
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regiodivergent sulfonylarylation of 1,3-enynes via nickel/photoredox dual catalysis†
Ya
Chen‡
a,
Kun
Zhu‡
ab,
Qingqin
Huang
ab and
Yixin
Lu
 *ab
*ab
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore, 117543, Singapore. E-mail: chmlyx@nus.edu.sg
bJoint School of National University of Singapore and Tianjin University, International Campus of Tianjin University, Binhai New City, Fuzhou, Fujian 350207, China
First published on 20th September 2021
Abstract
Catalytic difunctionalization of 1,3-enynes represents an efficient and versatile approach to rapidly assemble multifunctional propargylic compounds, allenes and 1,3-dienes. Controlling selectivity in such addition reactions has been a long-standing challenging task due to multiple reactive centers resulting from the conjugated structure of 1,3-enynes. Herein, we present a straightforward method for regiodivergent sulfonylarylation of 1,3-enynes via dual nickel and photoredox catalysis. Hinging on the nature of 1,3-enynes, diverse reaction pathways are feasible: synthesis of α-allenyl sulfones via 1,4-sulfonylarylation, or preparation of (E)-1,3-dienyl sulfones with high chemo-, regio- and stereoselectivity through 3,4-sulfonylarylation. Notably, this is the first example that nickel and photoredox catalysis are merged to achieve efficient and versatile difunctionalization of 1,3-enynes.
Introduction
Owing to their ambident reactivity and ready availability, 1,3-enynes have been exploited as versatile building blocks to access various highly useful molecular architectures,1 including propargylic compounds,2 allenes3 and dienes.4 Transition-metal catalyzed difunctionalization of unactivated 1,3-enynes represents one of the most powerful approaches.5,6 In this context, radical 1,4-difunctionalization of 1,3-enynes has drawn much attention recently,7 and reports from the groups of Liu,7a Bao,7b–d Liu,7e and Wang,7h among others, demonstrated practicality and great potential of such synthetic strategies. Despite all the impressive advances that have been made to date, a number of notable limitations exist: (1) most reports are focused on alkyl or perfluoroalkyl radicals, and N-, Si-, P-, and S-radicals are virtually not explored. (2) Reaction conditions are often harsh, and excess oxidants, bases, or high temperatures are often required. (3) Radical 3,4-addition of 1,3-enynes remains elusive.The merger of photoredox and transition metal catalysis has recently become a powerful synthetic strategy for the discovery of novel transformations.8 In this regard, dual nickel/photoredox catalysis evolved tremendously, finding remarkable applications in carbon–carbon bond forming reactions under mild conditions.9 Notably, a number of investigations are focused on 1,2-difunctionalization of conventional alkenes and alkynes (Fig. 1A).10,11 In contrast, difunctionalization of 1,3-enynes via Ni/photoredox dual catalysis is unknown (Fig. 1B). 1,3-Enynes contain conjugated carbon–carbon double bonds and triple bonds, thus controlling regioselectivities of such substrates in radical reactions is synthetically very challenging. If the radical addition takes place at the double bond site, functionalized propargylic compounds and allenes may be formed, via 1,2-addition and 1,4-addition pathways, respectively. Yet, another possibility is that the radical may add to the carbon–carbon triple bond, forming 1,3-dienes via a 3,4-addition pathway. Moreover, the presence of the carbon–carbon double bond in 3,4-difunctionalization products would naturally mean the existence of E/Z isomers, making this difficult synthetic task even more intimidating. Apparently, unproductive substrate cross-coupling poses another potential problem. Very recently, Glorius et al. reported radical carbonyl propargylation between 1,3-enynes and aldehydes by synergistic chromium and photoredox catalysis.12 Later, we developed the copper/photoredox dual-catalyzed decarboxylative 1,4-carbocyanation of 1,3-enynes, providing access to tetra-substituted allenes.13 Encouraged by these results, we hypothesized that by merging visible light photoredox and nickel catalysis, riding on the powerful single electron transfer of various nickel species, and with careful selection of radical precursors and 1,3-enyne substrates, regiodivergent difunctionalization of 1,3-enynes may be feasible.
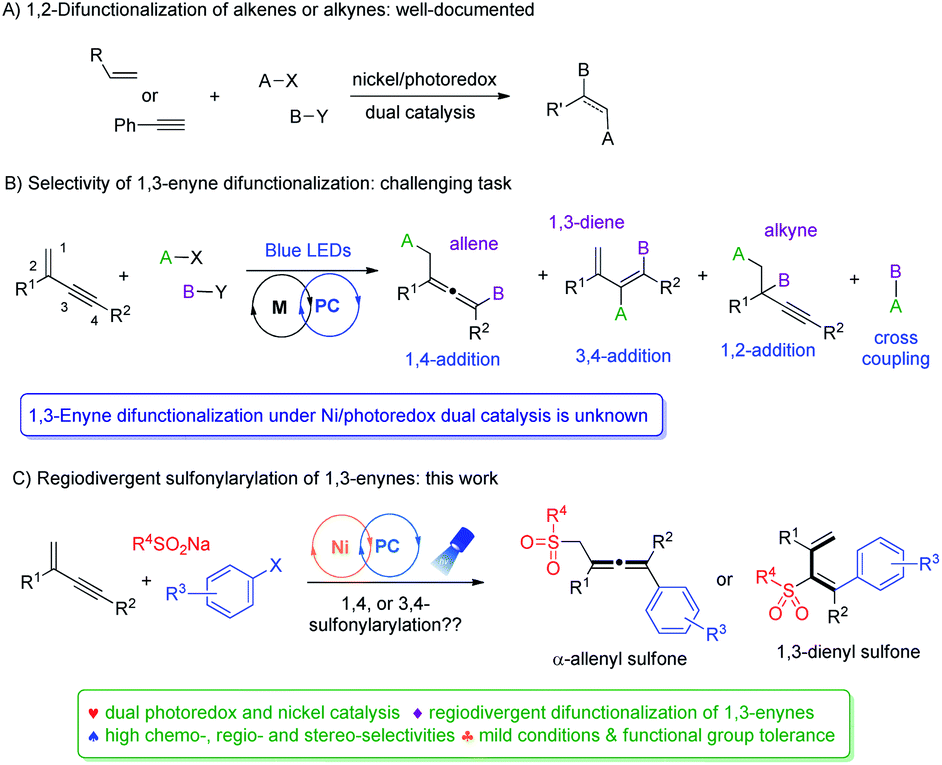 | ||
| Fig. 1 Difunctionalization of unsaturated bonds enabled by photoredox/nickel dual catalysis. | ||
Sulfone-containing molecules possess unique electronic properties and structural features, and consequently, they have played prominent roles in medicinal chemistry, materials science, and organic synthesis.14,15 Conventionally, sulfones are prepared through oxidation of sulfides, aromatic sulfonylation, and alkylation/arylation of sulfinates.16 These methods however do suffer from some serious drawbacks, e.g. the necessity of using foul-smelling thiols and employment of strong oxidants, and the requirement of harsh acidic conditions and high reaction temperatures. In this context, the addition of the sulfonyl radical to unsaturated bonds represents an attractive green approach to the efficient synthesis of various sulfone molecules. Allenic and 1,3-dienyl sulfones have shown to be versatile in conjugate addition, as well as in cyclization and cycloaddition reactions,17–19 yet, efficient synthesis of these molecules is under-developed. We therefore targeted their efficient preparation by employing readily available and cheap sulfonating agents, ideally via divergent synthetic pathways utilizing the same types of starting materials. Herein, we disclose divergent creation of α-allenyl sulfones and 1,3-dienyl sulfones by merging photoredox and nickel catalysis. Hinging on the alkyne terminal substituent, either 1,4-difunctionalization or 3,4-difunctionalization of 1,3-enynes took place smoothly under mild conditions, forming α-allenyl sulfones or 1,3-dienyl sulfones, respectively, and all the reactions proceeded with high chemo-, regio- and stereoselectivity (Fig. 1C).
Results and discussion
We started our investigation by evaluating the reaction of 1,3-enyne 1a, 4-bromobenzaldehyde 2a and sodium 4-methylbenzenesulfinate 3a in the presence of NiCl2·glyme, the dtbbpy ligand and Ru(bpy)3Cl2·6H2O under blue-light irradiation (Table 1). To our delight, regiospecific 1,4-addition was observed, and no 1,2- or 3,4-addition product could be detected. The molar ratio of reactants was found to be crucial for achieving high chemoselectivity. The employment of excess 1,3-enyne 1a significantly favored the 1,4-sulfonylarylation product 4a, and inhibited the undesired coupling between bromide 2a and sulfinate 3a (see ESI, Table S1†). It is noteworthy that the solvent also markedly influenced the reactivity and DMSO was the optimal solvent. Several common photocatalysts were subsequently examined, and inexpensive organic photosensitizer 4CzIPN (1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene) was found to be the best (Table 1, entries 1–4). Screening of nickel precatalysts and ligands was followed, and NiCl2·glyme (5 mol%) and diOMebpy (7 mol%) were identified to be the best combination, leading to the formation of sulfonylated allene product 4a in 72% isolated yield in a regiospecific manner (entries 5–10, for a complete screening, see ESI, Table S2†). We also performed a number of control experiments, showing that the photocatalyst, nickel precatalyst, ligand, light, and inert nitrogen were all essential, without which the reaction would not take place (entries 11–15).|
|
||||
|---|---|---|---|---|
| Entry | PC | [Ni] | Ligand | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.1 mmol) and 3a (0.12 mmol) in DMSO (1.0 mL), photocatalyst (1 mol%), nickel-precatalyst (10 mol%), ligand (14 mol%), at room temperature, 30 W blue LEDs, 20 h. b Isolated yield. c NiCl2·glyme (5 mol%), diOMebpy (7 mol%). d No light. e Reaction was performed in air. | ||||
| 1 | Ru(bpy)3Cl2·6H2O | NiCl2·glyme | dtbbpy | 55 |
| 2 | Ru(bpy)3(PF6)2 | NiCl2·glyme | dtbbpy | 45 |
| 3 | [Ir(dFCF3ppy)2(dtbbpy)]PF6 | NiCl2·glyme | dtbbpy | 62 |
| 4 | 4CzIPN | NiCl2·glyme | dtbbpy | 64 |
| 5 | 4CzIPN | NiCl2 | dtbbpy | 35 |
| 6 | 4CzIPN | NiCl2·6H2O | dtbbpy | 11 |
| 7 | 4CzIPN | NiCl2·glyme | diOMebpy | 73 |
| 8 | 4CzIPN | NiCl2·glyme | bpy | 62 |
| 9 | 4CzIPN | NiCl2·glyme | Phen | 0 |
| 10c | 4CzIPN | NiCl2·glyme | diOMebpy | 72 |
| 11 | — | NiCl2·glyme | diOMebpy | 0 |
| 12 | 4CzIPN | — | diOMebpy | 0 |
| 13 | 4CzIPN | NiCl2·glyme | — | 0 |
| 14d | 4CzIPN | NiCl2·glyme | diOMebpy | 0 |
| 15e | 4CzIPN | NiCl2·glyme | diOMebpy | 0 |
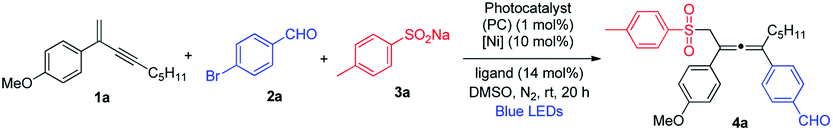
With the optimized reaction conditions in hand, we investigated the substrate scope for the 1,4-sulfonylarylation of 1,3-enynes, first focusing on the variation of 1,3-enyne structures, and the results are summarized in Scheme 1. For the alkene C2-aryl substituent, aryls containing both electron-donating and electron-withdrawing groups were well tolerated, and the corresponding tetra-substituted allenes were obtained in good yields (4a–4e). The reaction was applicable to aryls with different substitution patterns (4f and 4g). 1,3-Enyne 1h with a 2-naphthyl substituent was also found to be a suitable substrate, forming the corresponding allene in a satisfactory yield (4h). The C2-substituents for 1,3-enynes were not limited to aryls, 1,3-enynes bearing a 2-methyl group could also be employed, and the allene products were obtained in moderate to high yields (4i–4m). The alkynyl moiety in the 1,3-enyne structures could also be varied; the C4-substituent could be linear alkyl chains of different lengths (4n and 4o), or branched alkyl groups (4p and 4q), and consistently good yields were obtained. Furthermore, when 1,3-enyne substrates with structurally diverse C4-linear alkyl chains, i.e. bearing a chloride, a phenyl group, and a remote alkene function, were employed, the desired allene products were formed in acceptable yields (4r–4v). Interestingly, when sterically highly demanding 1,3-enyne 1w bearing a C4-tert-butyl substituent was utilized, we were able to isolate the very congested tetra-substituted allene 4w, albeit in low yield. To our surprise, when 1,3-enyne containing an internal alkene was subjected to the reaction, the allene product 4x was obtained with excellent diastereoselectivity (dr > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
 | ||
| Scheme 1 Variation of 1,3-enynes. aConditions: 1 (0.4 mmol), 2a (0.2 mmol) and 3 (0.24 mmol) in DMSO (2.0 mL), 4CzIPN (1 mol%), NiCl2·glyme (5 mol%), diOMebpy (7 mol%), at room temperature, 30 W blue LEDs, 20 h. Isolated yield. bNiCl2·glyme (10 mol%), diOMebpy (14 mol%). | ||
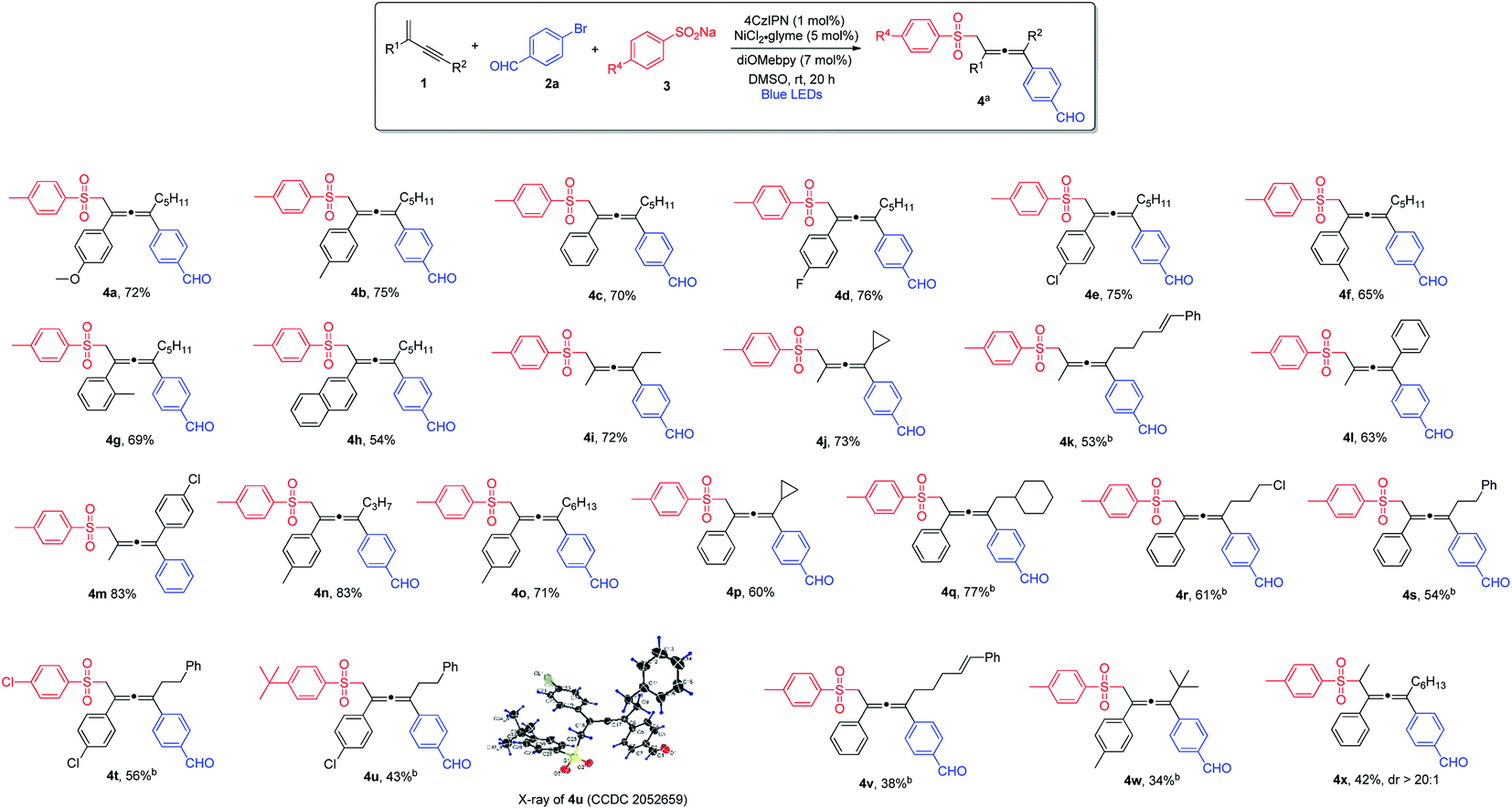
Next, we explored the scope of aryl halides (Scheme 2). In general, aryl bromides and aryl iodides bearing electron-withdrawing substituents were found to be the most suitable substrates, whereas aryl halides with electron-donating substituents tended to deliver the products in lower chemical yields. Notably, this mild synthetic protocol was compatible with a wide range of functional groups and structural moieties, including ketone (5a and 5h), nitrile (5b and 5n), trifluoromethyl (5c), aldehyde (5g) and ester (5l). Moreover, the allene products containing chlorine or fluorine on the aromatic ring were obtained with equal efficiency (5d, 5i and 5k), and such halogen atoms could be engaged for further structural elaborations when necessary. Disubstituted aryl halides and bicyclic substrates including phthalide- and naphthyl-derived halides proved to be good substrates, and the corresponding products were obtained in excellent yields (5q, 5j and 5r). In addition, thiophene-derived iodide worked well in this three-component cross-coupling reaction (5s). However, ortho-substituted aryl halides were found to be unsuitable substrates.
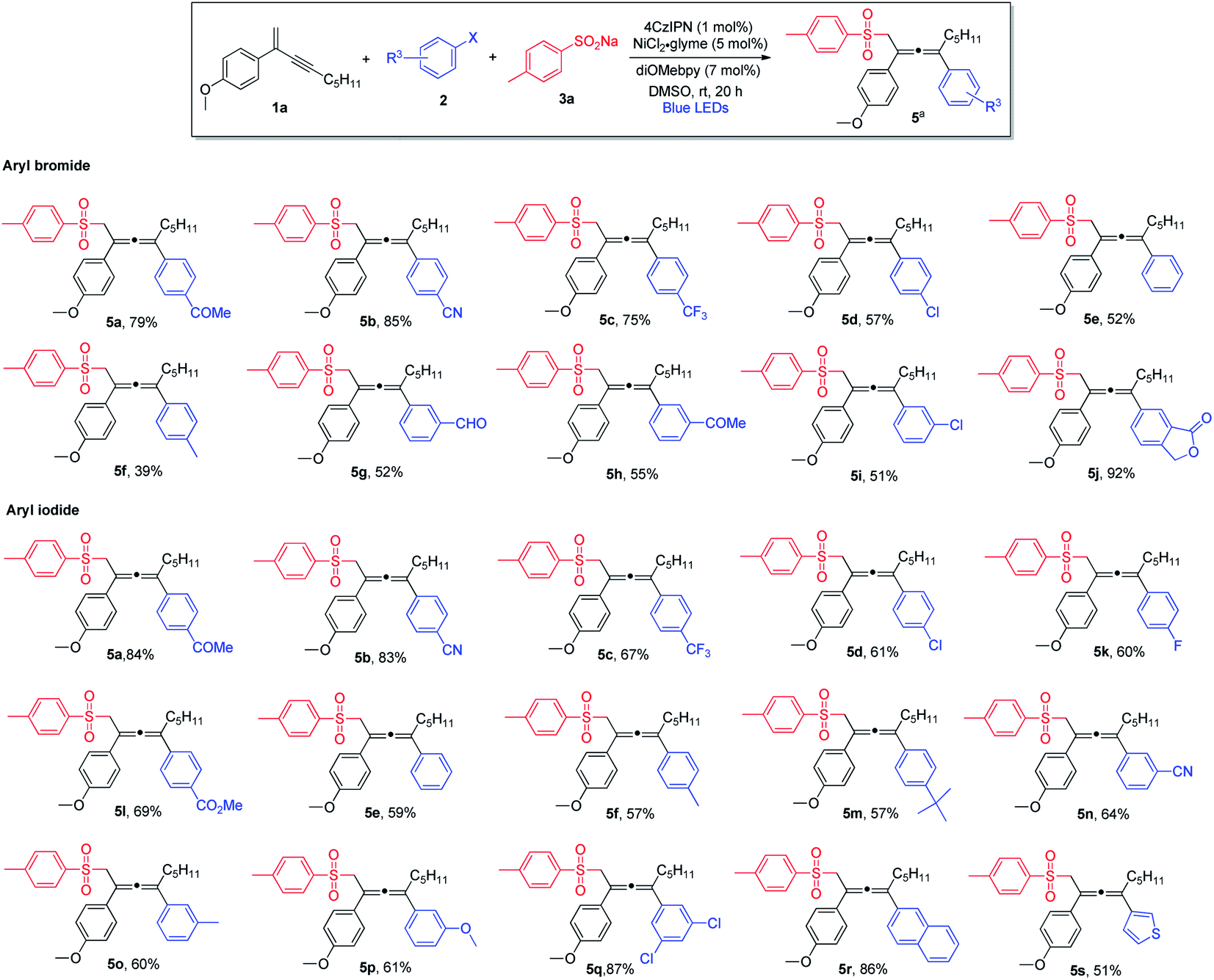 | ||
| Scheme 2 Variation of aryl halides. aConditions: 1a (0.4 mmol), 2 (0.2 mmol) and 3a (0.24 mmol) in DMSO (2.0 mL), 4CzIPN (1 mol%), NiCl2·glyme (5 mol%), diOMebpy (7 mol%), at room temperature, 30 W blue LEDs, 20 h. Isolated yield. | ||
We subsequently investigated the generality of the radical precursors, and accordingly, a series of sodium aryl sulfinates were examined (Scheme 3). Employment of benzenesulfinate and sodium sulfinate containing t-butyl-substituted phenyl led to the formation of the allene products in high yields (6a and 6b). Aryl sulfinates containing fluorine, chlorine, trifluoromethyl, and methyl groups were well tolerated, and the corresponding products were obtained in decent yields (6c–6f). Interestingly, sterically hindered sulfinate was shown to be an excellent substrate as well, and the cross-coupling product 6g was formed in 81% yield. Moreover, the reaction was found to be compatible with sodium naphthyl and thienyl sulfinates (6h and 6i). It is noteworthy that sodium alkyl sulfinate could also be employed, albeit having lower reactivity, and consequently a lower chemical yield was obtained (6j).
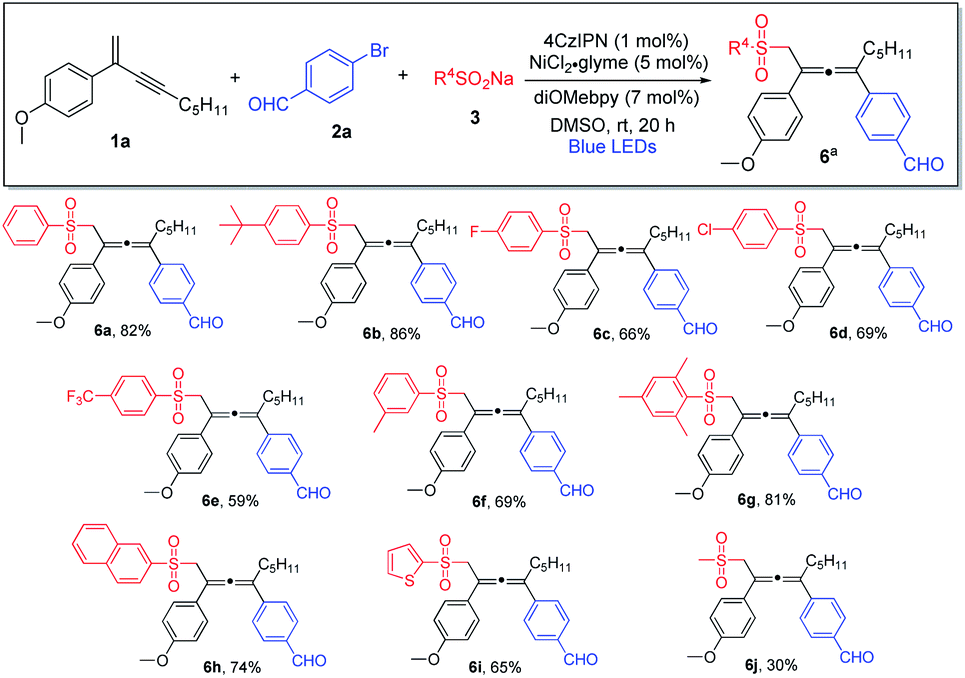 | ||
| Scheme 3 Variation of sulfinates. aConditions: 1a (0.4 mmol), 2a (0.2 mmol) and 3 (0.24 mmol) in DMSO (2.0 mL), 4CzIPN (1 mol%), NiCl2·glyme (5 mol%), diOMebpy (7 mol%), at room temperature, 30 W blue LEDs, 20 h. Isolated yield. | ||
In order to develop regiodivergent sulfonylation of 1,3-enynes, we hypothesized that small structural variation of 1,3-enynes may provide a straightforward solution: the presence of an alkyl or aryl substituent versus a hydrogen atom may affect the stability of radical species formed during the reaction, which may also lead to certain elementary organometallic steps, e.g. migratory insertion, which are more favorable, thus leading to different regiodivergent pathways. To test the above hypothesis, we chose terminal 1,3-enyne 1a′ and examined its reaction under the optimal reaction conditions. Gratifyingly, 2-sulfonyl 1,3-diene 7a was obtained in 70% yield as a single isomer, which was formed via the 3,4-addition pathway (see ESI, Table S4†). We suspected that the E/Z isomerization may take place via the energy transfer process when photocatalysts possessing different triplet state energies are employed. Indeed, when [Ir(dFCF3ppy)2(dtbbpy)]PF6 (ET = 59.4 kcal mol−1)11b or Ir(dFppy)3 (ET = 60.1 kcal mol−1)20 was utilized, the E/Z ratio of product 7a decreased drastically (Table S4†). Similarly, control experiments confirmed that the photocatalyst, nickel catalyst, ligand, and light are indispensable for the observed 3,4-sulfonylarylation of 1,3-enynes (see ESI, Table S5†).
The scope of 3,4-sulfonylarylation of 1,3-enynes for the assembly of (E)-1,3-dienyl sulfones was also found to be broad, which is summarized in Scheme 4. The addition of aryl sulfinates 3 and aryl halides 2 across the triple bond of 1,3-enyne 1′ took place in an anti-fashion to furnish the corresponding 1,3-diene products 7 in moderate to good yields. For terminal 1,3-enynes, aryls bearing both electron-withdrawing and electron-donating groups were found to be suitable, and excellent regio- and stereo-selectivities were attainable (7a–7e). The substitution pattern of aromatic rings was also examined, and 1,3-enynes containing either ortho- or meta-substituted aryl rings appeared to be less ideal substrates (7f–7h). When dioxole-derived 1,3-enyne was employed, the cross-coupling product was obtained in a moderate yield (7i). With regard to the sulfinate substrate, sodium aryl sulfinates bearing electron-rich or electron-deficient aryl groups were equally good, and the products were obtained in pure isomeric forms (7j–7m). Moreover, sodium naphthyl and thienyl sulfinates were both amenable to the reaction (7n and 7o). When different aryl bromides and aryl iodides bearing electron-withdrawing groups were utilized, regardless of the substitution pattern, the 1,3-diene products were obtained in satisfactory yields (7p–7u). However, electron-rich aryl halides failed to form the desired products in reasonable yields. In contrast to the inability of applying ortho-substituted aryl halides to the formation of allene products via 1,2-addition, the 3,4-sulfonylarylation of 1,3-enynes was applicable to ortho-substituted aryl halides (7t and 7u). When aryl halide 2v bearing both iodine and chlorine atoms was subjected to this cross-coupling reaction, chemoselectivity was observed and the reaction took place solely at the iodide site (7v). The employment of the bicyclic substrate was also well tolerated, and the product was obtained in a good yield (7w).
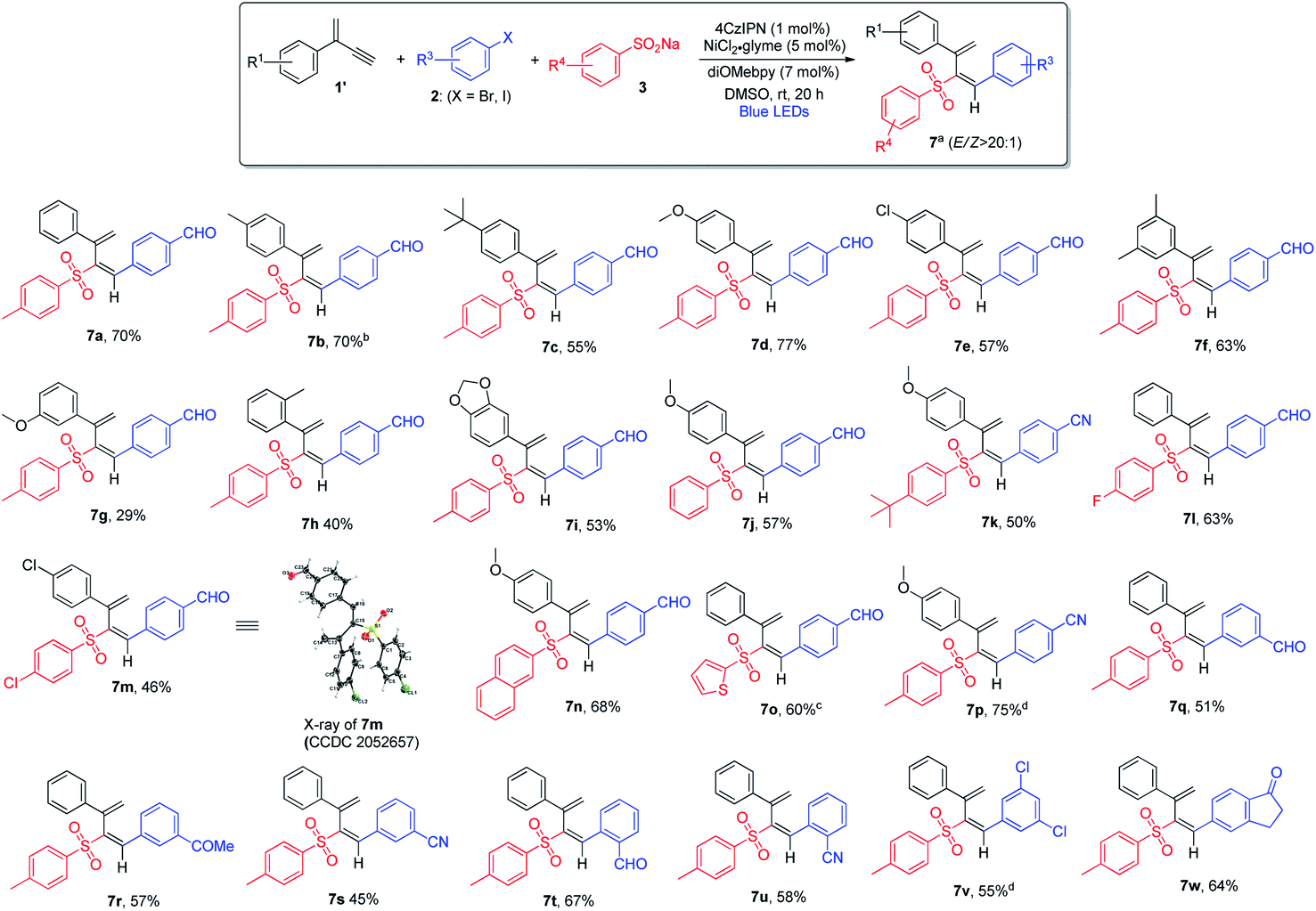 | ||
| Scheme 4 Variation of 3,4-addition of 1,3-enynes. aConditions: 1′ (0.4 mmol), aryl bromide 2 (0.2 mmol) and 3 (0.24 mmol) in DMSO (2.0 mL), 4CzIPN (1 mol%), NiCl2·glyme (5 mol%), diOMebpy (7 mol%), at room temperature, 30 W blue LEDs, 20 h. Isolated yield. Z/E selectivity was determined by 1H NMR analysis. bThe E/Z ratio of product 7b is 14:1. cThe E/Z ratio of product 7o is 11:1. dAryl iodide 2 (0.2 mmol) was utilized. | ||
To highlight the practicability and robustness of our methodology, we carried out gram-scale synthesis of diverse products (Scheme 5). Both allene product 4bvia 1,4-sulfonylarylation of 1,3-enyne, and 1,3-dienyl sulfone 7a through 3,4-sulfonylarylation of 1,3-enyne, were obtained in good yields at the gram-scale. When allene 4b was treated under acidic conditions, 3-sulfonylmethyl 1H-indene 8, which contains the synthetically valuable allylic sulfone moiety,21 was readily prepared in an excellent yield. Iodine, or N-iodosuccinimide (NIS)-mediated cyclization of allene 4a occurred smoothly to deliver different indenyl iodide products 9 or 10. Under palladium catalysis, desulfonylation of allene sulfone 4a and subsequent isomerization led to the formation of 1,3-diene 11 (Scheme 5A). Our new strategy may potentially be extended to include the utilization of other radical precursors. Using TMEDA as a sacrificial electron donor, the employment of tert-butyl bromide led to the formation of the corresponding allene 12 in a good yield (Scheme 5B). We also performed a number of transformations on the 1,3-dienyl sulfone product 7a (Scheme 5C). We first carried out regioselective epoxidation of 7a, as epoxy sulfones are known to be valuable in organic synthesis/medicinal chemistry.22 Treating 7a with m-CPBA led to the formation of unsaturated β,γ-epoxy sulfone 14 in a good yield, while α,β-epoxy sulfone 15 was obtained when t-BuOONa was used as a nucleophilic oxidant. When dienyl sulfone 13 reacted with the Grignard reagent under palladium catalysis, the cross-coupling reaction took place and diene 16 was obtained in 96% yield. Moreover, the two double bonds in 1,3-dienyl sulfones were easily differentiated, and under standard Pd/C conditions, regioselective hydrogenation of 13 formed vinyl sulfone 17 in a moderate yield.
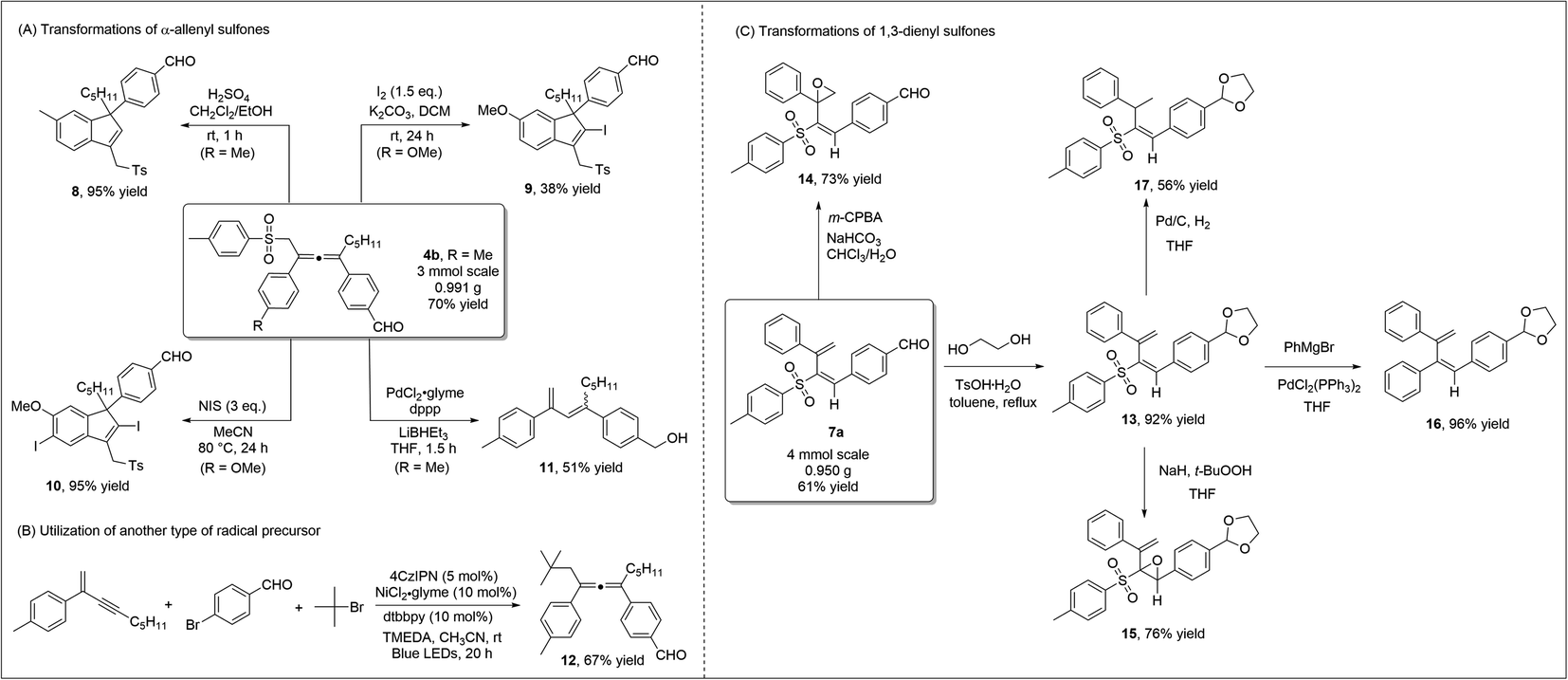 | ||
| Scheme 5 Scale-up reactions and synthetic applications. | ||
To gain insights into the mechanistic pathways leading to 1,4- and 3,4-sulfonylarylation of 1,3-enynes, we performed a number of experiments. Stern–Volmer quenching experiments of all three starting materials were conducted, which revealed that the excited state of 4CzIPN was only quenched by sodium 4-methylbenzenesulfinate, suggesting the involvement of sulfonyl radical species in the reaction processes (Fig. S1–S3 in the ESI†). The radical trapping experiments were also carried out. With the addition of one molar equivalence of radical scavenger TEMPO, the cross-coupling reaction of enyne 1a, bromide 2a, and sulfinate 3a was inhibited, 1,3-diene 18 isomerized from TEMPO-captured allene 19 was isolated in 12% yield, and the allene product 4a was not detected. Similarly, the addition of TEMPO to a mixture of 1,3-enyne 1a′, bromide 2a, and sulfinate 3a under standard reaction conditions completely inhibited the radical reaction pathways, and 1,3-dienyl sulfone 7a was not detected (Fig. 2A). These experiments suggested that both 1,4- and 3,4-difunctionalization reactions of 1,3-enynes most likely proceed via radical reaction pathways. Moreover, the isolation of 1,3-diene 18 provided solid evidence to support the existence of allenyl radicals in the 1,4-sulfonylarylation process. The following experiments were performed to understand the role of active nickel species involved in the reaction. When a mixture of 1,3-enyne 1a and sodium 4-methylbenzenesulfinate 3a was exposed to a stoichiometric amount of the Ni-A complex, the corresponding allene product 5c was not formed. However, treatment of 1,3-enyne 1a, bromide 2c, and sulfinate 3a in the presence of 10 mol% Ni-A led to the formation of desired product 5c in 58% yield (Fig. 2B). The above results imply that the aryl halide-derived Ar–Ni(II) complex may not be the reactive species in the catalytic cycle.
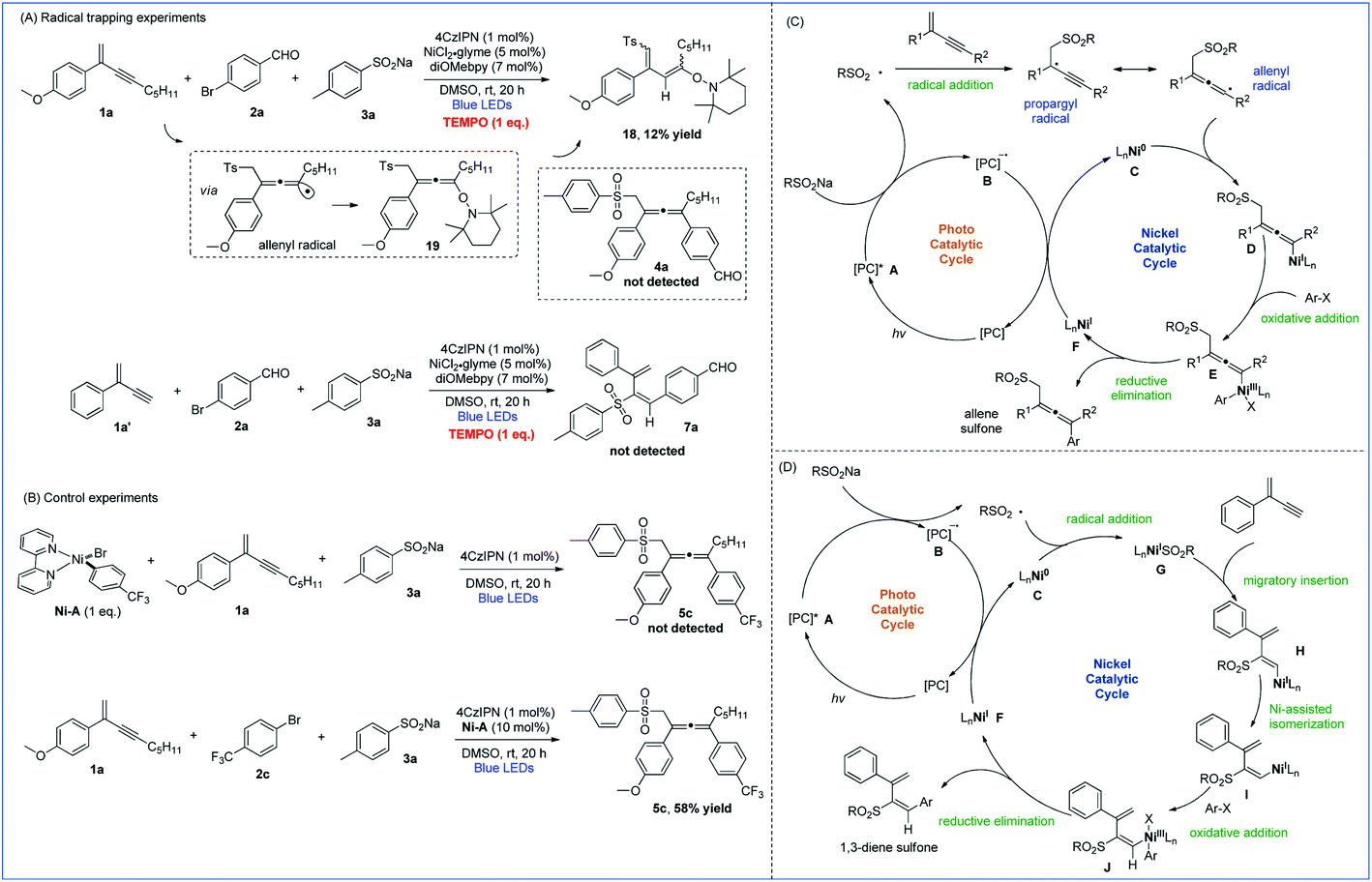 | ||
| Fig. 2 Mechanistic studies. (A) Radical trapping experiments. (B) Control experiments. (C) Proposed mechanism of 1,4-sulfonylarylation. (D) Proposed mechanism of 3,4-sulfonylarylation. | ||
On the basis of the above mechanistic investigations, as well as related literature reports,10b,i mechanisms of regiodivergent 1,4- and 3,4-sulfonylarylation of 1,3-enynes are proposed. In the photocatalytic cycle, excitation of photocatalyst (PC) 4CzIPN (E1/2 [*PC/PC− = + 1.35 V versus SCE in CH3CN])23 leads to its active state A, which then oxidizes sodium sulfinate (TsNa, E1/2 = + 0.45 V versus SCE in CH3CN)24 to generate the sulfonyl radical along with the reduced form B of the PC. When 1,3-enynes with a terminal substituent (R2) are used, the addition of the sulfonyl radical to the double bond of 1,3-enyne creates a propargyl radical, the resonance form of which is a relatively well-stabilized allenyl radical species. The interception of the allenyl radical by Ni0 species C then forms NiI intermediate D. Subsequently, oxidative addition of aryl halide to D furnishes NiIII species E, which undergoes reductive elimination to yield the 1,4-sulfonylarylated allene product and NiI species F. Finally, single-electron-transfer (SET) with PC reduced form B then returns the NiI species to the Ni0 catalyst and regenerates ground-state 4CzIPN (Fig. 2C). On the other hand, when terminal 1,3-enynes are used (R2 = H), compared with pathways of generating propargyl/allenyl radical species, the trapping of the sulfonyl radical by Ni0 specie C becomes more favorable. The sulfonyl NiI species G undergoes 1,2-migratory insertion into the triple bond of 1,3-enyne to generate intermediate H, which transforms to the intermediate Ivia Ni-assisted syn/anti isomerization. The subsequent oxidative addition of aryl halide produces NiIII species J, which readily reductively eliminates to deliver the 3,4-sulfonylarylated 1,3-diene product, and the resulting NiI returns to the Ni0 catalyst in a similar fashion as described in the 1,4-addition pathway (Fig. 2D).
Conclusions
In conclusion, the merger of nickel and photoredox catalysis enables the successful development of a practical catalytic strategy for regiodivergent sulfonylarylation of 1,3-enynes. By utilizing 1,3-enynes with subtle structural variation, efficient synthesis of α-allenyl sulfones or 1,3-dienyl sulfones was accomplished, via 1,4-sulfonylarylation or 3,4-sulfonylarylation of 1,3-enynes, respectively. The reactions proceeded with excellent chemo-, regio- and stereo-selectivities, mild conditions, broad substrate scopes and wide functional group tolerance. Moreover, the protocols described herein could be readily scaled-up, and the products were shown to be versatile in organic synthesis. Preliminary mechanistic investigations suggest the involvement of radical species in the difunctionalization of 1,3-enynes. We anticipate that our approach will stimulate more interest in multi-functionalization of 1,3-enynes and related compounds via photoredox and metal dual catalysis pathways.Data availability
All experimental procedures, characterization, copies of NMR spectra for all new compounds related to this article can be found in the ESI.†Author contributions
Y. C. and K. Z. designed and carried out the experiments, and prepared the ESI† Q. H. participated in the synthesis of partial substrates. Y. C. and Y. L. initiated the project and wrote the paper. Y. L supervised the project overall. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y.L. acknowledges the Singapore National Research Foundation, Prime Minister's Office for the NRF Investigatorship Award (R-143-000-A15-281). Financial support from the National University of Singapore (C-141-000-092-001) is also gratefully acknowledged.Notes and references
- B. M. Trost and J. T. Masters, Chem. Soc. Rev., 2016, 45, 2212 RSC.
- (a) K. Lauder, A. Toscani, N. Scalacci and D. Castagnolo, Chem. Rev., 2017, 117, 14091 CrossRef CAS; (b) H. Qian, D. Huang, Y. Bi and G. Yan, Adv. Synth. Catal., 2019, 361, 3240 CrossRef CAS.
- (a) N. Krause and A. S. K. Hashmi, Modern Allene Chemistry, Wiley-VCH, Weinhein, 2004 CrossRef; (b) A. Hoffmann-Röder and N. Krause, Angew. Chem., Int. Ed., 2004, 43, 1196 CrossRef PubMed; (c) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074 CrossRef CAS.
- N. Wachter-Jurcsak and K. A. Conlon, Synthetic Applications of Dienes and Polyenes, Excluding Cycloadditions, in PATAI'S Chemistry of Functional Groups, John Wiley & Sons, Hoboken, 2009 Search PubMed.
- L. Fu, S. Greßies, P. Chen and G. Liu, Chin. J. Chem., 2020, 38, 91 CrossRef CAS.
- Q. Dherbassy, S. Manna, F. J. T. Talbot, W. Prasitwatcharakorn, G. J. P. Perry and D. J. Procter, Chem. Sci., 2020, 11, 11380 RSC.
- (a) F. Wang, D. Wang, Y. Zhou, L. Liang, R. Lu, P. Chen, Z. Lin and G. Liu, Angew. Chem., Int. Ed., 2018, 57, 7140 CrossRef CAS; (b) X. Zhu, W. Deng, M.-F. Chiou, C. Ye, W. Jian, Y. Zeng, Y. Jiao, L. Ge, Y. Li, X. Zhang and H. Bao, J. Am. Chem. Soc., 2019, 141, 548 CrossRef CAS; (c) C. Ye, Y. Li, X. Zhu, S. Hu, D. Yuan and H. Bao, Chem. Sci., 2019, 10, 3632 RSC; (d) Y. Zeng, M.-F. Chiou, X. Zhu, J. Cao, D. Lv, W. Jian, Y. Li, X. Zhang and H. Bao, J. Am. Chem. Soc., 2020, 142, 18014 CrossRef CAS PubMed; (e) X.-Y. Dong, T.-Y. Zhan, S.-P. Jiang, X.-D. Liu, L. Ye, Z.-L. Li, Q.-S. Gu and X.-Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 2160 CrossRef CAS; (f) Y. Song, S. Song, X. Duan, X. Wu, F. Jiang, Y. Zhang, J. Fan, X. Huang, C. Fu and S. Ma, Chem. Commun., 2019, 55, 11774 RSC; (g) J. Terao, F. Bando and N. Kambe, Chem. Commun., 2009, 7336 RSC; (h) K.-F. Zhang, K.-J. Bian, C. Li, J. Sheng, Y. Li and X.-S. Wang, Angew. Chem., Int. Ed., 2019, 58, 5069 CrossRef CAS.
- For selected reviews, see: (a) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 52 CrossRef CAS; (b) D. C. Fabry and M. Rueping, Acc. Chem. Res., 2016, 49, 1969 CrossRef CAS PubMed; (c) M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Acc. Chem. Res., 2016, 49, 2261 CrossRef CAS PubMed; (d) A. Hossain, A. Bhattacharyya and O. Reiser, Science, 2019, 364, 450 CrossRef PubMed; (e) A. Lipp, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2021, 60, 1714 CrossRef CAS PubMed; (f) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035 CrossRef CAS PubMed; (g) Y. Chen, L.-Q. Lu, D.-G. Yu, C.-J. Zhu and W.-J. Xiao, Sci. China: Chem., 2019, 62, 24 CrossRef CAS; (h) H.-H. Zhang, H. Chen, C. Zhu and S. Yu, Sci. China: Chem., 2020, 63, 637 CrossRef CAS; (i) W. Zhou, Y. Jiang, L. Chen, K. Liu and D.-G. Yu, Chin. J. Org. Chem., 2020, 40, 3697 CrossRef; (j) M. Zhou, P. Qin, L. Jing, J. Sun and H. Du, Chin. J. Org. Chem., 2020, 40, 598 CrossRef; (k) F.-D. Lu, G.-F. He, L.-Q. Lu and W.-J. Xiao, Green Chem., 2021, 23, 5379 RSC.
- (a) J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2019, 58, 6152 CrossRef CAS PubMed; (b) S. O. Badir and G. A. Molander, Chem, 2020, 6, 1327 CrossRef CAS PubMed; (c) C. Zhu, H. Yue, L. Chu and M. Rueping, Chem. Sci., 2020, 11, 4051 RSC.
- For selected examples on difunctionalization of alkenes, see: (a) L. Guo, H. Y. Tu, S. Zhu and L. Chu, Org. Lett., 2019, 21, 4771 CrossRef CAS PubMed; (b) A. Garcia-Dominguez, R. Mondal and C. Nevado, Angew. Chem., Int. Ed., 2019, 131, 12414 CrossRef; (c) M. W. Campbell, J. S. Compton, C. B. Kelly and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 20069 CrossRef CAS PubMed; (d) R. S. Mega, V. K. Duong, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2020, 59, 4375 CrossRef CAS; (e) S.-Z. Sun, Y. Duan, R. S. Mega, R. J. Somerville and R. Martin, Angew. Chem., Int. Ed., 2020, 59, 4370 CrossRef CAS; (f) Z. Zhang and X. Hu, ACS Catal., 2019, 10, 777 CrossRef; (g) S. Zheng, Z. Chen, Y. Hu, X. Xi, Z. Liao, W. Li and W. Yuan, Angew. Chem., Int. Ed., 2020, 59, 17910 CrossRef CAS PubMed; (h) S. Xu, H. Chen, Z. Zhou and W. Kong, Angew. Chem., Int. Ed., 2021, 60, 7405 CrossRef CAS; (i) L. Huang, C. Zhu, L. Yi, H. Yue, R. Kancherla and M. Rueping, Angew. Chem., Int. Ed., 2020, 59, 457 CrossRef CAS PubMed; (j) L. Guo, M. Yuan, Y. Zhang, F. Wang, S. Zhu, O. Gutierrez and L. Chu, J. Am. Chem. Soc., 2020, 142, 20390 CrossRef CAS PubMed; (k) H. Jiang, X. Yu, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 14399 CrossRef CAS.
- For selected examples on difunctionalization of alkynes, see: (a) L. Guo, F. Song, S. Zhu, H. Li and L. Chu, Nat. Commun., 2018, 9, 4543 CrossRef PubMed; (b) C. Zhu, H. Yue, B. Maity, I. Atodiresei, L. Cavallo and M. Rueping, Nat. Catal., 2019, 2, 678 CrossRef CAS; (c) H. Yue, C. Zhu, R. Kancherla, F. Liu and M. Rueping, Angew. Chem., Int. Ed., 2020, 59, 5738 CrossRef CAS PubMed.
- H.-M. Huang, P. Bellotti, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2021, 60, 2464 CrossRef CAS PubMed.
- Y. Chen, J. Wang and Y. Lu, Chem. Sci., 2021, 12, 11316 RSC.
- (a) G. H. Whitham, Organosulfur Chemistry, Oxford University Press, Oxford, 1995 Search PubMed; (b) X. Jiang, Sulfur Chemistry, Springer, Berlin, 2019 CrossRef.
- (a) N. S. Simpkins, Sulphones in Organic Synthesis, Elsevier, Amsterdam, 2013 Search PubMed; (b) E. Schaumann, Sulfur-Mediated Rearrangements II, Springer, Berlin, 2007 Search PubMed; (c) M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200 CrossRef CAS PubMed.
- N.-W. Liu, S. Liang and G. Manolikakes, Synthesis, 2016, 48, 1939 CrossRef CAS.
- J.-E. Bäckvall, R. Chinchilla, C. Nájera and M. Yus, Chem. Rev., 1998, 98, 2291 CrossRef PubMed.
- T. G. Back, K. N. Clary and D. Gao, Chem. Rev., 2010, 110, 4498 CrossRef CAS PubMed.
- D. Yadav and R. S. Menon, Org. Biomol. Chem., 2020, 18, 365 RSC.
- A. Singh, K. Teegardin, M. Kelly, K. S. Prasad, S. Krishnan and J. D. Weaver, J. Organomet. Chem., 2015, 776, 51 CrossRef CAS.
- A. El-Awa, M. N. Noshi, X. M. du Jourdi and P. L. Fuchs, Chem. Rev., 2009, 109, 2315 CrossRef CAS PubMed.
- (a) T. Durst, K.-C. Tin, F. D. Reinach-Hirtzbach, J. M. Decesare and M. D. Ryan, Can. J. Chem., 1979, 57, 258 CrossRef CAS; (b) C. Nájera and J. M. Sansano, Tetrahedron, 1990, 46, 3993 CrossRef; (c) D. Dana, T. K. Das, I. Kumar, A. R. Davalos, K. J. Mark, D. Ramai, E. J. Chang, T. T. Talele and S. Kumar, Chem. Biol. Drug Des., 2012, 80, 489 CrossRef CAS PubMed.
- J. Luo and J. Zhang, ACS Catal., 2016, 6, 873 CrossRef CAS.
- H. Yue, C. Zhu and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 1371 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2052657 and 2052659. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04320j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |