
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA single-atom Cu–N2 catalyst eliminates oxygen interference for electrochemical sensing of hydrogen peroxide in a living animal brain†
Xiaolong
Gao‡
ac,
Wenjie
Ma‡
ac,
Junjie
Mao‡
d,
Chun-Ting
He
 e,
Wenliang
Ji
a,
Zheng
Chen
d,
Wenxing
Chen
f,
Wenjie
Wu
a,
Ping
Yu
ac and
Lanqun
Mao
*abc
e,
Wenliang
Ji
a,
Zheng
Chen
d,
Wenxing
Chen
f,
Wenjie
Wu
a,
Ping
Yu
ac and
Lanqun
Mao
*abc
aBeijing National Laboratory for Molecular Sciences, Key Laboratory of Analytical Chemistry for Living Biosystems, Institute of Chemistry, The Chinese Academy of Sciences (CAS), Beijing, 100190, China. E-mail: lqmao@bnu.edu.cn
bCollege of Chemistry, Beijing Normal University, Xinjiekouwai Street 19, Beijing, 100875, China
cUniversity of Chinese Academy of Sciences, Beijing, 100049, China
dKey Laboratory of Functional Molecular Solids, Ministry of Education, College of Chemistry and Materials Science, Anhui Normal University, Wuhu, 241002, China
eMOE Key Laboratory of Functional Small Organic Molecule, College of Chemistry and Chemical Engineering, Jiangxi Normal University, Nanchang, 330022, China
fBeijing Key Laboratory of Construction Tailorable Advanced Functional Materials and Green Applications, School of Materials Science and Engineering, Beijing Institute of Technology, Beijing, 100081, China
First published on 2nd November 2021
Abstract
Hydrogen peroxide (H2O2) plays essential roles in various physiological and pathological processes. The electrochemical hydrogen peroxide reduction reaction (HPRR) has been recognized as an efficient approach to H2O2 sensing; however, the HPRR has always suffered from low tolerance against the oxygen reduction reaction (ORR), resulting in poor selectivity of the HPRR-based sensing platform. In this study, we find that the electrochemical HPRR occurs preferentially compared to the ORR when isolated Cu atoms anchored on carbon nitride (Cu1/C3N4) are used as a single-atom electrocatalyst, which is theoretically attributed to the lower energy barrier of the HPRR than that of the ORR on a Cu1/C3N4 single-atom catalyst (SAC). With the Cu1/C3N4 SAC as the electrocatalyst, we fabricated microsensors that have a good response to H2O2, but not to O2 or other electroactive neurochemicals. When implanted into a living rat brain, the microsensor shows excellent in vivo sensing performance, enabling its application in real-time quantitative investigation of the dynamics of H2O2 production induced by mercaptosuccinate and glutathione monoethyl ester in a living animal brain.
Introduction
In physiological and pathological processes, H2O2 plays important roles, for example, as a biochemical mediator and signaling pathway modulator.1–7 In aerobic organisms, O2 is the sole source for H2O2 production which is accomplished by one-electron reduction of O2 to form superoxide (˙O2−) followed by the disproportionation of ˙O2− mediated by superoxide dismutase (SOD).6–8 As a representative of reactive oxygen species (ROS), the level of H2O2 can be regulated by antioxidants such as glutathione peroxidase, catalase, thioredoxins and thiols to maintain the cellular redox homeostasis.9,10 In addition, H2O2 with a low physiological level can serve as an intracellular signaling molecule to regulate metabolism, enabling cellular adaption to the changes of stress and the environment.11,12 In contrast, overproduction of H2O2 can break the redox balance and thus cause oxidative stress, which is closely related with a variety of pathological events such as aging and progressive neurodegenerative disorders.13–15 Moreover, H2O2 is produced in enzymatic reactions with O2 as an electron receptor, which can thus serve as an indicator of the enzymatic activity and the level of related substrates.16,17 Therefore, the information on the kinetics of H2O2 would largely advance these studies.18–20Electrochemical oxidation or reduction of H2O2 has been used to build a platform for H2O2 sensing;21 however, the oxidation of H2O2 often bears interference from some reductive biomolecules,22 and the reduction process always suffers from interference from O2 with almost all electrocatalysts.23–26 Although the HPRR is more favorable than the ORR thermodynamically (eqn (1) and (2)), it often proceeds more slowly than the ORR under practical conditions due to the sluggish kinetics of the HPRR and low mass transport efficiency of H2O2,23,24 resulting in the occurrence of the HPRR at more negative potentials than the ORR, which makes it difficult to selectively sense H2O2 free from the interference from O2 when potential-controlled amperometry is used. Therefore, the design and fabrication of high-performance electrocatalysts where the HPRR occurs prior to the ORR are very essential for selective H2O2 sensing in vivo.
| H2O2 + 2H+ + 2e− → 2H2O E0′ = 1.151 V vs. Ag/AgCl at pH 7.0 | (1) |
| O2 + 4H+ + 4e− → 2H2O E0′ = 0.617 V vs. Ag/AgCl at pH 7.0 | (2) |
Single-atom catalysts (SACs)27–32 with almost 100% atom utilization efficiency and uniform catalytic centers have recently received considerable attention in industrial chemical processing33–35 and electro/photochemical energy conversion36–39 such as the ORR,40–43 hydrogen evolution reaction (HER),44–46 oxygen evolution reaction (OER),47–49 and carbon dioxide reduction reaction (CO2RR).50–54 SACs exhibit unique selectivity because of the homogeneous and well-defined structure compared with nanoparticle-based catalysts.55–57 In addition, the structure of SACs can be flexibly regulated by changing the active atom species and/or the support and by tuning the coordination environment to achieve a high catalytic performance.58–61 Such unique characteristics and the inner-sphere electron-transfer features of the HPRR and ORR suggest that the use of SACs may offer priority to one of these two reactions because transition metal single atoms with different coordination environments embedded in a well-defined carbon support have shown high capacity in tuning the binding strength with O species,62–64 providing a great opportunity for the design and fabrication of selective electrocatalysts for prioritizing the HPRR over the ORR and thus enabling amperometric sensing of H2O2 free from interference from O2.
In this work, we report the design and synthesis of a SAC with atomically dispersed Cu sites anchored on mesoporous graphitic carbon nitride (Cu1/C3N4) to realize selective electrochemical HPRR against the ORR. The Cu1/C3N4 SAC synthesized by an impregnation method with a mesoporous structure exhibits a high electrocatalytic performance toward the HPRR with an onset potential at about 0.20 V (vs. Ag/AgCl) in neutral media. In contrast, the ORR commences at a more negative potential at around 0.0 V (vs. Ag/AgCl), demonstrating that the occurrence of the electrochemical HPRR prior to the ORR can be successfully achieved through rational design and fabrication of SACs. Density functional theory calculation (DFT) reveals that the priority given to the HPRR over the ORR on the Cu1/C3N4 SAC is due to the synergetic effect of the Cu–N2 site and its adjacent carbon site on the adsorption state of the *OH intermediate, resulting in a lower energy barrier of the HPRR on the SAC. This finding not only offers a new electrocatalyst for promoting the HPRR over the ORR, but also provides a novel avenue for selective in vivo monitoring of molecular events in a living rat brain with SACs as the electrocatalysts.
Results and discussion
To synthesize SACs, C3N4 with a mesoporous structure was prepared by the thermally induced self-condensation of cyanamide with colloidal silica as a hard template and was used as the support, enabling a large surface area with accessible active sites.65 In order to find an electrocatalyst with selectivity for the HPRR over the ORR, a series of transition metal SACs (denoted as M1/C3N4, M = Fe, Co, Cu, Mn) were prepared by calcining C3N4 absorbed with metal ions (Fig. 1a). Fig. 1b and S1† show the transmission electron microscopy (TEM) images of the as-synthesized samples, retaining the mesoporous structure with ca. 10 nm pores of C3N4 (Fig. S2†), which is beneficial to the mass transport of H2O2. No clusters or nanoparticles were observed in the TEM images, showing that metal atoms exist as single atoms dispersed on the support in all the prepared catalysts. X-ray photoelectron spectra (XPS) suggest a similar structure of the four samples and the existence of M–Nx species positioned at around 398.9 eV (Fig. S3 and S4†).66 Among the synthetic SACs, Cu1/C3N4 possesses striking electrocatalytic performance toward H2O2 reduction in terms of onset potential and current intensity (Fig. S5†), demonstrating the vital role of the single-atom metal centers in tuning the reaction activity. Fourier transform infrared (FT-IR) spectra of the as-prepared C3N4 and Cu1/C3N4 demonstrate the formation of extended CN heterocycles (adsorption bands between 1200 cm−1 and 1650 cm−1) and the introduction of Cu atoms does not change the support structure obviously (Fig. S6†).67 Besides, the corresponding energy-dispersive X-ray spectroscopy (EDS) demonstrates the existence of Cu, N and C elements in Cu1/C3N4 (Fig. S7†) and the corresponding mapping confirms that Cu, N, and C elements are uniformly distributed over the entire C3N4 skeleton (Fig. 1c). The N2 adsorption/desorption isotherms and pore size distribution plots of C3N4 and Cu1/C3N4 show a mesoporous architecture with a large BET surface area (Fig. S8†), which means that more active sites are available to reactant molecules (Table S1†). X-ray powder diffraction (XRD) patterns indicate that the crystal structure of C3N4 is maintained after the introduction of Cu (Fig. S9†). In addition, Cu1/C3N4 exhibits no characteristic peaks related with Cu, demonstrating that no Cu clusters or nanoparticles exist in the sample. To further verify the isolated single-atom form of Cu, high-resolution TEM and aberration corrected high-angle annular dark-field scanning TEM (AC HAADF-STEM) were performed (Fig. 1d and e). The bright dots with atomic size distributed on the C3N4 support were recognized to be single Cu atoms. Different areas were examined carefully to confirm the absence of Cu clusters or nanoparticles (Fig. S10†), revealing that only individual Cu single atoms are present in Cu1/C3N4. | ||
| Fig. 1 Synthesis and characterization of the Cu1/C3N4 SAC. (a) Illustration of the synthetic route to M1/C3N4. (b) TEM image of Cu1/C3N4. (c) HAADF-STEM and corresponding elemental mapping images of Cu1/C3N4 showing the uniform distribution of Cu (yellow), N (green) and C (red). (d and e) High-resolution TEM image (d) and AC HAADF STEM image (e) of Cu1/C3N4 (single Cu atoms are marked with red circles). | ||
X-ray absorption spectroscopy (XAS) measurements at the Cu K-edge were performed to explore the electronic structure and coordination configuration of Cu species in Cu1/C3N4. The X-ray absorption near-edge structure (XANES) spectrum of the as-prepared Cu1/C3N4 is illustrated in Fig. 2a, with Cu foil and CuO as references. The adsorption-edge of Cu1/C3N4 is located between those of Cu foil and CuO, showing that the valence of the Cu atom is between 0 and +2, which originates from the strong interaction between Cu and C3N4. Furthermore, the high-resolution Cu 2p XPS spectrum of Cu1/C3N4 (Fig. S11†) reveals the existence of only one kind of Cu species. The main peaks located at 932.2 eV and 952.1 eV are characteristic peaks of Cu+, consistent with the XANES analysis.66 No Cu0 signals were observed from the Cu 2p spectrum, indicating that no Cu particles or clusters exist. Fig. 2b shows the Fourier transformed (FT) extended X-ray absorption fine structure (EXAFS) spectra of Cu1/C3N4 and the references. Only one prominent peak at 1.5 Å similar to that of CuPc was observed for Cu1/C3N4, which was ascribed to the first coordination shell of Cu–N. No Cu–Cu coordination peak at 2.2 Å like for Cu foil was observed, suggesting that Cu species are atomically dispersed in Cu1/C3N4. In order to further explore the atomic distribution of Cu in Cu1/C3N4, wavelet transform (WT) analysis of Cu K-edge EXAFS was performed. As shown in Fig. 2c, only one intensity maximum at 3.3 Å−1 related to Cu–N coordination was observed from the WT plots of Cu1/C3N4, which is different from the WT plot of Cu foil with one intensity maximum corresponding to the Cu–Cu contribution at 6.8 Å−1, showing that no Cu–Cu bond is present in Cu1/C3N4, but isolated Cu atoms exist. EXAFS fitting was carried out to obtain the atomic structure parameters of Cu in Cu1/C3N4. According to the fitting curves (Fig. 2d and e, S12 and S13†) and the fitting parameters in Table S2,† the coordination number of Cu in Cu1/C3N4 was calculated to be 2.2 and the mean bond length of Cu–N was 1.97 Å, suggesting that one Cu atom is coordinated by two N atoms to form a Cu–N2 structure. The atomic structure model of Cu1/C3N4 was constructed and is illustrated as the inset in Fig. 2e.
 | ||
| Fig. 2 XAS analysis of Cu1/C3N4. (a and b) Normalized XANES spectra (a) and FT-EXAFS spectra (b) at the Cu K-edge of Cu1/C3N4 (red line), Cu foil (black line), CuO (blue line) and CuPc (purple line). (c) Wavelet transforms of the Cu K-edge EXAFS spectra for Cu foil, CuO, CuPc and Cu1/C3N4. (d and e) Corresponding fitting curves of the EXAFS spectra of Cu1/C3N4 in k space (d) and R space (e). The inset in (e) is the schematic structure of Cu1/C3N4. | ||
The electrocatalytic performance of C3N4 and Cu1/C3N4 towards the HPRR in artificial cerebrospinal fluid (aCSF) was investigated by cyclic voltammetry (CV). In order to improve the electrical conductivity of Cu1/C3N4 used for the HPRR, carbon nanotubes (CNTs) were incorporated with Cu1/C3N4 because of the semiconducting properties of C3N4. Electrochemical impedance spectra (EIS) (Fig. S14†) reveal a much lower charge transfer resistance after mixing Cu1/C3N4 with CNTs, which facilitates electron transfer to the active Cu sites. Fig. 3a and b show the CV curves of the Cu1/C3N4/CNT catalyst (CuSAC) towards the HPRR and ORR, respectively. Obviously, the CuSAC shows a high performance toward the HPRR with a high onset potential of ca. 0.20 V vs. Ag/AgCl, at which C3N4, CNTs and the C3N4/CNT composite exhibit no obvious current change (Fig. 3a and S15–S17†), implying that the single Cu atoms on the C3N4 substrate serve as active sites for the HPRR. In contrast, the ORR starts more negatively at ca. 0.0 V vs. Ag/AgCl (Fig. 3b). The activities of the catalyst toward the HPRR and ORR were further evaluated by linear sweep voltammetry (LSV) with comparable concentrations of H2O2 and O2 (Fig. 3c). Similarly, the HPRR commences at ca. 0.20 V vs. Ag/AgCl (red curve), which is obviously more positive than the onset potential of the ORR (0.0 V vs. Ag/AgCl) (blue curve), again revealing that the HPRR occurs prior to and is thus discriminated from the ORR at the CuSAC-based electrode.
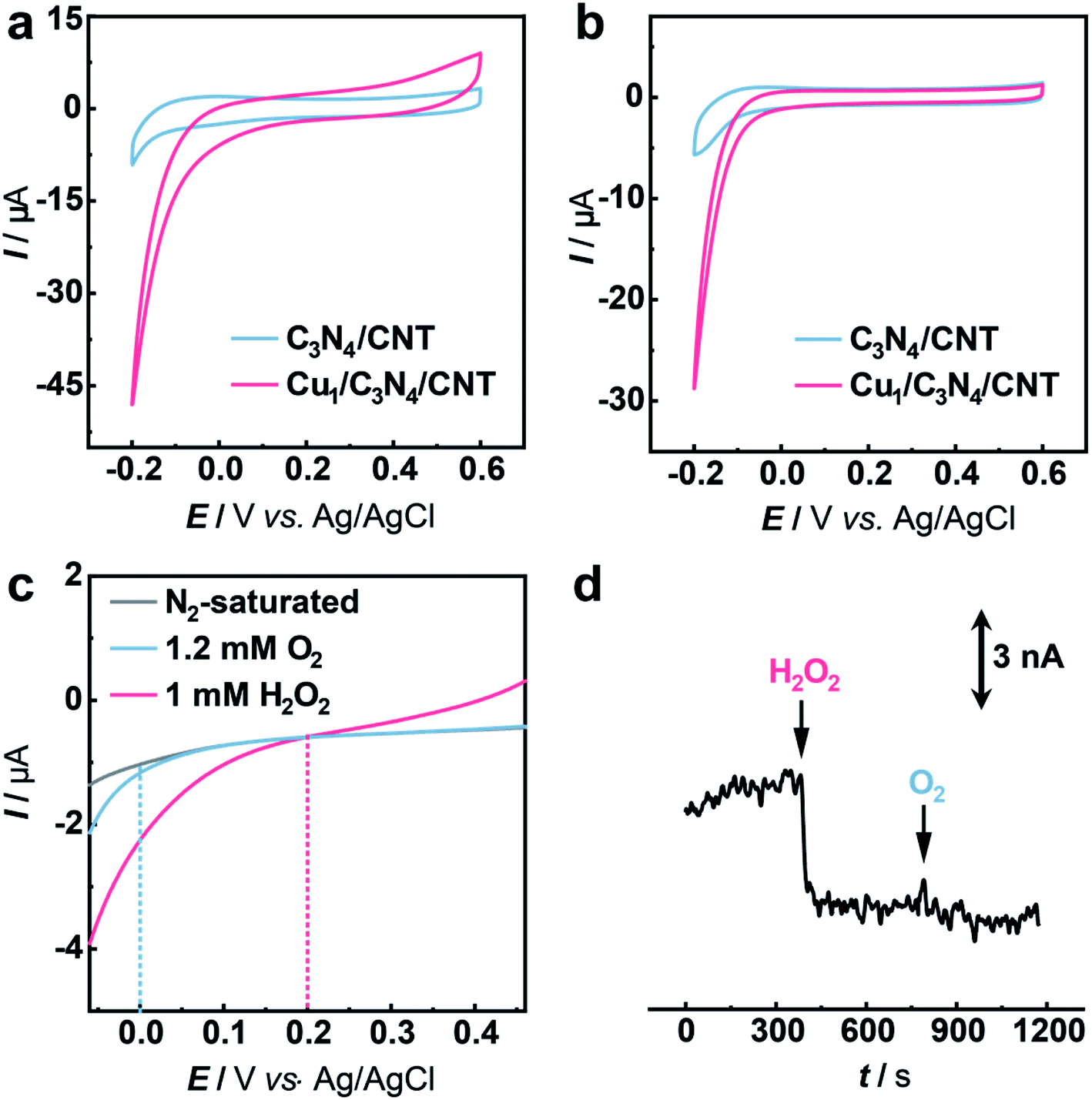 | ||
| Fig. 3 Electrochemical study of the HPRR over the ORR. (a and b) CVs obtained with glassy carbon (GC) electrodes modified with C3N4/CNT (blue curve) and Cu1/C3N4/CNT (red curve) in aCSF (pH 7.4) in the presence of 5 mM H2O2 (a) or under an O2 atmosphere (ca. 1.2 mM O2) (b). Scan rate: 50 mV s−1. (c) LSVs obtained with the CuSAC-based electrode in aCSF without (gray curve) and with 1.2 mM O2 (blue curve) or 1 mM H2O2 (red curve) in aCSF under a N2 atmosphere. Scan rate: 50 mV s−1. (d) Amperometric response recorded at the SAC-based electrode with the addition of 5 μM H2O2 and 50 μM O2 in aCSF. Applied potential: 0.0 V vs. Ag/AgCl. | ||
To further demonstrate the CuSAC-enabled priority of the HPRR over the ORR, potential-controlled amperometry was conducted with the CuSAC-based electrode. As displayed in Fig. 3d, the reduction current was observed after adding 5 μM H2O2 into the solution, while almost no current change was recorded with the addition of 50 μM O2, indicating that H2O2 is preferentially reduced compared with O2. This finding is remarkable because, in almost all electrochemical systems, H2O2 reduction normally occurs at about the same or an even more negative potential than that of O2 reduction, in spite of the more positive formal potential of the HPRR than that of the ORR in neutral pH. The use of SACs as the electrocatalyst successfully accelerates the HPRR and enables its occurrence prior to the ORR. Furthermore, after the HPRR test, the used Cu1/C3N4 SAC retained the mesoporous structure (Fig. S18a†) with Cu, N, and C elements uniformly dispersed on the substrate (Fig. S18b†), without obvious clusters or nanoparticles observed in the high-resolution TEM images (Fig. S18c and S18d†), showing that the Cu species were still atomically dispersed. Meanwhile, the Cu 2p XPS spectrum of the used Cu1/C3N4 also exhibits characteristic peaks of Cu+ at 932.5 eV and 952.3 eV (Fig. S19†), verifying the negligible valence changes of Cu atoms. All the results demonstrate a high stability of Cu1/C3N4.
To gain insights into the priority of the HPRR over the ORR, we carried out DFT calculations to investigate the reaction mechanism on the Cu1/C3N4 SAC. Fig. 4a and b show the calculated free energy diagrams of the two reactions on the Cu1/C3N4 SAC, and the atomic configurations of the intermediates for the two reactions are illustrated in Fig. S20 and S21.† For H2O2 reduction, the H2O2* dissociates into two OH* spontaneously without electron transfer, which subsequently adsorb on the Cu atom and its adjacent C atom, respectively. The last OH* desorption step on the C atom was regarded as the rate-determining step (RDS) owing to the endothermic nature with a free energy of 1.31 eV (Fig. 4a). For O2 reduction, only OH* desorption on the Cu atom is thermodynamically uphill with a free energy of 1.44 eV (Fig. 4b), which was determined to be the RDS. Due to the different adsorption sites of OH*, the last OH* desorption (i.e., RDS) of the two reactions gives an energy difference of 0.13 eV, indicating that the HPRR occurs prior to the ORR. Therefore, the Cu–N2 site and the surrounding C atom play important roles in intermediate adsorption and charge transfer for the HPRR. In contrast, only the Cu–N2 site acts as an active center participating in the ORR. These results suggest that the synergism between the Cu–N2 site and its adjacent C atom on C3N4 leads to a different pathway for the HPRR, enabling priority of the HPRR over the ORR on the Cu1/C3N4 SAC.
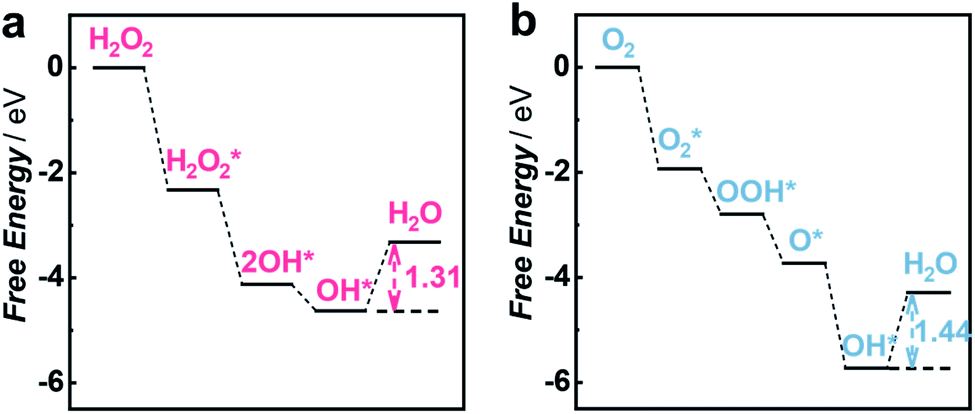 | ||
| Fig. 4 DFT calculations for the HPRR and ORR on the Cu1/C3N4 SAC. (a and b) Free energy diagrams of the HPRR (a) and ORR (b) at pH 7.0 on the active site of the Cu1/C3N4 SAC. | ||
Having demonstrated the excellent electrocatalytic performance of the Cu1/C3N4 SAC in the HPRR, especially over the ORR, we next applied this catalyst to develop a microsensor for in vivo selective sensing of H2O2 in a living rat brain. To do this, a carbon fiber electrode (CFE) with a diameter of 7 μm was prepared and used for fabrication of the microsensor.68,69 The microsensor exhibits high electrocatalytic performance towards H2O2 reduction (Fig. S22 and S23†). To evaluate the priority of the HPRR over the ORR, potential-controlled amperometry was performed with the microsensor toward H2O2 and O2. As shown in Fig. 5a, there is negligible current change upon the addition of 50 μM O2, while an obvious current decrease was observed for 5 μM H2O2, suggesting that H2O2 reduction occurs at a potential more positive than that of O2 reduction. In addition, the microsensor responds well to H2O2 with a good linearity between current response and H2O2 concentration (I (nA) = −5.454–0.155 C (μM), R2 = 0.992) (Fig. 5b and S24†). Considering that there are other redox-active species coexisting in the cerebral system, interference from these neurochemicals was studied. Compared with H2O2, the species studied here give rise to negligible current responses (Fig. 5c and S25†), indicating high selectivity of the microsensor for the detection of H2O2. Moreover, the microsensor shows a high stability toward H2O2 detection over 5 h (Fig. 5d), which is beneficial for in vivo monitoring H2O2 in living animals.
 | ||
| Fig. 5 Selective sensing of H2O2 with the CuSAC-based microsensor. (a) Amperometric responses recorded with the microsensor in aCSF toward the addition of 50 μM O2 and 5 μM H2O2. Applied potential: 0.0 V vs. Ag/AgCl. (b) Calibration curve for amperometric H2O2 detection obtained at the microsensor in aCSF toward successive addition of 5 μM H2O2. Applied potential: 0.0 V vs. Ag/AgCl. (c) Amperometric responses recorded with the microsensor in aCSF toward 10 μM DA, 10 μM DOPAC, 10 μM 5-HT, 10 μM NE, 10 μM UA, 50 μM O2 or 5 μM H2O2. Applied potential: 0.0 V vs. Ag/AgCl. (d) Long-time amperometric i–t curve recorded with the microsensor in aCSF toward 5 μM H2O2. Applied potential: 0.0 V vs. Ag/AgCl. | ||
To study the possibility of using the microsensor for in vivo sensing of H2O2 in a rat brain, the microsensor was implanted into the cortex of a guinea pig and the current response to the locally microinfused H2O2 was recorded (Fig. 6a). As illustrated in Fig. 6b, local infusion of 100 μM H2O2 solution into the rat cortex results in a fast amperometric response that can recover quickly after stopping the infusion, while no current response was observed with infusion of pure aCSF. These results show that the sensing platform with an SAC as the catalyst can be used for selective monitoring of H2O2 fluctuation in vivo.
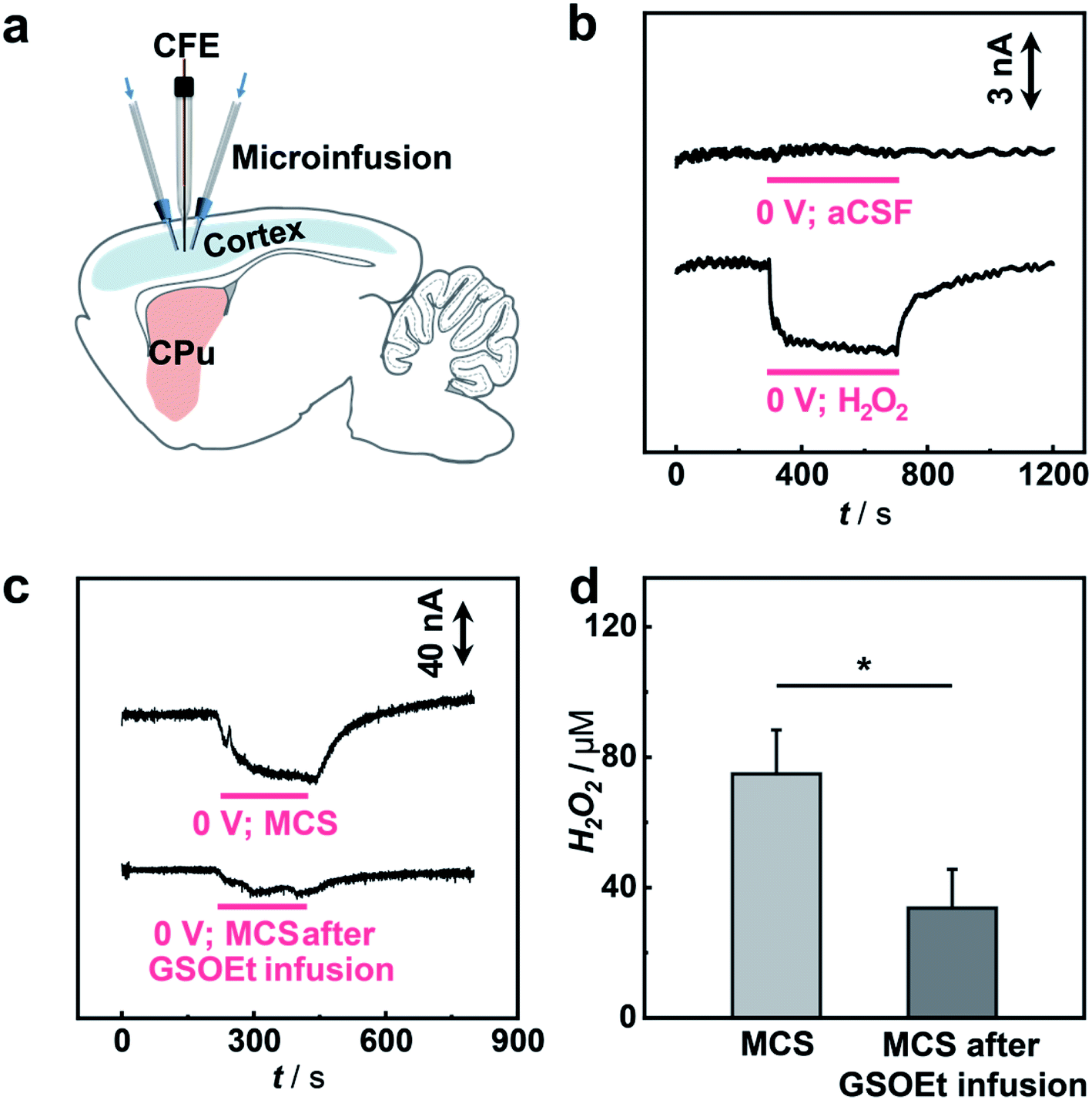 | ||
| Fig. 6 In vivo sensing of H2O2 with the SAC-based microsensor. (a) Scheme of the sensing platform for in vivo monitoring of H2O2. (b) Typical current responses at the microsensor implanted in the cortex of anesthetized guinea pigs upon local infusion (4 min, indicated by a red line) of aCSF without (top) and with (bottom) 100 μM H2O2 at 0.0 V vs. Ag/AgCl. (c) Typical current responses upon the local injection of 10 mM MCS (4 min, 1.0 μL min−1 indicated by a red line) without (top) and with (bottom) the pre-infusion of 10 mM GSOEt (15 min, 1.0 μL min−1). (d) Histogram of the H2O2 change calculated from (c). Error bars indicate the standard error of mean of 3 independent tests. Paired Student's t-test, *, p <0.05. | ||
Having shown the validity of our microsensor for in vivo sensing, we then performed quantitative analysis of H2O2-related biochemical processes induced by mercaptosuccinate (MCS) and glutathione monoethyl ester (GSOEt). As one kind of glutathione peroxidase inhibitor, MCS can increase the levels of H2O2 in the brain, while GSOEt can suppress H2O2 production with the generation of antioxidant glutathione.70,71 However, these biochemical processes still necessitate a quantitative study at a living animal level. To in vivo study the dynamics of H2O2 fluctuation in these biochemical processes induced by the two drugs in a living rat brain, we locally perfused MCS and GSOEt into the rat cortex and real-time tracked the level of H2O2 with the SAC-based microsensor developed here. As typically shown in Fig. 6c and d, the local microinjection of MCS into the cortex led to an increase in the current response of the microsensor (n = 3), corresponding to the increase of the H2O2 level in the brain. Quantitatively, microinjection of 10 mM MCS (4 μL) gives rise to an increase of ca. 75.0 ± 13.5 μM H2O2. In the other experiments, GSOEt was locally infused 30 min before MCS infusion and the current change induced by MCS was recorded. We found that pre-infusion of GSOEt to some extent suppressed the increase of current for H2O2, indicating that the H2O2 production induced by MCS was inhibited. The pre-infusion of 10 mM GSOEt (15 μL) results in the decrease of H2O2 production induced by MCS to only ca. 33.7 ± 11.9 μM (Fig. 6c and d). These quantitative description of H2O2 dynamics in a living rat brain provides a direct basis for the study of biochemical processes associated with H2O2 chemistry.
Conclusions
In summary, we have discovered a single-atom Cu catalyst (Cu1/C3N4) that gives priority to the electrochemical HPRR over the ORR and successfully established an O2-interference-free electrochemical sensing platform for selective detection of H2O2in vivo. The isolated Cu–N2 site and surrounding C atom from the C3N4 support are responsible for the preferable HPRR over the ORR through synergistic adsorption of the intermediates during the reaction, enabling highly selective H2O2 detection. In addition, the microsensor with the Cu1/C3N4 SAC responds quickly to H2O2 fluctuation in vivo and is successfully used for the quantitative study of H2O2-related biochemical processes. This study not only provides a new solution to the long-standing challenge in the electrochemical HPRR prior to the ORR, but also provides a novel sensing platform for understanding neurochemical processes by real-time monitoring of molecular events involved in physiological and pathological processes.Data availability
All experimental and computational data is available in the ESI.†Author contributions
L. M., J. M. and W. M. came up with the idea. X. G. conducted the sample preparation, physical characterization and electrochemical measurements. C. H. helped with the theoretical calculation and discussion. W. J. helped to conduct the animal experiment and discussion. W. C. helped with the analysis of XAS measurements. Z. C. and W. W. contributed to the synthesis experiment. P. Y. contributed to the discussion of the electrocatalysis part. X. G. and W. M. wrote the initial draft of this manuscript, and L. M. revised and finalized the manuscript. All the authors participated in the discussion of the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All in vivo experimental procedures complied with the guidelines of the Animal Advisory Committee at the State Key Laboratory of Cognitive Neuroscience and Learning, and were approved by the Institutional Animal Care and Use Committee at Beijing Normal University. This study was supported by the National Key Research and Development Project (Grant No. 2018YFE0200800), National Natural Science Foundation of China (Grant No. 21790390, 21790391 and 22134002 for L. M., 21971002 for J. M., 21705155 and 21790392 for W. M.), and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB30000000). We acknowledge the photoemission end stations at beamline BL14W1 in the Shanghai Synchrotron Radiation Facility for helpful characterization.Notes and references
- B. Morgan, K. V. Laer, T. N. E. Owusu, D. Ezeriņa, D. Pastor-Flores, P. S. Amponsah, A. Tursch and T. P. Dick, Nat. Chem. Biol., 2016, 12, 437–443 CrossRef CAS PubMed.
- G. S. Shadel and T. L. Horvath, Cell, 2015, 163, 560–569 CrossRef CAS PubMed.
- M. C. Sobotta, W. Liou, S. Stöcker, D. Talwar, M. Oehler, T. Ruppert, A. N. D. Scharf and T. P. Dick, Nat. Chem. Biol., 2015, 11, 64–70 CrossRef CAS PubMed.
- D. Peralta, A. K. Bronowska, B. Morgan, É. Dóka, K. V. Laer, P. Nagy, F. Gräter and T. P. Dick, Nat. Chem. Biol., 2015, 11, 156–163 CrossRef CAS.
- J. Noh, B. Kwon, E. Han, M. Park, W. Yang, W. Cho, W. Yoo, G. Khang and D. Lee, Nat. Commun., 2015, 6, 6907–6915 CrossRef CAS PubMed.
- S. E. Weinberg and N. S. Chandel, Nat. Chem. Biol., 2015, 11, 9–15 CrossRef CAS PubMed.
- B. C. Dickinson and C. J. Chang, Nat. Chem. Biol., 2011, 7, 504–511 CrossRef CAS PubMed.
- A. J. Shuhendler, K. Pu, L. Cui, J. P. Uetrecht and J. Rao, Nat. Biotechnol., 2014, 32, 373–380 CrossRef CAS PubMed.
- Y. Cheng, J. Dai, C. Sun, R. Liu, T. Zhai, X. Lou and F. Xia, Angew. Chem., Int. Ed., 2018, 57, 3123–3127 CrossRef CAS.
- R. Wang, C. Yan, H. Zhang, Z. Guo and W.-H. Zhu, Chem. Sci., 2020, 11, 3371–3377 RSC.
- E. W. Miller, O. Tulyathan, E. Y. Isacoff and C. J. Chang, Nat. Chem. Biol., 2007, 3, 263–267 CrossRef CAS PubMed.
- S. G. Rhee, Science, 2006, 312, 1882–1883 CrossRef.
- A. K. Nath, A. Ghatak, A. Dey and S. G. Dey, Chem. Sci., 2021, 12, 1924–1929 RSC.
- Z. Wang, X. Ai, Z. Zhang, Y. Wang, X. Wu, R. Haindl, E. K. L. Yeow, W. Drexler, M. Gao and B. Xing, Chem. Sci., 2020, 11, 803–811 RSC.
- D. Pramanik, C. Ghosh and S. G. Dey, J. Am. Chem. Soc., 2011, 133, 15545–15552 CrossRef CAS PubMed.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed.
- C. Martin, M. Trajkovic and M. W. Fraaije, Angew. Chem., Int. Ed., 2020, 59, 4869–4872 CrossRef CAS PubMed.
- S. Ye, N. Hananya, O. Green, H. Chen, A. Q. Zhao, J. Shen, D. Shabat and D. Yang, Angew. Chem., Int. Ed., 2020, 59, 14326–14330 CrossRef CAS PubMed.
- W. Li, M. Khan, L. Lin, Q. Zhang, S. Feng, Z. Wu and J. Lin, Angew. Chem., Int. Ed., 2020, 59, 9282–9287 CrossRef CAS PubMed.
- X.-W. Zhang, A. Oleinick, H. Jiang, Q.-L. Liao, Q.-F. Qiu, I. Svir, Y.-L. Liu, C. Amatore and W.-H. Huang, Angew. Chem., Int. Ed., 2019, 58, 7753–7756 CrossRef CAS PubMed.
- H. Zhu, A. Sigdel, S. Zhang, D. Su, Z. Xi, Q. Li and S. Sun, Angew. Chem., Int. Ed., 2014, 53, 12508–12512 CAS.
- X. Xu, S. Jiang, Z. Hu and S. Liu, ACS Nano, 2010, 4, 4292–4298 CrossRef CAS PubMed.
- Y. Qu, Z. Li, W. Chen, Y. Lin, T. Yuan, Z. Yang, C. Zhao, J. Wang, C. Zhao, X. Wang, F. Zhou, Z. Zhuang, Y. Wu and Y. Li, Nat. Catal., 2018, 1, 781–786 CrossRef CAS.
- C. H. Choi, W. S. Choi, O. Kasian, A. K. Mechler, M. T. Sougrati, S. Brüller, K. Strickland, Q. Jia, S. Mukerjee, K. J. J. Mayrhofer and F. Jaouen, Angew. Chem., Int. Ed., 2017, 56, 8809–8812 CrossRef CAS PubMed.
- R. Li, X. Liu, W. Qiu and M. Zhang, Anal. Chem., 2016, 88, 7769–7776 CrossRef CAS PubMed.
- Y. Lin, K. Liu, P. Yu, L. Xiang, X. Li and L. Mao, Anal. Chem., 2007, 79, 9577–9583 CrossRef CAS PubMed.
- Z. Li, S. Ji, Y. Liu, X. Cao, S. Tian, Y. Chen, Z. Niu and Y. Li, Chem. Rev., 2020, 120, 623–682 CrossRef CAS PubMed.
- L. Jiao, W. Xu, Y. Wu, H. Yan, W. Gu, D. Du, Y. Lin and C. Zhu, Chem. Soc. Rev., 2021, 50, 750–765 RSC.
- L. Li, X. Chang, X. Lin, Z.-J. Zhao and J. Gong, Chem. Soc. Rev., 2020, 49, 8156–8178 RSC.
- W. Ma, J. Mao, X. Yang, C. Pan, W. Chen, M. Wang, P. Yu, L. Mao and Y. Li, Chem. Commun., 2019, 55, 159–162 RSC.
- J. Zhang, C. Zheng, M. Zhang, Y. Qiu, Q. Xu, W.-C. Cheong, W. Chen, L. Zheng, L. Gu, Z. Hu, D. Wang and Y. Li, Nano Res., 2020, 13, 3082–3087 CrossRef.
- X. Liu, H. Pang, X. Liu, Q. Li, N. Zhang, L. Mao, M. Qiu, B. Hu, H. Yang and X. Wang, The Innovation, 2021, 2, 100076 CrossRef PubMed.
- R. Lang, X. Du, Y. Huang, X. Jiang, Q. Zhang, Y. Guo, K. Liu, B. Qiao, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 11986–12043 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Science, 2012, 335, 1209–1212 CrossRef CAS PubMed.
- Y. Xiong, J. Dong, Z.-Q. Huang, P. Xin, W. Chen, Y. Wang, Z. Li, Z. Jin, W. Xing, Z. Zhuang, J. Ye, X. Wei, R. Cao, L. Gu, S. Sun, L. Zhuang, X. Chen, H. Yang, C. Chen, Q. Peng, C.-R. Chang, D. Wang and Y. Li, Nat. Nanotechnol., 2020, 15, 390–397 CrossRef CAS PubMed.
- W. Zhu, L. Zhang, S. Liu, A. Li, X. Yuan, C. Hu, G. Zhang, W. Deng, K. Zang, J. Luo, Y. Zhu, M. Gu, Z.-J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2020, 59, 12664–12668 CrossRef CAS PubMed.
- D. Yang, H. Yu, T. He, S. Zuo, X. Liu, H. Yang, B. Ni, H. Li, L. Gu, D. Wang and X. Wang, Nat. Commun., 2019, 10, 3844 CrossRef PubMed.
- C. Fang and W. An, Nano Res., 2021, 14, 4211–4219 CrossRef CAS.
- L. Jiao, R. Zhang, G. Wan, W. Yang, X. Wan, H. Zhou, J. Shui, S.-H. Yu and H.-L. Jiang, Nat. Commun., 2020, 11, 2831 CrossRef CAS PubMed.
- H. Shang, Z. Jiang, D. Zhou, J. Pei, Y. Wang, J. Dong, X. Zheng, J. Zhang and W. Chen, Chem. Sci., 2020, 11, 5994–5999 RSC.
- W. Ma, F. Wu, P. Yu and L. Mao, Chem. Sci., 2021, 12, 7908–7917 RSC.
- W. Wu, Y. Liu, D. Liu, W. Chen, Z. Song, X. Wang, Y. Zheng, N. Lu, C. Wang, J. Mao and Y. Li, Nano Res., 2021, 14, 998–1003 CrossRef CAS.
- W. Fu, Y. Wang, W. Tian, H. Zhang, J. Li, S. Wang and Y. Wang, Angew. Chem., Int. Ed., 2020, 59, 23791–23799 CrossRef CAS PubMed.
- J. Mao, C.-T. He, J. Pei, W. Chen, D. He, Y. He, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 4958–4965 CrossRef PubMed.
- C. Zhou, J. Y. Zhao, P. F. Liu, J. Chen, S. Dai, H. G. Yang, P. Hu and H. Wang, Chem. Sci., 2021, 12, 10634–10642 RSC.
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang, H. Sun, P. An, W. Chen, Z. Guo, C. Lee, D. Chen, I. Shakir, M. Liu, T. Hu, Y. Li, A. I. Kirkland, X. Duan and Y. Huang, Nat. Catal., 2018, 1, 63–72 CrossRef CAS.
- P. Li, M. Wang, X. Duan, L. Zheng, X. Cheng, Y. Zhang, Y. Kuang, Y. Li, Q. Ma, Z. Feng, W. Liu and X. Sun, Nat. Commun., 2019, 10, 1711–1721 CrossRef PubMed.
- K. Kamiya, Chem. Sci., 2020, 11, 8339–8349 RSC.
- J. Gu, C.-S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS PubMed.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- J. Zhang, W. Cai, F. X. Hu, H. Yang and B. Liu, Chem. Sci., 2021, 12, 6800–6819 RSC.
- Z. Wang, C. Wang, Y. Hu, S. Yang, J. Yang, W. Chen, H. Zhou, F. Zhou, L. Wang, J. Du, Y. Li and Y. Wu, Nano Res., 2021, 14, 2790–2796 CrossRef CAS.
- Z. Li, R. Wu, L. Zhao, P. Li, X. Wei, J. Wang, J. S. Chen and T. Zhang, Nano Res., 2021, 14, 3795–3809 CrossRef CAS.
- M. Zhou, Y. Jiang, G. Wang, W. Wu, W. Chen, P. Yu, Y. Lin, J. Mao and L. Mao, Nat. Commun., 2020, 11, 3188–3196 CrossRef CAS.
- W. Wu, L. Huang, E. Wang and S. Dong, Chem. Sci., 2020, 11, 9741–9756 RSC.
- Z. Guo, Y. Xie, J. Xiao, Z.-J. Zhao, Y. Wang, Z. Xu, Y. Zhang, L. Yin, H. Cao and J. Gong, J. Am. Chem. Soc., 2019, 141, 12005–12010 CrossRef CAS PubMed.
- F. Luo, A. Roy, L. Silvioli, D. A. Cullen, A. Zitolo, M. T. Sougrati, I. C. Oguz, T. Mineva, D. Teschner, S. Wagner, J. Wen, F. Dionigi, U. I. Kramm, J. Rossmeisl, F. Jaouen and P. Strasser, Nat. Mater., 2020, 19, 1215–1223 CrossRef CAS PubMed.
- L. Jiao, W. Yang, G. Wan, R. Zhang, X. Zheng, H. Zhou, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2020, 59, 20589–20595 CrossRef CAS PubMed.
- X. Li, H. Rong, J. Zhang, D. Wang and Y. Li, Nano Res., 2020, 13, 1842–1855 CrossRef CAS.
- H. Jing, Z. Zhao, C. Zhang, W. Liu, D. Wu, C. Zhu, C. Hao, J. Zhang and Y. Shi, Nano Res., 2021, 14, 4025–4032 CrossRef CAS.
- F. Wu, C. Pan, C.-T. He, Y. Han, W. Ma, H. Wei, W. Ji, W. Chen, J. Mao, P. Yu, D. Wang, L. Mao and Y. Li, J. Am. Chem. Soc., 2020, 142, 16861–16867 CrossRef CAS PubMed.
- Y. Wang, G. Jia, X. Cui, X. Zhao, Q. Zhang, L. Gu, L. Zheng, L.-H. Li, Q. Wu, D. J. Singh, D. Matsumura, T. Tsuji, Y.-T. Cui, J. Zhao and W. Zheng, Chem, 2021, 7, 1–14 Search PubMed.
- X. Chen, L. Zhao, K. Wu, H. Yang, Q. Zhou, Y. Xu, Y. Zheng, Y. Shen, S. Liu and Y. Zhang, Chem. Sci., 2021, 12, 8865–8871 RSC.
- A. Savateev, N. V. Tarakina, V. Strauss, T. Hussain, K. Brummelhuis, J. M. S. Vadillo, Y. Markushyna, S. Mazzanti, A. P. Tyutyunnik, R. Walczak, M. Oschatz, D. M. Guldi, A. Karton and M. Antonietti, Angew. Chem., Int. Ed., 2020, 59, 15061–15068 CrossRef CAS PubMed.
- A. Bakandritsos, R. G. Kadam, P. Kumar, G. Zoppellaro, M. Medved', J. Tuček, T. Montini, O. Tomanec, P. Andrýsková, B. Drahoš, R. S. Varma, M. Otyepka, M. B. Gawande, P. Fornasiero and R. Zbořil, Adv. Mater., 2019, 31, 1900323 CrossRef PubMed.
- S. Tian, Z. Wang, W. Gong, W. Chen, Q. Feng, Q. Xu, C. Chen, C. Chen, Q. Peng, L. Gu, H. Zhao, P. Hu, D. Wang and Y. Li, J. Am. Chem. Soc., 2018, 140, 11161–11164 CrossRef CAS PubMed.
- H. Hou, Y. Jin, H. Wei, W. Ji, Y. Xue, J. Hu, M. Zhang, Y. Jiang and L. Mao, Angew. Chem., Int. Ed., 2020, 59, 18996–19000 CrossRef CAS PubMed.
- J. Jin, W. Ji, L. Li, G. Zhao, W. Wu, H. Wei, F. Ma, Y. Jiang and L. Mao, J. Am. Chem. Soc., 2020, 142, 19012–19016 CrossRef CAS PubMed.
- M. V. Avshalumov and M. E. Rice, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11729–11734 CrossRef CAS PubMed.
- V. P. Wellner, M. E. Anderson, R. N. Puri, G. L. Jensen and A. Meister, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 4732–4735 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc04755h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |