
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Flash chemistry enables high productivity metalation-substitution of 5-alkyltetrazoles†
Jeff Y. F.
Wong
a,
Christopher G.
Thomson
a,
Filipe
Vilela
 ab and
Graeme
Barker
*ab
ab and
Graeme
Barker
*ab
aInstitute of Chemical Sciences, Heriot-Watt University, Riccarton, Edinburgh, EH14 4AS, UK. E-mail: graeme.barker@hw.ac.uk
bContinuum Flow Lab, Heriot-Watt University, Riccarton, Edinburgh, EH14 4AS, UK
First published on 15th September 2021
Abstract
Tetrazoles play a prominent role in medicinal chemistry due to their role as carboxylate bioisosteres but have largely been overlooked as C–H functionalisation substrates. We herein report the development of a high-yielding and general procedure for the heterobenzylic C–H functionalisation of 5-alkyltetrazoles in up to 97% yield under batch conditions using a metalation/electrophilic trapping strategy. Through the use of thermal imaging to identify potentially unsafe exotherms, a continuous flow procedure using a flash chemistry strategy has also been developed, allowing products to be accessed in up to 95% yield. This enabled an extremely high productivity rate of 141 g h−1 to be achieved on an entry-level flow system.
Introduction
Tetrazoles are important structural motifs in medicinal chemistry, displaying bioisosterism with carboxylates.1 In addition to a similar size, electronic distribution and pKa,2 tetrazoles display increased metabolic stability vs. the carboxylate equivalent.3 Tetrazole has been shown to be the sixth most common heteroaromatic ring found in drugs listed in the FDA orange book that were commercially available before 2013,4 as is evident from their presence in multiple drugs including Losartan (an antihypertensive),5 Cilostazol (antithrombotic),6 Tomelukast (a leukotriene D4 receptor antagonist),7 and BMS-317180 (a growth hormone secretagogue),8 shown in Fig. 1.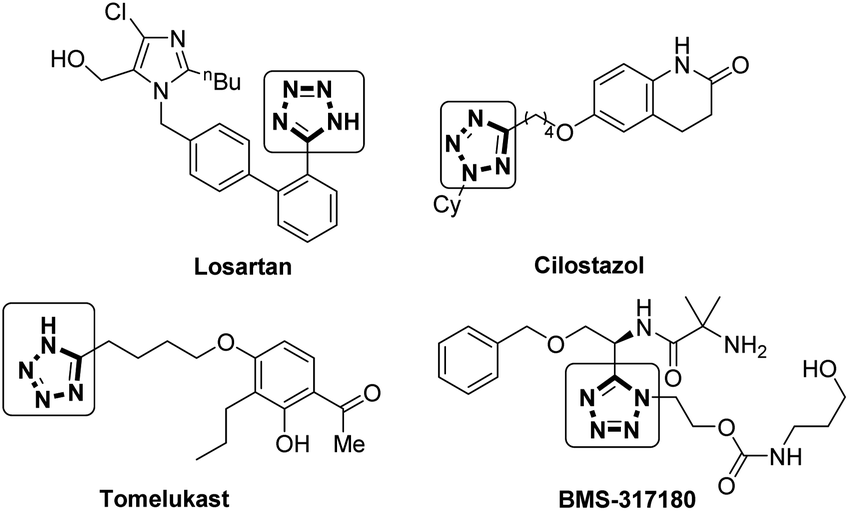 | ||
| Fig. 1 Tetrazole containing drugs. | ||
Despite this privileged position in medicinal chemistry, the α-functionalisation of 5-alkyltetrazoles (analogous to α-functionalisation of carboxylates) remains relatively unexplored.9 Previously reported functionalisations of tetrazoles have focused on alkylation and arylation at the N1- and N2- positions, and require high temperatures and transition metal catalysts.10–12 Ugi-azide reactions are often employed to synthesise highly functionalised tetrazoles, typically utilizing toxic and explosive TMSN3 as the azide source.13
Examples of C–H functionalisation of tetrazole 5-substituents are much rarer.14 Flippin has reported ortho-lithiation of 5-phenyltetrazoles,15 and an early example of lithiation-substitution of N-methyl-5-alkyltetrazoles at the α-position was reported by Thomas et al.,16 however pyrophoric t-BuLi and cryogenic temperatures were required and methods for N-methyl deprotections have yet to be identified, precluding the products from use as carboxylate bioisosteres. Later, a more convenient lithiation-substitution of trityl protected tetrazoles using n-BuLi was reported by Huff et al. (Scheme 1).17 However, in this case deprotection was challenging, requiring the use of gaseous HCl. In both cases, the substrate and electrophile scopes were extremely limited and industrially inconvenient cryogenic (−78 °C) reaction temperatures are required. C–H functionalisations of tetrazole derivatives under continuous flow conditions have yet to be reported.
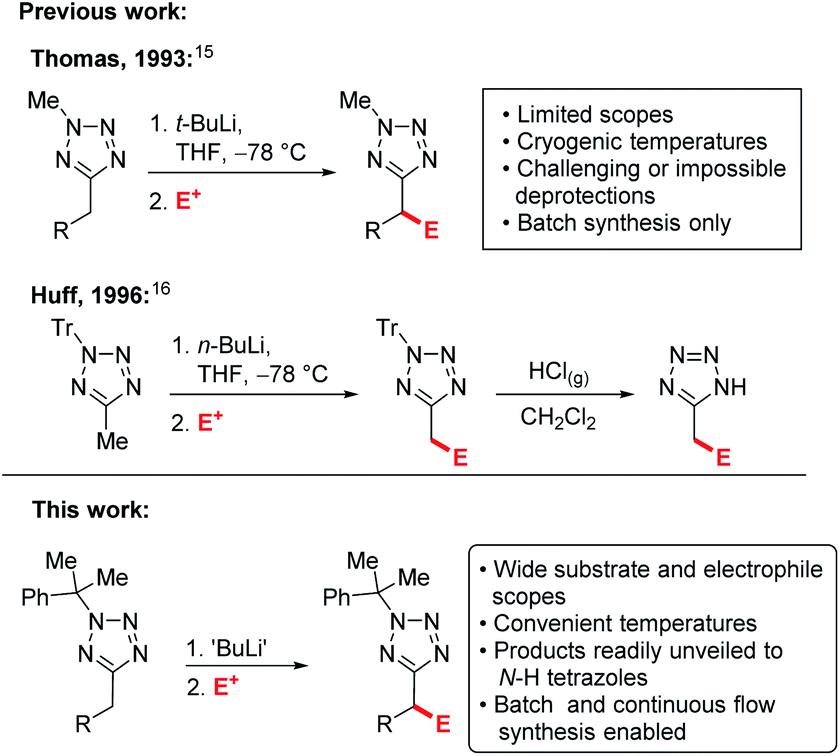 | ||
| Scheme 1 Previous alkyltetrazole lithiations vs. this work. | ||
Recently, our group has developed a lithiation-substitution protocol for 5-benzyltetrazoles using n-BuLi at an industrially convenient temperature.18 Unfortunately, 5-alkyltetrazoles proved not to be amenable to derivatisation using this approach, considerably limiting the substrate scope, and continuous flow conditions could not be optimised due to precipitation of an insoluble metalated intermediate, and the metalation-substitution of unprotected or readily deprotected 5-alkyltetrazoles in the α-position has not previously been described. Herein, we report a general and high-yielding strategy for the C–H functionalisation of 5-alkyltetrazoles under both batch and continuous flow conditions.
Results and discussion
To begin our study, we investigated convenient protecting groups for lithiation-substitution of 5-alkyltetrazoles (Table 1). N-Acyl-5-alkyltetrazoles readily undergo rearrangement to form the corresponding 1,3,4-oxadiazoles,19–22 so the pivaloyl protecting group was discounted. We have found N-Boc tetrazoles challenging to purify and store without slow decomposition, so instead we started with N-trityl-5-propyltetrazole 1, similar to protection employed by Huff et al. for the lithiation-substitution of 5-methyltetrazole.17 Treatment of 1 with n-BuLi at 0 °C for 1 hour followed by addition of acetone as an electrophile gave full recovery of starting material with no evidence for the formation of trapping product 2 (entry 1). Switching base to more reactive s-BuLi at 0 °C gave full consumption of starting material, however no identifiable species were observed by 1H NMR spectroscopy of the crude reaction mixture, suggesting the lithiated species might be unstable at 0 °C (entry 2). Repeating the lithiation at −78 °C gave trapping product 2 in excellent (84%) yield after trapping with acetone (entry 3).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | PG | x | RLi | Time | Temp. | R | Yielda |
| a Isolated yield. | |||||||
| 1 | Trityl | 1.5 | n-BuLi | 1 h | 0 °C | Me | No reaction |
| 2 | Trityl | 1.5 | s-BuLi | 1 h | 0 °C | Me | Complex mixture |
| 3 | Trityl | 1.6 | s-BuLi | 30 min | −78 °C | Me | 84% |
| 4 | CMePh2 | 2.3 | n-BuLi | 15 min | 0 °C | Me | Complex mixture |
| 5 | CMePh2 | 2.3 | n-BuLi | 15 min | −78 °C | Me | No reaction |
| 6 | CMePh2 | 2.3 | s-BuLi | 15 min | −78 °C | Me | 79% |
| 7 | Cumyl | 1.5 | n-BuLi | 1 h | 0 °C | Me | 63% |
| 8 | Cumyl | 2.1 | n-BuLi | 1 h | 0 °C | Me | 60% |
| 9 | Cumyl | 2.1 | n-BuLi | 1 h | −30 °C | Me | 76% |
| 10 | Cumyl | 2.1 | n-BuLi | 1 h | −78 °C | Me | No reaction |
| 11 | Cumyl | 1.6 | s-BuLi | 15 min | −78 °C | Me | 97% |
| 12 | Cumyl | 1.5 | n-BuLi | 15 min | 0 °C | Ph | 68% |
| 13 | Cumyl | 2.3 | n-BuLi | 15 min | 0 °C | Ph | 85% |
| 14 | Cumyl | 1.5 | LDA | 15 min | 0 °C | Me | No reaction |
| 15 | Cumyl | 1.5 | LDA | 15 min | −78 °C | Me | No reaction |
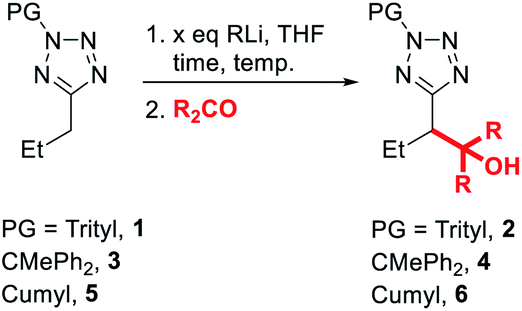
Since we aimed to develop reaction conditions without the need for cryogenic temperatures, we continued to screen other protecting groups. The use of a diphenylethyl protecting group led to no significant changes in results, with efficient lithiation-trapping of 3 observed only with s-BuLi at −78 °C (entries 4, 5 and 6). To our delight, changing the protecting group to cumyl, which has previously been used by Flippin et al. for the nucleophilic aromatic substitution of 5-aryltetrazoles,23 dramatically changed the reactivity. The cumyl group is readily installed in high yield using α-methylstyrene and TFA (see ESI† for full details). We note that α-methylstyrene is a cost efficient source of this protecting group (£53.60/2.5 L, Sigma–Aldrich, 2021) and that Flippin has outlined two complementary procedures for high-yielding deprotections of N-cumyl tetrazoles.23 A modest yield of 6 was obtained when 5 was treated with n-BuLi at 0 °C followed by electrophilic trapping with acetone (entry 7). Neither increasing the equivalents of base (entry 8) nor lowering the temperature to −30 °C (entry 9) gave significant improvement to the yield of product 6. Once again, no lithiation was observed using n-BuLi at −78 °C (entry 10), whilst switching the base to s-BuLi gave excellent yield (97%) of product 6 (entry 11). We speculated that the decrease in yield of 6 when the lithiation was carried out at 0 °C was due to the lithiated 5 acting as a base to deprotonate acetone instead of reacting as a nucleophile. Thus, the lithiation-trapping of 5 was attempted with benzophenone as the electrophile. Treatment of 5 with 1.5 equivalents of n-BuLi at 0 °C gave product 6 in modest (68%) yield after trapping with benzophenone (entry 12). Increasing the loading of n-BuLi to 2.3 equivalents gave 6 in an excellent 85% yield (entry 13). We note that this requirement for an extra equivalent of base over the strict stoichiometric requirements during lithiation of tetrazoles appears to be a common issue, and has been encountered by our group during the lithiation of benzyltetrazoles, as well as Flippin during the ortho-lithiation of 5-aryltetrazoles.15,18 Since no lithiation was observed when milder LDA was used as base (entries 14 and 15), the conditions in entries 11 and 13 were taken as optimal. It is hypothesised that the drop in yield at 0 °C when lengthening reaction time from 15 min to 1 h (entry 13 vs. 8) may arise from instability of the lithiated intermediate.
We next explored the electrophile scope under our optimised conditions (Scheme 2). We were pleased to find that almost all electrophiles were compatible with lithiation at 0 °C (conditions A). Alcohol 9 (75%) and 10 (76%) were isolated in good yield and moderate diastereoselectivities after trapping with benzaldehyde and cyclohexanone respectively. A slight drop in yield was observed with benzylation with BnBr (11, 61%) and silylation with TBDMSCl (12, 66%) with conditions A, however the yields of 12 could be improved to 80% by employing low temperature lithiation conditions with s-BuLi (conditions B). TBDMSCl is generally regarded as a sow-trapping electrophile, and this may allow time for partial decomposition of lithiated 5 at 0 °C, accounting for the lower yield under conditions A. Alkylation could be achieved in good yield with Me2SO4 (13, 77%), iso-pentyl bromide (14, 90%) and an epoxide (17, 56%, 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 dr) with conditions A. Finally, allylation and acylation could be achieved in excellent yields with allyl bromide (15, 90%) and a Weinreb amide (16, 81%). Unfortunately, attempted trapping with iodine resulted in no desired product.
:30 dr) with conditions A. Finally, allylation and acylation could be achieved in excellent yields with allyl bromide (15, 90%) and a Weinreb amide (16, 81%). Unfortunately, attempted trapping with iodine resulted in no desired product.
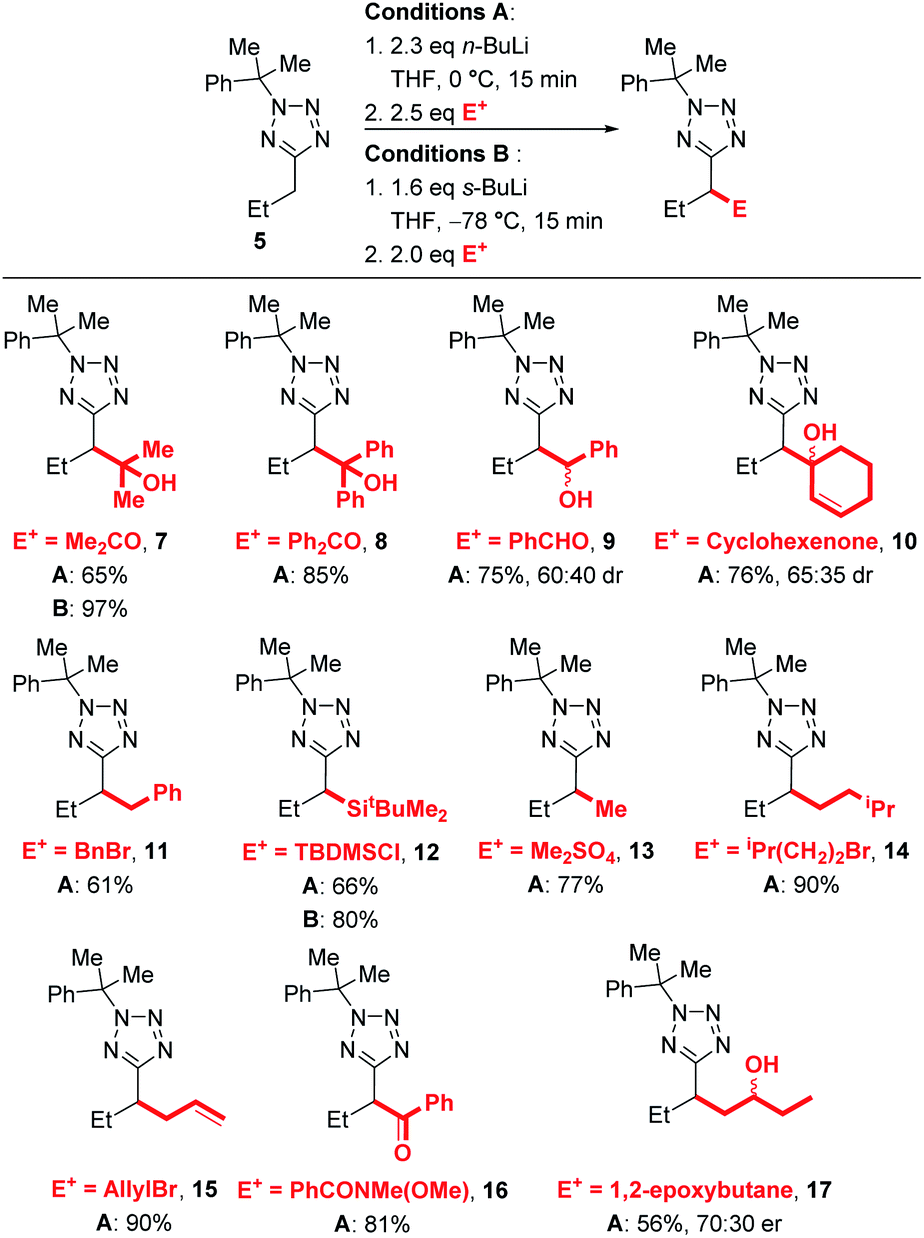 | ||
| Scheme 2 Electrophile scope. | ||
During our previously reported lithiation-substitution of alkyl-1,3,4-oxadiazoles, we observed that di-substituted products may be obtained when trapping with chloroformates, a common phenomenon with this class of electrophile in lithiation chemistry.24,25 We found that di-carbamoylation of 5 to give 18 could be achieved in modest (50%) yield using our optimised conditions A and trapping with an iso-cyanate, while diester 19 could be isolated in 46% yield using methyl chloroformate by increasing the base and electrophile to 3.5 and 4.0 equivalents, respectively (Scheme 3).
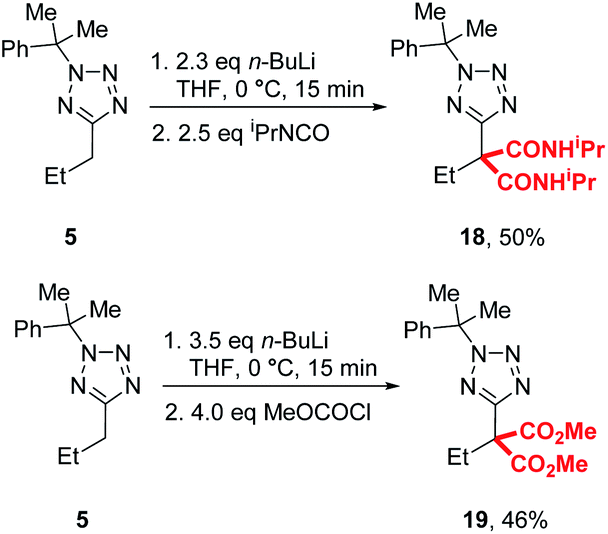 | ||
| Scheme 3 Di-substitution with chloroformates and isocyanates. | ||
Next, we turned our attention to exploring the substrate scope (Scheme 4). Efficient lithiation-substitution was observed with shortening the chain to an ethyl group (20, 70%). 21, bearing a longer alkyl chain, and MOM-protected alcohol 22 were isolated in moderate yield under conditions A (60% and 65%) after trapping with allyl bromide; yields were improved to 98% and quantitative respectively using conditions B. A phenyl group is also well tolerated, with the lithiation-substitution to form 23 in 83% after trapping with benzophenone at 0 °C. Trifluoromethyl (24, 76%) and N-trityl amino (25, 70%) motifs were also well tolerated under conditions A. In the presence of a methoxy group, an inseparable mixture of desired product 26 and cumyl ring lithiation-substitution product was observed when conditions A were employed. Competing ring lithiation could be avoided under conditions B and 26 was isolated in 79% yield. Acetal-containing product 27 was obtained in 50% yield under conditions B. Our previously reported C–H functionalisations of benzyltetrazoles were not amenable to continuous flow chemistry, so for a direct comparison with an attempted flow experiment, a benzyl substrate was reacted under modified conditions A to give 28 in 76% yield – in this case, reduced base was required to avoid disubstitution (see ESI†).
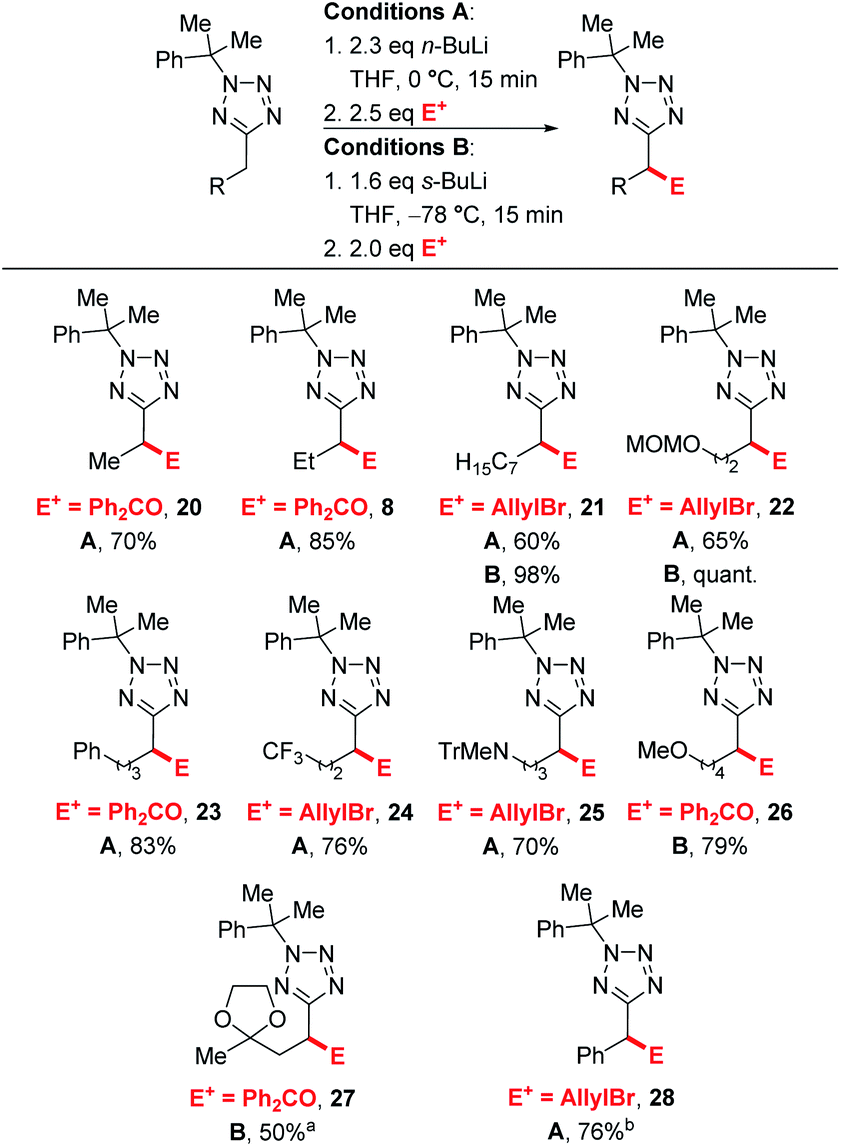 | ||
| Scheme 4 Substrate scope. aUsing 3.2 eq. s-BuLi.LiCl. bUsing 1.5 eq. n-BuLi. | ||
Interestingly, when chlorophenyl derivatives were subjected to our standard lithiation conditions, a mixture of heterobenzylic and benzylic trapping products 30 and 31 were obtained, a result which was not observed when reacting purely phenyl substrates (see 23, Scheme 4). Fortunately, expected product 30 was isolated in 53% vs. 11% for 31. The structure of each product was confirmed by NOESY NMR (see ESI†) and an X-ray crystal structure of 30 (Scheme 5).
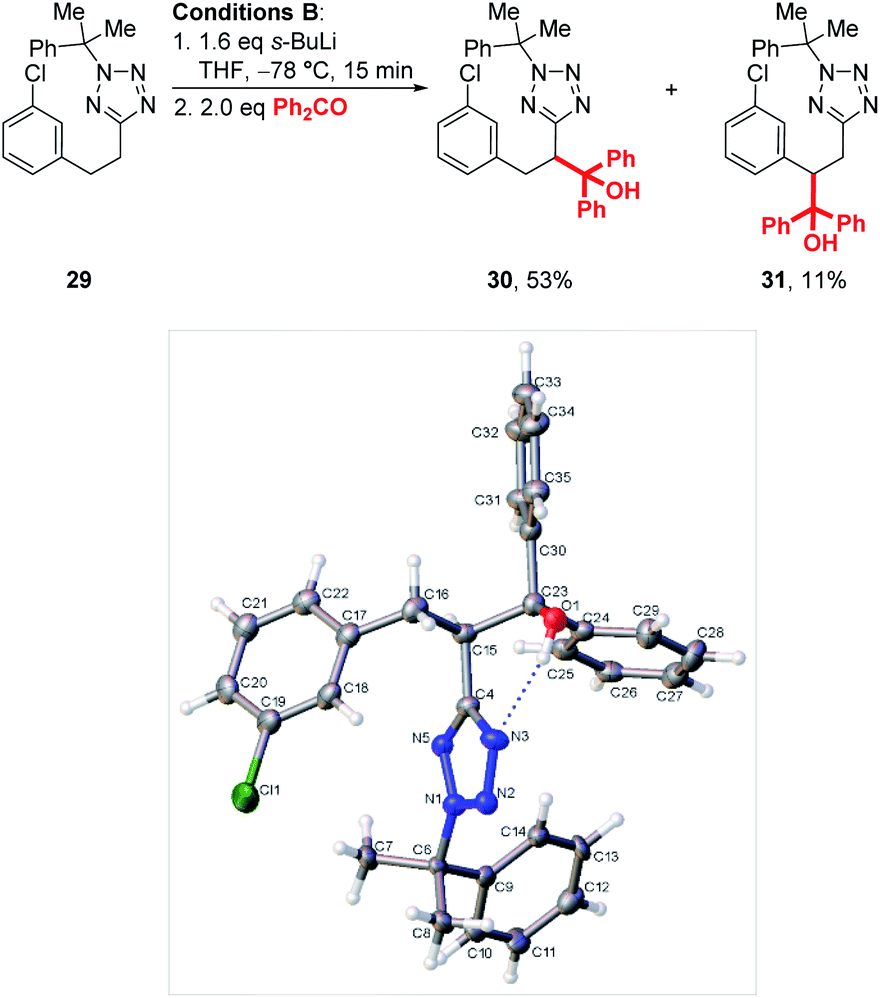 | ||
| Scheme 5 Lithiation-substitution of chlorophenyl substrate 29 and crystal structure of 30. | ||
While investigating the lithiation-substitution of substrates bearing protected amines, we found that N-Boc substrates 32–34 underwent internal electrophilic trapping to give tetrazolyl lactams. Thus, we were able to optimise this synthesis by omitting an external electrophile, and 5-, 6-, and 7-member lactams 35, 36 and 37 were obtained in 80%, 94% and 62% yield respectively (Scheme 6).26
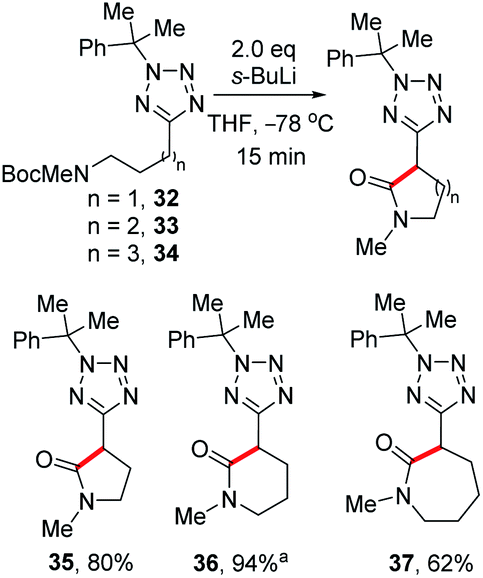 | ||
| Scheme 6 Synthesis of tetrazolyl lactams.aWith 3.2 eq. s-BuLi. | ||
Enantioselective lithiation-substitution protocols mediated by the chiral diamine sparteine are well established for a range of substrate classes,27–29 and we next attempted stereoselective functionalisations of our tetrazoles under the control of a (+)-sparteine/s-BuLi complex in Et2O at −78 °C before electrophilic trapping with acetone (Scheme 7). Unfortunately, product 7 was obtained in only 41% yield and 64:36 er. Investigation of a range of other chiral ligands known to promote enantioselective lithiation substitutions, including a trans-cyclohexane-1,2-diamine pioneered by Alexakis and dipeptides used by Beak and Coldham similarly failed to provide products in satisfactory enantioenrichments.29–44 We hypothesised that this may be due to configurational instability of the lithiated tetrazole intermediate – a Hoffman test using racemic and (S)-2-(dibenzylamino)-1,3-diphenylpropan-1-one as electrophiles confirmed that this is the case (for full details of ligand screen and Hoffman test, see ESI†).45
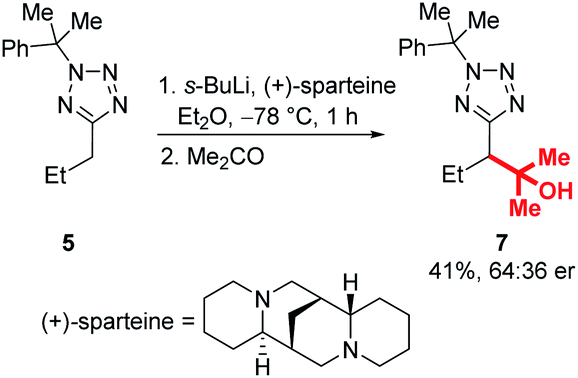 | ||
| Scheme 7 Attempted enantioselective lithiation-substitution. | ||
With a high-yielding batch procedure in hand, we next turned our attention to optimising a continuous flow procedure. Flow chemistry offers significant safety advantages as well as superior control of reaction mixing and thermal transfer,46,47 and as such the use of organolithiums under continuous flow conditions as been well reviewed in recent years.48–52 Flash chemistry, developed by Yoshida and co-workers,53–58 refers to synthesis in which “extremely fast reactions are conducted in a highly controlled manner to produce desired compounds with high selectivity”;54 in addition to intrinsically high productivity rates, impressive transformations via unstable intermediates (e.g. organolithiums bearing highly reactive electrophilic functional groups) may be achieved.51,59–79 Despite these advantages, generation of organolithium species in flow is dominated by halogen-lithium exchanges, with C–H metalations (and particularly Csp3–H metalations) comparatively underexplored.24,80–86
Recently, we reported a continuous flow lithiation-substitutions of alkyl-1,3,4-oxadiazoles with a 1.3 s reactor residence time for lithiation before in-flow electrophilic trapping.24 Quantitative yields were obtained at rt which were not replicable under comparable conditions in a batch reactor, and we were therefore inspired to investigate C–H functionalisations of 5-alkyltetrazoles in flow. We began by attempting to replicate our batch synthesis conditions (modified for room temperature) under continuous flow (Table 2).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | X (M) | Y (M) | Flow ratea (mL min−1) | Residence time (s) | Yieldb | SMb,c |
| a Flow rate from each pump; after junction, flow rate is doubled. b As determined by 1H NMR spectroscopy in the presence of 1,3,5-trimethoxybenzene. c Remaining starting material. | ||||||
| 1 | 2.5 | 1.1 | 5 | 2.6 | 35% | 50% |
| 2 | 2.5 | 0.55 | 5 | 2.6 | Trace | 35% |
| 3 | 1.75 | 0.76 | 5 | 2.6 | 48% | 48% |
| 4 | 1.25 | 0.54 | 5 | 2.6 | 12% | 75% |
| 5 | 0.75 | 0.33 | 5 | 2.6 | 12% | 71% |
| 6 | 0.75 | 0.33 | 1 | 13 | 12% | 56% |
| 7 | 0.75 | 0.33 | 1 | 10 min | 14% | 57% |
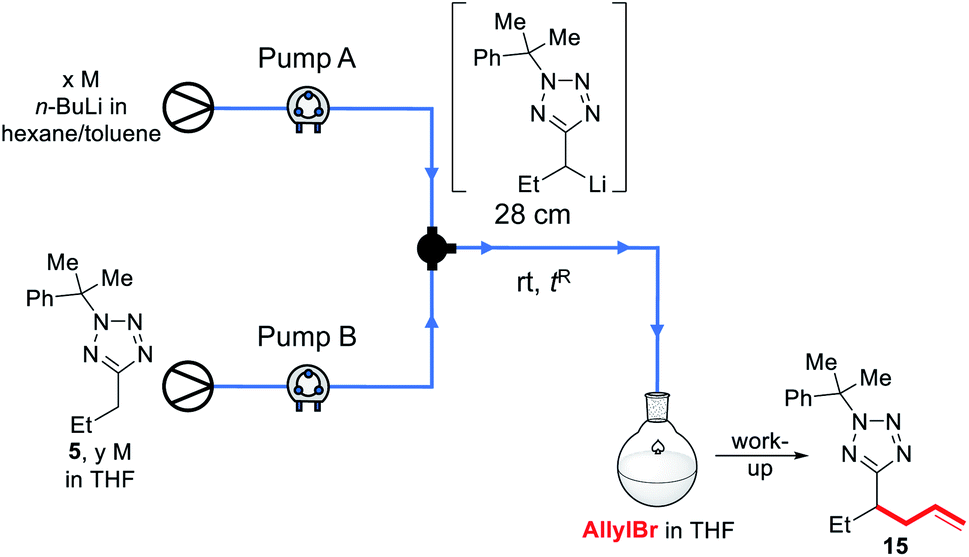
First, a 2.5 M solution of n-BuLi in hexanes/toluene and a 1.1 M solution of 5 in THF were reacted in flow in a 28 cm length of tubing at rt for 2.6 s before being pumped into a stirred solution of allyl bromide in THF at rt; after work up, a 35% conversion to 15 was observed by 1H NMR of the crude reaction mixture (entry 1). Decreasing the concentration of the starting material feedstock solution to 0.55 M resulted in a loss of conversion (entry 2). A 48% conversion was achieved with a 1.75 M concentration of n-BuLi, and 0.76 M of 5 (entry 3). Unfortunately, further dilution of the base and starting material solutions, as well as a lower flow rate led to a decrease in yield (entries 4–7). Before further optimisation could be conducted, however, we observed that under the conditions shown in entries 1 and 3, the solvent appeared to be boiling in the reactor, with bubbles seen to be far in excess of that which could be accounted for by evolution of n-butane. Alarmed by the safety implications of an exothermic reaction resulting in the boil-off of an ethereal solvent, we resolved to investigate further.
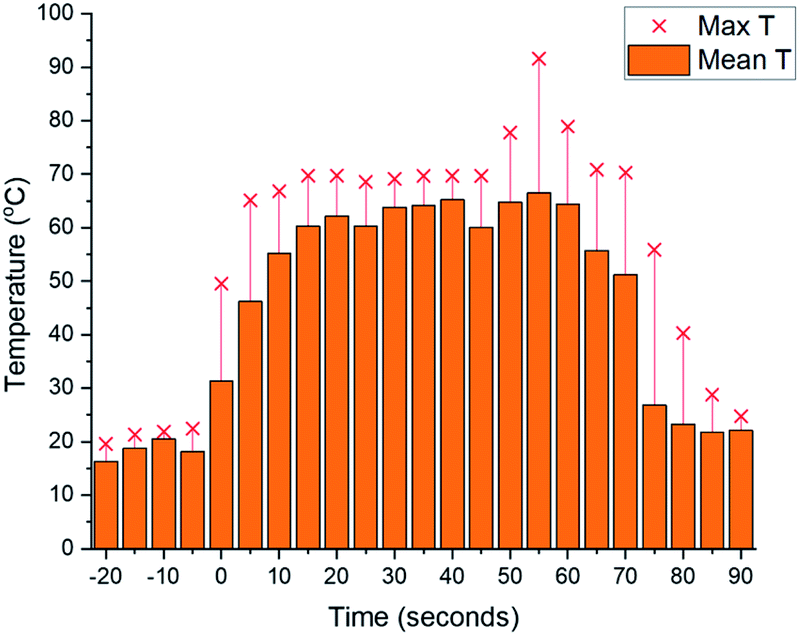
As we aimed to develop a continuous flow process with a short reactor residence time, direct temperature measurements of our flow reaction were judged to potentially impact on the mixing of reaction components and/or residence times, and we turned instead to thermal imaging of the reactor. This strategy has previously been successfully employed by Ley et al. to monitor the exothermic synthesis and decarboxylative dibromination of a glyoxylic acid oxime,87 and by Williams and co-workers to monitor a RANEY® nickel catalyst fixed bed reactor,88 but to our knowledge has not previously been used to study metalation processes or those with reactor residence times under 1 min. Thermal imaging of a flow reaction representative of the conditions shown in Table 2, entry 3 revealed a significant exotherm during the reaction, with an increase from rt while pumping solvent alone to ∼55 °C (averaged over the whole reactor) or ∼70 °C (hottest position on reactor) while the reaction was running, validating our concerns that the reaction was at or near the THF boiling point (Fig. 2).
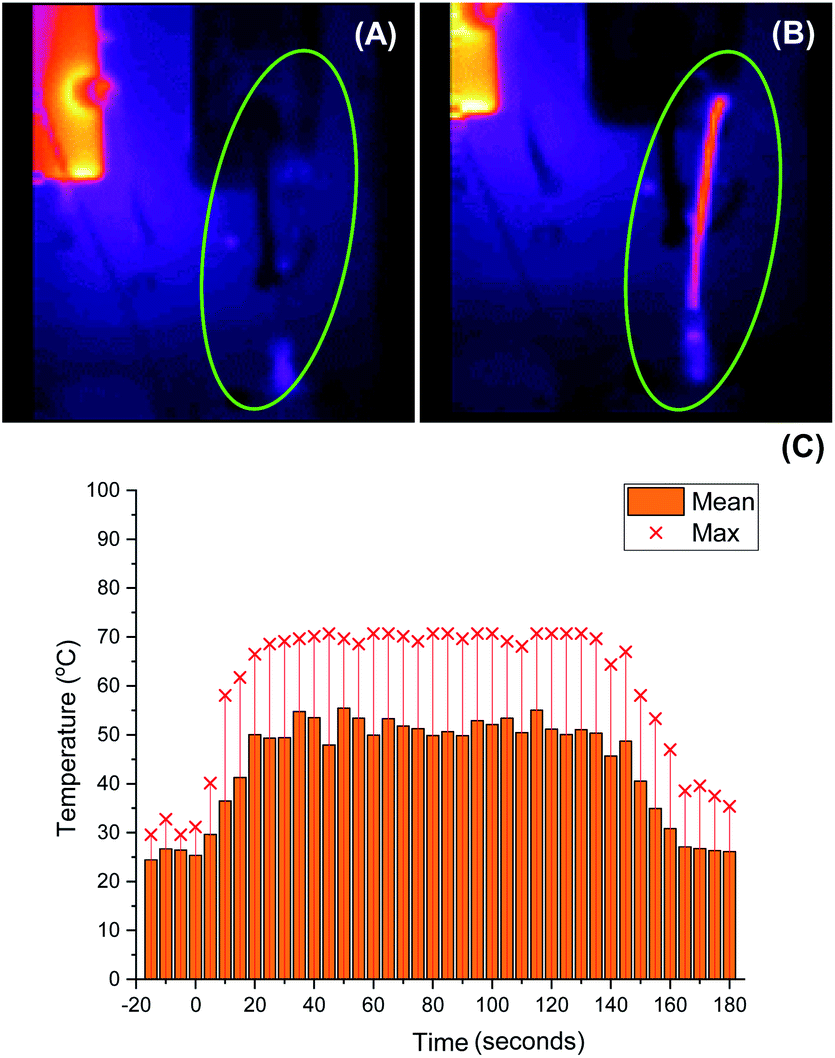 | ||
| Fig. 2 Thermal imaging of flow metalation in THF with the reactor tubing circled in green. (A) While pumping solvent at rt. (B) While reaction is running. (C) Graph of mean and maximum reactor temperatures over time. Only solvent at rt is pumping until 0 s, at which base and starting material solutions (both at rt) are pumped to the reactor. Pumping is switched back to solvent only at rt at 140 s. | ||
We decided instead to re-optimise our continuous flow reaction using a higher boiling and non-ethereal solvent, toluene. Since alkyllithiums are known to display increased reactivity in coordinating solvents such as THF,89–91 TMEDA was added to the feedstock solution of starting material 5 in toluene to achieve the same effect.92 Condition screening results are shown below in Table 3.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | X (M) | Toluene:TMEDA |
Flow ratea (mL min−1) | Residence time (min) | Yieldb | SMb,c |
| a Flow rate from each pump; after junction, flow rate is doubled. b As determined by 1H NMR spectroscopy in the presence of 1,3,5-trimethoxybenzene. c Remaining starting material. d Using a 28 cm length of tubing as a reactor. e No TMEDA used. | ||||||
| 1d | 1.1 | N/Ae | 5 | 2.6 s | 0% | 65% |
| 2 | 1.1 | 4:1 |
1 | 5 | 62% | 8% |
| 3 | 1.1 | 4:1 |
3 | 1.67 | 60% | 29% |
| 4 | 1.1 | 4:1 |
5 | 1 | 59% | 29% |
| 5 | 1.1 | 4:1 |
10 | 0.5 | 49% | 40% |
| 6 | 0.8 | 4:1 |
5 | 1 | 46% | 55% |
| 7 | 1.1 | 1:1 |
5 | 1 | 73% | 14% |
| 8 | 1.1 | 1:1 |
3 | 1.67 | 70% | 10% |
| 9 | 1.1 |
1:1 |
10 | 0.5 | 70% | 8% |
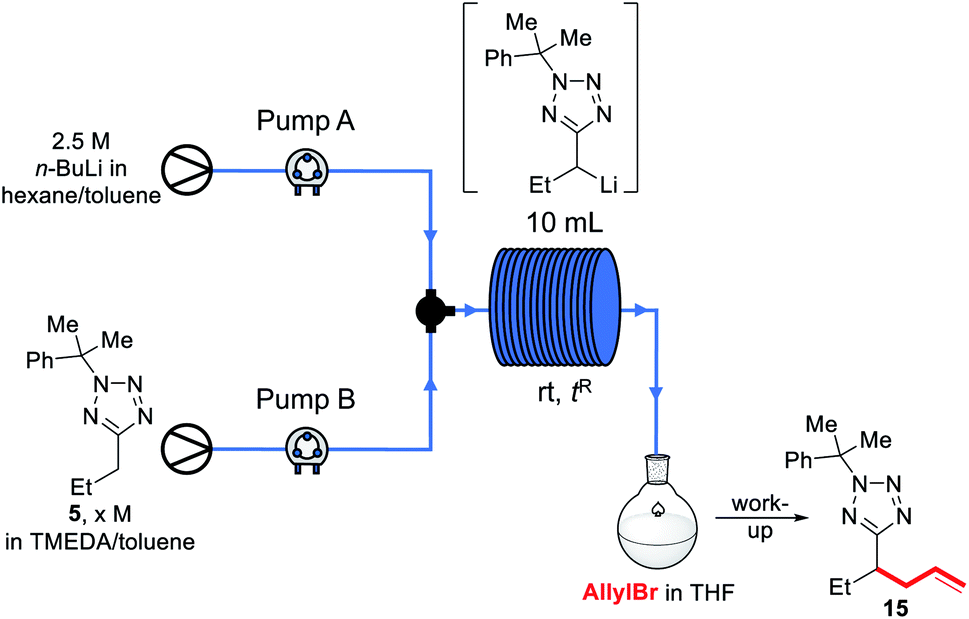
In each case, base and substrate were pumped to meet at a T-junction at the same rate. We initially replicated our THF continuous flow conditions as a control (entry 1) using a 28 cm length of tubing to give a 2.6 s reactor residence time and without TMEDA, however this did not lead to appreciable conversion to product 15, and we instead turned to the use of a 10 mL reactor to give longer residence times. Thus, pumping a 1.1 M solution of 5 in 4:1 toluene:TMEDA and base at 1 mL min−1 to give a residence time of 5 min before pumping into a stirred solution of allyl bromide in toluene gave a 62% conversion to 15, and 8% residual starting material (entry 2), indicating that the lithiated intermediate may be unstable over a 5 min timescale. Neither increasing the flow rate to reduce residence times (entries 3–5) nor reducing the concentration of the substrate solution (entry 6) increased the conversion, though more residual substrate was observed. Instead, the TMEDA:toluene ratio was increased to 1:1; this indeed led to an increase in yield (entries 7–9). As a higher flow rate corresponds to a higher productivity rate, the conditions shown in entry 9 were taken as optimal.
Before exemplifying our optimised flow conditions, we first replicated our thermal imaging experiments to probe any exotherms when using toluene (Fig. 3). Under the conditions show in Table 3, entry 9, during the reaction a mean reactor temperature of ∼65 °C, with a maximum temperature of ∼75 °C at the hottest point of the reactor was observed. We note that the differing solution structures and reactivities of organolithiums in THF and toluene are well documented and may account for this difference in exotherm magnitude and also note that the maximum temperature reached by the THF reaction corresponds to the solvent boiling point.89 While this represents a higher exotherm then when using THF, the reaction system was now free of ethereal solvent and vapourisation of the solvent within the tubing was not observed. We note that flash chemical C–H metalation processes are underexplored, possibly due to resultant exotherms and the difficulty of temperature monitoring. In this context, thermal imaging may supply a general strategy for overcoming current limitations in the field.
 | ||
| Fig. 3 Thermal imaging of flow metalation in toluene/TMEDA: graph of mean and maximum reactor temperatures over time. Only solvent at rt is pumping until 0 s, at which base and starting material solutions (both at rt) are pumped to the reactor. Pumping is switched back to solvent only at rt at 60 s. | ||
Flash chemistry is commonly asserted to operate under plug flow, in which there is little forward or backward mixing of components in the flow system. Usually, this is taken to mean that residence time distributions (RTDs) are limited to a small range, with a Péclet number (Pe) over 100.93,94 As we suspected that our lithiated intermediate may be unstable over longer reactor residence times (see Table 3, entry 2 vs. entries 3–5), we decided to investigate RTDs for our optimised system with a third pump added, corresponding to an in-flow electrophilic quenching step in order to determine whether our system was operating under plug flow during (a) metalation (pump A to position 2) and through (b) the metalation/electrophilic trapping sequence (pump A to position 3) representative of conducting the whole procedure in flow – as electrophilic trapping (while typically fast) is non-instantaneous, an unstable metalated intermediate could potentially still exhibit significant decomposition between positions 2 and 3 before trapping takes place (Fig. 4).
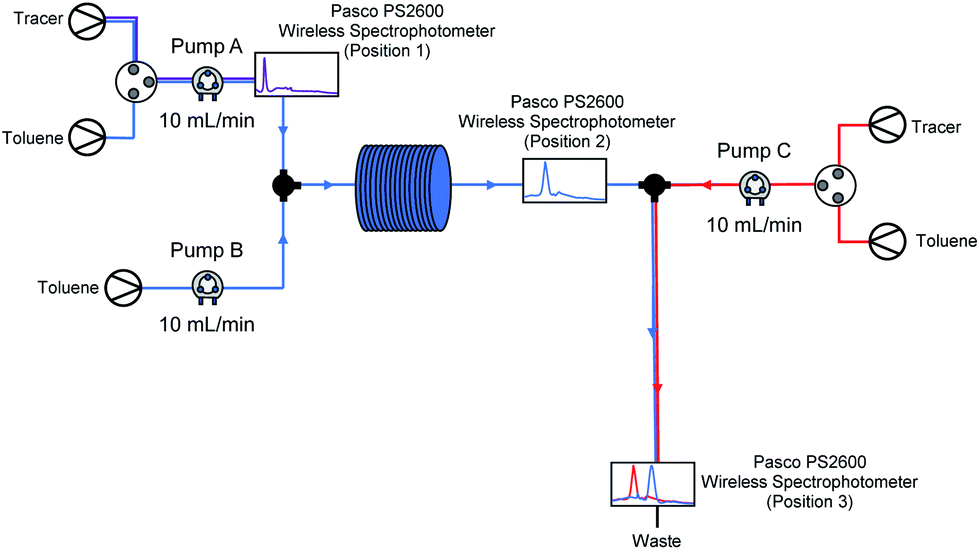 | ||
| Fig. 4 Flow system diagram for RTD determination. | ||
Using the method described by Sans and García-Verdugo,95 a tracer was injected into the system using a valve just after a pump, and detected downstream using a UV/Vis spectrometer. In each case, all three pumps A–C (corresponding to those pumping substrate, base and electrophile in a synthetic run respectively) are pumping at 10 mL min−1. Representative E(t) curves for pump A only (position 1), the metalation step with just pumps A and B running (position 2), the electrophilic trapping step (pump C to position 3) with all three pumps running and a pathway representative of the path taken by substrate through the lithiation-trapping sequence (pump A to position 3) can be found in the ESI.† After averaging results over several repeat experiments in each case, RTDs were found to be in line with those ascribed to perfect plug flow: an average Pe = 66.46 for pump A to position 1, Pe = 3170.11 for pump A to position 2, Pe = 140.99 for pump C to position 3 and Pe = 769.72 for pump A to position 3. The slightly lower Pe for pump C to position 3 may be due to dispersal at the T-junction where the 10 mL min−1 stream from pump C and the 20 mL min−1 stream from pumps A and B meet, however we note this is still over the value for plug flow. For a full breakdown of RTD determinations and Pe calculations, see ESI.†
With these results in hand, we were confident that an in-flow electrophilic trapping step could be added to our system. We first set out to determine the electrophile scope (Scheme 8).
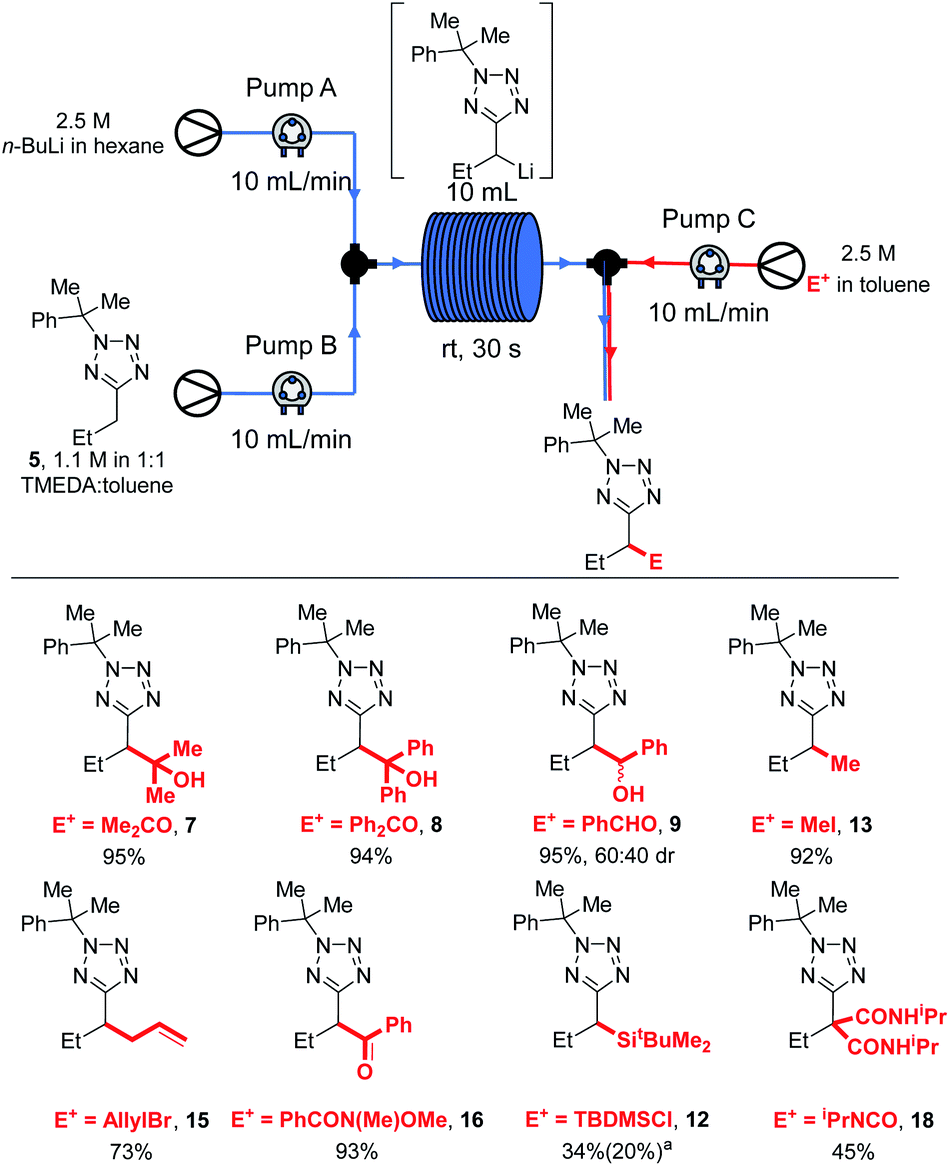 | ||
| Scheme 8 Electrophile scope in flow. a34% isolated yield of 12, 20% recovered 5. | ||
Most electrophiles investigated reacted to give the products in higher yields under our room temperature continuous flow conditions than were obtained in batch conditions. Thus, an 95% yield of 7 was isolated after trapping with acetone, compared to a 65% yield after reaction at 0 °C in batch (Scheme 2). Benzophenone and benzaldehyde gave 8 and 9 in 94% and 95% yields respectively (vs. 85% and 75% in batch). We found that lithiation-trapping reactions using Me2SO4 are incompatible with continuous flow conditions due to the formation of an insoluble by-product, however methylation to give 13 could be achieved in 92% yield using MeI. 15 and 16 were obtained in 73% and 93% yield after trapping with allyl bromide and a Weinreb amide respectively. A lower yield of 12 was obtained after trapping with TBDMSCl – 20% in flow vs. 80% isolated from a −78 °C reaction. This result is unsurprising as lithiated 5 is thought to be unstable at rt, and TBDMS reacts more slowly than the other electrophiles used. Finally, di-substituted product 18 was obtained in 45% yield after trapping with an isocyanate.
Methylated product 13 represents a cumyl-protected analogue of a compound patented in 2018 for use in anesthesia.96 To demonstrate the utility of our new synthetic protocol, 13 was deprotected via the protocol described by Flippin using palladium on carbon and KHCO2 to give 38 in 94% yield (Scheme 9).23
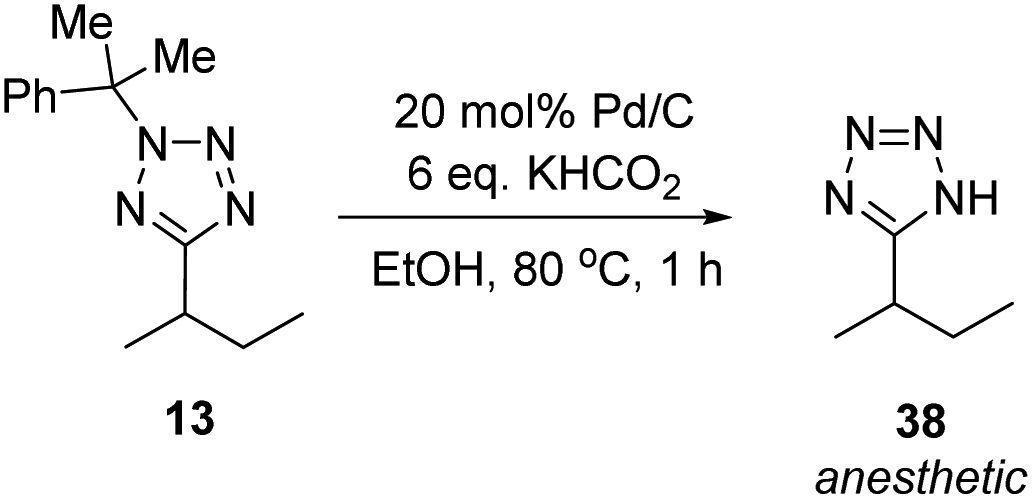 | ||
| Scheme 9 Synthesis of an anaesthetic agent. | ||
With the electrophile scope in hand, we next investigated the in-flow substrate scope (Scheme 10).
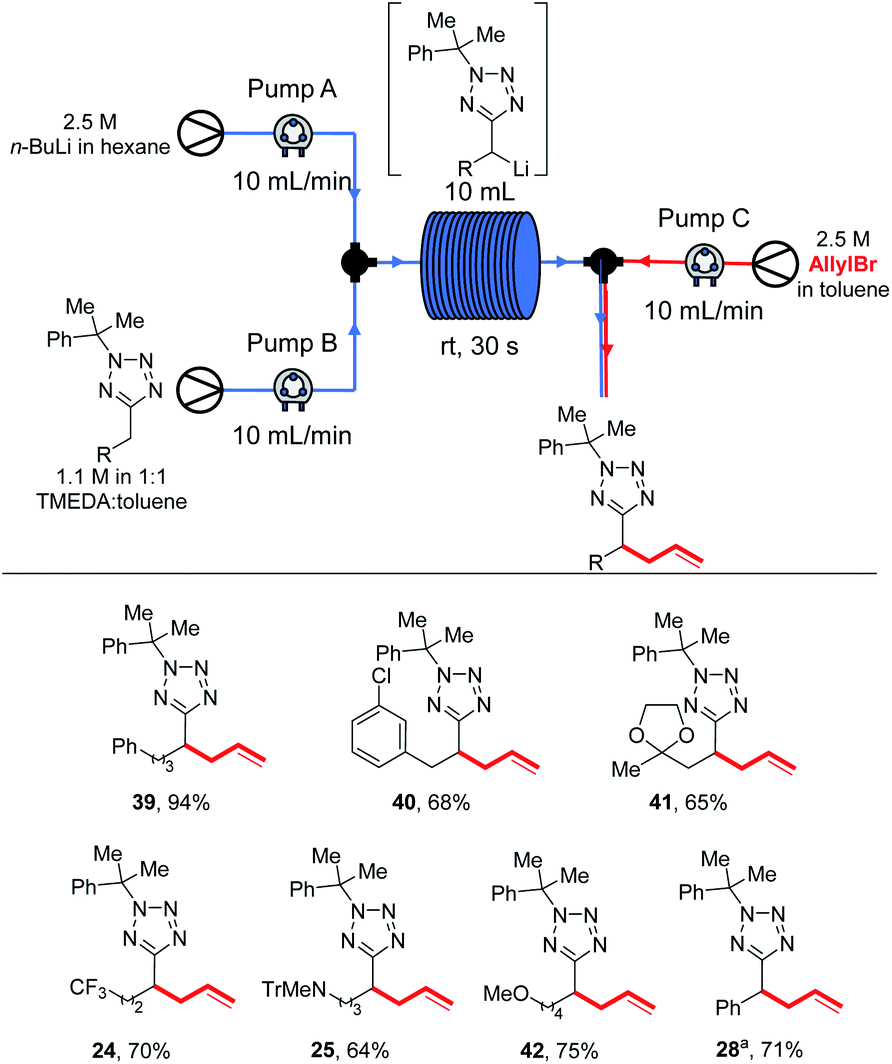 | ||
| Scheme 10 Substrate scope in flow. a1.7 M solution of substrate used. | ||
Again, for several substrates superior yields were obtained under continuous flow conditions at rt than under batch results: phenyl product 39 was obtained in 94% yield (vs. 83% from an allyl bromide-trapped 0 °C batch reaction), chlorophenyl 40 in 68% (vs. 53% from a −78 °C batch reaction trapped with Ph2CO) with no evidence of the regioisomer analogous to 31 observed. Acetal 41 was obtained in 65% yield (vs. 50% trapping with allyl bromide in batch at −78 °C). Trifluoromethyl, amino and alkoxy products 24, 25 and 42 were obtained in 70%, 64% and 75% yields respectively. During our previous study of benzyltetrazole lithiations we had been unable to develop a continuous flow protocol and were pleased to be able to isolate 28 in 71% yield.
With our new continuous flow procedure fully exemplified, the scalability was demonstrated with a longer run, and the productivity rate determined (Scheme 11).
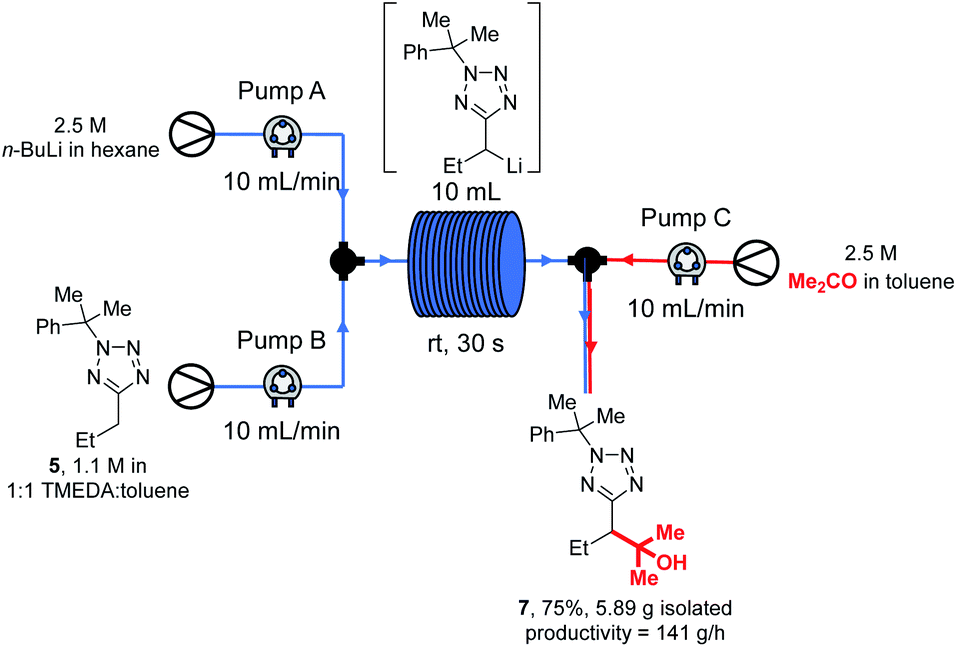 | ||
| Scheme 11 Large scale continuous flow lithiation-trapping. | ||
Thus, tetrazole 5, n-BuLi and acetone were reacted under our standard flow conditions for 2.5 min, giving 5.89 g of product 7 (75% yield) after work up and purification. This corresponds to a productivity rate of 141 g h−1, or alternatively 2.04 h would be required to produce 1 mole of 7, and 7.09 h would be needed to make 1 kg. This scale of synthesis is in line with that commonly achieved in the kilo lab, the first step in API synthesis scale up, as recently discussed by Casey et al.97 Precipitation resulting in blockage of continuous flow systems is occasionally observed over extended reaction times.98 In our case, careful inspection of the flow system and collection vessel revealed no evidence of precipitate after the large scale experiment. We note that in our experience, precipitates from organolithium reactions are often soluble in THF, and should other researchers encounter this problem a periodic valve-controlled automated flush of the flow system with dry THF may provide a solution.
Finally, we note that the requirement for 20 mol% palladium on carbon in the deprotection protocol originally reported by Flippin may be off-putting to some researchers, particularly those engaged in large-scale synthesis.23 In response, we decided to optimise this procedure for lower catalyst loadings (Table 4).
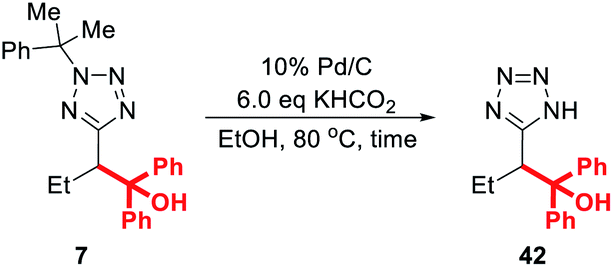
Thus, treatment of 7 with 20 mol% of 10% Pd/C and 6 eq. KHCO2 in ethanol at 80 °C for 1 h (the conditions reported by Flippin) gave an 80% yield of 42 after purification (entry 1). A comparable yield (78%) was obtained after dropping the catalyst loading to 10 mol% (entry 2). After further dropping the catalyst loading to 5 mol%, an extended reaction time of 4 h was required for complete conversion, and 42 was obtained in 75% yield. Any further reduction of the amount of Pd/C resulted in negligible conversion to the deprotected product.
Experimental
For full experimental details and analytical data, see ESI.†General optimised procedure for batch lithiation-trapping
Base (2.3 eq. n-BuLi or 1.6 eq. s-BuLi) was added dropwise to a stirred solution of N-cumyltetrazole (1.0 eq.) in THF at a specified temperature 0 °C (when using n-BuLi) or −78 °C (when using s-BuLi) under N2. The resulting solution was stirred at the specified temperature for 15 min. Then, electrophile (2.0 eq.) was added and the resulting solution was stirred at the specified temperature for 10 min and then allowed to warm to rt over 16 h. Saturated NH4Cl(aq) was then added and the two layers were separated, extracting the aqueous with Et2O (×3) The combined organic layers were dried (MgSO4) and evaporated under reduced pressure to give the crude product.General optimised procedure for continuous flow lithiation-trapping
A 1.1 M solution of N-cumyltetrazole (1.0 eq.) in 1:1 toluene/TMEDA (2.5 M) (0.5 mL) and a 2.5 M solution of n-BuLi in hexanes (0.5 mL) were driven through two separate peristaltic pumps at 10 mL min−1 at rt. The solutions were pumped through 1 mm I.D. PTFE tubing and mixed at a T-junction. The resulting solution was passed through a 10 mL PFA reactor (20 mL min−1, 10 mL, tR1 = 30 s) before mixing at a second T-junction with a 2.5 M solution of electrophile pumped at 10 mL min−1 through a third peristaltic pump. The resulting solution was passed through a second length of tubing (30 mL min−1, 0.47 mL, tR2 = 1.94 s) and collected as the product solution. A saturated solution of NH4Cl(aq) was added to the collected solution and the layers were separated, extracting the aqueous with Et2O (×3). The combined organic layers were dried (MgSO4) and evaporated under reduced pressure to give the crude product.
Conclusions
To conclude, we have developed a convenient and general heterobenzylic C–H functionalisation of readily deprotected 5-alkyltetrazoles, affording access to α-functionalised N–H tetrazolyl products which are bioisosteric with amino acid derived carboxylates bearing α-stereocentres. Under batch conditions, tetrazoles were metalated in THF using n-BuLi at 0 °C or s-BuLi at −78 °C before trapping with a wide range of electrophiles in up to 97% yield. Under continuous flow conditions, metalation was achieved in toluene/TMEDA using n-BuLi at room temperature at a high flow rate, short reactor residence time and corresponding narrow residence time distribution to avoid decomposition of an unstable organolithium intermediate. Products were obtained in up to 95% yield including in cases where batch synthesis resulted in modest yields, even with reaction cooling. The optimisation of continuous flow conditions was facilitated using thermal imaging to identify potentially unsafe exotherms, alerting us to re-optimise reaction conditions to avoid boiling ethereal solvent. We note that standardisation of flow processes remains a topic of discussion and while the 1 mm internal diameter tubing we have used is ubiquitous in research-scale flow chemistry, researchers wishing to replicate our chemistry with different reactor architectures may wish to re-optimize conditions. For researchers wishing to scale-up this work, we point to numbering-up as a convenient strategy for continuous flow chemistry in which simply using a larger reactor is unattractive.99,100 Given the requirement of flash chemistry to take place with short residence times and perfect plug flow, as well as the engineering challenges posed by flash protocols, we propose that thermal imaging has significant advantages over directly measuring reaction temperatures in cases where active thermal control is not being used. Our optimised continuous flow conditions facilitated the synthesis of a functionalised alkyltetrazole with a high productivity rate of 141 g h−1 – this has been achieved using an entry-level (Vapourtec E-series) flow system.Data availability
All experimental data and procedures are available in the ESI.†Author contributions
Author contributions are itemised using the CRediT system below: JYFW: data curation, formal analysis, investigation, methodology, visualisation, writing – original draft. CGT: data curation, formal analysis, investigation, methodology, visualisation. FV: funding acquisition, resources, supervision, writing – review & editing. GB: conceptualisation, data curation, funding acquisition, project administration, resources, supervision, visualisation, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Heriot-Watt University, the EPSRC CRITICAT Centre for Doctoral Training (PhD studentship for CGT, grant code: EP/LO14419/1), the Royal Society of Chemistry (grant code: R19-0308) and the Heriot-Watt Annual Fund for financial support. We further thank Dr David Ellis for assistance with NMR, Dr Georgina Rosair for X-ray crystallography and Prof. David Bucknall for loaning the thermal imaging camera as well as Tricia Cairns and Nichola Swann of SciChem Ltd. for lending the Pasco PS2600 spectrometer used for RTD analysis. Finally, we thank Dr Adilet Zhakeyev (HWU) for useful discussions on RTD analysis and Vapourtec for technical support.References
- R. J. Herr, Bioorg. Med. Chem., 2002, 10, 3379 Search PubMed
.
- J. Kaczmarek, H. Smagowski and Z. Grzonka, J. Chem. Soc., Perkin Trans. 2, 1979, 1670 RSC
- V. Subramanian, J. S. Knight, S. Parelkar, L. Anguish, S. A. Coonrod, M. J. Kaplan and P. R. Thompson, J. Med. Chem., 2015, 58, 1337 CrossRef CAS PubMed
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845 CrossRef CAS PubMed
- R. D. Larsen, A. O. King, C. Y. Chen, E. G. Corley, B. S. Foster, F. E. Roberts, C. Yang, D. R. Lieberman, R. A. Reamer, D. M. Tschaen, T. R. Verhoeven, P. J. Reider, Y. S. Lo, L. T. Rossano, A. S. Brookes, D. Meloni, J. R. Moore and J. F. Arnett, J. Org. Chem., 1994, 59, 6391 CrossRef CAS
- T. M. Chapman and K. L. Goa, Am. J. Cardiovasc. Drugs, 2003, 3, 117 CrossRef CAS PubMed
- Y. L. Guo, T. P. Kennedy, J. R. Michael, A. M. Sciuto, A. J. Ghio, J. N. F. Adkinson and G. H. Gurtner, J. Appl. Physiol., 1990, 69, 1615 CrossRef CAS PubMed
- J. Li, S. Y. Chen, J. J. Li, H. Wang, A. S. Hernandez, S. Tao, C. M. Musial, F. Qu, S. Swartz, S. T. Chao, N. Flynn, B. J. Murphy, D. A. Slusarchyk, R. Seethala, M. Yan, P. Sleph, G. Grover, M. A. Smith, B. Beehler, L. Giupponi, K. E. Dickinson, H. Zhang, W. G. Humphreys, B. P. Patel, M. Schwinden, T. Stouch, P. T. W. Cheng, S. A. Biller, W. R. Ewing, D. Gordon, J. A. Robl and J. A. Tino, J. Med. Chem., 2007, 50, 5890 CrossRef CAS PubMed
-
L. Yet, in Progress in Heterocyclic Chemistry, ed. G. W. Gribble and J. A. Joule, Elsevier, Amsterdam, 2021, vol. 32, pp. 279–323 Search PubMed
- S. Rajamanickam, C. Sah, B. A. Mir, S. Ghosh, G. Sethi, V. Yadav, S. Venkataramani and B. K. Patel, J. Org. Chem., 2020, 85, 2118 CrossRef CAS PubMed
- M. Seki and M. Nagahama, J. Org. Chem., 2011, 76, 10198 CrossRef CAS PubMed
- J. Roh, K. Vávrová and A. Hrabálek, Eur. J. Org. Chem., 2012, 2012, 6101 CrossRef CAS
- C. G. Neochoritis, T. Zhao and A. Dömling, Chem. Rev., 2019, 119, 1970 CrossRef CAS PubMed
- R. A. LaForge, A. D'Adamo, C. E. Cosgrove and C. R. Jacobson, J. Org. Chem., 1956, 21, 767 CrossRef CAS
- L. A. Flippin, Tetrahedron Lett., 1991, 32, 6857 CrossRef CAS
- E. W. Thomas and M. M. Cudahy, J. Org. Chem., 1993, 58, 1623 CrossRef CAS
- B. E. Huff, M. E. LeTourneau, M. A. Staszak and J. A. Ward, Tetrahedron Lett., 1996, 37, 3655 CrossRef CAS
- J. Y. F. Wong, A. Lewandowska, B. R. Trowse and G. Barker, Org. Lett., 2019, 21, 7069 CrossRef CAS PubMed
- R. Huisgen, J. Sauer and H. J. Sturm, Angew. Chem., 1958, 7, 272 CrossRef
- R. Huisgen, J. Sauer, H. J. Sturm and J. H. Markgraf, Chem. Ber., 1960, 93, 2106 CrossRef CAS
- J. Sauer, R. Huisgen and H. J. Sturm, Tetrahedron, 1960, 11, 241 CrossRef CAS
- L. Green, K. Livingstone, S. Bertrand, S. Peace and C. Jamieson, Chem.–Eur. J., 2020, 26, 14866 CrossRef CAS PubMed
- D. P. G. Norman, A. E. Bunnell, S. R. Stabler and L. A. Flippin, J. Org. Chem., 1999, 64, 9301 CrossRef CAS
- J. Y. F. Wong, J. M. Tobin, F. Vilela and G. Barker, Chem.–Eur. J., 2019, 25, 12439 CrossRef CAS PubMed
- G. Barker, P. O'Brien and K. R. Campos, Org. Lett., 2010, 12, 4176 CrossRef CAS PubMed
- P. Saiprathima, K. Srinivas, B. Sridhar and M. M. Rao, RSC Adv., 2013, 3, 7708 RSC
- H. Nozaki, T. Aratani, T. Toraya and R. Noyori, Tetrahedron, 1971, 27, 905 CrossRef CAS
- D. Hoppe, F. Hintze and P. Tebben, Angew. Chem., Int. Ed. Engl., 1990, 29, 1422 CrossRef
- P. Beak, S. T. Kerrick, S. Wu and J. Chu, J. Am. Chem. Soc., 1994, 116, 3231 CrossRef CAS
- D. Stead, P. O'Brien and A. Sanderson, Org. Lett., 2008, 10, 1409 CrossRef CAS PubMed
- J.-C. Kizirian, J.-C. Caille and A. Alexakis, Tetrahedron Lett., 2003, 44, 8893 CrossRef CAS
- J.-C. Kizirian, N. Cabello, L. Pinchard, J.-C. Caille and A. Alexakis, Tetrahedron, 2005, 61, 8939 CrossRef CAS
- N. Cabello, J.-C. Kizirian, S. Gille, A. Alexakis, G. Bernardinelli, L. Pinchard and J.-C. Caille, Eur. J. Org. Chem., 2005, 2005, 4835 CrossRef
- S. Gille, N. Cabello, J.-C. Kizirian and A. Alexakis, Tetrahedron: Asymmetry, 2006, 17, 1045 CrossRef CAS
- Q. Perron and A. Alexakis, Tetrahedron: Asymmetry, 2007, 18, 2503 CrossRef CAS
- D. J. Gallagher, S. Wu, N. A. Nikolic and P. Beak, J. Org. Chem., 1995, 60, 8148 CrossRef CAS
- I. Coldham, J. J. Patel and G. Sanchez-Jimenez, Chem. Commun., 2005, 3083 RSC
- I. Coldham, S. Dufour, T. F. N. Haxell, J. J. Patel and G. Sanchez-Jimenez, J. Am. Chem. Soc., 2006, 128, 10943 CrossRef CAS PubMed
- I. Coldham, P. O'Brien, J. J. Patel, S. Raimbault, A. J. Sanderson, D. Stead and D. T. E. Whittaker, Tetrahedron: Asymmetry, 2007, 18, 2113 CrossRef CAS
- I. Coldham, J. J. Patel, S. Raimbault and D. T. E. Whittaker, Chem. Commun., 2007, 4534 RSC
- I. Coldham, S. Raimbault, P. T. Chovatia, J. J. Patel, D. Leonori, N. S. Sheikh and D. T. E. Whittaker, Chem. Commun., 2008, 4174 RSC
- I. Coldham, S. Raimbault, D. T. E. Whittaker, P. T. Chovatia, D. Leonori, J. J. Patel and N. S. Sheikh, Chem.–Eur. J., 2010, 16, 4082 CrossRef CAS PubMed
- S. P. Robinson, N. S. Sheikh, C. A. Baxter and I. Coldham, Tetrahedron Lett., 2010, 51, 3642 CrossRef CAS
- I. Coldham and D. Leonori, J. Org. Chem., 2010, 75, 4069 CrossRef CAS PubMed
-
R. W. Hoffmann, in Stereochemical Aspects of Organolithium Compounds, ed. R. E. Gawley, John Wiley & Sons, Chichester, 2010, pp. 165–188 Search PubMed
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796 CrossRef CAS PubMed
- L. Malet-Sanz and F. Susanne, J. Med. Chem., 2012, 55, 4062 CrossRef CAS PubMed
- M. Power, E. Alcock and G. P. McGlacken, Org. Process Res. Dev., 2020, 24, 1814 CrossRef CAS
- P. Plouffe, A. Macchi and D. M. Roberge, Org. Process Res. Dev., 2014, 18, 1286 CrossRef CAS
- A. Hafner, P. Filipponi, L. Piccioni, M. Meisenbach, B. Schenkel, F. Venturoni and J. Sedelmeier, Org. Process Res. Dev., 2016, 20, 1833 CrossRef CAS
- M. Colella, A. Nagaki and R. Luisi, Chem.–Eur. J., 2020, 26, 19 CrossRef CAS PubMed
- L. Degennaro, C. Carlucci, S. De Angelis and R. Luisi, J. Flow Chem., 2016, 6, 136 CrossRef CAS
- J.-i. Yoshida, A. Nagaki and T. Yamada, Chem.–Eur. J., 2008, 14, 7450 CrossRef CAS PubMed
-
J.-i. Yoshida, in Flash Chemistry, ed. J.-i. Yoshida, John Wiley & Sons, Chichester, 2008, pp. 19–21 Search PubMed
-
H. Kim and J.-i. Yoshida, in Flow Chemistry in Organic Synthesis, ed. T. F. Jamison and G. Kock, Georg Thieme Verlag, Stuttgart, 2018, pp. 223–242 Search PubMed
- J.-i. Yoshida, H. Kim and A. Nagaki, J. Flow Chem., 2017, 7, 60 CrossRef CAS
- J.-i. Yoshida, H. Kim and A. Nagaki, J. Flow Chem., 2017, 7, 60 CrossRef CAS
- J.-i. Yoshida, Y. Takahashi and A. Nagaki, Chem. Commun., 2013, 49, 9896 RSC
- A. Nagaki, Y. Ashikari, M. Takumi and T. Tamaki, Chem. Lett., 2021, 50, 485 CrossRef CAS
- A. Nagaki, Tetrahedron Lett., 2019, 60, 150923 CrossRef CAS
- M. Seto, S. Masada, H. Usutani, D. G. Cork, K. Fukuda and T. Kawamoto, Org. Process Res. Dev., 2019, 23, 1420 CrossRef CAS
- P. Musci, M. Colella, A. Sivo, G. Romanazzi, R. Luisi and L. Degennaro, Org. Lett., 2020, 22, 3623 CrossRef CAS PubMed
- M. Colella, A. Tota, Y. Takahashi, R. Higuma, S. Ishikawa, L. Degennaro, R. Luisi and A. Nagaki, Angew. Chem., Int. Ed., 2020, 59, 10924 CrossRef CAS PubMed
- L. Degennaro, F. Fanelli, A. Giovine and R. Luisi, Adv. Synth. Catal., 2015, 357, 21 CrossRef CAS
- K. Okano, Y. Yamane, A. Nagaki and A. Mori, Synlett, 2020, 31, 1913 CrossRef CAS
- Y. Ashikari, T. Kawaguchi, K. Mandai, Y. Aizawa and A. Nagaki, J. Am. Chem. Soc., 2020, 142, 17039 CrossRef CAS PubMed
- A. Nagaki, H. Yamashita, K. Hirose, Y. Tsuchihashi and J.-i. Yoshida, Angew. Chem.,
Int. Ed., 2019, 58, 4027 CrossRef CAS PubMed
- A. Nagaki, Y. Jiang, H. Yamashita, N. Takabayashi, Y. Takahashi and J.-i. Yoshida, Chem. Eng. Technol., 2019, 42, 2113 CrossRef CAS
- A. Nagaki, H. Yamashita, Y. Tsuchihashi, K. Hirose, M. Takumi and J.-i. Yoshida, Chem.–Eur. J., 2019, 25, 13719 CrossRef CAS PubMed
- D. Ichinari, Y. Ashikari, K. Mandai, Y. Aizawa, J.-i. Yoshida and A. Nagaki, Angew. Chem., Int. Ed., 2020, 59, 1567 CrossRef CAS PubMed
- Y. Ishigaki, T. Iwai, Y. Hayashi, A. Nagaki, R. Katoono, K. Fujiwara, J.-i. Yoshida and T. Suzuki, Synlett, 2018, 29, 2147 CrossRef CAS
- T. Fujita, N. Konno, Y. Watabe, T. Ichitsuka, A. Nagaki, J.-i. Yoshida and J. Ichikawa, J. Fluorine Chem., 2018, 207, 72 CrossRef CAS
- H. Kim, Y. Yonekura and J.-i. Yoshida, Angew. Chem., Int. Ed., 2018, 57, 4063 CrossRef CAS PubMed
- H.-J. Lee, H. Kim, J.-i. Yoshida and D.-P. Kim, Chem. Commun., 2018, 54, 547 RSC
- A. Nagaki, K. Hirose, Y. Moriwaki, K. Mitamura, K. Matsukawa, N. Ishizuka and J. Yoshida, Catal. Sci. Technol., 2016, 6, 4690 RSC
- A. Nagaki, Y. Takahashi and J.-i. Yoshida, Angew. Chem., Int. Ed., 2016, 55, 5327 CrossRef CAS PubMed
- A. Nagaki, Y. Tsuchihashi, S. Haraki and J.-i. Yoshida, Org. Biomol. Chem., 2015, 13, 7140 RSC
- A. Nagaki, C. Matsuo, S. Kim, K. Saito, A. Miyazaki and J.-i. Yoshida, Angew. Chem., Int. Ed., 2012, 51, 3245 CrossRef CAS PubMed
- A. Nagaki, H. Kim, Y. Moriwaki, C. Matsuo and J.-i. Yoshida, Chem.–Eur. J., 2010, 16, 11167 CrossRef CAS PubMed
- A. Nagaki, E. Takizawa and J.-i. Yoshida, Chem.–Eur. J., 2010, 16, 14149 CrossRef CAS PubMed
- F. Venturoni, N. Nikbin, S. V. Ley and I. R. Baxendale, Org. Biomol. Chem., 2010, 8, 1798 RSC
- E. Takizawa, A. Nagaki and J.-i. Yoshida, Tetrahedron Lett., 2012, 53, 1397 CrossRef CAS
- A. Giovine, B. Musio, L. Degennaro, A. Falcicchio, A. Nagaki, J.-i. Yoshida and R. Luisi, Chem.–Eur. J., 2013, 19, 1872 CrossRef CAS PubMed
- A. Mambrini, D. Gori, C. Kouklovsky, H. Kim, J.-i. Yoshida and V. Alezra, Eur. J. Org. Chem., 2018, 2018, 6754 CrossRef CAS
- T. von Keutz, F. J. Strauss, D. Cantillo and C. O. Kappe, Tetrahedron, 2018, 74, 3113 CrossRef CAS
- J.-P. Kestemont, J. R. Frost, J. Jacq, P. Pasau, F. Perl, J. Brown and M. Tissot, Org. Process Res. Dev., 2021 DOI:10.1021/acs.oprd.1c00092
- C. Battilocchio, F. Bosica, S. M. Rowe, B. L. Abreu, E. Godineau, M. Lehmann and S. V. Ley, Org. Process Res. Dev., 2017, 21, 1588 CrossRef CAS
- A. Carangio, L. J. Edwards, E. Fernandez-Puertas, J. F. Hayes, M. M. Kucharski, G. W. Rutherford, K. M. P. Wheelhouse and G. D. Williams, Org. Process Res. Dev., 2020, 24, 1909 CrossRef CAS
-
J. Clayden, in Organolithiums: Selectivity for Synthesis, ed. J. E. Baldwin and R. M. Williams, Pergamon, Oxford, 2002 Search PubMed
- R. W. Hoffmann and B. Kemper, Tetrahedron Lett., 1981, 22, 5263 CrossRef CAS
- C. G. Screttas and I. C. Smonou, J. Organomet. Chem., 1988, 342, 143 CrossRef CAS
- D. B. Collum, Acc. Chem. Res., 1992, 25, 448 CrossRef CAS
-
O. Levenspiel, in Chemical Reaction Engineering, John Wiley & Sons, Chichester, 1999 Search PubMed
- M. Moreau, N. Di Miceli Raimondi, N. Le Sauze, M. Cabassud and C. Gourdon, Chem. Eng. Process., 2015, 95, 54 CrossRef CAS
- V. Sans, N. Karbass, M. I. Burguete, E. García-Verdugo and S. V. Luis, RSC Adv., 2012, 2, 8721 RSC
-
J. M. Sonner and J. L. Krstenansky, US Pat., US 20200375958, 2018 Search PubMed
-
M. Casey, J. Hamm, M. Miller, T. Ramsey, R. Schild, A. Stewart and J. Tom, in Chemical Engineering in the Pharmaceutical Industry, ed. D. J. Am Ende and M. T. Am Ende, John Wiley & Sons, Chichester, 2019, pp. 1011–1036 Search PubMed
- P. Filipponi, B. Guelat, J. Haber, S. Mostarda, R. O'Meadhra, L. Piccioni, J. Polenk, B. Schenkel, S. Schoenebeck, A. Streit, R. Suremann, F. Venturoni and S. Wegmann, Chimia, 2019, 73, 809 CrossRef CAS PubMed
- Y. Su, K. Kuijpers, V. Hessel and T. Nöel, React. Chem. Eng., 2016, 1, 73 RSC
- Z. Dong, Z. Wen, F. Zhao, S. Kuhn and T. Nöel, Chem. Eng. Sci.: X, 2021, 10, 100097 CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2088099. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04176b |
| This journal is © The Royal Society of Chemistry 2021 |