
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemically selective double C(sp2)–X (X = S/Se, N) bond formation of isocyanides†
Zhipeng
Guan‡
a,
Shuxiang
Zhu‡
a,
Yankai
Yang
a,
Yanlong
Liu
a,
Siyuan
Wang
a,
Faxiang
Bu
a,
Hengjiang
Cong
 a,
Hesham
Alhumade
de,
Heng
Zhang
*a and
Aiwen
Lei
*abc
a,
Hesham
Alhumade
de,
Heng
Zhang
*a and
Aiwen
Lei
*abc
aInstitute for Advanced Studies (IAS), College of Chemistry and Molecular Sciences, Engineering Research Center of Organosilicon Compounds & Materials, Ministry of Education, Wuhan University, Wuhan, Hubei 430072, People's Republic of China. E-mail: aiwenlei@whu.edu.cn; hengzhang@whu.edu.cn
bNational Research Center for Carbohydrate Synthesis, Jiangxi Normal University, Nanchang 330022, Jiangxi, P. R. China
cKing Abdulaziz University, Jeddah, Saudi Arabia
dDepartment of Chemical and Materials Engineering, Faculty of Engineering, King Abdulaziz University, Jeddah, Saudi Arabia
eCenter of Research Excellence in Renewable Energy and Power Systems, King Abdulaziz University, Jeddah, Saudi Arabia
First published on 1st October 2021
Abstract
The construction of C(sp2)–X (X = B, N, O, Si, P, S, Se, etc.) bonds has drawn growing attention since heteroatomic compounds play a prominent role from biological to pharmaceutical sciences. The current study demonstrates the C(sp2)–S/Se and C(sp2)–N bond formation of one carbon of isocyanides with thiophenols or disulfides or diselenides and azazoles simultaneously. The reported findings could provide access to novel multiple isothioureas, especially hitherto rarely reported selenoureas. The protocol showed good atom-economy and step-economy with only hydrogen evolution and theoretical calculations accounted for the stereoselectivity of the products. Importantly, the electrochemical reaction could exclusively occur at the isocyano part regardless of the presence of susceptible radical acceptors, such as a broad range of arenes and alkynyl moieties, even alkenyl moieties.
The construction of C(sp2)–X (X = B, N, O, Si, P, S, Se, etc.) bonds has drawn increased attention from researchers since heteroatomic compounds play a prominent role in various fields.1 Traditionally, the transition-metal-catalyzed cross-coupling of a nucleophile and an electrophile is an important method for the formation of C(sp2)–X bonds.2 Obviously, the direct cross-coupling of C(sp2)–H/X–H is a time-, effort-, and resource-economical process to construct C(sp2)–X bonds as it avoids the pre-functionalization of substrates.3 Alternatively, radical chemistry provides greener and more mild strategies for the formation of C(sp2)–X bonds, and a variety of high added-value compounds can be afforded successfully.4 Despite the numerous advances, these reported reactions are limited as they involve only single C(sp2)–X bond formation. Access to double C(sp2)–X bonds formation of one carbon remains still a room for improvement stimultaneously.
As an ideal connector, isocyanides could be inserted into metal–carbon and metal–heteroatom bonds to construct double C(sp2)–X bonds.5 On the other hand, isocyanides could transform into heteroatomic molecules with electrophiles, nucleophiles and radicals.6 Although many elegant studies have been reported, the above methods are largely limited by the pre-functionalization of the substrate, the toxicity of metals or tedious synthesis steps. From the point of synthetic efficiency and economy, exploiting a novel, efficient, and diverse radical strategy to access double C(sp2)–X bonds under mild conditions and with broad functional group tolerance is of paramount importance and in urgent demand.
Radical transformation processes are ubiquitous throughout synthesis chemistry and provide new ideas for forging new bonds and novel molecules, especially valuable product motifs.7 As a complementary strategy to conventional radical-based reactions, electrochemistry, effectively transferring electrons from the electrode surface to the substance, has been developed into a powerful synthetic technique toward radical chemistry.8 This electrochemical strategy further stimulated a resurgence of interest in radical chemistry with good atom-economy and step-economy.9 Myriad electrochemical-induced radical reactions have been reported via single-electron oxidation/reduction with a plethora of radicals and radical acceptors. As reported, alkenes, alkynes, and cyano and aromatic compounds could be considered to be favorable radical acceptors in diverse transformations, such as cross-coupling, cyclization and difunctionalization reactions.10 However, the application of isocyanides as radical acceptors is limited in utility due to electrochemical tandem cyclization.11 Therefore, on the basis of our research on electrochemical oxidative functionalization of isocyanides,12 we continued to explore the properties of isocyanides as radical acceptors under electrochemical conditions.
Initially, we commenced our investigations by using abundant and inexpensive phenyl disulfide (1), ethyl 2-isocyanoacetate (2) and 1H-benzo[d][1,2,3]triazole (3) as model substrates in a single operation via a three-component cross-coupling. After systematic optimization, the isothiourea 21 could been obtained in 90% isolated yield. To gain insights into the reaction process, we continued to carry out a series of control experiments as shown in Scheme 1. The control experiment demonstrated that electricity is indispensable. Under standard conditions, addition of 2.0 equivalents of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) showed no traces of product. Similarly, the reaction was suppressed in the presence of butylated hydroxytoluene (BHT). The P(OEt)3-trapping product could be detected by gas chromatography mass spectrometry (GCMS). Simultaneously, a S-centred radical signal (g = 2.0070, AN = 13.40 G, AH = 14.88 G) was quickly trapped by 5,5-dimethyl-1-pyrroline N-oxide (DMPO) via electron paramagnetic resonance (EPR) experiments. The reported results revealed that this reaction might proceed via S-radical pathways.
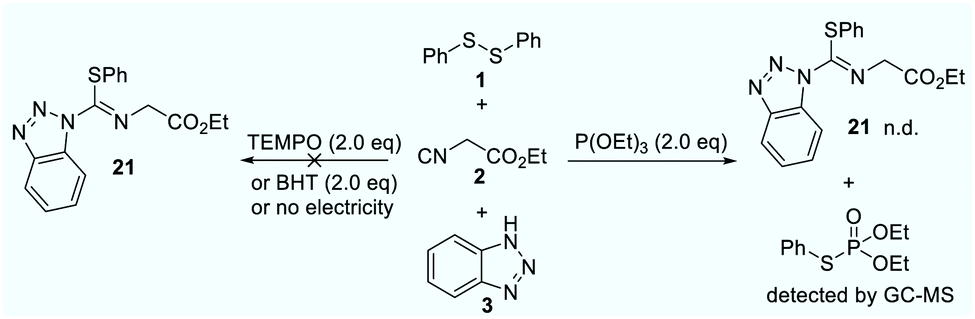 | ||
| Scheme 1 Control experiments. | ||
For deeper research of the priority of interaction for isocyanides vs. other radical acceptors with radicals, we sought to conduct robustness screening of a wide range of radical acceptors as shown in Scheme 2.13 The addition of an aryl olefin led to the complete shutdown of product formation (21) and the products of difunctionalization of alkenes were also not detected. To our delight, the alkenyl moiety did not affect the reaction efficiency under the electrochemical conditions. Unlike the aryl olefin, the addition of phenylacetylene did not inhibit product formation (21), delivering a yield of 93%. Similarly, ethyl 2-isocyanoacetate could be transformed into product (21) smoothly with 1-heptyne. The addition of mesitylene preserved the yield, while the yield decreased slightly with anisole. With the addition of furan and thiophene, an excellent yield of product (21) could still be obtained. Beside electron-donating arenes, we continued to add electron-withdrawing arenes to the reaction system. As expected, a satisfactory yield was obtained with 4-cyanopyridine. The results revealed that the electrochemical reaction exclusively occurred at the isocyano part regardless of the presence of electron-withdrawing arenes, electron-donating arenes and the alkynyl moiety, even the alkenyl moiety, which are also susceptible to radical conditions.
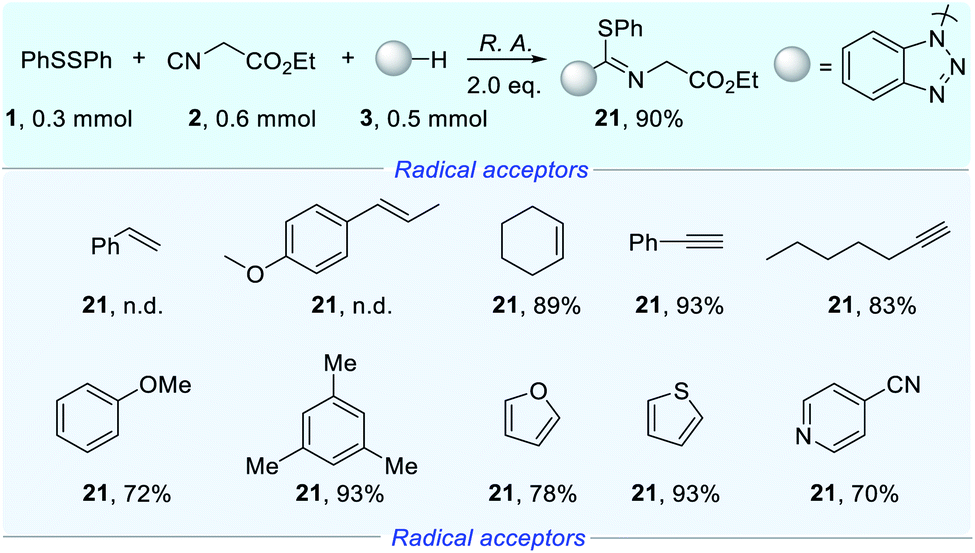 | ||
| Scheme 2 Competitive experiments of isocyanides vs. other radical acceptors with radicals. Conditions: 1 (0.15 mmol), 2 (0.6 mmol), 3 (0.5 mmol), R. A. = radical acceptors (0.6 mmol), nBu4NBF4 (0.5 mmol), MeCN (6 mL), cloth anode, Pt cathode, undivided cell, constant current = 10 mA, room temperature, N2, 2.25 h. 1HNMR yield, dibromomethane as an internal standard. | ||
In order to further investigate the compatibility, we firstly examined the substrate scope of alkyl disulfides for the synthesis of isothioureas with ethyl 2-isocyanoacetate (2) and 1H-benzo[d][1,2,3]triazole (3) as shown in Scheme 3. To our delight, the yields did not decrease significantly with the increase of the carbon chain from methyl to decyl in this transformation (4–7). Isopropyl, isobutyl, cyclopentyl and cyclohexyl were well tolerated with good to excellent yields (8–11). Substances, containing sensitive benzylic C–H bonds, were tolerated to access novel isothioureas in this electrochemical induced-radical process (12–14, 34). Afterwards, we continued to explore the applicability from alkyl disulfides to thiophenol and a series of molecule isothioureas were obtained successfully with satisfactory yield. Thiophenol, containing electron-neutral (F, Cl, Br, H), electron-withdrawing (OCF3, CF3, and CO2Me) and electron-donating (Me, Et, tBu, OMe, and SMe) groups, could smoothly transform to realize this process with high stereoselectivity under mild conditions (15–26). The structure of Z-conformer (16) was confirmed by X-ray crystallographic analysis. In addition, this protocol was successfully applied to thiophenols bearing diverse groups at different positions with good yields, indicating that steric hindrance has no obvious effect (20, 27–31). With curiosity, we tried to evaluate the tolerance of diselenides to construct novel Se–C–N bonds simultaneously. Unexpectedly, regardless of alkyl diselenides (32–33), benzyl diselenide (34) or aryl diselenide (35), all were well tolerated under electrochemical conditions, providing a series of unprecedented isoselenoureas, which were inaccessible by conventional methods. The compatibility of sensitive functional groups provides an opportunity for further molecular modification. The sensitive functional architectures including but not limited to ester (36), benzyloxy (37), allyl and allyloxy as well as endene (38, 41–44), labile alkyl chloride (39) and alkyl bromide (40), propargyl (44), and thiophene (45), all remained intact, enriching the diversity of heteroatom compounds. The merit of this methodology was further demonstrated by elaboration of a wide gamut of functional molecules and drug molecules to diverse isothioureas. Natural products including L-menthol and geraniol (46–47), food additives including isopulegol and furfuryl alcohol (48–49), and pharmaceuticals including ibuprofen (50) were apt to give rise to the corresponding isothioureas in 50–78% yields.
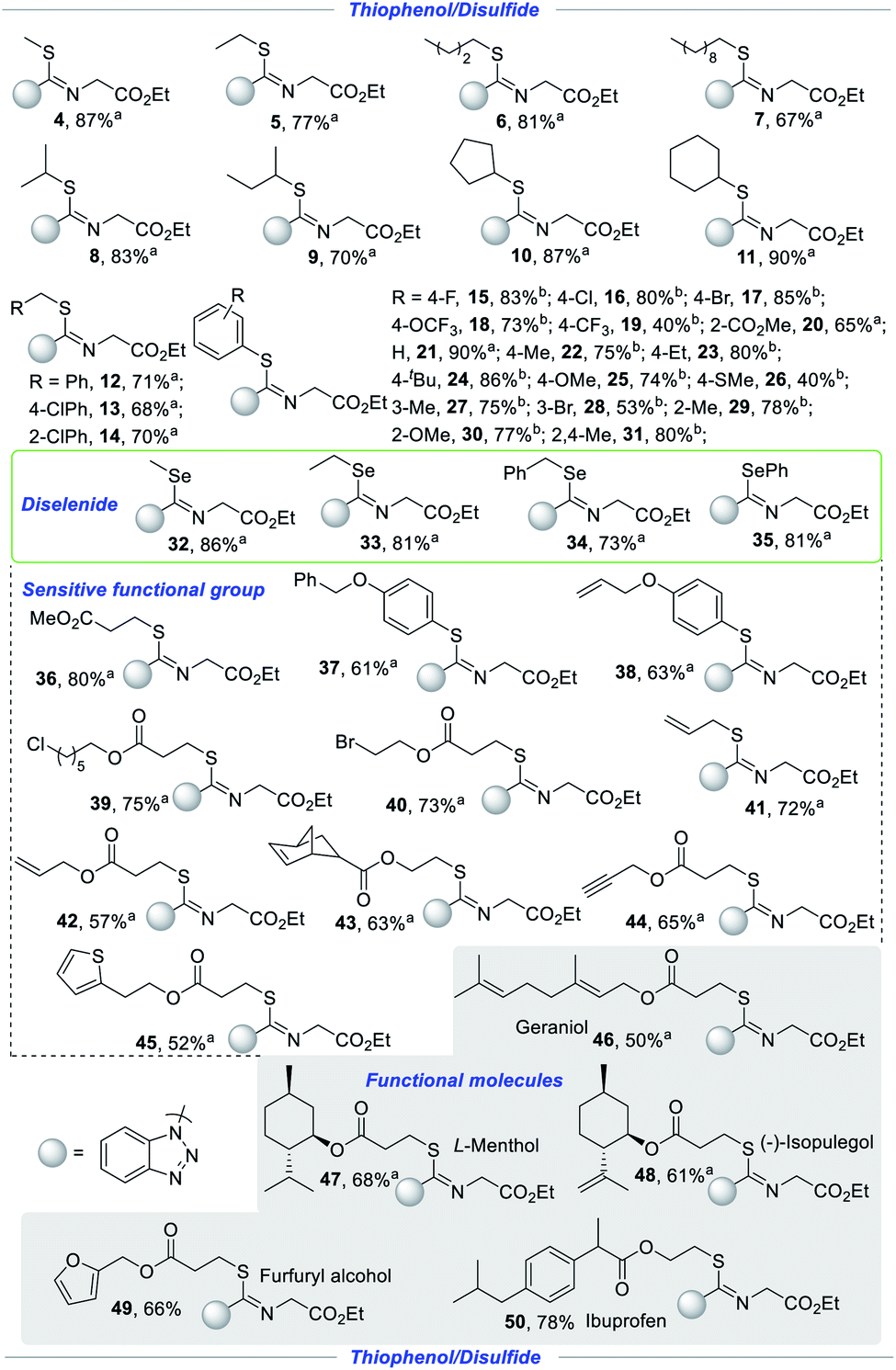 | ||
| Scheme 3 Scope of thiophenols/disulfides/diselenides. Conditions: a disulfides/diselenides (0.15 mmol) or b thiophenols (0.3 mmol), isocyanides (0.6 mmol), 1H-benzotriazole (0.5 mmol), nBu4NBF4 (0.5 mmol), MeCN (6 mL), cloth anode, Pt cathode, undivided cell, constant current = 10 mA, room temperature, N2, 2.25 h. Isolated yield. | ||
After defining the scope of thiophenols and disulfides, we turned our attention to explore the substrate scope with respect to nucleophiles in Scheme 4. Benzotriazole, with mono-substitution or multi-substitution, could be efficiently converted to the corresponding skeletons in 55–75% yields (51–54). Under standard conditions, pyrazole also showed great reactivity with phenyl disulfide or ethyl disulfide (55–56). The introduction of a halogen group in pyrazolylcycle, especially a subtle C–I bond, was compatible with satisfactory results (57–59, 64–65). 4-Methyl-1H-pyrazole as a coupling partner also realized the aminosulfenylation of isocyanide, albeit less efficiently (62). Of note is that the yield was increased to 70% from 40% by replacing methyl with phenyl (63). In addition to benzotriazole and pyrazole derivatives, other triazoles and tetrazole were proved to be viable coupling partners as well, enabling access to the target products in synthetically useful yields (66–70). Subsequently, with this established-optimized condition, we set out to investigate substrates for isocyanide coupling partners. With methyl isocyanoacetate (71), a similar effect was observed in an electrochemical difunctionalization reaction. As expected, both tosylmethyl isocyanide and benzyl isocyanide were demonstrated to be competent substrates (72–73). Cyclohexyl isocyanide, as a representative of secondary isocyanides, converted into the corresponding product (74). Gratifyingly, isocyanides, regardless of the steric effect, could engage with thiophenols to give the corresponding adducts in good yields (75–76). Aryl isocyanides however afforded the desired products in low yields under standard reaction conditions (77–78).
 | ||
| Scheme 4 Scope of azazoles. Conditions: a disulfides/diselenides (0.15 mmol) or b thiophenols (0.3 mmol), isocyanides (0.6 mmol), azazoles (0.5 mmol), nBu4NBF4 (0.5 mmol), MeCN (6 mL), cloth anode, Pt cathode, undivided cell, constant current = 10 mA, room temperature, N2, 2.25 h. Isolated yield. | ||
To evaluate the feasibility of this electrochemical protocol, we monitored the reaction on a 4.5 mmol scale as shown in Scheme 5. Under similar conditions, by prolonging the reaction time to 34 h, a good isolated yield of 72% was obtained, which provided an opportunity for further synthetic manipulations.
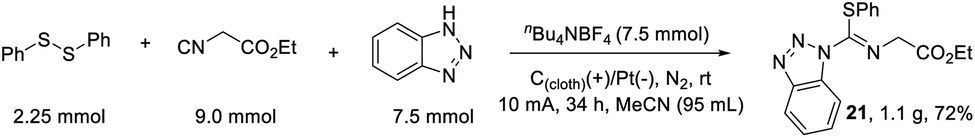 | ||
| Scheme 5 Gram-scale synthesis. | ||
In order to account for the stereoselectivity of products, theoretical calculations were performed. The result demonstrated that the free energy of the Z-conformer of the product 16 is 2.3 kcal mol−1 smaller than that of the E-conformer.
Based on a previously reported mechanistic study14 and these above observations, a synthetically possible mechanism for the electrochemical intermolecular difunctionalization reaction is depicted in Scheme 6. A sulfur radical was formed via single-electron-transfer (SET) reduction of the disulfide or SET oxidation of the thiophenol. Subsequently, the exclusive capture of the S-radical by the isocyano part yields the imine C-radical I. Further SET oxidation of this radical intermediate to the corresponding imine carbocation II, followed by nucleophilic trapping intermolecularly, affords the final isothioureas.
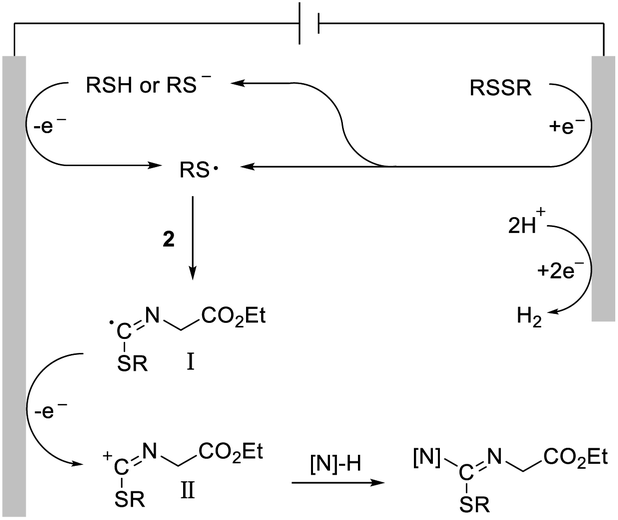 | ||
| Scheme 6 Proposed reaction mechanism. | ||
Conclusions
In summary, we have developed an efficient and sustainable strategy for the double C(sp2)–X (S/Se, N) bond formation of isocyanides simultaneously. A series of novel isothio/selenoureas were obtained via a three-component cross-coupling. The electrochemical reaction could exclusively occur at the isocyano part regardless of the presence of susceptible radical acceptors, such as arenes and an alkynyl moiety, even an alkenyl moiety. The strategy showed high atom- and step-economy as well as gram-scale synthesis demonstrating potential application in molecular engineering and materials science.Data availability
The ESI† include experimental detail, NMR data, HRMS data, crystallography data and DFT data.Author contributions
Z. Guan and S. Zhu designed and carried out most of the chemical reactions and analyzed the data. S. Zhu, Y. Yang, and Y. Liu, S. Wang performed the synthetic experiments. F. Bu analysed EPR data. H. Cong analysed the X-ray data. H. Zhang carried out theoretical calculations. A. Lei and Z. Guan designed and supervised the project. Z. Guan, H. Zhang, H. Alhumade and A. Lei wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22031008) and Science Foundation of Wuhan (2020010601012192). The Deanship of Scientific Research (DSR) at King Abdulaziz University, Jeddah, Saudi Arabia has funded this project, under grant no. (FP-123-43). Therefore, the authors gratefully acknowledge technical and financial support from the Ministry of Education and King Abdulaziz University, DSR, Jeddah, Saudi Arabia.Notes and references
- (a) F. Liu, Z. Zhang, H.-y. Diao and Z.-j. Shi, ChemCatChem, 2021, 13, 1475–1497 CrossRef CAS; (b) J. Bariwal and E. Van der Eycken, Chem. Soc. Rev., 2013, 42, 9283–9303 RSC; (c) Q. Lu, H. Yi and A. Lei, Acta Chim. Sin., 2015, 73, 1245 CrossRef CAS.
- W. Shi, C. Liu and A. Lei, Chem. Soc. Rev., 2011, 40, 2761–2776 RSC.
- (a) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138–12204 CrossRef CAS PubMed; (b) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236–10254 CrossRef CAS PubMed; (c) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef CAS; (d) K.-J. Jiao, Y.-K. Xing, Q.-L. Yang, H. Qiu and T.-S. Mei, Acc. Chem. Res., 2020, 53, 300–310 CrossRef CAS PubMed.
- C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed.
- (a) J. W. Collet, T. R. Roose, E. Ruijter, B. U. W. Maes and R. V. A. Orru, Angew. Chem., Int. Ed., 2020, 59, 540–558 CrossRef CAS PubMed; (b) T. Vlaar, E. Ruijter, B. U. W. Maes and R. V. A. Orru, Angew. Chem., Int. Ed., 2013, 52, 7084–7097 CrossRef CAS PubMed; (c) V. P. Boyarskiy, N. A. Bokach, K. V. Luzyanin and V. Y. Kukushkin, Chem. Rev., 2015, 115, 2698–2779 CrossRef CAS PubMed; (d) B. Song and B. Xu, Chem. Soc. Rev., 2017, 46, 1103–1123 RSC.
- (a) A. V. Gulevich, A. G. Zhdanko, R. V. A. Orru and V. G. Nenajdenko, Chem. Rev., 2010, 110, 5235–5331 CrossRef CAS PubMed; (b) B. Zhang and A. Studer, Chem. Soc. Rev., 2015, 44, 3505–3521 RSC; (c) P. Mampuys, Y. Zhu, T. Vlaar, E. Ruijter, R. V. A. Orru and B. U. W. Maes, Angew. Chem., Int. Ed., 2014, 53, 12849–12854 CrossRef CAS PubMed; (d) A. V. Lygin and A. de Meijere, Angew. Chem., Int. Ed., 2010, 49, 9094–9124 CrossRef CAS PubMed.
- (a) K. J. Romero, M. S. Galliher, D. A. Pratt and C. R. J. Stephenson, Chem. Soc. Rev., 2018, 47, 7851–7866 RSC; (b) U. Wille, Chem. Rev., 2013, 113, 813–853 CrossRef CAS PubMed; (c) B. Chen, L.-Z. Wu and C.-H. Tung, Acc. Chem. Res., 2018, 51, 2512–2523 CrossRef CAS PubMed; (d) X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed; (e) H.-M. Huang, P. Bellotti and F. Glorius, Chem. Soc. Rev., 2020, 49, 6186–6197 RSC; (f) H. Jiang and A. Studer, Chem. Soc. Rev., 2020, 49, 1790–1811 RSC; (g) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58–102 CrossRef CAS PubMed; (h) L. Pitzer, J. L. Schwarz and F. Glorius, Chem. Sci., 2019, 10, 8285–8291 RSC.
- (a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (b) J.-i. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265–2299 CrossRef CAS PubMed; (c) B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC; (d) S. H. Shi, Y. Liang and N. Jiao, Chem. Rev., 2021, 121, 485–505 CrossRef CAS PubMed; (e) L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC; (f) R. Francke and R. D. Little, ChemElectroChem, 2019, 6, 4373–4382 CrossRef CAS; (g) M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC; (h) T. Wu and K. D. Moeller, Angew. Chem., Int. Ed., 2021, 60, 12883–12890 CrossRef CAS PubMed.
- (a) S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27–45 CrossRef CAS; (b) X. Dong, J. L. Roeckl, S. R. Waldvogel and B. Morandi, Science, 2021, 371, 507 CrossRef CAS PubMed; (c) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed; (d) Y. Jiang, K. Xu and C. Zeng, Chem. Rev., 2018, 118, 4485–4540 CrossRef CAS PubMed; (e) H. Wang, M. Yu, P. Zhang, H. Wan, H. Cong and A. Lei, Sci. Bull., 2021 DOI:10.1016/j.scib.2021.07.004.
- (a) G. M. Martins, B. Shirinfar, T. Hardwick, A. Murtaza and N. Ahmed, Catal. Sci. Technol., 2019, 9, 5868–5881 RSC; (b) P. Xiong and H.-C. Xu, Acc. Chem. Res., 2019, 52, 3339–3350 CrossRef CAS PubMed; (c) J. C. Siu, N. Fu and S. Lin, Acc. Chem. Res., 2020, 53, 547–560 CrossRef CAS PubMed; (d) Y. Yuan and A. Lei, Acc. Chem. Res., 2019, 52, 3309–3324 CrossRef CAS PubMed; (e) J. H. Qin, M. J. Luo, D. L. An and J. H. Li, Angew. Chem., Int. Ed., 2021, 60, 1861–1868 CrossRef CAS PubMed; (f) J. Ke, W. Liu, X. Zhu, X. Tan and C. He, Angew. Chem., Int. Ed., 2021, 60, 8744–8749 CrossRef CAS PubMed; (g) X. Zhang, T. Cui, X. Zhao, P. Liu and P. Sun, Angew. Chem., Int. Ed., 2020, 59, 3465–3469 CrossRef CAS PubMed; (h) M. Findlater, S. Zhang, L. Li, J. Li, J. Shi, K. Xu, W. Gao, L. Zong and G. Li, Angew. Chem., Int. Ed., 2021, 60, 7275–7282 CrossRef PubMed; (i) X. Hu, L. Nie, G. Zhang and A. Lei, Angew. Chem., Int. Ed., 2020, 59, 15238–15243 CrossRef CAS PubMed; (j) K.-Y. Ye, G. Pombar, N. Fu, G. S. Sauer, I. Keresztes and S. Lin, J. Am. Chem. Soc., 2018, 140, 2438–2441 CrossRef CAS PubMed.
- M. Lübbesmeyer, D. Leifert, H. Schäfer and A. Studer, Chem. Commun., 2018, 54, 2240–2243 RSC.
- Z. Guan, S. Zhu, S. Wang, H. Wang, S. Wang, X. Zhong, F. Bu, H. Cong and A. Lei, Angew. Chem., Int. Ed., 2021, 60, 1573–1577 CrossRef CAS PubMed.
- M. Teders, A. Gómez-Suárez, L. Pitzer, M. N. Hopkinson and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 902–906 CrossRef CAS PubMed.
- (a) P. Wang, S. Tang, P. Huang and A. Lei, Angew. Chem., Int. Ed., 2017, 56, 3009–3013 CrossRef CAS PubMed; (b) P. Huang, P. Wang, S. Tang, Z. Fu and A. Lei, Angew. Chem., Int. Ed., 2018, 57, 8115–8119 CrossRef CAS PubMed; (c) D. Li, S. Li, C. Peng, L. Lu, S. Wang, P. Wang, Y.-H. Chen, H. Cong and A. Lei, Chem. Sci., 2019, 10, 2791–2795 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2077984. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04475c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |