
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn enzyme-mediated bioorthogonal labeling method for genome-wide mapping of 5-hydroxymethyluracil†
Cheng-Jie
Ma‡
a,
Lin
Li‡
 b,
Wen-Xuan
Shao
a,
Jiang-Hui
Ding
a,
Xiao-Li
Cai
c,
Zhao-Rong
Lun
c,
Bi-Feng
Yuan
*ad and
Yu-Qi
Feng
ad
b,
Wen-Xuan
Shao
a,
Jiang-Hui
Ding
a,
Xiao-Li
Cai
c,
Zhao-Rong
Lun
c,
Bi-Feng
Yuan
*ad and
Yu-Qi
Feng
ad
aSauvage Center for Molecular Sciences, Department of Chemistry, Wuhan University, Wuhan 430072, China. E-mail: bfyuan@whu.edu.cn
bSchool of Pharmacy, Shanghai Jiao Tong University, Shanghai 200240, P. R. China
cCenter for Parasitic Organisms, State Key Laboratory of Biocontrol, School of Life Sciences, Sun Yat-Sen University, Guangzhou 510275, P. R. China
dSchool of Public Health, Wuhan University, Wuhan 430071, China
First published on 4th October 2021
Abstract
DNA 5-hydroxymethyluracil (5hmU) is a thymine modification existing in the genomes of various organisms. The post-replicative formation of 5hmU occurs via hydroxylation of thymine by ten-eleven translocation (TET) dioxygenases in mammals and J-binding proteins (JBPs) in protozoans, respectively. In addition, 5hmU can also be generated through oxidation of thymine by reactive oxygen species or deamination of 5hmC by cytidine deaminase. While the biological roles of 5hmU have not yet been fully explored, determining its genomic location will highly assist in elucidating its functions. Herein, we report a novel enzyme-mediated bioorthogonal labeling method for selective enrichment of 5hmU in genomes. 5hmU DNA kinase (5hmUDK) was utilized to selectively install an azide (N3) group or alkynyl group into the hydroxyl moiety of 5hmU followed by incorporation of the biotin linker through click chemistry, which enabled the capture of 5hmU-containing DNA fragments via streptavidin pull-down. The enriched fragments were applied to deep sequencing to determine the genomic distribution of 5hmU. With this established enzyme-mediated bioorthogonal labeling strategy, we achieved the genome-wide mapping of 5hmU in Trypanosoma brucei. The method described here will allow for a better understanding of the functional roles and dynamics of 5hmU in genomes.
Introduction
A number of modified nucleobases have been identified in genomes among a variety of organisms besides the four canonical nucleobases of adenine, thymine, cytosine, and guanine.1,2 It has been considered that DNA modifications play sophisticated roles in regulating spatiotemporal gene expression.3,4 DNA cytosine methylation (5-methylcytosine, 5 mC) is the most extensively characterized epigenetic modification in mammals.5–7 In addition to 5 mC, many other types of base modifications have been discovered in DNA, such as 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5 fC), 5-carboxycytosine (5caC), N6-methyladenosine (6 mA), 5-hydroxymethyluracil (5hmU), β-D-glucosyl-5-hydroxymethyluracil (base J), and 5-glyceryl-methylcytosine (5gmC).1,8–135hmU is a thymine base modification present in the genomes of diverse organisms ranging from bacteriophages to mammals.14 5hmU has long been known as a DNA lesion formed from the oxidation of thymine by reactive oxygen species.15,16 Enzyme-mediated replicative incorporation of 5hmU into bacteriophage genomes indicates that 5hmU has functional importance.17 It has been reported that thymine in genomes of some bacteriophages and dinoflagellates is fully or partially replaced by 5hmU.18 It is worth noting that the loss of nucleotide salvage factor DNPH1 can give rise to aberrant incorporation of 5-hmdU into human cells.19 The post-replicative formation of 5hmU occurs via hydroxylation of thymine, which can be mediated by ten-eleven translocation (TET) dioxygenases in mammals20 and J-binding proteins (JBPs) in protozoan genomes,21,22 respectively. Another proposed mechanism for the formation of 5hmU is through the deamination of 5hmC by activation-induced cytidine deaminase (AID) or apolipoprotein B mRNA-editing catalytic polypeptide-like (APOBEC) family enzymes.23 Hydroxylation of thymine to 5hmU could generate a 5hmU:A base pair, while deamination of 5hmC would give rise to a 5hmU:G mispair.
It has been demonstrated that the majority of 5hmU within mouse embryonic stem cells (mESCs) is produced by mammalian TET dioxygenases, while only minor 5hmU is generated through 5hmC deamination or reactive oxygen species.20 Thus, the majority of 5hmU in the genome is matched (5hmU:A) but not mismatched (5hmU:G). The level of 5hmU was found to be dynamic throughout mESC differentiation, suggesting that 5hmU may have functional importance.20 Although it was reported that 5hmU could affect biological processes such as protein–DNA interactions and transcription factor binding,24,25 the consequences of 5hmU formation in genomes have not been fully explored.
Revealing the functions of 5hmU relies on the sensitive and precise detection and mapping of 5hmU within genomes. In recent years, several strategies have been developed to map 5hmU in DNA. A method through KRuO4 oxidation of 5hmU to generate 5fU followed by biotinylation using a hydrazide-biotin probe was developed to localize 5hmU.26,27 However, other aldehyde groups present, such as 5 fC and abasic sites (AP sites) in genomic DNA, may also react with the hydrazide-biotin probe, which could interfere with the accurate mapping of 5hmU. The chemical conversion of 5hmU to 5fU by KRuO4 oxidation could induce partial T-to-C base transition in polymerase extension owing to the ability of 5fU to form a 5fU:G mispair, which was then employed as the signature to map original 5hmU.28 The applicability of this method was demonstrated using synthetic oligonucleotides and part of the genome of a eukaryotic pathogen. However, only ∼40% of reads were cytosine at the 5hmU sites even under optimized conditions, which therefore required a sophisticated algorithm to analyze the sequencing data and identify the sites of 5hmU in DNA. In addition, β-glycosyltransferase (β-GT) was applied to tag a modified N3-glucose onto the hydroxyl group of 5hmU followed by incorporation of the biotin linker through click chemistry.29 After the capture of 5hmU-containing fragments with streptavidin-coupled beads, the enriched DNA fragments can be applied to deep sequencing to map the distribution of 5hmU. However, β-GT only works on mismatched 5hmU:G but not matched 5hmU:A.29 Thus, this method is not applicable to map 5hmU derived from thymine.
Herein, we report a novel enzyme-mediated bioorthogonal labeling method to selectively enrich genomic regions containing 5hmU. 5hmU DNA kinase (5hmUDK) was utilized to selectively install an azide (N3) group or alkynyl group into the hydroxyl group of 5hmU using γ-(2-azidoethyl)-adenosine 5′-triphosphate (N3-ATP) or γ-[(propargyl)-imido]-adenosine 5′-triphosphate (alkynyl-ATP) as the cofactor. The N3 group or alkynyl group in 5hmU was then utilized to incorporate the biotin linker through click chemistry. The enrichment of 5hmU-containing DNA fragments was performed via streptavidin pull-down. The enriched fragments were applied to deep sequencing to map the distribution of 5hmU (Fig. 1). With this established enzyme-mediated bioorthogonal labeling strategy, we achieved the genome-wide mapping of 5hmU in Trypanosoma brucei (T. brucei).
 | ||
| Fig. 1 Schematic illustration of the enzyme-mediated bioorthogonal labeling strategy for genome-wide mapping of 5hmU. 5hmUDK is used to selectively transfer the N3 group from N3-ATP to 5hmU. Biotin is then incorporated into 5hmU through click chemistry between the N3 group and the DBCO group in DBCO-SS-biotin. The biotin labeled DNA is enriched using streptavidin-coupled beads followed by the release with DTT treatment. The resulting DNA fragments are then subjected to high-throughput sequencing. | ||
Experimental section
Materials and reagents
5hmU-containing DNA (5hmU-DNA) was purchased from Takara Biotechnology Co., Ltd. (Dalian, China). Other oligonucleotides were purchased from Sangon Biotech (Shanghai, China). The sequences of these oligonucleotides are listed in Table S1 in ESI.†Shrimp alkaline phosphatase (SAP), calf intestine alkaline phosphatase (CIAP), E. coli C75 alkaline phosphatase (EAP), and TB Green® Premix Ex Taq™ II (Tli RNaseH Plus) were purchased from Takara Biotechnology Co., Ltd. (Dalian, China). γ-(2-Azidoethyl)-adenosine 5′-triphosphate (N3-ATP) and γ-[(propargyl)-imido]-adenosine 5′-triphosphate (alkynyl-ATP) were bought from Sigma-Aldrich (Beijing, China). DBCO-SS-biotin was bought from Confluore Biological Technology Co., Ltd. (Xi'an, China). 5-Hydroxymethyluridine DNA kinase (5hmUDK), NcoI-HF, hSMUG1 (single-strand-selective monofunctional uracil-DNA glycosylase 1), and NEBNext® Multiplex Oligos for Illumina® (Index Primers Set 1) were purchased from New England Biolabs (Beijing, China).
Trypanosome and DNA extraction
Trypanosoma brucei AnTat1.1 was used to infect mice, and parasites were isolated from the infected blood by using DEAE cellulose (DE-52) as previously described30 when the parasitaemia was up to 5 × 108. Trypanosomes were collected and washed twice with 5 mL phosphate buffered saline glucose (PSG) by centrifugation at 1000g for 10 min. After the last centrifugation, the supernatant PSG was discarded and the trypanosome sediment was used for DNA extraction. Purified trypanosomes were resuspended and lysed in SNET buffer (100 mM NaCl, 10 mM Tris–HCl, 25 mM EDTA, 0.5% SDS, pH 8.0) with 200 ng μL−1 proteinase K at 56 °C for 3 h, followed by phenol–chloroform extraction and ethanol precipitation. After air drying, DNA was dissolved in TE buffer (10 mM Tris–HCl, 1 mM EDTA, pH 8.0) and stored at −20 °C until use.Phosphorylation of 5hmU by 5hmUDK
Single-stranded or duplex T-DNA, U-DNA and 5hmU-DNA (sequences of these oligonucleotides are listed in Table S1 in the ESI†) were incubated with 20 U of 5hmUDK in 20 μL reaction buffer (50 mM Tris–HCl, pH 7.0, 10 mM DTT, 10 mM MgCl2, and 1 mM ATP, or N3-ATP, or alkynyl-ATP) at 37 °C for different times. The resulting DNA was purified using the Oligo Clean & Concentrator kit (Zymo Research). The phosphorylation of 5hmU was confirmed by NcoI restriction enzyme digestion as well as hSMUG1 cleavage assay.As for the NcoI restriction enzyme digestion, the resulting DNA was incubated with 10 U NcoI-HF in 1× CutSmart buffer at 37 °C for 1 h followed by polyacrylamide gel electrophoresis analysis. As for the hSMUG1 cleavage assay, the resulting DNA was incubated with 5 U hSMUG1 in 1× NEBuffer 1 at 37 °C for 2 h. Then the DNA was treated with 100 mM NaOH at 95 °C for 10 min followed by polyacrylamide gel electrophoresis analysis. The gel was visualized using a Tanon fluorescence imager (Shanghai, China).
Bioorthogonal labeling of 5hmU
The N3-labeled 5hmU-DNA was incubated with DBCO-SS-biotin (1 mM) at 37 °C for 2 h in 10% DMSO (v/v) solution. The alkynyl-labeled 5hmU-DNA was incubated with azide–biotin (1 mM), Cu–TBTA (0.5 mM) and freshly prepared ascorbic acid (0.5 mM) at 25 °C for 12 h. The excess free DBCO-SS-biotin or N3-biotin was removed using the Oligo Clean & Concentrator kit (Zymo Research). The resulting DNA was dissolved in TE buffer.Evaluation of the enrichment of 5hmU-containing DNA
A mixture of 0.1 ng of 60-bp 5hmU-DNA, 0.1 ng of 129-bp control DNA (Table S1 in the ESI†) and 10 μg of fragmented HEK293T genomic DNA was used to evaluate the enrichment of 5hmU-containing DNA. The mixture was processed sequentially with 5hmUDK treatment (using N3-ATP), bioorthogonal labeling (with DBCO-SS-biotin), and streptavidin pull-down.Real-time qPCR was carried out to evaluate the enrichment efficiency. Briefly, the enriched DNA was dissolved in 20 μL H2O. Then 2 μL of enriched DNA, 1 μL of the forward primer (10 μM), 1 μL of the reverse primer (10 μM), and 6 μL of H2O were added into a Takara TB Green Premix Ex Taq™ II qPCR mix (10 μL), to give a final volume of 20 μL. The real-time qPCR program was performed for 45 cycles at 95 °C for 10 s, 54 °C for 30 s and 72 °C for 1 min.
The enrichment efficiency was calculated using the following equations.
| ΔCt input = Ct (input 5hmU-DNA) − Ct (input control DNA), |
| ΔCt enrich = Ct (enriched 5hmU-DNA) − Ct (enriched control DNA), |
| ΔΔCt = ΔCt enrich − ΔCt input, |
| Enrichment fold = 2−ΔΔCt |
To evaluate the PCR amplification efficiencies, the standard curves for amplification of 60-bp 5hmU-DNA and 129-bp control DNA were generated. Briefly, a series of 60-bp 5hmU-DNA and 129-bp control DNA with different concentrations were prepared and used as templates for real-time qPCR. The PCR amplification efficiency was calculated using the following equation.
| Amplification efficiency = (10−1/slope − 1) × 100%. |
Enrichment of 5hmU-containing DNA fragments
T. brucei genomic DNA was fragmented to obtain 300 to 500 bp DNA by using a JY92-II N ultrasonic homogenizer (Scientz) with the following settings: 130 W peak incident power for 48 cycles (1 cycle = 5 s on and 5 s off). Then 2 μg of fragmented DNA was applied for the enrichment of 5hmU-containing DNA. Briefly, the fragmented DNA was incubated with 5hmUDK (20 U) in a 20 μL reaction buffer (50 mM Tris–HCl, pH 7.0, 10 mM DTT, 10 mM MgCl2, and 1 mM N3-ATP) at 37 °C for 30 min. The resulting DNA was purified using the DNA Clean & Concentrator kit (Zymo Research). The N3-labeled 5hmU-containing fragments were incubated with DBCO-SS-biotin (10 mM) at 37 °C for 2 h in 10% DMSO (v/v) solution. Then the mixture was purified by ethanol precipitation and dissolved in 50 μL water.For the streptavidin pull-down assay, streptavidin-coupled beads were pre-washed three times with 1× binding buffer (5 mM Tris–HCl, pH 7.0, 0.5 mM EDTA, 1 M NaCl, and 0.05% Tween 20) and resuspended in 50 μL of 2× binding buffer (10 mM Tris–HCl, pH 7.0, 1 mM EDTA, 2 M NaCl, and 0.1% Tween 20). The biotin-labeled DNA was added into resuspended streptavidin-coupled beads and incubated at 25 °C for 25 min with gentle rotation. Then the beads were washed five times with 1× binding buffer. To release the biotin labeled DNA, 50 mM freshly prepared dithiothreitol (DTT) was added into the beads and incubated at 37 °C for 2 h. Then the supernatant was collected, and DNA was purified by ethanol precipitation. The enriched DNA was subjected to library preparation for high-throughput sequencing.
Library preparation and high-throughput sequencing
The enriched DNA fragments or the DNA fragments without enrichment (input) were end repaired and dA-tailed using the Hieff NGS® Fast-Pace End Repair/dA-Tailing module (YEASEN Biotechnology Co., Ltd., Shanghai, China) according to the manufacturer's recommended protocol. As for the adapter ligation, 60 μL of end-repaired DNA, 5 μL of NEBNext adapter (15 μM), 5 μL of Fast-Pace DNA ligase and 30 μL of Fast-Pace DNA ligation enhancer (YEASEN Biotechnology Co., Ltd., Shanghai, China) were incubated at 20 °C for 30 min in a 100 μL solution. Then 3 U (1 U μL−1) USER enzyme (New England Biolabs) was added to the ligation products (100 μL) and incubated at 37 °C for 15 min to linearize the hairpin adaptor. The resulting DNA was purified with KAPA Pure beads (1.0× volume) (Kapa biosystems Pty Ltd., Cape Town, South Africa) to remove the excess adapter. The purified DNA was then amplified by PCR. The PCR solution (50 μL) included 20 μL of purified DNA, 25 μL of Q5 high-fidelity master mix (New England Biolabs), 1 μL of P7 primer (10 μM), 1 μL of P5 primer (10 μM), and 3 μL of H2O. The amplification program started at 98 °C for 30 s, followed by 15 cycles at 98 °C for 10 s, 60 °C for 30 s, and 72 °C for 30 s, and ended with 1 cycle at 72 °C for 5 min. The PCR products were sequentially purified using KAPA Pure beads (Kapa Biosystems Pty Ltd., Cape Town, South Africa) and agarose gel using an agarose gel extraction kit (Zymo Research). Library quality was assessed on an Agilent Bioanalyzer 2100 system. The library was sequenced on an Illumina Hiseq platform and 150-bp paired-end reads were generated.Data analysis
Raw data from Illumina Hiseq sequencing were analyzed using FastQC (v 0.11.5). Low quality reads and the adapter were removed to obtain clean data using the Trimmomatic software. The T. brucei (TREU927) reference genomes were downloaded from GenBank. Clean reads were aligned to the T. brucei reference genomes using BWA (v 0.7.12-r1039). Peak calling was performed with MACS2 (v.2.1.1). The IGV (v 2.9.4) software was used to visualize the mapping results. MEME-ChIP (v 4.11.2) was used for motif enrichment analysis.31 For peak annotation, the T. brucei transcripts were downloaded in the GTF format from the NCBI database.Results and discussion
Specific phosphorylation of 5hmU in DNA
5hmUDK could selectively transfer the γ-phosphate in ATP to the hydroxymethyl moiety of 5hmU in duplex DNA to form 5-phosphomethyluridine (5pmU) (Fig. 2A). We used a 5′-FAM labeled 34-bp duplex 5hmU-DNA that contains one 5hmU site to evaluate the phosphorylation of 5hmU (Fig. 2B). 5hmU was placed in the recognition and cleavage site (CCATGG, T is replaced by 5hmU) of NcoI restriction enzyme. Phosphorylation of 5hmU by 5hmUDK to form 5pmU could prevent the cleavage by NcoI enzyme (Fig. 2B). Duplex T-DNA and U-DNA that have the same sequence contexts as 5hmU-DNA except 5hmU being replaced by thymine or uracil were used as controls. The results showed that both T-DNA and U-DNA could be cleaved by NcoI to produce the short fragments with or without 5hmUDK treatment (Fig. 2C, lanes 2–5). However, phosphorylation of 5hmU by 5hmUDK fully resisted the cleavage by NcoI enzyme (Fig. 2C, lanes 6 and 7), indicating the efficient phosphorylation of 5hmU. The duplex T-DNA and 5hmU-DNA were also mixed at different ratios followed by treatment with 5hmUDK. The results showed that the cleaved fractions of 5hmU-DNA were proportional to the percentages of 5hmU-DNA in the mixture (Fig. S1 in the ESI†), suggesting that the level of 5hmU-DNA in the DNA mixture can be quantitatively obtained by 5hmUDK assay.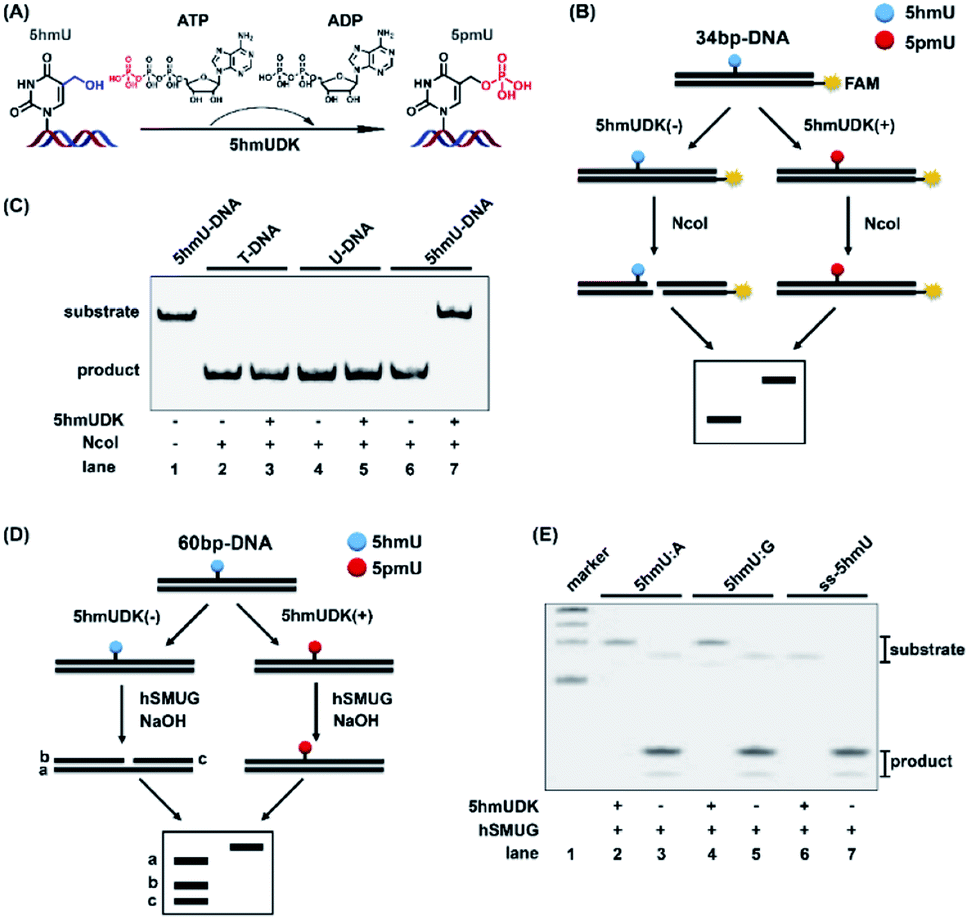 | ||
| Fig. 2 Specific phosphorylation of 5hmU in DNA. (A) 5hmUDK selectively transfers the γ-phosphate of ATP to 5hmU using ATP as the cofactor. (B) Workflow of restriction endonuclease cleavage assay to verify the phosphorylation of 5hmU. NcoI is used to cleave the 34 bp-DNA with or without 5hmUDK treatment. Phosphorylation of 5hmU to form 5pmU can resist the cleavage by NcoI. (C) Analysis of duplex T-DNA, U-DNA and 5hmU-DNA after incubation with or without 5hmUDK at 37 °C for 30 min according to the workflow shown in (B). (D) Workflow of hSMUG cleavage assay to verify the phosphorylation of 5hmU. (E) Analysis of the phosphorylation of 5hmU in duplex DNA with a 5hmU:A base pair or 5hmU:G mispair, and 5hmU in single-stranded DNA (ss-5hmU) by hSMUG cleavage assay. All three types of DNA samples were incubated with or without 5hmUDK. The resulting DNA was treated with 100 mM NaOH at 95 °C for 10 min followed by polyacrylamide gel electrophoresis analysis. | ||
hSMUG can specifically catalyze the hydrolysis of the N-glycosidic bond of 5hmU to form an AP site that can be broken to generate a gap by alkaline hydrolysis at high temperature.32 We also employed the properties of hSMUG to examine the phosphorylation of 5hmU. A duplex 60-bp 5hmU-DNA was processed with 5hmUKD followed by sequential hSMUG and NaOH treatment (Fig. 2D). The results showed that phosphorylation of 5hmU in duplex 5hmU-DNA by 5hmUDK could prevent the hydrolysis of the N-glycosidic bond of 5hmU by hSMUG (Fig. 2E, lane 2). However, the 60-bp 5hmU-DNA without 5hmUDK treatment was cleaved by sequential hSMUG and NaOH treatment (Fig. 2E, lane 3). These results further confirmed the successful phosphorylation of 5hmU. It was reported that 5hmU could form a 5hmU:A base pair (herein 5hmU was generated from oxidation of thymine by ROS or TETs) and 5hmU:G mispair (herein 5hmU was generated from deamination of 5hmC).23 Using the same assay, we found that 5hmU in both 5hmU:A base pair and 5hmU:G mispair can be phosphorylated by 5hmUDK and thus resisted the subsequent hydrolysis by hSMUG and NaOH treatment (Fig. 2E, lanes 2 and 4). Interestingly, the hSMUG assay demonstrated that 5hmU in ssDNA could also be phosphorylated by 5hmUDK (Fig. 2E, lanes 6 and 7).
Functionalization of 5hmU in DNA
The specific phosphorylation of 5hmU in DNA inspired us to further functionalize 5hmU with a suitable moiety that could be utilized for the enrichment of 5hmU-containing DNA. In this respect, we evaluated the phosphorylation of 5hmU in DNA using ATP analogues such as N3-ATP and alkynyl-ATP instead of ATP (Fig. 3A). We aimed to tag the N3 group or alkynyl group to 5hmU (Fig. 3B). These results showed that the use of N3-ATP or alkynyl-ATP to replace ATP led to the similar resistance to NcoI cleavage (Fig. 3C), suggesting that 5hmUDK could recognize both ATP analogues and add N3 or alkynyl groups onto 5hmU. When 5hmU-ssDNA was used as the substrate, N3-ATP was more efficiently transferred to 5hmU by 5hmUDK than alkynyl-ATP (Fig. S2 in the ESI†). | ||
| Fig. 3 5hmUDK selectively transfers the alkynyl group or N3 group to 5hmU using alkynyl-ATP or N3-ATP as the cofactor. (A) The chemical structures of ATP, alkynyl-ATP, and N3-ATP. (B) The reaction for the addition of the alkynyl group or N3 group to 5hmU by 5hmUDK using alkynyl-ATP or N3-ATP as the cofactor. (C) Analysis of the phosphorylation of 5hmU in DNA by NcoI cleavage assay using ATP, alkynyl-ATP, or N3-ATP as the cofactor. (D) Analysis of the phosphorylation of 5hmU in DNA using ATP, alkynyl-ATP, or N3-ATP as the cofactor for different reaction times. | ||
The reaction for phosphorylation of 5hmU was also carried out for different times (0.5 h and 12 h). It can be seen that complete conversion of 5hmU to alkynyl-5hmU or N3-5hmU could be achieved within 0.5 h (Fig. 3D). In addition, the reaction at 37 °C for 12 h didn't lead to the obvious degradation of alkynyl- or N3-labeled DNA (Fig. 3D), suggesting that the alkynyl- or N3-labeled DNA was relatively stable. The treatment of 5pmU-DNA by calf intestine alkaline phosphatase (CIAP) and E. coli C75 alkaline phosphatase (EAP) resulted in the cleavage of 5pmU-DNA (Fig. S3 in the ESI†), indicating that the phosphate group in 5hmU could be removed by CIAP or EAP. Unlike CIAP and EAP, shrimp alkaline phosphatase (SAP) showed weak dephosphorylation activity toward 5pmU (Fig. S3 in the ESI†). However, all these three alkaline phosphatases showed weak dephosphorylation activity toward alkynyl-5hmU and no dephosphorylation activity toward N3-5hmU (Fig. S3 in the ESI†).
The selective labeling of 5hmU with N3 or alkynyl groups endows the 5hmU-containing DNA with appropriate groups for the bioorthogonal reaction, which can be employed to incorporate the biotin linker for subsequent enrichment. To this end, we evaluated the bioorthogonal reaction between N3-5hmU and DBCO-Cy3. The results showed that the reaction between N3-5hmU DNA and DBCO-Cy3 led to a slow shift compared to the unlabeled DNA (Fig. S4 in the ESI†), indicating the successful bioorthogonal reaction between N3-5hmU DNA and DBCO-Cy3.
Enrichment of 5hmU-containing DNA
The specific phosphorylation of 5hmU followed by bioorthogonal labeling enables the selective enrichment of 5hmU-containing DNA, which can be utilized for genome-wide mapping of 5hmU by high-throughput sequencing.Since the N3 or alkynyl group could be successfully added to 5hmU, we used two reagents of N3-biotin and DBCO-SS-biotin to react with alkynyl-5hmU and N3-5hmU, respectively (Fig. 4A). The results showed that the reaction of N3-5hmU with DBCO-SS-biotin or alkynyl-5hmU with N3-biotin led to the slow shift in gel electrophoresis (Fig. 4B, lanes 8 and 9), indicating the successful bioorthogonal reaction. In addition, the liquid chromatography-ultraviolet detection also confirmed the successful installation of the N3-phosphate group on 5hmU and the bioorthogonal labelling of DBCO-SS-biotin (Fig. S5 in the ESI†). In contrast, neither the T-DNA nor U-DNA substrate was labeled by N3-biotin or DBCO-SS-biotin (Fig. 4B, lanes 1–6). The bioorthogonal reaction between alkynyl-5hmU and N3-biotin required a 12 h incubation to achieve complete labeling. However, the bioorthogonal reaction between N3-5hmU and DBCO-SS-biotin required only a 2 h incubation to achieve the complete labeling. Thus, we chose N3-ATP to label 5hmU and incorporated the biotin tag with DBCO-SS-biotin in the subsequent experiments. We also used a mixture of ATP and N3-ATP with different ratios to label 5hmU-DNA followed by biotinylation with DBCO-SS-biotin. The results demonstrated that N3-ATP could be proportionally transferred to 5hmU-DNA (Fig. S6 in the ESI†), indicating that 5hmUDK has no significant preference to ATP over N3-ATP.
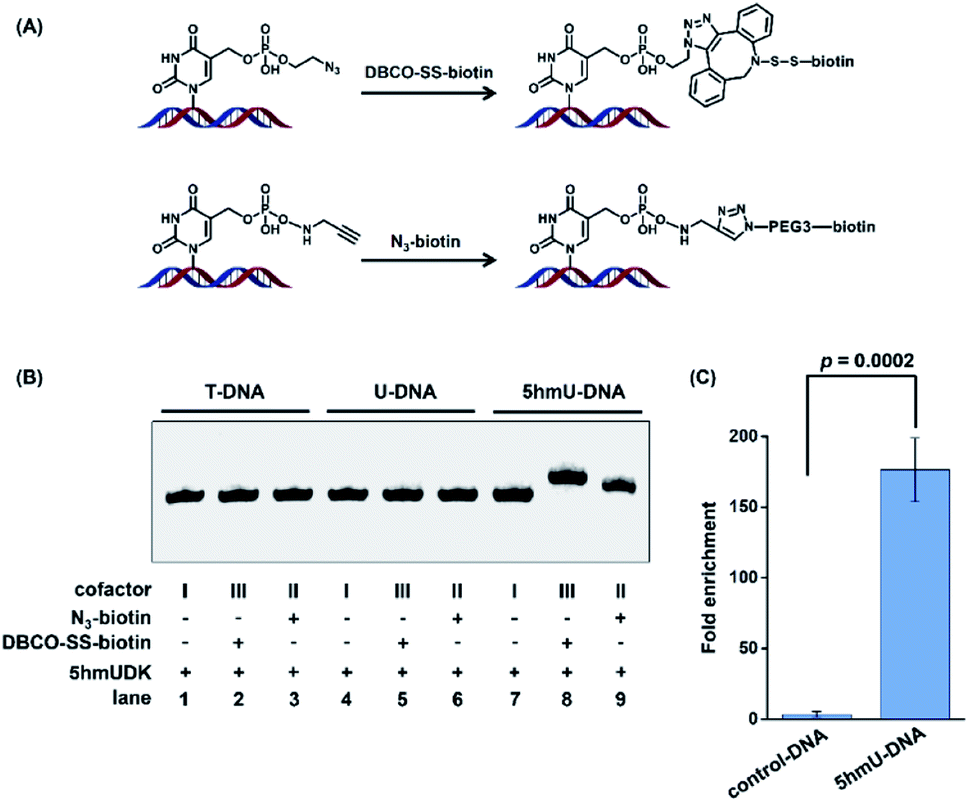 | ||
| Fig. 4 5hmUDK-mediated bioorthogonal labeling assay and the selective enrichment of 5hmU-containing DNA. (A) The bioorthogonal reactions of N3-5hmU with DBCO-SS-biotin and alkynyl-5hmU with N3-biotin. (B) Analysis of the duplex T-DNA, U-DNA and 5hmU-DNA by 5hmUDK-mediated bioorthogonal labeling. (C) Evaluation of the enrichment efficiency toward 5hmU-DNA by 5hmUDK-mediated bioorthogonal labeling assay. The mixture of 60-bp 5hmU-DNA, 129-bp control DNA, and fragmented HEK293T genomic DNA was processed with the 5hmUDK-mediated bioorthogonal labeling and enrichment procedure. | ||
We further used synthesized DNA (5hmU-containing 60-bp duplex DNA and 129-bp control duplex DNA, Table S1 in the ESI†) to evaluate the enrichment efficiency by the 5hmUDK-mediated bioorthogonal labeling assay. A mixture of 0.1 ng of 60-bp duplex 5hmU-DNA, 0.1 ng of 129-bp duplex control DNA, and 10 μg of fragmented HEK293T genomic DNA was processed with the 5hmUDK-mediated N3 incorporation and DBCO-SS-biotin bioorthogonal reaction. The resulting biotin-labeled DNA was enriched using streptavidin-coupled beads and then amplified using primers specific for 5hmU-containing 60-bp duplex DNA or 129-bp control duplex DNA. The real-time qPCR amplification efficiencies were evaluated with the constructed standard curves. The results showed that each standard curve had an R2 value of at least 0.99 and the amplification efficiencies were between 90% and 103% (Fig. S7 in the ESI†), which are suitable for the enrichment evaluation. The quantification results showed that a 170-fold enrichment for 5hmU-DNA was obtained (Fig. 4C), demonstrating that 5hmU-DNA could be efficiently enriched by the 5hmUDK-mediated bioorthogonal labeling assay.
Genome-wide mapping of 5hmU in T. brucei
The biosynthesis of base J in T. brucei includes the hydroxylation of thymidine to form 5hmU by JBPs and then the glycosylation of 5hmU by base J glucosyltransferase.21 Here, we employed the aforementioned 5hmUDK-mediated bioorthogonal labeling assay for genome-wide mapping of 5hmU in T. brucei. The isolated T. brucei genomes were first fragmented and the 5hmU-containing DNA fragments were enriched by the 5hmUDK-mediated bioorthogonal labeling assay. The enriched 5hmU-containing DNA fragments were processed for library construction and high-throughput sequencing analysis (Fig. S8 and S9 in the ESI†).We found 2348 peaks in the two pull-down replicates in comparison with the input samples, and an overlap of 679 peaks between two pull-down replicates (Fig. 5A and S10 in the ESI†). The results showed that 62.15% peaks were located in the gene regions, 27.69% peaks in the intergenic regions, 9.57% peaks in the promoter regions, and 0.59% peaks in the downstream regions (Fig. 5B). These peaks were mainly enriched between the transcription start sites (TSS) and transcription end sites (TES) (Fig. 5C). We used real-time qPCR to verify the 5hmU-rich region obtained by high-throughput sequencing. The results showed that all 7 examined regions were enriched compared to the control (Fig. S11A and S12 in the ESI†), indicating that these regions should contain 5hmU modification. In contrast, no enrichment was observed for the vicinal regions of these peaks (Fig. S11B in the ESI†). Moreover, use of ATP instead of N3-ATP in the assay led to no enrichment of 5hmU-containing DNA fragments (Fig. S13 in the ESI†), further confirming that the enriched DNA fragments should contain 5hmU. We could also clearly observe the fragment enrichment of 5hmU in comparison with that of the input sample (Fig. 5D and S12 in the ESI†). The motif analysis showed that 5hmU preferentially occurred at the sequence of AATATGCCA (Fig. 5E).
 | ||
| Fig. 5 Genome-wide mapping of 5hmU in T. brucei by 5hmUDK-mediated bioorthogonal labeling, enrichment, and high-throughput sequencing. (A) The overlap of 5hmU peaks between two pull-down replicates. (B) The percentages of 5hmU peaks in different genomic regions of T. brucei. (C) The location of 5hmU peaks relative to the TSS site and TES site. (D) Representative IGVtool view of 5hmU peaks in pull-down samples compared to input sample. (E) The sequence motif identified from 5hmU mapping. | ||
Given that 5hmUDK specifically catalyzes the phosphorylation of 5hmU, DNA pull-down is enriched with 5hmU and is ready for high-throughput sequencing. Based on the 5hmUDK-mediated bioorthogonal labeling and enrichment procedure presented here, we provide the first map of 5hmU in the T. brucei genome. While 5hmUDK can modify both matched (5hmU:A) and mismatched 5hmU (5hmU:G), the method should be useful in mapping 5hmU from both sources. Taken together, our study highlights the application of this method in mapping the genomic distribution of 5hmU and provides a useful tool for probing the functions of 5hmU. Future experiments will utilize this method for 5hmU profiling in other organisms, such as mESC genomic DNA.
Conclusions
In the current study, we developed a 5hmUDK-mediated bioorthogonal labeling strategy for the enrichment of 5hmU-containing DNA. 5hmUDK was utilized to selectively add an N3 group into the hydroxyl moiety of 5hmU followed by incorporation of the biotin linker through the DBCO-SS-biotin bioorthogonal reaction. The biotin labeled 5hmU-containing DNA fragments were then successfully captured and enriched via streptavidin pull-down followed by high-throughput sequencing. Application of this strategy to the T. brucei genome showed that 5hmU sites were mainly located at gene regions in the T. brucei genome, suggesting that 5hmU might play roles in RNA transcription. The method presented here provides a useful tool to address the biological functions of 5hmU.Data availability
Sequence date is available in the Gene Expression Omnibus (GEO) database at NCBI with accession number GSE179809.Author contributions
C. J. M. and B. F. Y. designed the experiments and interpreted the data. C. J. M., W. X. S. and J. H. D. performed the bioorthogonal labeling of 5hmU, evaluation of the enrichment of 5hmU-containing DNA, and library preparation and high-throughput sequencing. X. L. C. and Z. R. L. cultured Trypanosoma brucei and extracted genomic DNA. L. L. and Y. Q. F. analysed the sequencing data. C. J. M. and B. F. Y. wrote the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The work was supported by the National Natural Science Foundation of China (22074110, 21635006, and 21721005).References
- A. Hofer, Z. J. Liu and S. Balasubramanian, J. Am. Chem. Soc., 2019, 141, 6420–6429 CrossRef CAS PubMed.
- Y. Feng, N. B. Xie, W. B. Tao, J. H. Ding, X. J. You, C. J. Ma, X. Zhang, C. Yi, X. Zhou, B. F. Yuan and Y. Q. Feng, CCS Chem., 2020, 2, 994–1008 Search PubMed.
- K. Chen, B. S. Zhao and C. He, Cell Chem. Biol., 2016, 23, 74–85 CrossRef CAS PubMed.
- M. V. C. Greenberg and D. Bourc'his, Nat. Rev. Mol. Cell Biol., 2019, 20, 590–607 CrossRef CAS PubMed.
- C. Luo, P. Hajkova and J. R. Ecker, Science, 2018, 361, 1336–1340 CrossRef CAS PubMed.
- Y. Dai, B. F. Yuan and Y. Q. Feng, RSC Chem. Biol., 2021, 2, 1096–1114 RSC.
- M. D. Lan, B. F. Yuan and Y. Q. Feng, Chin. Chem. Lett., 2019, 30, 1–6 CrossRef CAS.
- T. Carell, M. Q. Kurz, M. Muller, M. Rossa and F. Spada, Angew. Chem., Int. Ed. Engl., 2018, 57, 4296–4312 CrossRef CAS PubMed.
- B. F. Yuan, Chem. Res. Toxicol., 2020, 33, 695–708 Search PubMed.
- W. Y. Lai, J. Z. Mo, J. F. Yin, C. Lyu and H. L. Wang, TrAC, Trends Anal. Chem., 2019, 110, 173–182 CrossRef CAS.
- Q. Wang, J. H. Ding, J. Xiong, Y. Feng, B. F. Yuan and Y. Q. Feng, Chin. Chem. Lett., 2021 DOI:10.1016/j.cclet.2021.05.020.
- Y. Feng, J. J. Chen, N. B. Xie, J. H. Ding, X. J. You, W. B. Tao, X. Zhang, C. Yi, X. Zhou, B. F. Yuan and Y. Q. Feng, Chem. Sci., 2021, 12, 11322–11329 RSC.
- J. Xiong, T. T. Ye, C. J. Ma, Q. Y. Cheng, B. F. Yuan and Y. Q. Feng, Nucleic Acids Res., 2019, 47, 1268–1277 CrossRef CAS PubMed.
- J. H. Gommers-Ampt and P. Borst, FASEB J., 1995, 9, 1034–1042 CrossRef CAS PubMed.
- Z. Djuric, D. A. Luongo and D. A. Harper, Chem. Res. Toxicol., 1991, 4, 687–691 Search PubMed.
- D. W. Potter and Z. Djuric, Adv. Exp. Med. Biol., 1991, 283, 801–803 CrossRef CAS PubMed.
- H. Witmer, J. Virol., 1981, 39, 536–547 CrossRef CAS PubMed.
- P. M. Rae, Proc. Natl. Acad. Sci. U. S. A., 1973, 70, 1141–1145 CrossRef CAS PubMed.
- K. Fugger, I. Bajrami, M. Silva Dos Santos, S. J. Young, S. Kunzelmann, G. Kelly, G. Hewitt, H. Patel, R. Goldstone, T. Carell, S. J. Boulton, J. MacRae, I. A. Taylor and S. C. West, Science, 2021, 372, 156–165 CrossRef CAS PubMed.
- T. Pfaffeneder, F. Spada, M. Wagner, C. Brandmayr, S. K. Laube, D. Eisen, M. Truss, J. Steinbacher, B. Hackner, O. Kotljarova, D. Schuermann, S. Michalakis, O. Kosmatchev, S. Schiesser, B. Steigenberger, N. Raddaoui, G. Kashiwazaki, U. Muller, C. G. Spruijt, M. Vermeulen, H. Leonhardt, P. Schar, M. Muller and T. Carell, Nat. Chem. Biol., 2014, 10, 574–581 CrossRef CAS PubMed.
- P. Borst and R. Sabatini, Annu. Rev. Microbiol., 2008, 62, 235–251 CrossRef CAS PubMed.
- S. Liu, D. Ji, L. Cliffe, R. Sabatini and Y. Wang, J. Am. Soc. Mass Spectrom., 2014, 25, 1763–1770 CrossRef CAS PubMed.
- R. Olinski, M. Starczak and D. Gackowski, Mutat. Res., Rev. Mutat. Res., 2016, 767, 59–66 CrossRef CAS.
- D. K. Rogstad, P. Liu, A. Burdzy, S. S. Lin and L. C. Sowers, Biochemistry, 2002, 41, 8093–8102 CrossRef CAS PubMed.
- J. R. Greene, L. M. Morrissey and E. P. Geiduschek, J. Biol. Chem., 1986, 261, 12828–12833 CrossRef CAS.
- F. Kawasaki, D. Beraldi, R. E. Hardisty, G. R. McInroy, P. van Delft and S. Balasubramanian, Genome Biol., 2017, 18, 23 CrossRef PubMed.
- C. B. Qi, J. H. Ding, B. F. Yuan and Y. Q. Feng, Chin. Chem. Lett., 2019, 30, 1618–1626 CrossRef CAS.
- F. Kawasaki, S. Martinez Cuesta, D. Beraldi, A. Mahtey, R. E. Hardisty, M. Carrington and S. Balasubramanian, Angew. Chem., Int. Ed. Engl., 2018, 57, 9694–9696 CrossRef CAS PubMed.
- M. Yu, C. X. Song and C. He, Methods, 2015, 72, 16–20 CrossRef CAS PubMed.
- S. M. Lanham and D. G. Godfrey, Exp. Parasitol., 1970, 28, 521–534 CrossRef CAS PubMed.
- T. L. Bailey and C. Elkan, Proc. Int. Conf. Intell. Syst. Mol. Biol., 1994, 2, 28–36 CAS.
- H. Hashimoto, S. Hong, A. S. Bhagwat, X. Zhang and X. Cheng, Nucleic Acids Res., 2012, 40, 10203–10214 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Tables S1–S3; Fig. S1–S13. See DOI: 10.1039/d1sc03812e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |