
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible light-induced oxidative N-dealkylation of alkylamines by a luminescent osmium(VI) nitrido complex†
Jing
Xiang
 *a,
Min
Peng
a,
Yi
Pan
b,
Li-Juan
Luo
a,
Shun-Cheung
Cheng
b,
Xin-Xin
Jin
a,
Shek-Man
Yiu
b,
Wai-Lun
Man
c,
Chi-Chiu
Ko
b,
Kai-Chung
Lau
*b and
Tai-Chu
Lau
*b
*a,
Min
Peng
a,
Yi
Pan
b,
Li-Juan
Luo
a,
Shun-Cheung
Cheng
b,
Xin-Xin
Jin
a,
Shek-Man
Yiu
b,
Wai-Lun
Man
c,
Chi-Chiu
Ko
b,
Kai-Chung
Lau
*b and
Tai-Chu
Lau
*b
aCollege of Chemistry and Environmental Engineering, Yangtze University, Jingzhou 434020, Hubei, P. R. China. E-mail: xiangjing@yangtzeu.edu.cn
bDepartment of Chemistry, City University of Hong Kong, Tat Chee Avenue, Kowloon Tong, Hong Kong, China. E-mail: bhtclau@cityu.edu.hk; kaichung@cityu.edu.hk
cDepartment of Chemistry, Hong Kong Baptist University, Kowloon Tong, Hong Kong 999077, People's Republic of China
First published on 9th October 2021
Abstract
N-Dealkylation of amines by metal oxo intermediates (M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) is related to drug detoxification and DNA repair in biological systems. In this study, we report the first example of N-dealkylation of various alkylamines by a luminescent osmium(VI) nitrido complex induced by visible light.
O) is related to drug detoxification and DNA repair in biological systems. In this study, we report the first example of N-dealkylation of various alkylamines by a luminescent osmium(VI) nitrido complex induced by visible light.
High-valent metal oxo (M
O) species play key roles in many chemical and biological oxidation processes.1 They are versatile oxidants that can perform oxidation of substrates via a variety of pathways, including electron transfer, H-atom transfer, hydride transfer and O-atom transfer. In principle, high-valent metal nitrido (M![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) complexes should also function as versatile oxidants similar to MO. Although there have been significant advances in MN oxidation chemistry in recent years, the reactivity of MN is still rather limited in scope compared to MO.2 MN is intrinsically less oxidizing than MO due to the stronger electron donating property of the N3− ligand than the O2− ligand. Attempts to increase the oxidizing power of MN by increasing the oxidation state or by using less electron-donating ancillary ligands often led to decomposition of the complexes, mainly due to facile coupling of the nitrido ligands to yield N2 (2MN → 2M + N2).3 One appealing strategy to enhance the reactivity of MN is photochemical excitation. We have recently designed an osmium(VI) nitrido complex [OsVI(N)(L)(CN)3]− (NO2-OsN, HL = 2-(2-hydroxy-5-nitrophenyl)benzoxazole) that is strongly luminescent in the solid state and in fluid solutions.4 It readily absorbs visible light to generate a long-lived and highly oxidizing excited state with a redox potential of ca. 1.4 V. The excited state of this complex also possesses [OsN˙] nitridyl characteristics that enable it to readily abstract H-atoms from inert organic substrates.5
N) complexes should also function as versatile oxidants similar to MO. Although there have been significant advances in MN oxidation chemistry in recent years, the reactivity of MN is still rather limited in scope compared to MO.2 MN is intrinsically less oxidizing than MO due to the stronger electron donating property of the N3− ligand than the O2− ligand. Attempts to increase the oxidizing power of MN by increasing the oxidation state or by using less electron-donating ancillary ligands often led to decomposition of the complexes, mainly due to facile coupling of the nitrido ligands to yield N2 (2MN → 2M + N2).3 One appealing strategy to enhance the reactivity of MN is photochemical excitation. We have recently designed an osmium(VI) nitrido complex [OsVI(N)(L)(CN)3]− (NO2-OsN, HL = 2-(2-hydroxy-5-nitrophenyl)benzoxazole) that is strongly luminescent in the solid state and in fluid solutions.4 It readily absorbs visible light to generate a long-lived and highly oxidizing excited state with a redox potential of ca. 1.4 V. The excited state of this complex also possesses [OsN˙] nitridyl characteristics that enable it to readily abstract H-atoms from inert organic substrates.5
We report herein the visible-light induced N-dealkylation of various alkylamines by NO2-OsN. Iron oxo species have been used by heme and nonheme enzymes to carry out N-dealkylation reactions of tertiary amines, which are important processes involved in detoxification and DNA repair.6 A number of synthetic iron(IV) oxo complexes are also able to carry out such N-dealkylation reactions.7 Mechanistic studies using cytochrome P450 and synthetic iron oxo complexes indicate that there are two possible mechanisms for N-dealkylation of amines, namely hydrogen-atom transfer (HAT) and electron transfer–proton transfer (ET–PT) (Fig. 1).8 In this work we report the first example of N-dealkylation of various aromatic as well as aliphatic tertiary amines by a nitrido complex upon visible light excitation. We also provide unambiguous evidence that these reactions occur via an ET/PT mechanism.
 | ||
| Fig. 1 Two possible mechanisms for N-demethylation of tertiary amines by cytochrome P450 and synthetic Fe(IV) oxo complexes (P = porphyrin). | ||
N-Dealkylation of amines
The reactions of NO2-OsN with various amines are summarized in Fig. 2. The reaction with N,N-dimethylaniline (DMA) is described in detail. Upon irradiation with blue light (λ > 460 nm), the light-yellow solution containing NO2-OsN and 10 equiv. of DMA in CH2Cl2 gradually turned red. Analysis of the product solution by GC/MS and GC/FID reviewed the formation of N-methylaniline in 71% yield (based on NO2-OsN consumed) (Table S1†), indicating that N-demethylation of DMA has occurred. The fate of the fragmented methyl group and the nature of the osmium product(s) were investigated by ESI/MS (Fig. S1†). The ESI/MS of the product solution exhibits a predominant peak at m/z 275.6, which can be assigned to [Os(L)(CN)4]2−, and this peak shifted to m/z 276.1 when the 15N-labeled nitrido complex (NO2-Os15N) was used, indicating that the extra CN− is derived from the addition of the fragmented CH3 group to the nitrido ligand, followed by internal redox (see the Proposed mechanism section below). The ESI/MS also shows a major peak at m/z 659, which is assigned to the amidine product [Os(L)(CN)3(NHDMA(–2H))]− (2), see Fig. S1.†.
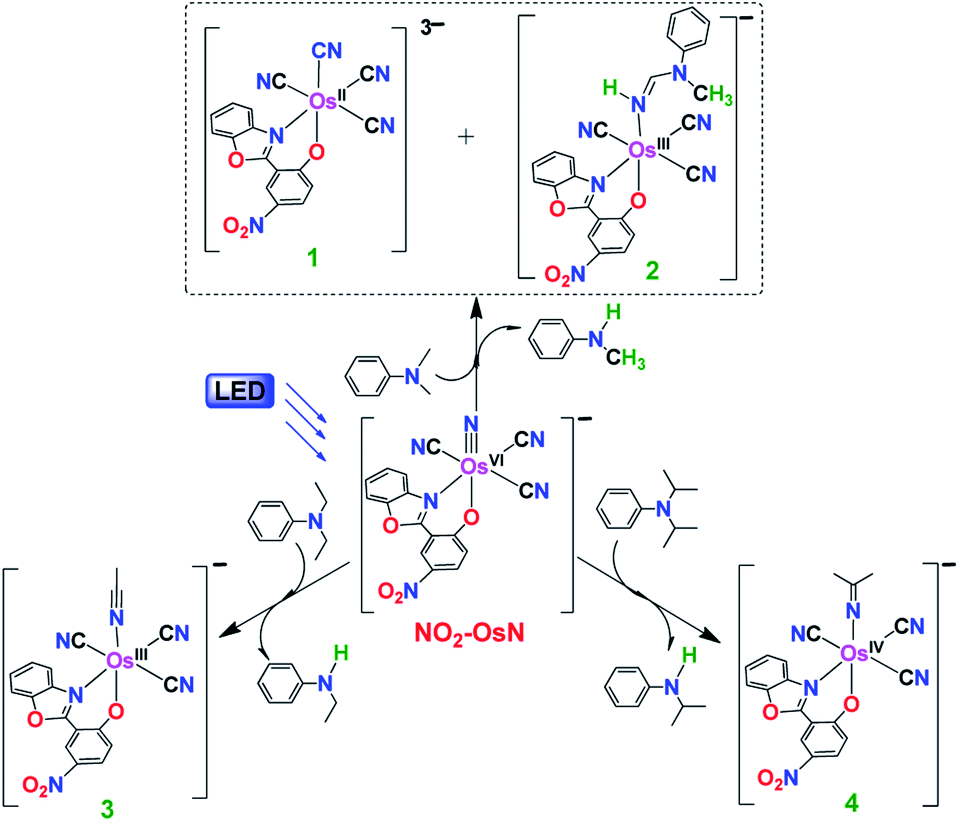 | ||
| Fig. 2 The photoreactions of NO2-OsN* with various amines. | ||
An attempt to isolate complex 1 by extracting the product residue with H2O followed by the addition of PPh4Cl resulted in a PPh4+ salt of [OsII(L)(CN)4]3− (1) mixed with a small amount of [OsIII(L)(CN)4]2− (1′). However, pure (PPh4)21′ could be isolated as a dark red microcrystalline solid in ca. 68% yield by slow evaporation of a MeOH/H2O solution of the mixture under air. Both 1 and 1′ show the same peak at m/z 275.6 in ESI/MS. On the other hand, complex 2 could be readily separated from the reaction mixture by column chromatography and isolated as the PPh4+ or nBu4N+ salt in ca. 16% yield. (PPh4)21′ and (PPh4)2 were characterized by IR, UV/vis, cyclic voltammetry (CV) and ESI/MS (Fig. S2–S6†). The IR spectrum of 1′ shows three v(CN) stretches at 2085, 2038 and 1995 cm−1, while that of 2 shows two v(CN) stretches at 2113 and 2088 cm−1.
N-Dealkylation also occurs in the reactions of NO2-OsN* with N,N-diethylaniline (DEA) and N,N-diisopropylaniline (DPA), resulting in 87% of N-ethylaniline and 91% of N-isopropylaniline, respectively. Similar to the case of DMA, the fragmented alkyl groups in DEA and DPA are attached to the nitrido ligand, followed by internal redox to give [OsIII(L)(CN)3(NCCH3)]− (3) and [OsIV(L)(CN)3{NC(CH3)2}]− (4), respectively. 3 and 4 were isolated as the PPh4+ salts in 80% and 82% yields, respectively, and they were characterized by IR, UV/vis, ESI/MS and 1H NMR (Fig. S7–S9†). Besides the aromatic tertiary amines, dealkylation of the aliphatic tertiary amine Et3N by NO2-OsN* was also found, with diethylamine formed in 72% yield. In contrast to the case of DMA, <2% of amidine products were found for DEA and Et3N reactions, while no amidine product was found for DPA reaction. The UV/vis and ESI/MS collected at various time intervals for these photochemical reactions are summarized in Fig. S10 and S11.†
The molecular structures of (nBu4N)2, (PPh4)3 and (PPh4)4 were determined by X-ray crystallography. As shown in Fig. 3a, the Os center in 2 is 6-coordinated by three CN− ligands, a bidentate O^N ligand and a neutral amidine ligand in a distorted octahedral geometry. The Os–N6 bond length is 2.048(4) Å and the Os–N6–C17 bond angle is 126.3(3)°, consistent with a neutral amidine ligand. The C17–N6 (1.276(6) Å) and C17–N7 (1.338(6) Å) bond distances, and the N6–C17–N7 bond angle (127.8(5)o) are similar to those of reported amidine complexes.9 In 3, the Os–N5 and N5–C17 bond lengths of 2.017(4) and 1.135(7) Å, respectively, and the close to linear Os1–N5–C17 bond angle of 172.0(4)° are consistent with a neutral CH3CN ligand. Complex 4 features an anionic iminato ligand; the Os–N5 bond length is 1.849(9) Å, indicating a double bond character. The C5–N17 bond is 1.187(13) Å, typical of the CN double bond.
 | ||
| Fig. 3 Molecular structures of 2 (a), 3 (b) and 4 (c). Cations were omitted for simplicity. | ||
Quenching of the excited state of NO2-OsN by N,N-dimethylaniline (DMA) and N,N-diethylaniline (DEA) in CH2Cl2 was investigated. The bimolecular quenching rate constants (kq) obtained from Stern–Volmer plots are (8.5 ± 0.1) × 109 M−1 s−1 and (9.2 ± 0.3) × 109 M−1 s−1, respectively for DMA and DEA (Fig. 4a). kq for d6-DMA is (9.0 ± 0.1) × 109 M−1 s−1, indicating that there is no deuterium isotope effect. The observed near diffusion-controlled rate constants are consistent with the high excited state redox potential of NO2-OsN (ca. 1.4 V vs. NHE).4a Upon excitation of a mixture of NO2-OsN and DMA in CH2Cl2, a band at ca. 460 nm was observed in the nanosecond transient absorption (ns-TA) spectrum (Fig. 4b), which is similar to that of the DMA+˙ cation radical,10 indicating one-electron oxidation of DMA by NO2-OsN*.
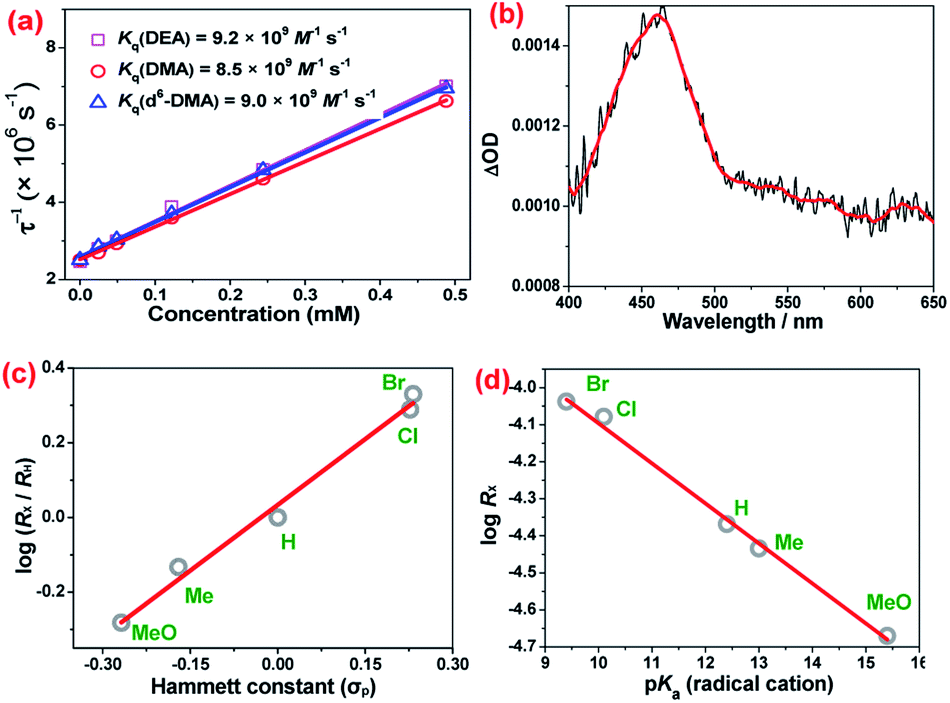 | ||
| Fig. 4 (a) Stern–Volmer plots for the quenching of NO2-OsN (2.46 × 10−5 M) by DEA, DMA and d6-DMA. (b) ns-Transient absorption spectrum of DMA taken immediately after 355 nm laser excitation. (c) Hammett plot of σpversus the relative initial rates log(Rx/RH) (y-intercept = 0.03 ± 0.01; slope = 1.17 ± 0.06; r2 = 0.989). (d) Plot of log(Rx) vs. pKa for the radical cation of para-substituted DMAs (y-intercept = −3.01 ± 0.08; slope = −(1.08 ± 0.06) × 10−1; r2 = 0.992). | ||
The electronic effects of various para-substituents on the aromatic ring of DMA have been investigated by the method of initial rates (Rx), which were obtained from the UV/vis spectral changes of the photoreactions of NO2-OsN with various DMAs (Fig. S12†). The rates were found to be accelerated by electron withdrawing groups, and the Hammett plot of log (Rx/RH) versus σp is linear with a positive ρ value of 1.17 (Fig. 4c). A linear relationship supports a common transition state (TS)/mechanism for the series of reactants, and a positive ρ value indicates that the TS is stabilized by electron-withdrawing substituents. A linear plot was also found for log(Rx) vs. pKa of the radical cations of the DMAs (Fig. 4d and Table S2†).11 These results are consistent with proton transfer from DMA+˙ to NO2-OsVN in the rate-limiting step, since the acidity of DMA+˙ is enhanced by electron-withdrawing substituents.12 The photoreaction of NO2-OsN with N-ethyl-N-methylaniline was also studied. The result shows that the N-demethylation product (72%) is greatly favored over that of N-deethylation (4%) (Fig. S13†), which further supports proton transfer as the rate-limiting step, since the acidity of the methyl proton in the radical cation is higher than that of the ethyl protons.13
The kinetic isotope effects (KIE) for the N-dealkylation reaction of DMA by NO2-OsN* were determined. Inter-molecular KIE was obtained by competition experiments using equimolar C6H5N(CH3)2 and C6H5N(CD3)2 as the substrate (Fig. S14†). A KIE value of 4.0 ± 0.5 was obtained from analysis of the products C6H5NHCH3 and C6H5NHCD3 by GC/FID and GC/MS. The inter-molecular KIE for amidine formation was also investigated by analysis of the products by ESI/MS, and a KIE of ∼4.5 was estimated from the ratio of the most intense peaks at m/z 659 and m/z 664 for the protio- and deutero-osmium(IV) amidine species, respectively, assuming that the spraying and ionization efficiencies of the two ions are similar (Fig. S15†). Intra-molecular KIE was also determined by using 4-BrC6H4N(CH3)(CD3) as the substrate and a KIE value of 4.9 ± 0.5 was obtained from analysis of the products 4-BrC6H5NHCH3 and 4-BrC6H5NHCD3 (Fig. S16†). Similar KIE values for the N-dealkylation and amidine formation suggest that these two pathways occur via a common intermediate.
Proposed mechanism
The experimental results are consistent with an ET/PT mechanism in the N-dealkylation of various tertiary amines with NO2-OsN*, as shown in Fig. 5 using DMA as an example. The first step is electron transfer from DMA to NO2-OsN* to generate DMA+˙ and NO2-OsVN, which occurs at the near diffusion-controlled rate. This step is supported by the observation of the transient DMA+˙ species. Rate-limiting proton transfer then occurs from DMA+˙ to NO2-OsVN, followed by rapid N-rebound to give an osmium(IV) amido intermediate, a species that is analogous to the carbinolamine species proposed in cytochrome P450 catalyzed N-dealkylation of amines,6 except that in this case the intermediate amide remains bound to the metal center. The rate-limiting proton transfer step is supported by a large KIE of 4.5, a +ve Hammett ρ value of 1.17, a linear dependence of log(Rx) on pKa and a high preference for N-demethylation over N-deethylation in the photoreaction of N-ethyl-N-methylaniline. The osmium(IV) amido species further decomposes via two parallel pathways. In pathway A, spontaneous C–N cleavage occurs to give the dealkylated product N-methylaniline and an Os(IV) iminato complex, the latter species then undergoes internal redox to afford 1. In pathway B, the osmium(IV) amido intermediate undergoes H-atom abstraction by another NO2-OsN* followed by internal redox to give the osmium(III) amidine product. This proposed step is supported by the observed formation of around 1/3 equiv. of NO2-OsIIINH3: NO2-OsVIN + 3H → NO2-OsIIINH3, by UV-vis spectrophotometry. We and others have previously shown that OsN readily abstracts H-atoms from various substrates to give the ammine complex OsIII-NH3.4a,14 Similar mechanisms are proposed for the other substrates (Fig. S17–S19†). Pathway B is much less significant for the other substrates as it does not result in a stable, conjugated amidine ligand as 2.
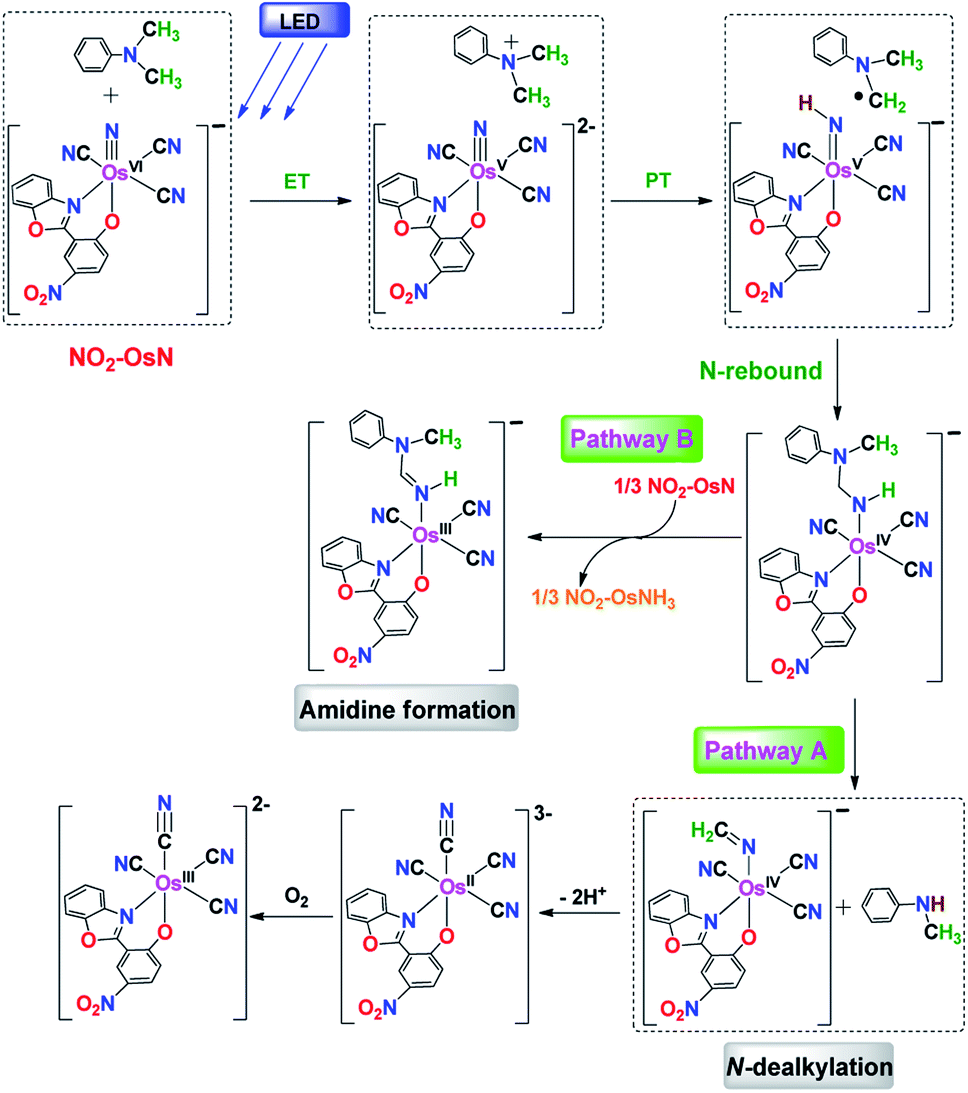 | ||
| Fig. 5 Proposed mechanism for the reaction of NO2-OsN* with DMA. | ||
DFT calculations
Our proposed mechanism for the oxidative dealkylation of tertiary amines by NO2-OsN is also supported by DFT calculations (see ESI, Computational details†). The potential energy surface for the reaction of NO2-OsVN with DMA+, that is generated from electron transfer between DMA and NO2-OsN*, is shown in Fig. 6; and insets are the structures of the intermediates and transition states. 3INT1 is formed by binding NO2-OsVN with DMA+via a weak hydrogen bond. A rapid proton transfer from DMA+ to NO2-OsVN occurs via3TS1 with a barrier height (ΔG‡298) of 9.8 kcal mol−1, with a simultaneous N-rebound step to generate a stable Os(IV) amido intermediate (3INT2). The Os–N bond is elongated from 1.739 Å in 3INT1 to 1.766 Å in 3TS1 to 1.923 Å in 3INT2. This is followed by a second proton transfer step from Os(IV) amido to the nitrogen of aniline via3TS2 (ΔG‡298 = −1.8 kcal mol−1) to give an osmium(IV) species (3INT3) via3TS2 with a four-member-ring structure. The weak HN…CH2 bond (∼1.581 Å) in 3INT3 is then broken to afford the products N-methylaniline and osmium(IV) iminato species (3INT4) via 3TS3; this step is almost barrierless (0.1 kcal mol−1 relative to 3INT3). Based on our calculations, the rate-determining step is the simultaneous proton transfer and N-rebound step via TS1. | ||
| Fig. 6 The potential energy surface for N-dealkylation of DMA+ by [OsV(N)(CN)3(L)]2− in CH3CN at the B3LYP-D3(BJ)/def2-TZVP level with the PCM solvent effect. Relative 298 K Gibbs free energies in acetonitrile are given in kcal mol−1. Bond lengths are in angstrom (Å). | ||
In conclusion, we have shown that the strongly luminescent osmium(VI) nitrido complex [OsVI(N)(L)(CN)3]− undergoes facile N-dealkylation of aromatic and aliphatic tertiary amines upon irradiation with visible light. We have provided definitive evidence that these reactions occur via an ET/PT mechanism. Our results should contribute to a significant advance in metal nitrido chemistry.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
J. X. and T.-C. L. designed the experiments, analysed the data and wrote the manuscript. S. M. Y. and W.-L. M. solved the X-ray structures. S.-C. C. and C.-C. K investigated the photophysical properties. M. P., L. J. L. and X. X. J. carried out experiments and analysed the data. Y. P. and K. C. L. did the DFT calculations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21771026), the Hubei Provincial Natural Science Foundation of China (2018CFA047), and the Financial support from the Research Grants Council of Hong Kong (CityU 11301618 and N_CityU111/20).References
-
(a)
W. A. Nugent and J. M. Mayer, Metal–Ligand Multiple Bonds, Wiley-Inter Science, New York, 1988 Search PubMed
; (b) A. Gunay and K. H. Theopold, Chem. Rev., 2010, 110, 1060–1081 CrossRef CAS PubMed
-
(a) T. J. Meyer and M. H. V. Huynh, Inorg. Chem., 2003, 42, 8140–8160 CrossRef CAS PubMed
-
(a) R. M. Clarke and T. Storr, J. Am. Chem. Soc., 2016, 138, 15299–15302 CrossRef CAS PubMed
-
(a) J. Xiang, X. X. Jin, Q. Q. Su, S. C. Cheng, C. C. Ko, W. L. Man, M. Y. Xue, L. L. Wu, C. M. Che and T. C. Lau, Commun. Chem., 2019, 2, 40 CrossRef
-
(a) M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev., 1996, 96, 2841–2888 CrossRef CAS PubMed
-
(a) H. C. Aspinall, J. Bacsa, A. C. Jones, J. S. Wrench, K. Black, P. R. Chalker, P. J. King, P. Marshall, M. Werner, H. O. Davies and R. Odedra, Inorg. Chem., 2011, 50, 11644–11652 CrossRef CAS PubMed
-
(a) F. P. Guengerich, C. H. Yun and T. L. Macdonald, J. Biol. Chem., 1996, 271, 27321–27329 CrossRef CAS PubMed
- Q.-Q. Su, K. Fan, X.-D. Huang, J. Xiang, S.-C. Cheng, C.-C. Ko, L.-M. Zheng, M. Kurmood and T.-C. Lau, Dalton Trans., 2020, 49, 4084–4092 RSC
-
(a) P. J. Bailey and S. Pace, Coord. Chem. Rev., 2001, 214, 91–141 CrossRef CAS
- H. Kandori, K. Kemnitz and K. Yoshihara, J. Phys. Chem., 1992, 96, 8042–8048 CrossRef CAS
-
(a) A. Barbieri, M. De Gennaro, S. Di Stefano, O. Lanzalunga, A. Lapi, M. Mazzonna, G. Olivo and B. Ticconi, Chem. Commun., 2015, 51, 5032–5035 RSC
- S. Sumalekshmy and K. R. Gopidas, Chem. Phys. Lett., 2005, 413, 294–299 CrossRef CAS
-
(a) O. Okazaki and F. P. Guengerich, J. Biol. Chem., 1993, 268, 1546–1552 CrossRef CAS PubMed
- G. Berger, A. Wach, J. Sá and J. Szlachetko, Inorg. Chem., 2021, 60, 6663–6671 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2083709–2083711. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04369b |
| This journal is © The Royal Society of Chemistry 2021 |