
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible light-mediated radical fluoromethylation via halogen atom transfer activation of fluoroiodomethane†
Patrick J.
Deneny
 ,
Roopender
Kumar
and
Matthew J.
Gaunt
*
,
Roopender
Kumar
and
Matthew J.
Gaunt
*
Yusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: mjg32@cam.ac.uk
First published on 8th September 2021
Abstract
Incorporation of the fluoromethyl group can profoundly influence the physicochemical properties of organic molecules, offering a promising strategy for the discovery of novel pharmaceutical agents. Direct fluoromethylation of unfunctionalized C(sp2) centres can be achieved using fluoromethyl radicals, but current methods for their generation usually rely on the activation of non-commercial or expensive radical precursors via inefficient single electron transfer pathways, which limits their synthetic application. Here we report the development of a fluoromethylation strategy based on the generation of fluoromethyl radicals from commercially available fluoroiodomethane via halogen atom transfer. This mode of activation is orchestrated by visible light and tris(trimethylsilyl)silane, which serves as both a hydrogen- and halogen atom transfer reagent to facilitate the formation of C(sp3)–CH2F bonds via a radical chain process. The utility of this metal- and photocatalyst-free transformation is demonstrated through the multicomponent synthesis of complex α-fluoromethyl amines and amino acid derivatives via radical addition to in situ-formed iminium ions, and the construction of β-fluoromethyl esters and amides from electron-deficient alkene acceptors. These complex fluoromethylated products, many of which are inaccessible via previously reported methods, may serve as useful building blocks or fragments in synthetic and medicinal chemistry both in academia and industry.
Introduction
The prevalence of fluoroalkyl groups in pharmaceutical and agrochemical agents reflects their profound influence over the physicochemical and pharmacological properties of small organic molecules.1,2 Fluorine incorporation enables fine-tuning of lipophilicity, solubility, basicity, membrane permeability and metabolic stability,3–5 which has led to the broad application of fluoroalkyl groups as versatile bioisosteres in lead optimization.6 Moreover, rapid screening of fluorinated, sp3-rich fragment libraries via19F NMR spectroscopy is an emerging strategy in fragment-based drug discovery.7–10 Novel synthetic methods for fluoroalkylation are, therefore, valuable tools for exploring new areas of fluorinated chemical space.11,12 In this context, diverse strategies to achieve trifluoromethylation13–21 and, increasingly, difluoromethylation22–28 have been established, while analogous monofluoromethylation29–31 processes remain under-developed. Nonetheless, the fluoromethyl (CH2F) group is recognized as a pharmaceutically important motif, as evidenced by its presence in a number of lead compounds and approved drugs (Fig. 1a).29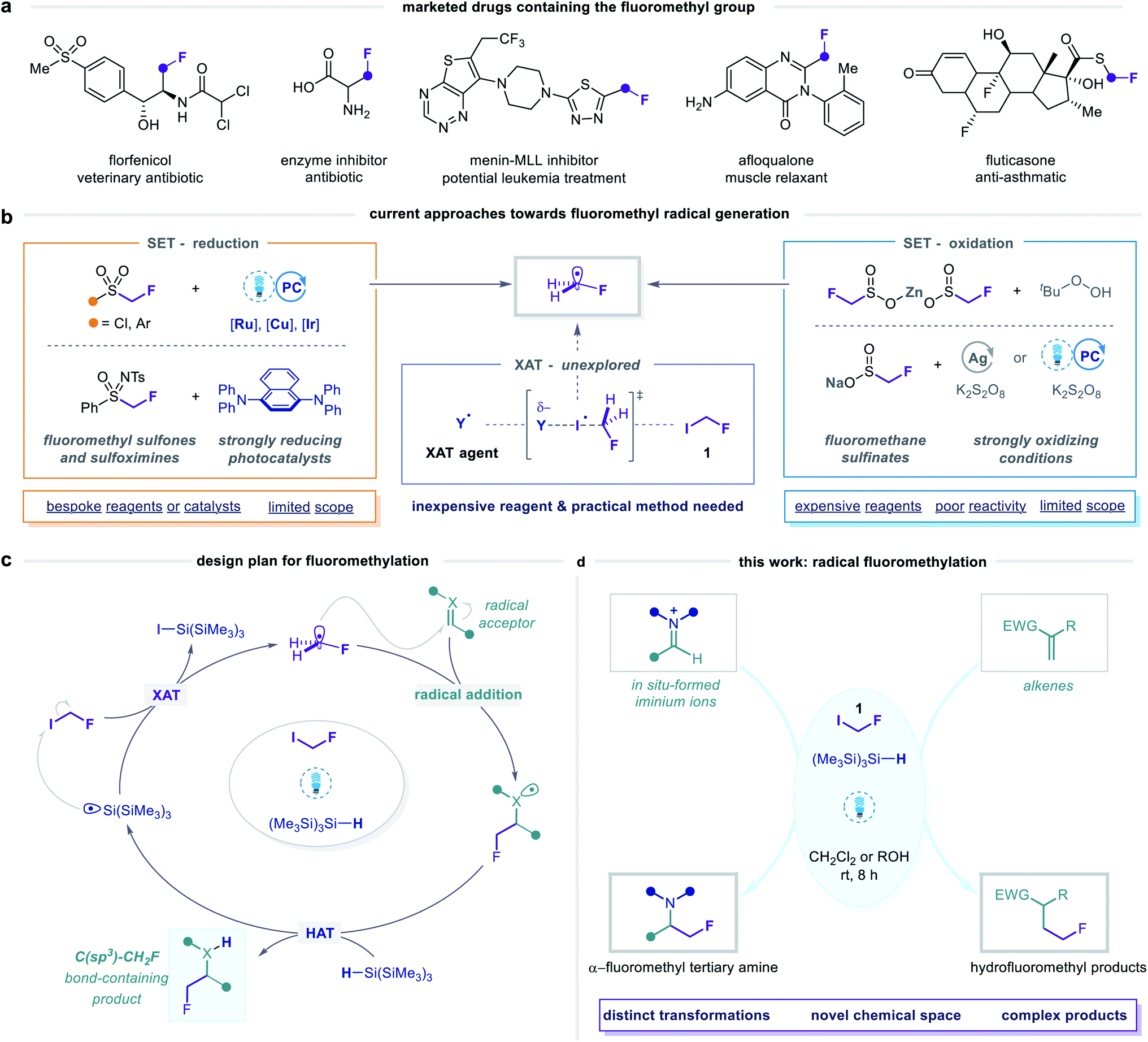 | ||
Fig. 1 Design plan for radical fluoromethylation via halogen atom transfer (XAT) activation of fluoroiodomethane. (a) The fluoromethyl motif is prevalent in pharmaceutical compounds. (b) Existing methods for radical fluoromethylation require expensive radical precursors, and strong oxidants or reductants, and are limited in scope. (c) Proposed XAT activation of fluoroiodomethane. (d) This work: application of XAT activation of fluoroiodomethane towards radical monofluoromethylation of unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X bonds. X bonds. | ||
The fluoromethyl unit is primarily used as a bioisosteric replacement for methyl and hydroxymethyl groups,6,32 whereby the high electronegativity of fluorine and the strength of the carbon–fluorine bond can improve oral bioavailability, binding affinity, and metabolic stability. Alternatively, the α-fluoromethyl amine group, present in a class of amino acid-derived enzyme inhibitors (so-called suicide inhibitors), serves as a source of a fluoride leaving group during their mechanism of action.33,34 Considering the diverse and beneficial properties conferred by the fluoromethyl motif, exploration of novel fluoromethylated molecules constitutes a promising strategy for the discovery of new biologically active agents. To this end, the development of general and operationally simple methods for selective fluoromethylation remains a significant challenge in organofluorine chemistry.29
In this context, various methods for nucleophilic, electrophilic, and metal-mediated fluoromethylation have been reported.29,35 Limitations of these reactions, however, include the use of auxiliary groups that require additional incorporation and removal steps, high loadings of metallic reagents, strong bases, and pre-functionalized substrates. Alternative and arguably more attractive strategies are centred on the generation of fluoromethyl radicals (Fig. 1b), which can directly engage in substitution,36 addition,37–39 or cascade cyclization40–43 reactions at unfunctionalized C(sp) and C(sp2) centres. A pervasive feature of these methods is the use of radical precursors in which the fluoromethyl group is bound to a sulfur atom, from which fluoromethyl radicals are generated via single-electron transfer (SET) pathways involving either reduction or oxidation at sulfur.44 While this approach is analogous to that of many successful radical di- and trifluoromethylation strategies, its wider application for monofluoromethylation is limited by the higher cost, lower availability, and, in particular, reduced reactivity of the corresponding fluoromethylated reagents. The single-electron reduction of fluoromethyl sulfonyl derivatives is challenging due to their highly negative reduction potentials [Ered as low as −2.43 V versus saturated calomel electrode (SCE)].37,39–41 Therefore, methods employing these reagents require strong reductants, elevated temperatures, or bespoke radical precursors, and exhibit limited scope. On the other hand, fluoromethanesulfinate salts undergo single-electron oxidation to generate fluoromethyl radicals,36,38,42,43 but display similarly limited reactivity, purportedly due to slow or poorly controlled radical generation from these species.38,45
Given the limitations of these redox-active reagents, we sought to establish a radical fluoromethylation strategy comprising (i) a readily available radical precursor, (ii) a distinct mode of activation to generate fluoromethyl radicals, and (iii) the construction of previously intractable C(sp3)–CH2F bond-containing products. For this purpose, we considered fluoroiodomethane 1, a commercially available and easily handled liquid (bp 53 °C) that serves as a versatile source of the fluoromethyl group in many two-electron processes.46 Despite these attractive features, the use of 1 as fluoromethyl radical precursor had been, until very recently, limited to a solitary report from 1975 in which gas-phase mixtures of 1 and simple olefins were photolyzed using ultraviolet (UV) light to form complex mixtures of substitution products in low conversions.47 The lack of synthetic application of 1 using single-electron (photo)redox chemistry reflects its highly negative reduction potential (Ered < −2 V versus SCE) which would necessitate the use of strongly reducing reagents or photocatalysts. To circumvent this inherent redox requirement of radical generation via SET, we proposed that homolytic cleavage of the carbon–iodine bond in 1 could be achieved via a halogen atom transfer (XAT) pathway (Fig. 1b and c). We hypothesised that visible light irradiation of 1 and a suitable XAT-agent precursor, namely tris(trimethylsilyl)silane [(Me3Si)3Si–H],48 would orchestrate the generation of fluoromethyl radicals and facilitate their polarity-matched addition to electron-deficient C(sp2) centres (Fig. 1c and d). A key feature of this proposed strategy is the multi-faceted role played by (Me3Si)3Si–H: following fluoromethyl radical addition to the acceptor, (Me3Si)3Si–H intercepts the intermediate radical in a hydrogen atom transfer (HAT) step that forms both the desired fluoromethylated product and a (Me3Si)3Si˙ radical; subsequent XAT between this silicon-centred radical and a molecule of 1 generates another fluoromethyl radical to establish a chain process. During the preparation of this manuscript, Gouverneur and co-workers reported a similar visible light-mediated fluoromethylation process, using 1, to affect the corresponding variant of the Giese reaction (Fig. 2).49 Here, we report the successful realization of our design concept through the complementary development and implementation of a visible light-mediated strategy for the radical fluoromethylation of iminium ions to form α-fluoromethyl alkylamines. The multicomponent carbonyl fluoromethylative amination reaction affords complex fluoromethylated amine products that occupy previously under-explored areas of chemical space, and which should serve as valuable fluorinated building blocks to practitioners of synthetic and medicinal chemistry. We also detail that our protocol enables addition of the fluoromethyl group to electron-deficient alkenes to form β-fluoromethyl esters and amides, which demonstrates the wider capability of the visible light-mediated activation mode for the fluoromethyl radical.
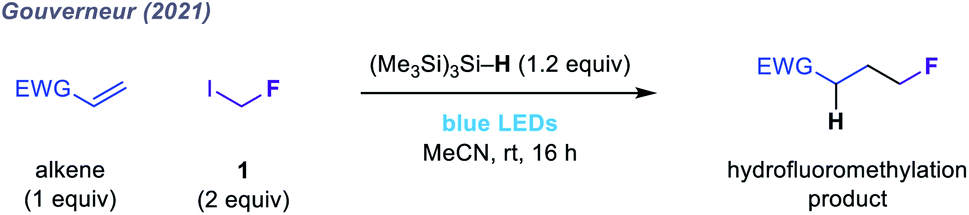 | ||
| Fig. 2 Hydrofluoromethylation of electron-deficient olefins using 1.49 | ||
Results and discussion
Our group recently reported a general method for the multicomponent synthesis of α-branched tertiary alkylamines, comprising the addition of alkyl radicals to iminium ions generated in situ via condensation of aldehydes and secondary amines.50 Further development of this carbonyl alkylative amination (CAA) strategy using fluoromethyl radicals would enable the modular construction of α-fluoromethyl tertiary alkylamines, a pharmaceutically relevant class of amines for which no direct, general synthetic strategies have been reported. Our initial investigations to address this synthetic need focused on the carbonyl fluoromethylative amination reaction between fluoroiodomethane 1, piperidine 2a and hydrocinnamaldehyde 3a (Table 1).|
|
||||
|---|---|---|---|---|
| Entry | Initiation method | HAT reagent | Additive | Yielda (%) |
| a Yields determined by 1H NMR analysis using 1,1,2,2-tetrachloroethane as internal standard. Isolated yield given in parentheses. | ||||
| 1 | 40 W blue LED | (Me3Si)3Si–H | — | <5 |
| 2 | 40 W blue LED | (Me3Si)3Si–H | TMS–Cl | 30 |
| 3 | 40 W blue LED | (Me3Si)3Si–H | TMS–OTf | 94 |
| 4 | 40 W blue LED | (Me3Si)3Si–H | TBS–OTf | 95 (86) |
| 5 | 50 °C, air atm | (Me3Si)3Si–H | TBS–OTf | 0 |
| 6 | 70 °C, AIBN | (Me3Si)3Si–H | TBS–OTf | 77 |
| 7 | 70 °C, AIBN | Bu3Sn–H | TBS–OTf | 0 |
| 8 | 40 W blue LED | (Et3Si)3Si–H | TBS–OTf | 52 |
| 9 | 40 W blue LED | Et3Si–H | TBS–OTf | 0 |
| 10 | 40 W blue LED | Ph3Si–H | TBS–OTf | 0 |
| 11 | 40 W blue LED | PhSiH3 | TBS–OTf | 0 |
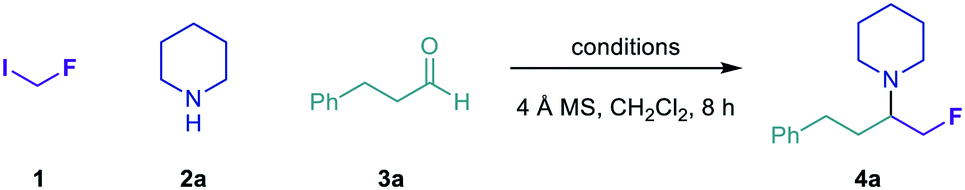
Visible light irradiation of 1 (3 equiv.), 2a (1 equiv.) and 3a (2 equiv.) in the presence of (Me3Si)3Si–H (2 equiv.) and 4 Å molecular sieves (MS) in CH2Cl2 for 8 h formed only trace amounts of the desired amine 4a, as determined by 1H NMR spectroscopy (entry 1). Evaluation of Lewis acid additives to promote formation of the iminium ion acceptor in high concentration revealed both tert-butyldimethylsilyl trifluoromethanesulfonate (TBS–OTf) and trimethylsilyl trifluoromethanesulfonate (TMS–OTf) to be more effective than trimethylsilyl chloride (TMS–Cl), affording 4a in near-quantitative assay yield (entries 2–4). We also assessed a number of alternative initiation methods and HAT reagents for the synthesis of 4a (entries 5–11).51 In all cases, these proved inferior to the combination of visible light and (Me3Si)3Si–H, although the reaction proceeded in good yield under thermal initiation using azobisisobutyronitrile (AIBN) and (Me3Si)3Si–H (entry 6). Ultimately, the conditions outlined in entry 4 were found to be optimal, affording 4a in 86% isolated yield.
Having identified suitable conditions for the carbonyl fluoromethylative amination reaction, we evaluated the scope of the transformation with respect to other cyclic secondary alkylamines (Fig. 3). A variety of functionalized cyclic and heterocyclic secondary alkylamines could be used in the reaction with aldehyde 3a, affording the α-fluoromethyl cyclic tertiary alkylamines 4a–4h in good yields. Notably, tertiary alkylamines of this type are not readily accessible via nucleophilic fluoromethylation strategies which require auxiliary-activated imines.52 Acyclic secondary alkylamines containing a variety of linear and branched alkyl substituents, as well as ester, nitrile, hydroxyl and heteroaromatic functionality, also reacted efficiently to give amines 4i–4s. In addition to these manipulable and medicinally important motifs, the N-benzyl group in amines 4k–4s provides a useful handle for their deprotection and further elaboration. For N-alkyl anilines and diarylamines, reductive amination was observed as a competing side reaction, although the desired α-fluoromethylated amine products 4t–4v could generally be obtained in synthetically useful yields.
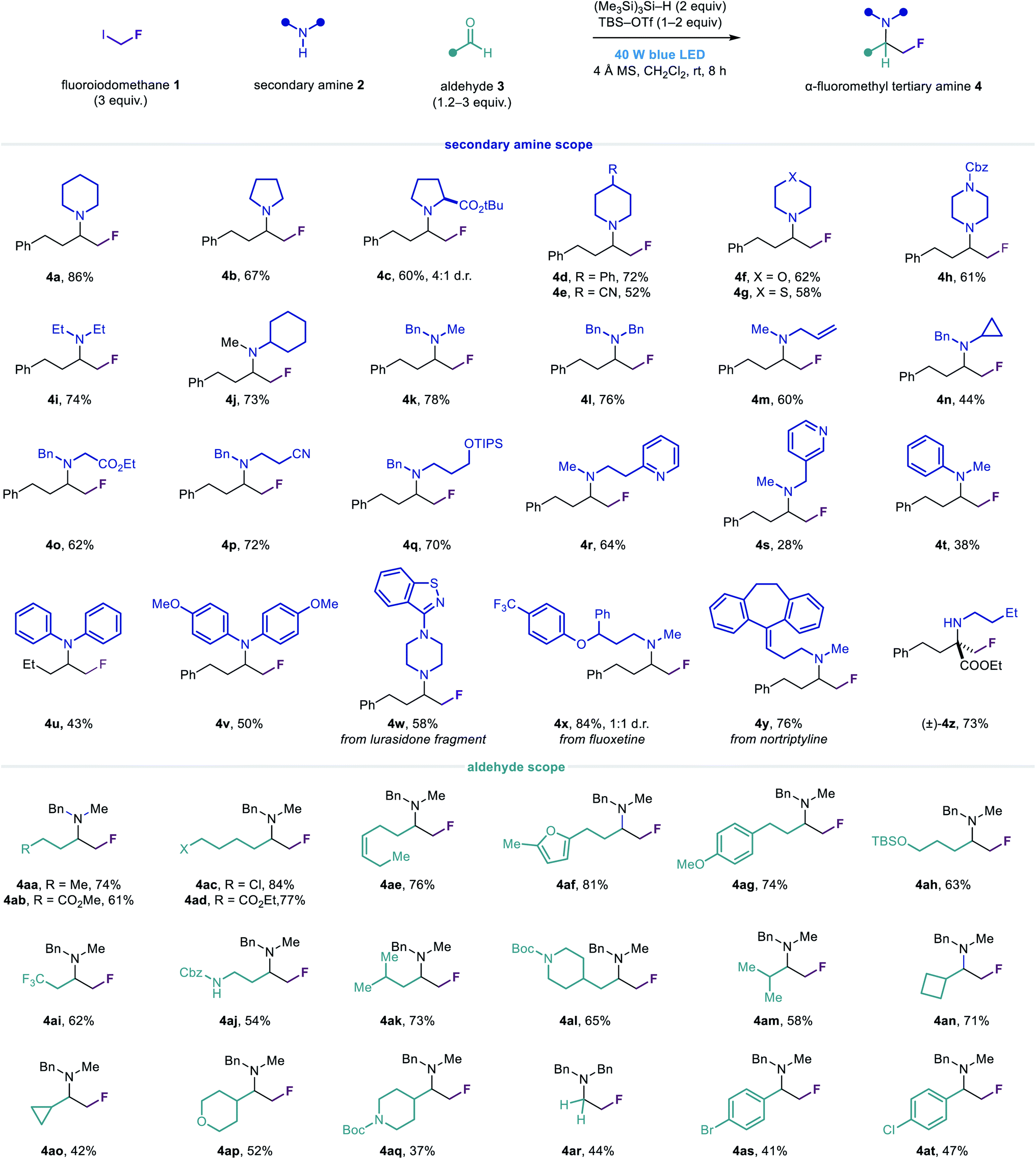 | ||
| Fig. 3 Scope of the carbonyl fluoromethylative amination reaction. | ||
The reaction was amenable to the functionalization of several secondary amine drugs and drug fragments, affording their α-fluoromethyl tertiary amine derivatives 4w–4y in moderate to excellent yields. Considering the significant and predictable effect that β-fluorine incorporation has on reducing the basicity of amines, this transformation may prove useful for the development of novel pharmaceutical agents and the modulation of their biological properties.
To further demonstrate the utility of our novel fluoromethylation strategy, we employed 1 with iminium ions derived from n-butylamine and ethyl 2-oxo-4-phenylbutanoate (an α-keto ester) to generate α-fluoromethyl α-tertiary amino acid derivative 4z in good yield.53 Compounds such as 4z are structural analogues of a class of biologically important α-fluoromethyl α-amino acid decarboxylase inhibitors (Fig. 1a), which are generally synthesized via linear, multi-step routes. Conversely, these α-fluoromethyl α-tertiary amino esters can easily be synthesized in one step from readily available precursors, exemplifying the rapid access to structural diversity that this modular approach can provide.
Next, we assessed the scope of the aldehyde component, using N-methylbenzylamine 2k and diversely functionalized linear and β-branched aldehydes to prepare the α-fluoromethyl tertiary alkylamines 4aa–4al. We found that the greater steric demand imposed by α-branched aldehydes generally led to slightly diminished yields of the corresponding amines 4am–4aq, although this could be mitigated by using additional equivalents of aldehyde in some cases. Notably, amine 2k exhibited sufficient reactivity in the absence of TBS–OTf to enable the synthesis of tertiary alkylamine products containing acid-sensitive functional groups such as the tert-butyloxycarbonyl (Boc) group (4al and 4aq). A reaction employing paraformaldehyde and dibenzylamine afforded unbranched tertiary fluoroethylamine 4ar in modest yield. Substituted benzaldehydes could also be tolerated, giving α-fluoromethyl tertiary amines 4as and 4at, albeit in low yields, which are suitably functionalized for downstream cross-coupling reactions.
In our efforts to develop a general fluoromethylation strategy, we sought to engage electrophilic species other than iminium ions in reactions with fluoromethyl radicals. We reasoned that our newly developed fluoromethylation strategy would be suitable for the direct hydrofluoromethylation of electron-deficient olefins via radical conjugate addition followed by rapid HAT from (Me3Si)3Si–H. Notably, although the addition of fluoromethyl radicals to acrylamides or acrylates is the elementary step in several previously reported fluoromethylation strategies (Fig. 1b), to the best of our knowledge, simple reduction of the resulting radical intermediates (via either HAT or SET/proton transfer) has not been reported; instead, these species are typically involved in subsequent migration, cyclization or atom transfer processes. Using slightly modified reaction conditions, a range of electron-deficient olefins were subjected to the novel hydrofluoromethylation reaction (Fig. 4). Simple acrylamides and acrylates, as well as maleimide 5g, reacted efficiently, affording the corresponding β-fluoromethyl carbonyl products 6a–6g. Phenyl vinyl sulfone was also readily functionalized, affording 6h in good yield. Vinyl phosphonate and vinyl ketone substrates were less well tolerated, affording 6i and 6j in modest assay yields. While less efficient, hydrofluoromethylation of phenyl propiolate and an unactivated terminal alkene (5l) to afford 6k and 6l demonstrated the hydrofluoromethylation of more diverse unsaturated acceptors.
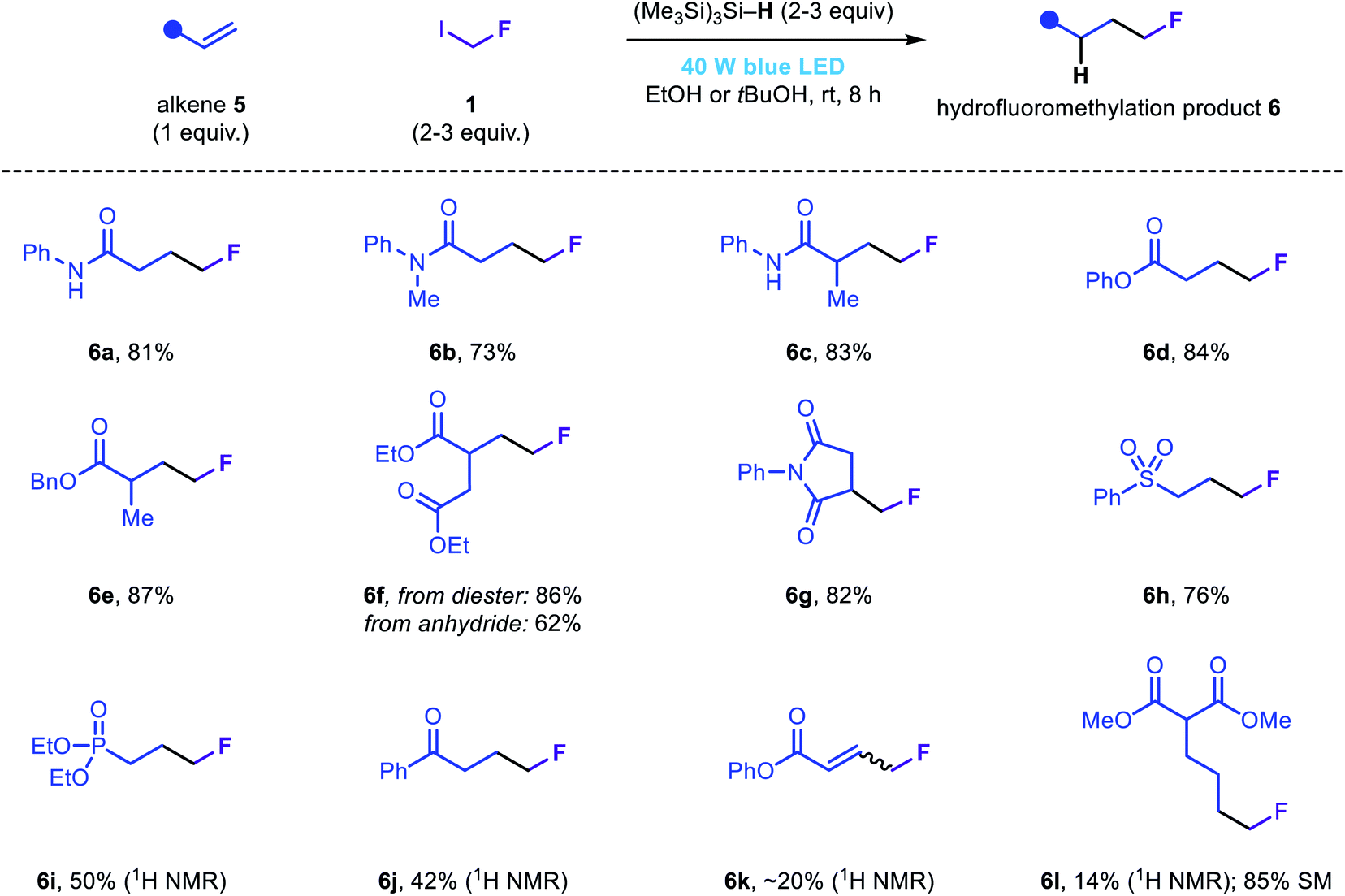 | ||
| Fig. 4 Scope of the hydrofluoromethylation of electron-deficient olefins. | ||
Conclusions
In summary, we have developed a novel visible light-mediated method for the monofluoromethylation of unsaturated carbon–carbon/heteroatom bonds (CX bond, X = carbon or nitrogen) by addition of fluoromethyl radicals. In contrast to previously reported methods, the fluoromethyl radical is generated via halogen atom abstraction from commercially available fluoroiodomethane by a stable silyl radical. This light-mediated process underpins a modular strategy in which commercially available amines, aldehydes and fluoroiodomethane are combined to access synthetically complex α-fluoromethylated tertiary amines. Critically, the reaction features a vast scope and excellent functional group tolerance, including highly elaborate drug scaffolds and other biologically relevant structures such as α-fluoromethyl α-tertiary amino esters. The synthetic utility of this new method is also demonstrated in a hydrofluoromethylation reaction of electron-deficient olefins to obtain C(sp3)–C(sp3) coupled fluoromethylated products. Given the intuitive retrosynthetic logic underpinning the carbonyl fluoromethylative amination and hydrofluoromethylation reactions, we hope they may be rapidly adopted in academic and industrial laboratories alike.
Data availability
Data associated with this article, including experimental procedures and compound characterization, are available in the ESI.†Author contributions
M. J. G., R. K. and P. J. D. conceived the project. P. J. D. and R. K. conducted the experiments. P. J. D., R. K. and M. J. G. analyzed the experiments. P. J. D., R. K. and M. J. G. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the EPSRC (P. J. D.) and the Swiss National Science Foundation (Grant P400P2_186730, (R. K.)) for funding and to the EPSRC (EP/S020292/1) and Royal Society for a Wolfson Merit Award (M. J. G.).Notes and references
- M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed.
- Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, iScience, 2020, 23, 101467 CrossRef CAS PubMed.
- B. M. Johnson, Y.-Z. Shu, X. Zhuo and N. A. Meanwell, J. Med. Chem., 2020, 63, 6315–6386 CrossRef CAS PubMed.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed.
- A. Vulpetti and C. Dalvit, ChemMedChem, 2013, 8, 2057–2069 CrossRef CAS PubMed.
- C. Dalvit and A. Vulpetti, J. Med. Chem., 2019, 62, 2218–2244 CrossRef CAS PubMed.
- A. Lingel, A. Vulpetti, T. Reinsperger, A. Proudfoot, R. Denay, A. Frommlet, C. Henry, U. Hommel, A. D. Gossert, B. Luy and A. O. Frank, Angew. Chem., Int. Ed., 2020, 59, 14809–14817 CrossRef CAS PubMed.
- N. S. Troelsen, E. Shanina, D. Gonzalez-Romero, D. Danková, I. S. A. Jensen, K. J. Śniady, F. Nami, H. Zhang, C. Rademacher, A. Cuenda, C. H. Gotfredsen and M. H. Clausen, Angew. Chem., Int. Ed., 2020, 59, 2204–2210 CrossRef CAS PubMed.
- T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
- C. Ni and J. Hu, Chem. Soc. Rev., 2016, 45, 5441–5454 RSC.
- X. Liu, C. Xu, M. Wang and Q. Liu, Chem. Rev., 2015, 115, 683–730 CrossRef CAS PubMed.
- R. P. Bhaskaran and B. P. Babu, Adv. Synth. Catal., 2020, 362, 5219–5237 CrossRef CAS.
- D. J. P. Kornfilt and D. W. C. MacMillan, J. Am. Chem. Soc., 2019, 141, 6853–6858 CrossRef CAS PubMed.
- J. A. Ma and D. Cahard, J. Fluorine Chem., 2007, 128, 975–996 CrossRef CAS.
- D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875–10877 CrossRef CAS PubMed.
- J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650–682 CrossRef CAS PubMed.
- C. Le, T. Q. Chen, T. Liang, P. Zhang and D. W. C. MacMillan, Science, 2018, 360, 1010–1014 CrossRef CAS PubMed.
- A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950–8958 CrossRef CAS PubMed.
- X. Pan, H. Xia and J. Wu, Org. Chem. Front., 2016, 3, 1163–1185 RSC.
- A. F. Garrido-Castro, A. Gini, M. C. Maestro and J. Alemán, Chem. Commun., 2020, 56, 3769–3772 RSC.
- J. Yang, S. Zhu, F. Wang, F. L. Qing and L. Chu, Angew. Chem., Int. Ed., 2021, 60, 4300–4306 CrossRef CAS PubMed.
- S. Q. Zhu, Y. L. Liu, H. Li, X. H. Xu and F. L. Qing, J. Am. Chem. Soc., 2018, 140, 11613–11617 CrossRef CAS PubMed.
- X.-J. J. Tang, Z. Zhang and W. R. Dolbier Jr, Chem.–Eur. J., 2015, 21, 18961–18965 CrossRef CAS PubMed.
- T. Koike and M. Akita, Chem, 2018, 4, 409–437 CAS.
- D. E. Yerien, S. Barata-Vallejo and A. Postigo, Chem.–Eur. J., 2017, 23, 14676–14701 CrossRef CAS PubMed.
- J. Rong, C. Ni and J. Hu, Asian J. Org. Chem., 2017, 6, 139–152 CrossRef CAS.
- M. Reichel and K. Karaghiosoff, Angew. Chem., Int. Ed., 2020, 59, 12268–12281 CrossRef CAS PubMed.
- T. Koike and M. Akita, Org. Biomol. Chem., 2019, 17, 5413–5419 RSC.
- J. Hu, W. Zhang and F. Wang, Chem. Commun., 2009, 7465–7478 RSC.
- K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- J. Kollonitsch, A. A. Patchett, S. Marburg, A. L. Maycock, L. M. Perkins, G. A. Doldouras, D. E. Duggan and S. D. Aster, Nature, 1978, 274, 906–908 CrossRef CAS PubMed.
- H. Mei, J. Han, K. D. Klika, K. Izawa, T. Sato, N. A. Meanwell and V. A. Soloshonok, Eur. J. Med. Chem., 2020, 186, 111826 CrossRef CAS PubMed.
- Q. Xie and J. Hu, in. Emerging Fluorinated Motifs, Wiley, 2020, pp. 119–133 Search PubMed.
- Y. Fujiwara, J. A. Dixon, F. O’hara, E. D. Funder, D. D. Dixon, R. A. Rodriguez, R. D. Baxter, B. Herlé, N. Sach, M. R. Collins, Y. Ishihara and P. S. Baran, Nature, 2012, 492, 95–99 CrossRef CAS PubMed.
- X.-J. Tang and W. R. Dolbier Jr, Angew. Chem., Int. Ed., 2015, 54, 4246–4249 CrossRef CAS PubMed.
- Z. He, P. Tan, C. Ni and J. Hu, Org. Lett., 2015, 17, 1838–1841 CrossRef CAS PubMed.
- N. Noto, T. Koike and M. Akita, ACS Catal., 2019, 9, 4382–4387 CrossRef CAS.
- X. J. Tang, C. S. Thomoson and W. R. Dolbier, Org. Lett., 2014, 16, 4594–4597 CrossRef CAS PubMed.
- J. Rong, L. Deng, P. Tan, C. Ni, Y. Gu and J. Hu, Angew. Chem., Int. Ed., 2016, 55, 2743–4747 CrossRef CAS PubMed.
- J. Fang, W. G. Shen, G. Z. Ao and F. Liu, Org. Chem. Front., 2017, 4, 2049–2053 RSC.
- W. Fu, Y. Sun and X. Li, Synth. Commun., 2020, 50, 388–398 CrossRef CAS.
- C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765–825 CrossRef CAS PubMed.
- A. G. O-Brien, A. Maruyama, Y. Inokuma, M. Fujita, P. S. Baran and D. G. Blackmond, Angew. Chem., Int. Ed., 2014, 53, 11868–11871 CrossRef CAS PubMed.
- S. Monticelli and V. Pace, Aust. J. Chem., 2018, 71, 473–475 CrossRef CAS.
- J. P. Sloan, J. M. Tedder and J. C. Walton, J. Chem. Soc., Perkin Trans. 2, 1975, 65, 1846–1850 RSC.
- C. Chatgilialoglu, C. Ferreri, Y. Landais and V. I. Timokhin, Chem. Rev., 2018, 118, 6516–6572 CrossRef CAS PubMed.
- S. M. Hell, C. F. Meyer, S. Ortalli, J. B. I. Sap, X. Chen and V. Gouverneur, Chem. Sci., 2021 10.1039/d1sc03421a.
- R. Kumar, N. J. Flodén, W. G. Whitehurst and M. J. Gaunt, Nature, 2020, 581, 415 CrossRef CAS PubMed.
- H. Jiang, J. R. Bak, F. J. López-Delgado and K. A. Jørgensen, Green Chem., 2013, 15, 3355–3359 RSC.
- G. Parisi, M. Colella, S. Monticelli, G. Romanazzi, W. Holzer, T. Langer, L. Degennaro, V. Pace and R. Luisi, J. Am. Chem. Soc., 2017, 139, 13648–13651 CrossRef CAS PubMed.
- J. Henry Blackwell, R. Kumar and M. J. Gaunt, J. Am. Chem. Soc., 2021, 143, 1598–1609 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc04554g |
| This journal is © The Royal Society of Chemistry 2021 |