1,3-coupling values of amide NHs of both syn and anti-forms are in the range 8–9 Hz (Tables S1 and S7†), suggesting an extended β-strand conformation in both syn and anti-forms as observed from 1H NMR and MD simulations.22,23 The chemical shifts of the α-protons for all amino acid residues of bispidine-linked compounds are downfield shifted with respect to the random coil structure indicating β-strand structures (Table S2†).15 The bispidine scaffold adopts a double chair conformation, with the distance between the nitrogen atoms at around 2.9 Å. The rotation of the carbonyl units can result in four conformers, two anti (a1 and a2′) and two syn (s1 and s2′) (Fig. S1†). The syn and anti-forms are readily observable in the 1H NMR spectra of bispidine conjugates.20 In CDCl3, the ratio of anti to syn was 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, while DMSO favored the structure with a high dipole moment, the syn conformer.24,25 The ROESY spectrum confirmed the presence of syn and anti-conformers in F3 (Fig. 3a and b).
:1, while DMSO favored the structure with a high dipole moment, the syn conformer.24,25 The ROESY spectrum confirmed the presence of syn and anti-conformers in F3 (Fig. 3a and b).
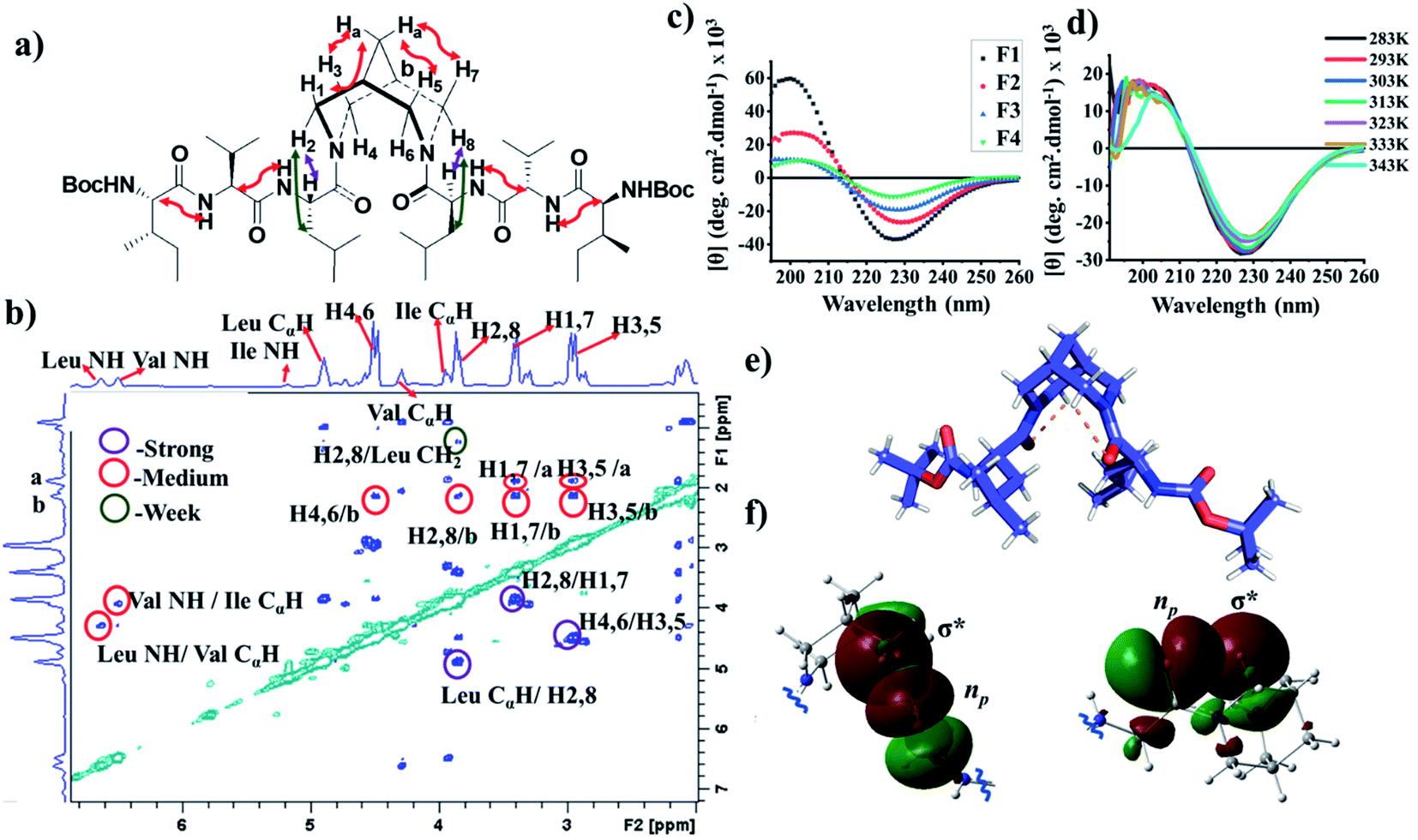 | ||
| Fig. 3 (a) The ROE observation in molecule F3. (b) A cross section of the ROESY spectrum of F3 in chloroform showing the correlation between the peptide chain and the bispidine linker. (c) Circular dichroism (CD) spectrum of compounds F1, F2, and F3 in methanol, and F4 in water. (d) Temperature-dependent CD of compound F3. (e) X-ray crystal structure of compound F1 showing intramolecular H-bonding between Leu carbonyl and bispidine equatorial hydrogen (C–H⋯O). (f) The orbital view of C5–hydrogen bonding in the crystal structure of F1. The lone pair containing p-type orbital of carbonyl oxygen interacts with the C–H σ* orbital. The two colors (red and green) show the two phases (positive and negative) of molecular orbitals, respectively. | ||
The exchange rate is obtained by performing selective and non-selective inversion recovery experiments26 under varying temperature ranges to get the thermodynamic parameters. The calculated rate constants were fitted using the Arrhenius and the Eyring equations resulting in an activation energy barrier (ΔE‡) and a standard enthalpy of activation (ΔH‡) between syn and anti-conformers equal to 110.08 kJ mol−1 and 107.67 kJ mol−1, respectively (Fig. S2a–c, Tables S4 and S5†). This molecular switching behavior of bispidine diamides is analogous to the peptide bond isomerization of proline-containing peptides. Prolyl isomerization plays a key role in protein folding and in the cell signaling process.27,28 The ΔE‡ and ΔH‡ values of bispidine compounds are comparable to the cis–trans isomerization of proline peptides, and hence bispidine could serve as a scaffold in the design of artificial functional switching peptide systems.
1H NMR studies revealed that all amide NHs appeared at <7.0 ppm in F2 and F3 indicating that they are non-hydrogen bonded, since the hydrogen-bonded amide NH usually appears around 8.0 ppm in CDCl3 solution.29,30 The VT-NMR studies on compound F3 in CDCl3 revealed Δδ/Δt in the range −0.004 to −0.006 ppm per K (Fig. S3†) for the amide NH, indicating the absence of intramolecular hydrogen bonding.31,32 Furthermore, the FT-IR spectrum of F2 and F3 in chloroform solution showed a band at 3438 and 3435 cm−1 respectively (Fig. S6c and d†), again indicating that the predominant form in solution is non-bonded NHs. The addition of a hydrogen bond accepting solvent like DMSO-d6 to CDCl3 solutions of peptides F2 and F3 resulted in significant chemical shift changes of amide NHs (Fig. S4†). All amide NHs in F2 and F3 are solvent-exposed as evident from the relatively high chemical shift values of NHs upon addition of DMSO-d6. These orthogonal measurements confirm the absence of intramolecular hydrogen bonds, as also corroborated by the MD simulation of F3 in CHCl3 wherein we obtained only 0.28 intramolecular H bonds (involving NHs) per molecule (Table S11†).33
The ROESY spectra of compounds F2 and F3 showed strong ROE between CαHs of Leu and equatorial hydrogens of bispidine (Fig. 3a, b, and S5a–g†), a significant observation that is discussed later. Furthermore, all NHs showed strong ROE with neighboring CαHs, supporting a β-strand structure. Additionally, the amide I stretching of F2 and F3 showed a band at 1637 and 1633 cm−1 respectively, indicating that it is predominantly in the β-strand conformation (Fig. S6a and b†).35,36 This is also evident from MD simulations as the inter-proton distances between neighboring amide hydrogens are under 3 Å indicative of a β-strand (Fig. S13†).37 Our simulations reveal that the stable solution structure of F3 in all three solvents used here, viz., chloroform, methanol, and water, has an extended conformation (Fig. 4a and Table S7†) with all backbone torsional angles having a high population in the β-sheet region (Fig. 4b and Table S8†).
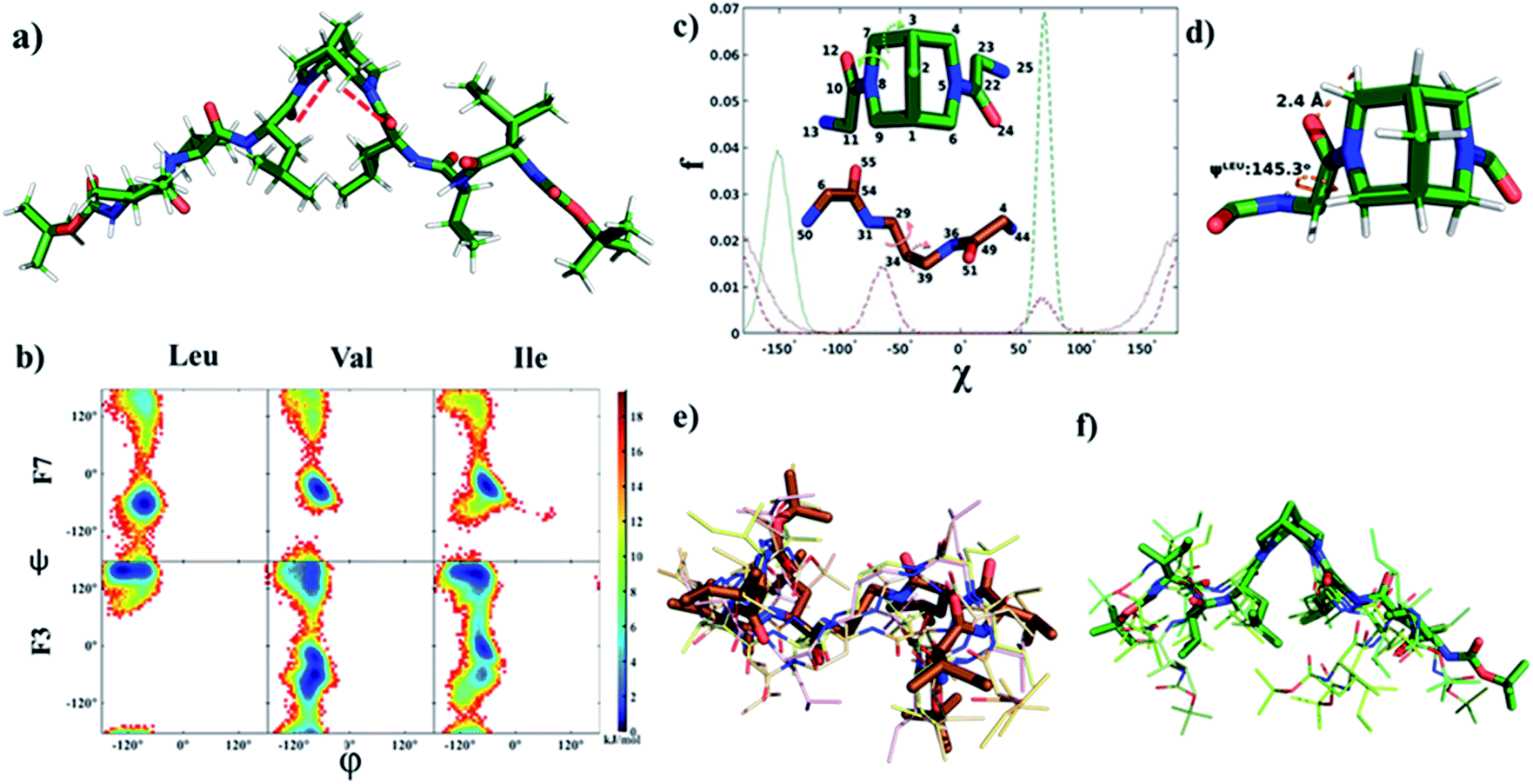 | ||
| Fig. 4 (a) Solution structure of F3 in chloroform obtained from MD simulation (intramolecular C–H⋯O bond between Leu carbonyl and bispidine equatorial hydrogen is shown by a dashed red line. (b) Density plot of the population (fraction f reported in the units of free energy −RTlnf) of main-chain torsion angles (φ and ψ) obtained from the MD simulation of a single molecule of F3 (consisting of a bispidine linker) or F7 (consisting of a 1,3 diaminopropane linker) in chloroform. A significant shift to the characteristic β-strand region (φ ∼−140° and ψ ∼130°) is seen for all residues of F3. (c) Comparison of the distribution of the two torsion angles (shown in the inset) capturing the linker conformational space: C22–N5–C6–C1 (solid green) and N5–C6–C1–C9 (dashed green) in F3, C54–N31–C29–C34 (solid brown) and N31–C29–C34–C39 (dashed brown) in F6. (d) A representative snapshot showing the main chain ψ dihedral of leucine, and the C–H⋯O bond between bispidine equatorial hydrogen (atom 77) and leucine carbonyl oxygen (atom 24) in F3. (e and f) Overlay of the central structures of the five most populated clusters of F7 and F3, respectively (cluster population in Table S9†), obtained from the equilibrium portion of MD trajectory using the Gromos algorithm34 with a backbone root mean squared deviation (RMSD) cut-off of 0.1 nm. | ||
The CD spectrum of compounds F1, F2, and F3 in methanol showed a positive maximum at ∼197 nm and a negative band around 226 nm (Fig. 3c), indicating the presence of a β-strand conformation in all three compounds. The shift from the typical β-sheet CD is not uncommon in synthetic peptides.38–40 A water-soluble version of the bispidine–peptide conjugate was also synthesized by deprotecting the terminal Boc groups. The water-soluble amine derivate F4 showed a similar CD spectrum as that of F2 and F3 (Fig. 3c and Table S8†). The temperature-dependent CD of F3 from 0 °C to 70 °C showed no significant changes (Fig. 3d and S7a†), indicating a highly robust secondary structure of these peptides.41 The β-strand conformation is devoid of extensive hydrogen bonds, and therefore, shows a weak temperature dependence. The representative conformation of F1, F2, F3, and F4 is a β-arch type structure.42 In this case, the two β-stands are isolated with bispidine at the middle, which acts as a turn. The angle between the two β-strands is 150°. The bispidine linker that connects two parallel β-strands could adopt a syn- or anti-form. The syn form keeps the parallel β-strands with their faces on the same side, while the anti-form keeps the faces of β-strands in opposite directions. A 2D potential energy scan on torsional angles O30–C2–N31–C19 and O29–C3–N23–C1 in the gas phase, obtained using quantum mechanical calculations with B3LYP/6-31G*, revealed the anti- and the syn-forms as the lowest energy states (anti more stable than syn) (Fig. S15†). The topological placement of β-strands is important, since one face or both faces of the β-strand could be involved in interaction with the target protein.43 The bispidine scaffold providing various topological arrangements of β-strands (Fig. S14†) is an added advantage. The β-arch motif is found in the amyloid-forming fibrillar assemblies and also in β-solenoid proteins.44
To evaluate the nucleating effect when bispidine is placed at the terminus, compound F6 was synthesized (Scheme S3†). The CD spectrum of F6 revealed the presence of a β-strand conformation (Fig. S7b†). MD simulations also show that the solution structure of F6 is essentially the same as that of F3: a β-strand conformation with main chain (φ and ψ) angles predominantly in the β region (Table S8†) and 3JHNHα values (>8 Hz) also equivalent to those in F3 (Table S7†). To understand the reason behind the nucleation of the β-strand by bispidine, a control compound F5 (tetrapeptide with 1,3-diaminopropane as the spacer) was synthesized (Scheme S2†), while a similar control compound F7 (hexapeptide with 1,3-diaminopropane as the spacer) was used for MD simulations. The unique role of bispidine as a β-strand inducer is evident from the following observations: the CD spectrum of F5 showed no characteristic secondary structure (Fig. S7c†), the chemical shift value of CαH in F5 is comparable to the random coil structure15 unlike bispidine-linked compounds (Table S3†), and the population of the main chain (φ and ψ) angles of Leu, Val, and Ile in the β-strand region was 98%, 30%, and 60% for F3, while it was only 14%, 8%, and 6% for F7, respectively (Fig. 4b and Table S8†).
The X-ray crystal structure of F1 gave an insight into the unique role of bispidine as a β-strand nucleator. The distance between Leu carbonyl and bispidine equatorial hydrogens (H4,6 in the anti-form, Fig. S5e;† H2,6 in the syn form, Fig. S5f†) was 2.4 Å with a donor and acceptor angle 〈CHO 〉 90° (Table S10†), thus satisfying the C–H⋯O hydrogen bond criterion (dH–O < 2.7 Å and 〈CHO〉 90°).45 This implies that bispidine could restrict the ψ angle of the connected residue (Leu) by C5-hydrogen bonding (Fig. 3e and Table S6†). These structural features were also present in the solution state structure of F3 (bispidine hexapeptide) and F6 (terminal bispidine tripeptide), as determined from MD simulations. The average distance between the carbonyl oxygen of Leu and equatorial hydrogens of bispidine was found to be ∼2.5 Å (Fig. 4d and Table S10†), and the average number of C–H⋯O bonds per tripeptide arm was found to be 0.77 in F3 and 0.71 in F6, while it was only 0.27 in F7 (Table S11 and Fig. S12†). F3 and F6 also show significantly higher preference than F7 for a β-strand geometry, consistent with the role of C5 hydrogen bonding in stabilizing the protein β-strand secondary structure.46
The orbital analysis of the X-ray structure of F1 was performed by using the Gaussian package,47,48 which showed the overlap of the p-type orbital of carbonyl oxygen with the C–H σ* orbital (Fig. 3f). We also analyzed X-ray crystal structures of several bispidine diamides that further supported similar orbital interactions (Fig. 3f). Computational analysis of the rotation of diamides such as simple diacetyl derivatives revealed syn- and anti-forms that are stabilized by intramolecular C–H⋯O hydrogen bonding (Table S12†). We envisioned that the C–H⋯O interaction could preorganize the first residue, thereby reducing the number of conformations that the polypeptide can adopt; hence decreasing the entropy cost of folding. Unlike the bispidine scaffold, the diaminopropane spacer itself has considerably higher conformational flexibility: Schlitter's entropy, calculated from the covariance matrix of position fluctuations for N–C–C–C–N atoms in the MD trajectory, was 0.24 J (mol K)−1 for F3 and 1.16 J (mol K)−1 for F7. This higher flexibility of F7 compared with F3 is evident from the sampling of multiple conformational substates of the linker (Fig. 4c) as well as a wider sampling of the whole molecule (Fig. 4e and f), which can be attributed to the lack of a well-defined secondary structure in F7. The initial preorganization provided by the scaffold through two C–H⋯O interactions could lead to cooperativity in folding. The four conformational states in bispidine diamides are stabilized by non-canonical intramolecular C–H⋯O hydrogen bonding, hence making many of the possible orientations of carbonyls unfavorable. A pair of non-canonical interactions between the scaffold and amino acid carbonyl preorganize the peptide strand.
The energy diagram of the rotation of amide carbonyls clearly shows the minima at syn- and anti-states (Fig. S15†). The two peptide strands separated by bispidine adopt an independent albeit identical conformation, and the different rotamer forms of bispidine (two anti and two syn) do not appear to affect the strand conformation. We obtained an equal number of C–H⋯O hydrogen bonds (2), a similar value for the Leu-ψ angle (153°–162°), and the same secondary structure assignment (β) for all four metastable conformations of F1 determined from quantum chemical calculations (Table S12†). Also, a backbone RMSD of 0.8 ± 0.2 Å was obtained between the peptide strands in the syn- and the anti-states of F3 in chloroform. The C5 hydrogen bonding was present in both syn- and anti-forms (Fig. S12a and b†), which in-turn could constrain the conformation of bispidine anchored peptides. Therefore, even though these rotamers are observed as a result of slow rotation on the NMR time scale, both conformers are identically locked by intramolecular H-bonding. The non-canonical tethering by the bispidine scaffold could control the ψ dihedral angle of the residue adjacent to it and this conformational preference is propagated to the remaining residues (Table S8†). Therefore, the lack of C5 hydrogen bonding in diaminopropane spacer-linked compounds could be directly linked to the absence of β-strand geometry in them.
The link between secondary and quaternary structures is the holy grail of biomolecular interactions. The coiled-coil structure of keratin and the assembly of poly-proline helices in collagen highlight how the secondary structure leads to a functional assembly.49,50 The engineering of the secondary structure itself is a surmountable challenge, while the assembly from an engineered secondary structure into a defined quaternary structure is a formidable task.
In order to investigate the self-assembly of the β-arch mimetic, detailed ultramicroscopic imaging such as scanning electron microscopy (SEM), transmission electron microscopy (TEM), and atomic force microscopy (AFM) is performed. At 0.25 mg ml−1 concentration of F2, single orifice cages are observed (Fig. 5a) with the size of hollow cages ranging from 150 to 400 nm (Fig. S9a†). This is similar to single-hole nanocages reported in mesoporous silver and polymeric materials.51 Molecular containers such as cucurbiturils52 and cyclodextrins53 were extensively used for the encapsulation of a variety of molecules and served as chemical models for studying enzyme mechanisms. In these molecular containers, two sides are open, while our self-assembled systems provide more possibilities for various topologies and dimensions.
 | ||
| Fig. 5 (a) SEM of F2 at 0.25 mg ml−1. (b) SEM of F2 at 0.5 mg ml−1. (c) TEM of F2 at 0.25 mg ml−1 (red arrows indicate the hole). (d) TEM of F2 at 0.5 mg ml−1. (e) AFM of F2 at 0.25 mg ml−1 (red arrows indicate the hole). (f) AFM of F2 at 0.5 mg ml−1. | ||
Self-assembled systems offer advantages in terms of controlling the size by engineering building blocks. In addition to that, the confined space could be used for performing chemical reactions. Self-assembled peptide nanotubes and peptide-based vesicles have several applications.54–56 For example, protein nanocages are versatile platforms for nanotechnology applications.57 Here, we found that increasing F2 concentration to 0.5 mg ml−1 resulted in the formation of vesicles with sizes ranging from 400 nm to 1200 nm (Fig. 5b and S9b†). The fine-tuning of self-assembled structures like single-hole cages and vesicles by changing the concentration will be useful in controlled encapsulation and delivery. The results obtained from SEM analysis are consistent with other measurements. For example, TEM of F2 also revealed single-hole submicron cages at a concentration of 0.25 mg ml−1 (Fig. 5c).
Increasing the concentration to 0.5 mg ml−1 resulted in complete vesicle formation (Fig. 5d). AFM imaging of F2 also showed single-hole submicron cages at a concentration of 0.25 mg ml−1 (Fig. 5e) and vesicles at 0.5 mg ml−1 (Fig. 5e, f, S8c and d†). Size distribution analysis using dynamic light scattering (DLS) based studies also reveals that the average size of single-hole cages lies in the range 200 to 600 nm at 0.25 mg ml−1 and the average size of vesicles lies in the range 400 to 900 nm in solution (Fig. S10a and b†). Similarly, F3 showed vesicular assembly in methanol at 0.5 mg ml−1 with sizes ranging from 200 nm to 800 nm (Fig. S8b and S9c†). The self-assembly of F5 was also investigated by SEM and it showed no vesicular assembly (Fig. S8a†), which confirms that bispidine has a significant role in the formation of vesicles via the self-assembly of β-strands.
Conclusions
By using integrative experimental structure determination techniques combined with MD simulations, we showed that using the scaffolding approach, short peptides could be folded into a well-defined and stable β-strand conformation. This forced folding of peptides using a bicyclic scaffold, bispidine, is a versatile approach. In this approach, the C5 hydrogen bonding between the scaffold and the first residue resulted in the restriction of the ψ angle that further propagated and controlled the conformation of the peptide. Our strategy, wherein, one could induce a β-strand in a peptide, will be a powerful strategy in protein engineering. The self-assembly of the beta strand-turn-beta strand secondary structure into higher-ordered structures such as single-hole submicron cages and vesicles is an additional aspect that is expected to find applications in drug delivery.Data availability
All synthetic procedures, MD simulations, and NMR data supporting this article are in the ESI.†Author contributions
This manuscript was written through the contributions of all authors. HS designed, synthesized, characterized, and studied the conformation and self-assembly of all the compounds under the supervision of VH. AC and JB performed the simulations under the supervision of GG. Dynamic NMR studies were performed by UG.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Science and Engineering Research Board SERB, New Delhi, Government of India (grant number EMR/2017/003192/OC). We thank the IITD HPC facility for providing the computational resources. AC thanks Prime Minister's Research Fellowship scheme (PMRF) for a fellowship. HS and UG thank the Council of Scientific and Industrial Research (CSIR), New Delhi for fellowships. JB thanks Tata Consultancy Services (TCS) for a fellowship. We thank Prof. Narayanan D Kurur for stimulating discussions. We thank Prof. Venugopalan Paloth, Punjab University, for help in the X-ray structure analysis.Notes and references
- M. H. Barbee, Z. M. Wright, B. P. Allen, H. F. Taylor, E. F. Patteson and A. S. Knight, Macromolecules, 2021, 54, 3585–3612 CrossRef CAS.
- W. S. Horne and T. N. Grossmann, Nat. Chem., 2020, 12, 331–337 CrossRef CAS PubMed.
- T. A. Martinek, A. Hetényi, L. Fülöp, I. M. Mándity, G. K. Tóth, I. Dékány and F. Fülöp, Angew. Chem., Int. Ed., 2006, 45, 2396–2400 CrossRef CAS PubMed.
- J. Eom, J. Gong, S. Kwon, A. Jeon, R. Jeong, R. W. Driver and H. Lee, Angew. Chem., Int. Ed., 2015, 54, 13204–13207 CrossRef CAS PubMed.
- L.-G. Milroy, T. N. Grossmann, S. Hennig, L. Brunsveld and C. Ottmann, Chem. Rev., 2014, 114, 4695–4748 CrossRef CAS PubMed.
- J. M. Davis, L. K. Tsou and A. D. Hamilton, Chem. Soc. Rev., 2007, 36, 326–334 RSC.
- V. Haridas, Eur. J. Org. Chem., 2009, 2009, 5112–5128 CrossRef.
- L. Cussol, L. Mauran-Ambrosino, J. Buratto, A. Y. Belorusova, M. Neuville, J. Osz, S. Fribourg, J. Fremaux, C. Dolain, S. R. Goudreau, N. Rochel and G. Guichard, Angew. Chem., Int. Ed., 2021, 60, 2296–2303 CrossRef CAS PubMed.
- H. N. Hoang, C. Wu, T. A. Hill, A. Dantas de Araujo, P. V. Bernhardt, L. Liu and D. P. Fairlie, Angew. Chem., Int. Ed., 2019, 58, 18873–18877 CrossRef CAS PubMed.
- V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Nat. Chem., 2013, 5, 161–173 CrossRef CAS PubMed.
- A. M. Watkins and P. S. Arora, ACS Chem. Biol., 2014, 9, 1747–1754 CrossRef CAS PubMed.
- R. Kaul, A. R. Angeles, M. Jäger, E. T. Powers and J. W. Kelly, J. Am. Chem. Soc., 2001, 123, 5206–5212 CrossRef CAS PubMed.
- A. G. Kreutzer, S. Yoo, R. K. Spencer and J. S. Nowick, J. Am. Chem. Soc., 2017, 139, 966–975 CrossRef CAS PubMed.
- H. Lingard, J. T. Han, A. L. Thompson, I. K. H. Leung, R. T. W. Scott, S. Thompson and A. D. Hamilton, Angew. Chem., Int. Ed., 2014, 53, 3650–3653 CrossRef CAS PubMed.
- F. Freire and S. H. Gellman, J. Am. Chem. Soc., 2009, 131, 7970–7972 CrossRef CAS PubMed.
- S. T. Phillips, M. Rezac, U. Abel, M. Kossenjans and P. A. Bartlett, J. Am. Chem. Soc., 2002, 124, 58–66 CrossRef CAS PubMed.
- P. N. Wyrembak and A. D. Hamilton, J. Am. Chem. Soc., 2009, 131, 4566–4567 CrossRef CAS PubMed.
- A. B. Smith, T. P. Keenan, R. C. Holcomb, P. A. Sprengeler, M. C. Guzman, J. L. Wood, P. J. Carroll and R. Hirschmann, J. Am. Chem. Soc., 1992, 114, 10672–10674 CrossRef CAS.
- A. M. C. Marcelino and L. M. Gierasch, Biopolymers, 2008, 89, 380–391 CrossRef CAS PubMed.
- V. Haridas, S. Sadanandan, Y. K. Sharma, S. Chinthalapalli and A. Shandilya, Tetrahedron Lett., 2012, 53, 623–626 Search PubMed.
- V. Haridas, S. Sadanandan, M. V. S. Gopalakrishna, M. B. Bijesh, R. P. Verma, S. Chinthalapalli and A. Shandilya, Chem. Commun., 2013, 49, 10980 RSC.
- K. Wüthrich, C. Spitzfaden, K. Memmert, H. Widmer and G. Wider, FEBS Lett., 1991, 285, 237–247 CrossRef.
- N. H. Andersen, J. W. Neidigh, S. M. Harris, G. M. Lee, Z. Liu and H. Tong, J. Am. Chem. Soc., 1997, 119, 8547–8561 CrossRef CAS.
- S. Levinger, Y. Sharabi-Ronen, A. Mainfeld and A. Albeck, J. Org. Chem., 2008, 73, 7793–7796 CrossRef CAS PubMed.
- V. A. Palyulin, S. V. Emets, V. A. Chertkov, C. Kasper and H.-J. Schneider, Eur. J. Org. Chem., 1999, 1999, 3479–3482 CrossRef.
- A. D. Bain, G. J. Duns, F. Rathgeb and J. Vanderkloet, J. Phys. Chem., 1995, 99, 17338–17343 CrossRef CAS.
- D. Kern, M. Schutkowski and T. Drakenberg, J. Am. Chem. Soc., 1997, 119, 8403–8408 CrossRef CAS.
- A. H. Andreotti, Biochemistry, 2003, 42, 9515–9524 CrossRef CAS PubMed.
- J. S. Nowick, E. M. Smith and G. Noronha, J. Org. Chem., 1995, 60, 7386–7387 Search PubMed.
- J. S. Nowick, D. L. Holmes, G. Mackin, G. Noronha, A. J. Shaka and E. M. Smith, J. Am. Chem. Soc., 1996, 118, 2764–2765 CrossRef CAS.
- T. Cierpicki and J. Otlewski, J. Biomol. NMR, 2001, 21, 249–261 CrossRef CAS PubMed.
- N. J. Baxter and M. P. Williamson, J. Biomol. NMR, 1997, 9, 359–369 CrossRef CAS PubMed.
- Y. Zhang, Y. Zhong, A. L. Connor, D. P. Miller, R. Cao, J. Shen, B. Song, E. S. Baker, Q. Tang, S. V. S. R. K. Pulavarti, R. Liu, Q. Wang, Z. Lu, T. Szyperski, H. Zeng, X. Li, R. D. Smith, E. Zurek, J. Zhu and B. Gong, J. Am. Chem. Soc., 2019, 141, 14239–14248 CrossRef CAS PubMed.
- X. Daura, K. Gademann, B. Jaun, D. Seebach, W. F. van Gunsteren and A. E. Mark, Angew. Chem., Int. Ed., 1999, 38, 236–240 CrossRef CAS.
- A. Dong, P. Huang and W. S. Caughey, Biochemistry, 1990, 29, 3303–3308 CrossRef CAS PubMed.
- A. M. Woys, A. M. Almeida, L. Wang, C.-C. Chiu, M. McGovern, J. J. de Pablo, J. L. Skinner, S. H. Gellman and M. T. Zanni, J. Am. Chem. Soc., 2012, 134, 19118–19128 CrossRef CAS PubMed.
- S. Pal, in Fundamentals of Molecular Structural Biology, Elsevier, 2020, pp. 119–147 Search PubMed.
- S. Chowdhury, G. Schatte and H.-B. Kraatz, Angew. Chem., Int. Ed., 2008, 47, 7056–7059 CrossRef CAS PubMed.
- D. E. Clarke, E. T. Pashuck, S. Bertazzo, J. V. M. Weaver and M. M. Stevens, J. Am. Chem. Soc., 2017, 139, 7250–7255 CrossRef CAS PubMed.
- L. Zhai, Y. Otani, Y. Hori, T. Tomita and T. Ohwada, Chem. Commun., 2020, 56, 1573–1576 RSC.
- K. H. Mayo, E. Ilyina and H. Park, Protein Sci., 1996, 5, 1301–1315 CrossRef CAS PubMed.
- E. Marcos, T. M. Chidyausiku, A. C. McShan, T. Evangelidis, S. Nerli, L. Carter, L. G. Nivón, A. Davis, G. Oberdorfer, K. Tripsianes, N. G. Sgourakis and D. Baker, Nat. Struct. Mol. Biol., 2018, 25, 1028–1034 CrossRef CAS PubMed.
- N. Sawyer, A. M. Watkins and P. S. Arora, Acc. Chem. Res., 2017, 50, 1313–1322 CrossRef CAS PubMed.
- J. Hennetin, B. Jullian, A. C. Steven and A. V. Kajava, J. Mol. Biol., 2006, 358, 1094–1105 CrossRef CAS PubMed.
- Z. S. Derewenda, L. Lee and U. Derewenda, J. Mol. Biol., 1995, 252, 248–262 CrossRef CAS PubMed.
- R. W. Newberry and R. T. Raines, Nat. Chem. Biol., 2016, 12, 1084–1088 CrossRef CAS PubMed.
- F. Weinhold and C. R. Landis, Valency and Bonding: A Natural Bond Orbital Donor-acceptor Perspective, Cambridge University, Cambridge, 2005 Search PubMed.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1, Gaussian, Pittsburg, PA, CT, 2003 Search PubMed.
- D. Voet and J. G. Voet, Biochemistry, J. Wiley & Sons, New York, 2nd ed., 1995 Search PubMed.
- C.-I. Brändén and J. Tooze, Introduction to protein structure, Garland Publishing, New York, 2nd ed., 1999 Search PubMed.
- X. Li, L. Zhou, Y. Wei, A. M. El-Toni, F. Zhang and D. Zhao, J. Am. Chem. Soc., 2015, 137, 5903–5906 CrossRef CAS PubMed.
- Y.-M. Jeon, J. Kim, D. Whang and K. Kim, J. Am. Chem. Soc., 1996, 118, 9790–9791 CrossRef CAS.
- C. O. Mellet, J. M. G. Fernández and J. M. Benito, Chem. Soc. Rev., 2011, 40, 1586–1608 RSC.
- D. T. Bong, T. D. Clark, J. R. Granja and M. R. Ghadiri, Self-Assembling Organic Nanotubes, Angew. Chem., Int. Ed. Engl., 2001, 40, 988–1011 CrossRef CAS PubMed.
- V. Haridas, Acc. Chem. Res., 2021, 54, 1934–1949 CrossRef CAS PubMed.
- A. Bhattacharya, R. J. Brea and N. K. Devaraj, Chem. Sci., 2017, 8, 7912–7922 RSC.
- W. M. Aumiller, M. Uchida and T. Douglas, Chem. Soc. Rev., 2018, 47, 3433–3469 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details on the synthesis and MD simulation. CCDC 2099286. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04860k |
| This journal is © The Royal Society of Chemistry 2021 |