
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElement coding based accurate evaluation of CRISPR/Cas9 initial cleavage†
Jianyu
Hu
a,
Rui
Liu
 b,
Jing
Zhou
a and
Yi
Lv
*ab
b,
Jing
Zhou
a and
Yi
Lv
*ab
aAnalytical & Testing Center, Sichuan University, Chengdu 610064, PR China. E-mail: lvy@scu.edu.cn
bKey Laboratory of Green Chemistry and Technology of Ministry of Education, College of Chemistry, Sichuan University, 29 Wangjiang Road, Chengdu 610064, PR China
First published on 7th September 2021
Abstract
As a powerful gene editing tool, the kinetic mechanism of CRISPR/Cas9 has been the focus for its further application. Initial cleavage events as the first domino followed by nuclease end trimming significantly affect the final on-target rate. Here we propose EC-CRISPR, element coding CRISPR, an accurate evaluation platform for initial cleavage that directly characterizes the cleavage efficiency and breaking sites. We benchmarked the influence of 19 single mismatch and 3 multiple mismatch positions of DNA-sgRNA on initial cleavage, as well as various reaction conditions. Results from EC-CRISPR demonstrate that the PAM-distal single mismatch is relatively acceptable compared to the proximal one. And multiple mismatches will not only affect the cleavage efficiency, but also generate more non-site #3 cleavage. Through in-depth research of kinetic behavior, we uncovered an abnormally higher non-#3 proportion at the initial stage of cleavage by using EC-CRISPR. Together, our results provided insights into cleavage efficiency and breaking sites, demonstrating that EC-CRISPR as a novel quantitative platform for initial cleavage enables accurate comparison of efficiencies and specificities among multiple CRISPR/Cas enzymes.
Introduction
As a revolutionary breakthrough in the field of genetic engineering, clustered regularly interspaced short palindromic repeats (CRISPR) have a wide range of innovative applications in biological research, gene therapy, human medicine, genomic loci imaging and genomics.1–7 Among CRISPR-associated (Cas) enzymes, CRISPR/Cas9 has become a research hotspot and ideal candidate for in vivo applications due to its accuracy and tunability in double-stranded DNA cleavage.8–10 The cleavage of a substrate gene by Cas9 can be generally divided into two processes, initial cleavage by HNH and RuvC active sites of Cas9 and the post-cleavage trimming (PCT) process by the RuvC domain.11–13 The study of the cleavage specificity of CRISPR/Cas9 is based on the accurate evaluation of the cleavage kinetic process, which demands separate identification of the initial cleavage and PCT process of Cas9.14–16 Cas9 specificity used to be speculated from the repair scars at break sites, on the basis of on and off-target cleavage which induces differential imprinting on the DNA substrate.17–19 Cas off-target direct identification in living cells and tissues was realized by using DISCOVER-seq.20 Nevertheless, direct profiling of Cas9-mediated initial cleavage is still lacking. Using evaluation strategies based on cleavage products, including in vitro next-generation sequencing (NGS) and PAGE, it is difficult to observe the initial cleavage site, and the kinetic process of repair, and it is not possible to eliminate the influence of the genetic context and cell cycle phase.21–24 Initial cleavage events as the mechanistic basis of Cas9-mediated catalytic DNA strand breaking vary with reaction conditions and DNA-sgRNA mismatches. Despite the fact that the subsequent nuclease end trimming process could partially recover these variations, the difference of initial cleavage sites is just like the first domino, which leads to the final gap of the on-target rate and gene editing result.11–13,16 Therefore, a novel analysis platform is required to accurately characterize the initial cleavage of Cas9.DNA tagging strategies directly capture the cleavage behaviors, including FRET labels for monitoring the conformational change and 31P radioactive labels for cleavage product detection.13,14 Inspired by successful application of the tagging strategy in CRISPR/Cas research, we developed an element coding based CRISPR/Cas9 strategy (EC-CRISPR) for accurate evaluation of the Cas9 initial cleavage, which is able to avoid the influence of the subsequent PCT process and directly read the result of initial cleavage. This unique label mechanism contributes to accurate benchmarking of initial cleavage under various reaction conditions and nucleic acid substrate mismatches. By invoking the calculated cleavage efficiency and non-site #3 cleavage (non-#3) rates, the complete cleavage kinetics of Cas9 can be described. We verified the influence of reaction temperature and complex concentrations on initial cleavage. Not surprisingly, the appropriate increase of temperature and Cas9/sgRNA concentrations does contribute to the cleavage efficiency, while the excess leads to a dramatic enhancement of non-#3 rates. As for the specificity, results from EC-CRISPR also demonstrate that the PAM-distal single mismatch is relatively acceptable compared to the proximal one.
Intriguingly, multiple mismatches inside the DNA/sgRNA complex would not only slow the cleavage efficiency, but more than two base mismatches would have a vast effect on the initial on-target rates. Through in-depth research of kinetic behavior, we uncovered the abnormally higher non-#3 proportion at the initial stage of cleavage by using the EC-CRISPR system, which might be caused by the unstabilized conformation. EC-CRISPR provided a novel quantitative platform for the initial cleavage of CRISPR/Cas enzymes and provided insights into the cleavage efficiency and site selection. Moreover, EC-CRISPR can be regarded as an evaluation framework for the accurate and sensitive comparison of efficiencies and specificities among multiple CRISPR/Cas enzymes.
Results and discussion
Synthesis and characterization of the EC-CRISPR system
Elemental tagging based bioassays made a series of advances in biomolecule quantification, benefiting from the ultra-sensitivity, wide dynamic range, and most importantly the multiplex absolute quantification ability guaranteed by inductively coupled plasma mass spectrometry (ICPMS) detection.25–28 Several multiplex bioassay platforms have been established based on stable isotope tagging and ICPMS detection, showing ideal accuracy and sensitivity.29,30 Thanks to separation-free detection, labeled nucleic acid could be analyzed directly through ICPMS. Therefore, we developed EC-CRISPR for the evaluation of Cas9 initial cleavage as displayed in Fig. 1a and b. DNA targeting by the Cas9/sgRNA complex is achieved by recognizing a protospacer adjacent motif (PAM) on the non-target DNA strand (ntDNA), followed by forming a heteroduplex between sgRNA and the target DNA strand (tDNA) as shown in Fig. 1b. Continuous activation of nuclease domains by coupled conformational kinetics then resulted in rapid cleavage of tDNA and ntDNA at the HNH and RuvC active sites, respectively.31 Based on existing evidence from product sequencing, it is widely accepted that Cas9 endonuclease cleavage is specifically focused at the site of three bases upstream of PAM on ntDNA.6 We fabricated six element labeling sites located at 2–7 bases upstream of PAM on ntDNA (color marked in part 1 of Fig. 1a). Each labeling site corresponds to a synthesized lanthanide element tag. Six labeled substrates comprise an element coding system. Following initial cleavage, the element coding substrate is divided into two parts: DNA products of various lengths carrying element labels in the supernatant fluid, and the other including the undigested substrate carrying the biotin label removed by streptavidin coated magnetic beads (SA-MBs) as shown in part 2 of Fig. 1a. With element detection in the supernate by ICPMS, each difference subtracted from the concentrations of adjacent element labels indicates the relative intensity of the corresponding cleavage site (part 3 of Fig. 1a).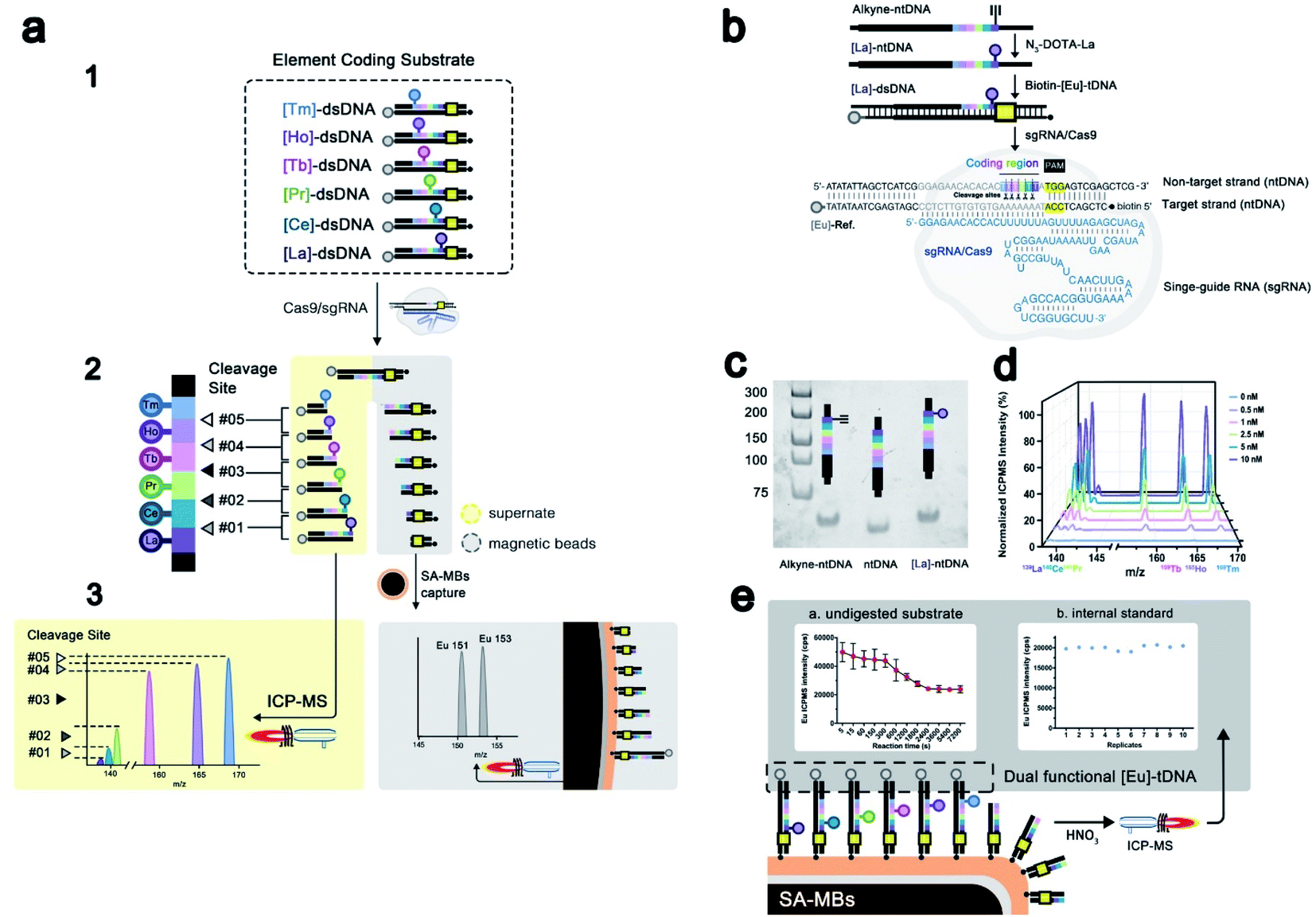 | ||
| Fig. 1 (a) Principle of EC-CRISPR detection of the initial cleavage of Cas9. Synthesis and characterization of the EC-CRISPR system. (b) Synthesis route to the element coding dsDNA substrate. (c) Native PAGE image of synthesized [La]-ntDNA: (1) DNA ladder. (2) Alkyne-ntDNA. (3) Label-free ntDNA. (4) [La]-ntDNA product. (d) ICP-MS detection result of various concentrations of the EC-CRISPR substrate (0, 0.5, 1, 2.5, 5 and 10 nM). (e) Dual functional [Eu] element label indicated an undigested substrate (part a) and internal standard isotope (part b). The intensity of 153Eu is used to verify the reliability of replicates. Reaction conditions: 300 nM Cas9/sgRNA pre-incubated for 30 min, followed by introducing the 30 nM mixed [Ln]-dsDNA substrate. After 2 h incubation at 37 °C, the cleavage reaction is quenched by EDTA at successive time intervals (5–7200 s). | ||
With 6 element coding bases, the relative intensities of 5 adjacent cleavage sites were obtained, which laid a foundation for further profiling of site specificity in initial cleavage. Second, the complementary tDNA was labeled with both the 3′-terminal dual functional Eu tag and 5′-terminal biotin which can be specific captured by SA-MBs. Through magnetic separation and acid digestion, the Eu signal can also be read by ICPMS (part 3 of Fig. 1a). To minimize the interference induced by element abundance, six lanthanide elements with an abundance close to 100% were selected to label ntDNA with N3-DOTA, while Eu was chosen as a dual functional label element (Fig. S1†). The ntDNA fragments with element tags were cleaved initially. The following degradation process caused by the PCT effect of the RuvC domain would change the length of the cleaved ntDNA fragments but would not affect the element tags carried. Therefore, EC-CRISPR based detection will directly provide information on the initial splicing site, supporting accurate detection of initial cleavage. To evaluate the initial cleavage, we invoked the calculated cleavage efficiency and non-#3 (non-site #3 initial cleavage) ratio for characterization in further studies (see the formula in the Experimental section). The calculated cleavage efficiency can be used to evaluate the kinetics of Cas9 cleavage. And the non-site #3 initial cleavage ratio can indicate the probability of cleavage at a non-recognized site, although this doesn't belong to off-target cleavage. With these two introduced factors, we can evaluate the initial cleavage from both efficiency and specificity channels under various conditions.
As shown in Fig. 1b, the labeling procedures of the element coding dsDNA substrate include two major steps: (1) alkyne-ntDNA, an intermediate functionalized with alkyne groups on the selected base, was specifically derivatized with the azido group of the pre-synthesized N3-DOTA–[Ln] complex as shown in Fig. S2.† (2) The obtained [Ln]-ntDNA from step 1 was further hybridized with pre-synthesized [Eu]-tDNA to form the element coding dsDNA substrate. The 153Eu signal serves as a dual functional label on tDNA as shown in Fig. 1e. Because the 3′-terminal (Eu3+ labeled) residue of the cleaved substrate has been left in the supernate, the 153Eu intensity on SA-MBs can be used to represent the undigested substrate. Besides, to increase the accuracy of EC-CRISPR, the 153Eu intensity on SA-MBs also plays a role as an internal standard. The intensity of 153Eu is used to verify the reliability of replicates. By elemental coding of La, Ce, Pr, Tb, Ho and Tm at 2–7 bases upstream of the PAM on ntDNA respectively, six separate [Ln]-ntDNAs were assembled. As shown in Fig. 1b, each kind of [Ln]-dsDNA possesses a single element label. All of [La]-dsDNA, [Ce]-dsDNA, [Pr]-dsDNA, [Tb]-dsDNA, [Ho]-dsDNA and [Tm]-dsDNA assembled the element coding substrate system. The experimental details of synthesis procedures and further characterization are listed in the Experimental section. The obtained [Ln]-ntDNA was further characterized by polyacrylamide gel electrophoresis (Fig. 1c). The increase in the molecular weight of ntDNA, alkyne-ntDNA and [La]-ntDNA resulted in a gradual slowdown in the movement in each lane. And the mixed EC-CRISPR probes were characterized by ICPMS and the energy dispersive spectrum (EDS) after direct capture by SA-MBs (Fig. 1d and S6†), respectively. Element analysis results demonstrated that six N3-DOTA–[Ln] were successfully coupled with alkyne-ntDNA. Based on this principle, we replace the traditional PAGE based assay with the proposed EC-CRISPR strategy to calculate the cleavage efficiency and non-#3 ratio. The ICPMS detection results of element labels play the same roles as the intensity of each band of electrophoresis (see the formula in the Experimental section). Utilizing the EC-CRISPR, we evaluated the initial cleavage based on the cleavage efficiency and missed target cleavage under different experimental conditions, including the reaction temperature, Cas9/sgRNA complex dosage, substrate mismatch and kinetic curve.
Concentration and temperature effects on CRISPR/Cas9 initial cleavage
The effect of lanthanide labeling on cleavage behavior was investigated first. We compared the cleavage products of label-free and lanthanide labeled ntDNA with native-PAGE, respectively. [Pr]-ntDNA was selected as the model substrate in this study. As shown in Fig. 2, lane 1 and lane 2 represent the uncleaved substrate. Lanthanide labeling increases the molecular weight of [Pr]-ntDNA, which corresponds to the slower movement of the band in lane 2. Lanes 3 and 4 represent the corresponding cleavage products. The comparison indicates that the product fragments without carrying the lanthanide label (i.e., 30-mer, 31-mer and 32-mer) remain the same between lanes 3 and 4. Besides, the lanthanide label-carrying fragments significantly moved slower in lane 4 (i.e., 33-mer, 34-mer and 18-mer). The change in the position of some bands is only due to the increase in molecular weight caused by the carrying of the lanthanide label. Hence, the cleavage pattern remains the same with the lanthanide labels.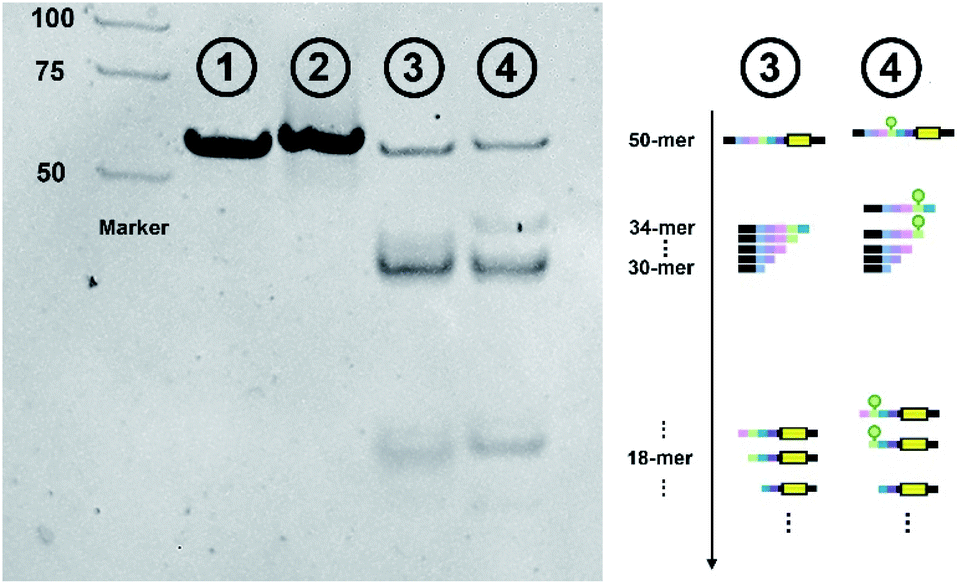 | ||
| Fig. 2 Native PAGE characterization of the effect of lanthanide labeling on cleavage behavior. Far left: DNA marker. Lane 1: uncleaved label-free ntDNA. Lane 2: uncleaved [Pr]-ntDNA. Lane 3: cleaved products of label-free ntDNA. Lane 4: cleaved products of [Pr]-ntDNA. Far right: schematic diagram of various cleavage products. | ||
We then investigated the effect of Cas9/sgRNA concentration and reaction temperature on the initial cleavage of CRISPR/Cas9. Normally, an increase of endonuclease concentration and temperature will have a promotion effect on the enzymatic reaction. However, previous research showed that higher temperature and enzyme concentration might amplify the off-target effect.32 To accurately capture the variation tendency of initial cleavage, we employed the proposed EC-CRISPR system to evaluate the same. Element concentration results are listed in Fig. 2d and 3a, with raw ICPMS intensity data shown in Fig. S17 and S18† respectively. The relative intensity of each cleavage site was processed as the gap between corresponding adjacent label elements. The Eu label intensity on SA-MBs represented the uncleaved substrate. As displayed in Fig. 3b, the processed curves showed that the cleavage efficiency was increasing until the concentration of Cas9/sgRNA was 10 times that of the dsDNA substrate.
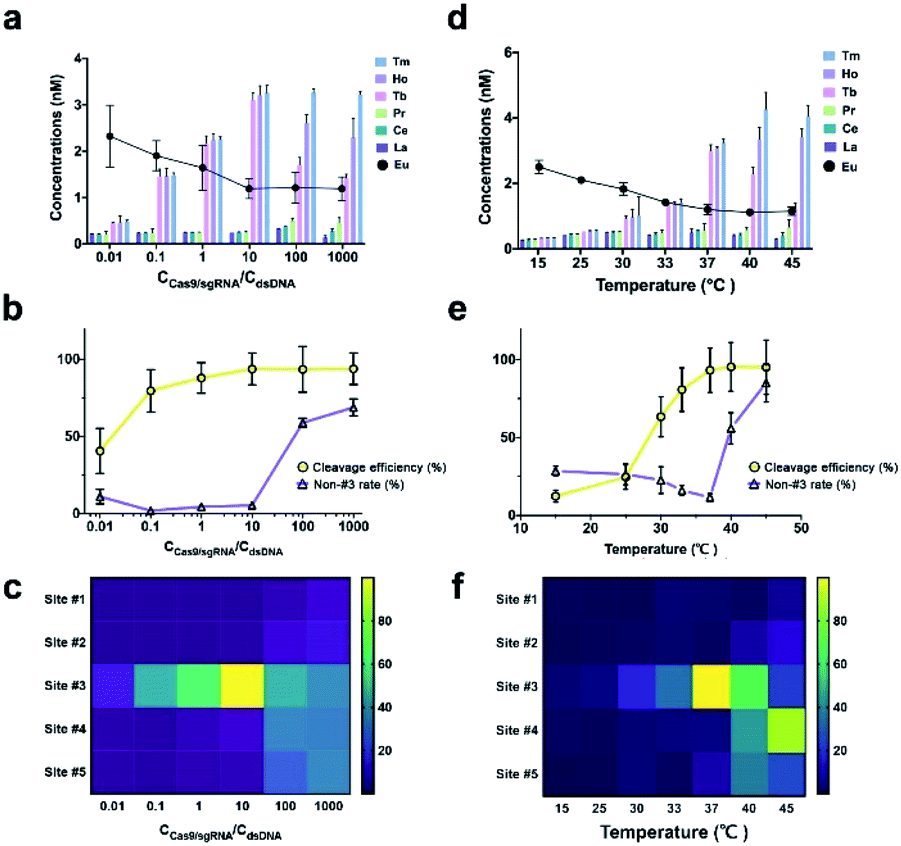 | ||
| Fig. 3 (a) [Ln] raw concentrations, (b) cleavage efficiency and non-#3 rate (calculated from raw data in (a)), (c) heatmap representation of various cleavage sites of different Cas9/sgRNA concentrations (aggregated from raw data in (a)). (d) [Ln] raw concentrations, (e) cleavage efficiency and non-#3 rate (calculated from (d)), (f) heatmap representation of various cleavage sites of different reaction temperatures (aggregated from (d)). Reaction conditions: Cas9 concentrations, Cas9/sgRNA pre-incubated for 30 min (0.3 nM, 3 nM, 30 nM, 300 nM, 3 μM and 30 μM), followed by introducing the 30 nM mixed [Ln]-dsDNA substrate. Then incubated for 2 h at 37 °C. Temperature, 300 nM Cas9/sgRNA pre-incubated for 30 min, followed by introducing the 30 nM mixed [Ln]-dsDNA substrate. Then incubated for 2 h at 15, 25, 30, 33, 37, 40 and 45 °C. After removal of biotin labeled residues, the acidulated supernate was directly analyzed by ICPMS to acquire the raw intensity of 139La, 140Ce, 141Pr, 159Tb, 165Ho and 169Tm, while the remaining SA-MBs were acid digested with 20% HNO3 and then analyzed by ICPMS to read the 153Eu label intensity. | ||
Beyond 10 times, the cleavage efficiency remained steady while the non-#3 ratio was significantly enhanced (purple curve). Also as shown in the heatmap in Fig. 3c, the initial cleavage site was dispersed from site #3 to site #5 at higher concentrations. A similar conclusion can be summarized from the PAGE result. As shown in Fig. S8,† when the [Cas9/sgRNA]/[dsDNA] ratio is increased to 10, more obvious cleaved bands are generated along with a higher ratio of the shorter product. And a larger concentration ratio will not significantly further increase the intensity of the products.
As for the temperature effect, the cleavage efficiency keeps increasing from 15 to 37 °C due to the growing enzyme activity, nevertheless the non-#3 ratio increased at higher temperature (Fig. 3e). Combined with the heatmap in Fig. 3f, it is found that the majority of initial cleavage sites transferred from site #3 to #4 at 45 °C, indicating that the accurate location and recognition of the PAM sequence by using the RuvC residue were affected. Because the noncomplementary segment in ntDNA is flexible within the active site of the RuvC residue, we speculated that the single strand ntDNA was twisted or deformed at higher temperature, which affected the accuracy of initial cleavage. The native-PAGE image also shows that 37 °C is the optimized reaction temperature (Fig. S9†). But limited by the PCT effect, the products at 37 °C to 45 °C exhibit similar lengths, which reflects the advantage of EC-CRISPR for accurate detection at the initial cleavage site. The control experiment was also performed to verify the effect of non-specific sgRNA. As shown in Fig. S7,† the results demonstrated that neither only Cas9 with no sgRNA nor Cas9 with non-specific sgRNA generated valid cleavage.
Nucleotide specificity of CRISPR/Cas9 initial cleavage
We further systematically evaluated the effect of base-pair mismatches between sgRNA and tDNA on the initial cleavage of CRISPR/Cas9. As shown in Fig. 4a, we systematically synthesized a series of sgRNAs (M-1 to M-19) containing single-nucleotide substitution at positions 1–19 next to NGG PAM in the sgRNA/tDNA complementary region, respectively (Fig. 4a and S11†). These single-mismatch sgRNAs were employed to evaluate the cleavage activity and site selection of the CRISPR/Cas9 system through the proposed EC-CRISPR strategy, along with comparison with the result of native-PAGE.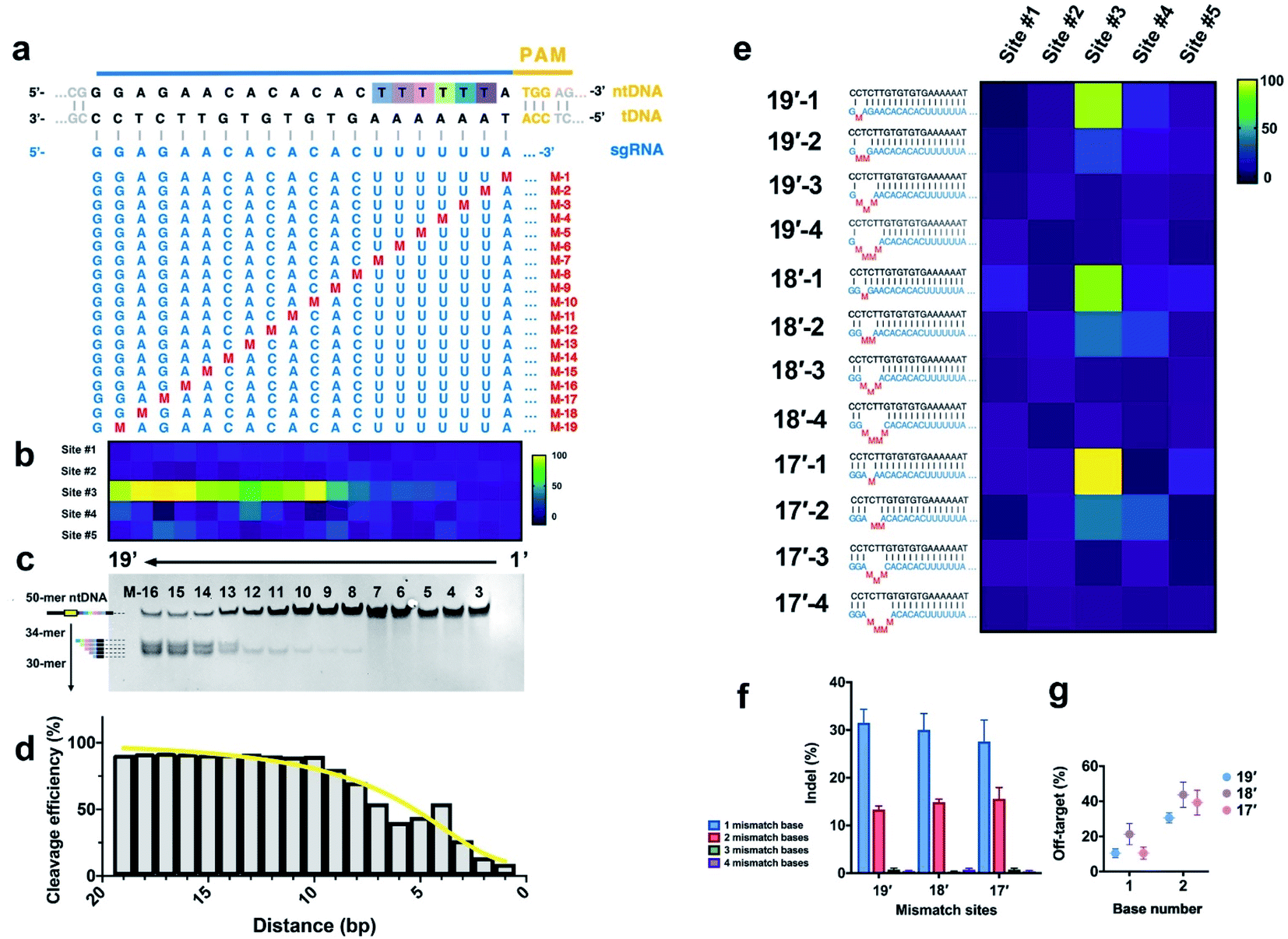 | ||
| Fig. 4 (a) Single mismatch loci of sgRNA. (b) Heatmap and (c) native-PAGE image of initial cleavage sites with various single mismatches in sgRNA (aggregated from Fig. S11†). (d) Cleavage efficiency variation with the mismatch distance to the PAM region (calculated from raw data in Fig. S11 and S19†). (e) Multiple mismatch loci of sgRNA. Effects of mismatch loci and numbers on (f) cleavage efficiency and (g) non-#3 rate. Reaction conditions: 300 nM sgRNA containing the mismatch base was pre-incubated with Cas9 for 30 min, followed by introducing the 30 nM mixed [Ln]-dsDNA substrate. Then incubated for 2 h at 37 °C, compiled from aggregate data from Fig. S12 and S20.† | ||
Previous research was built on the basis of the RuvC-catalyzed PCT effect, while we aim to establish an accurate specificity curve for the initial cleavage of the Cas9 endonuclease with EC-CRISPR. The results show that the cleavage efficiency of Cas9 is greatly affected by the position of a single-base mismatch, especially the distance to the PAM region. The cleavage efficiency curve and heatmap of multiple sites both show that the more proximal the distance to the PAM region, the greater the effect of single base mismatch on the cleavage efficiency (Fig. 3d and 4b), which is consistent with previous conclusions.33 Mismatches existing at 1–10 bp proximal to PAM (M-1 to M-10) present a greater impact on the cleavage efficiency, while the mismatches after 10 bp (M-11 to M-19) are relatively acceptable. It is worth noting that the single-base mismatch of sgRNA can only affect the efficiency instead of the specificity of cleavage sites. The initial cleavage mainly proceeded at site #3 with hardly any increase of the non-#3 ratio. The electrophoresis results (Fig. 4c) exhibit a similar result that the bands of cleavage products are much clearer with PAM-distal mismatches compared to PAM-proximal ones. Besides, the obvious PCT effect bands also appear after M-13. Despite no visible cleavage product band before M-8, we can capture the gradual increase from M-1 to M-10 by EC-CRISPR due to its higher sensitivity (Fig. 4d). Limited by the PCT effect and the accuracy, direct measurement of initial cleavage sites became a bottleneck in previous research. Now with the EC-CRISPR strategy, our conclusion shows that although the sgRNA with 2 consecutive mismatches seems to maintain an acceptable cleavage efficiency, the initial cleavage sites disperse to sites #3 and #4 (Fig. 3f and 4e). As summarized in Fig. 4g, the non-#3 ratio of initial cleavage compared with that of only 1 mismatch is remarkably increased. The specificity of CRISPR/Cas9 cleavage is drastically reduced. Catalyzed by the PCT effect of RuvC, ntDNA will eventually tend to form blunt ends. Therefore, the influence caused by initial cleavage at different sites is controllable.
Kinetics of initial cleavage versus 3′ → 5′ post-cleavage trimming
EC-CRISPR-based detection has provided undiscovered rules for the profiling of initial cleavage sites under different experimental conditions (temperature and Cas9 concentration) and DNA targeting specificity. Previous research creatively pointed out that the initial ntDNA cleavage by RuvC might generate multiple adjacent sites based on the single turnover assay.16 To accurately capture the kinetic behavior of initial cleavage, we proceeded with the single turnover assay with successive time intervals (5–7200 s) based on both label-free gel electrophoresis and EC-CRISPR detection. As shown in Fig. 5a, the initial cleavage occurred at sites #1 to #5 which corresponds to 34-mer to 30-mer 5′-terminal products, respectively. Besides, it is easy to find that the 46-mer biotin labeled undigested tDNA shows no band due to the removal by SA-MBs (Fig. 5b). The PAGE result reveals the fact that in the early stage (5–15 s), there exist bands of 32-mer to 34-mer products, which indicates that the early cleavage concentrates at multiple sites #1 to #3 close to PAM.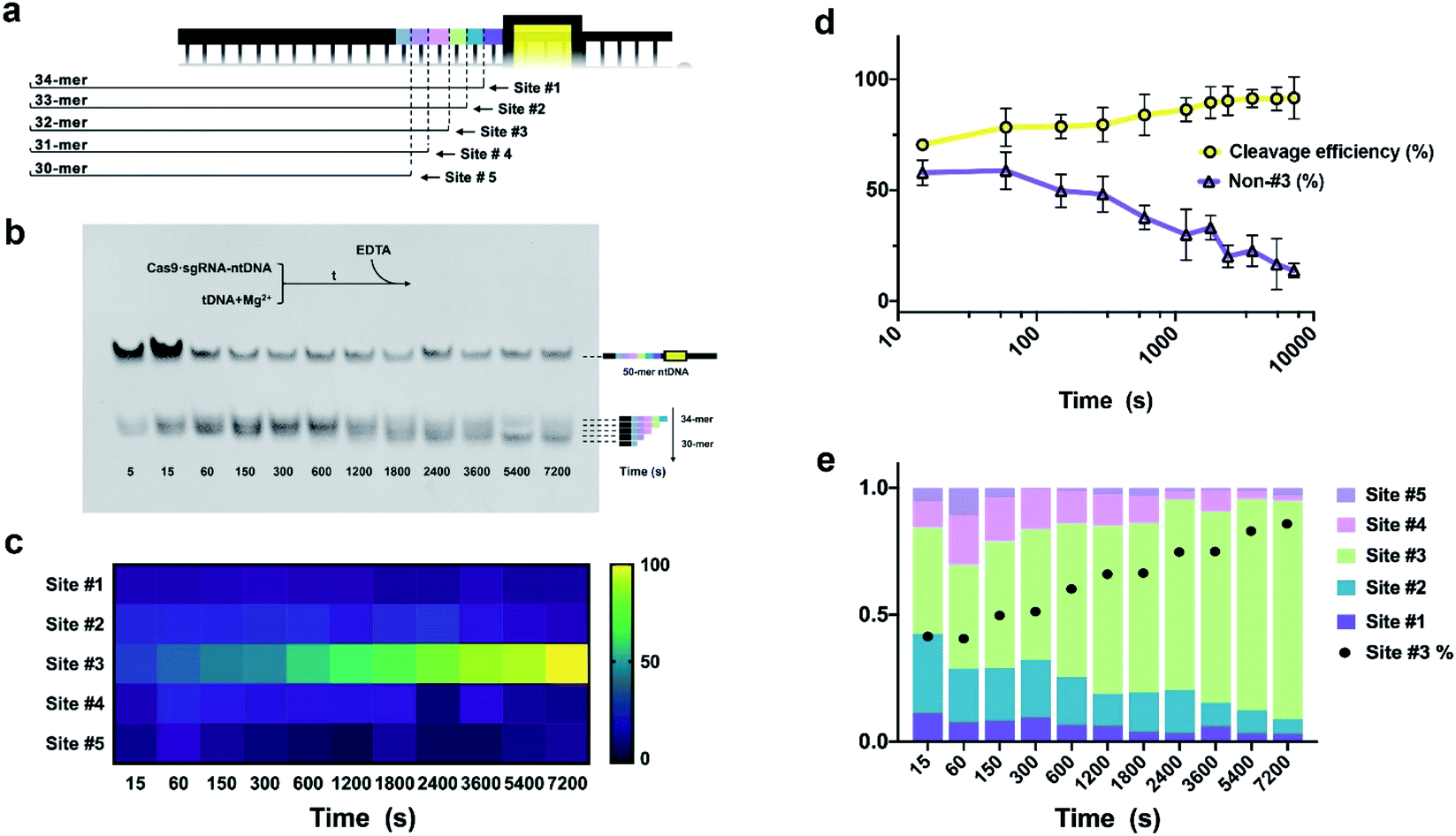 | ||
| Fig. 5 (a) Relationship between various cleavage sites and product lengths. (b) Native-PAGE image, and (c) heatmap at various reaction time intervals. (d) Variation tendency of cleavage efficiency and non-#3 rate with reaction time. (e) Distribution of cleavage sites with various reaction times, compiled from aggregate data from Fig. S13.† Reaction conditions: EC-CRISPR, 300 nM Cas9/sgRNA pre-incubated for 30 min, followed by introducing the 30 nM mixed [Ln]-dsDNA substrate. After 2 h incubation at 37 °C, the cleavage reaction is quenched by EDTA at successive time intervals (5–7200 s). Native-PAGE, element label-free ntDNA was pre-hybridized with 3′-biotin-tDNA as the dsDNA substrate. 3 μM Cas9/sgRNA was pre-incubated for 30 min, followed by introducing the 300 nM dsDNA substrate. All the above procedures were performed in the Mg2+ free reaction buffer. With 1 h incubation at 25 °C, 6 mM Mg2+ was rapidly introduced to trigger the cleavage at 37 °C. After the corresponding cleavage time, the reaction was quenched with 0.5 M EDTA. | ||
As the reaction time increases to 600 s, additional 30-mer and 31-mer bands can be clearly observed, while the 32-mer product is more inclined to be generated. This tendency shows that from 15 s to 600 s, the Cas9 endonuclease prefers to cleave at site #3, which is four bases upstream of PAM, consistent with previous research.16The effect of 3′ → 5′ post-cleavage bidirectional degradation appears at later time points (600 s to 2 h). Differing from initial cleavage, the 31-mer band shows remarkably stronger intensity while the rest faded due to the 3′ → 5′ PCT activity of RuvC.
Correspondingly, the EC-CRISPR strategy was then used to study the kinetic behavior. By processing the ICPMS intensities of six label elements, we obtained the relative cleavage intensity from sites #1 to #5 and displayed it as a heat map (Fig. 5c, aggregated from S13 and S21†), and calculated the cleavage efficiency and non-#3 ratio (Fig. 5d). This reveals the fact that as the Cas9 cleavage progresses, the proportion of initial cleavage at site #3 is remarkably concentrated. And as the reaction time increases, the cleavage efficiency continues to increase along with the decrease of the non-#3 ratio. In order to specifically study the relative distribution of different sites at each time interval, we normalized the EC-CRISPR results and summarized them in Fig. 5e. In the initial stage of the reaction, the proportion of site #3 marked in green remains relatively low (compared to the later stage) with a certain proportion of cleavage at sites #2 and #4, and even at #1 and #5. As time increases, additional cleavage is concentrated at site #3. Comparing the relative distribution of site #3 separately as the black dots (site #3 rate) in Fig. 5e, the proportion of site #3 also gradually increases, promoting site #3 as a selection with absolute advantage. Therefore, we believe that the cleavage of the CRISPR/Cas9 system is more inclined to gradually accumulate at site #3. Similar to the temperature effect, we speculated that the lower proportion of site #3 in the early stage might be because there exists a space displacement margin between the dsDNA substrate and Cas9/sgRNA complex and the drift of the splice site caused by the unstabilized conformation. This phenomenon also enhances the understanding of biological processes of CRISPR/Cas9. Compared with the results of electrophoresis, although the initial cleavage site is gradually concentrated to site #3, CRISPR/Cas9 still tends to form a blunt end product due to the PCT bidirectional degradation process. The results from EC-CRISPR kinetic behavior research provide solid evidence for the PCT bidirectional degradation process with direct detection of initial cleavage sites, compared to molecular dynamics simulation or product mixture PAGE.
Experimental
Preparing azido-monoamide-DOTA (N3-DOTA) chelated element tags
Six lanthanide chlorides were directly dissolved in NH4Ac buffer (500 mM, pH = 5.5) to 20 mM. Similarly, N3-DOTA was dissolved in the same NH4Ac buffer (500 mM, pH = 5.5) to 10 mM. Then, both lanthanide and N3-DOTA solutions were mixed in equal volumes. Chelation was completed in 30 min with gentle shaking at 37 °C. The product of chelation was further analyzed by ESI-MS (Fig. S3 and S4†), showing that lanthanide elements were successfully coupled to N3-DOTA. Six N3-DOTA–[Ln] were pre-synthesized as element tags for further use.Labeling alkyne-ntDNA with element tags
Six ntDNA with alkyne modification at six adjacent sites were purified and synthesized by Shanghai Sangon Biotechnology Co. Ltd. The alkyne-ntDNA labeling with the N3-DOTA–[Ln] element tag by the click chemistry reaction followed previous reports.34,35 First, 100 μM alkyne-ntDNA solution dissolved in PBS buffer (10 mM, pH = 7.4) was pre-heated to 95 °C for 2 min, and then rapidly cooled to 25 °C. After that, 20-fold excess N3-DOTA–[Ln] was introduced. The click reaction was catalyzed by using copper sulfate and ascorbate solution (1 mM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 mM). To hold catalyzed Cu(I) and promote the click reaction, 2 mM BTTAA (2-(4-((bis((1-(tert-butyl)-1H-1,2,3-triazol-4-yl)methyl)amino)-methyl)-1H-1,2,3-triazol-1-yl)acetic acid) was added. And PCR grade paraffin was introduced at last to achieve isolation from air. The whole click reaction is incubated overnight at 40 °C. And the final product, [Ln]-ntDNA, was purified by ultrafiltration (10k, Amicon Ultra-0.5 NMWL) and characterized by MALDI-TOF-MS and PAGE as shown in Fig. S5,† respectively.
:5 mM). To hold catalyzed Cu(I) and promote the click reaction, 2 mM BTTAA (2-(4-((bis((1-(tert-butyl)-1H-1,2,3-triazol-4-yl)methyl)amino)-methyl)-1H-1,2,3-triazol-1-yl)acetic acid) was added. And PCR grade paraffin was introduced at last to achieve isolation from air. The whole click reaction is incubated overnight at 40 °C. And the final product, [Ln]-ntDNA, was purified by ultrafiltration (10k, Amicon Ultra-0.5 NMWL) and characterized by MALDI-TOF-MS and PAGE as shown in Fig. S5,† respectively.
Labeling 5′-terminal biotin functional tDNA with the Eu tag
Different from alkyne-ntDNA, the Eu tag was labeled on tDNA with the thiol group (–SH) on the 3′-terminal of tDNA chelated with 1,4,7,10-tetraazacyclododecane-1,4,7-tris-acetic acid-10-maleimidoethylacetamide (MMA-DOTA). Similarly, Eu3+ was chelated with MMA-DOTA as in the above procedure. Then MMA-DOTA–[Eu] was incubated with SH-tDNA in NH4Ac buffer (500 mM, pH = 5.5) at 37 °C for 60 min, in which MMA-DOTA–[Eu] was in an excess of 25-fold. And the final product, [Eu]-tDNA, was purified by ultrafiltration (10k, Amicon Ultra-0.5 NMWL) and characterized by MALDI-TOF-MS as shown in Fig. S5.† All purified [Ln]-ntDNA and [Eu]-tDNA were redissolved in Tris–HCl buffer (10 mM, pH = 8.5, 100 mM NaCl, DEPC). Six times diluted [Ln]-ntDNA was mixed and hybridized together with [Eu]-tDNA to 9 μM as the element coding substrate ([Ln]-dsDNA) and characterized by ICPMS (Fig. 3c).In vitro transcription (IVT) preparation of sgRNA
The sequence of sgRNA was designed followed by the previous reported protocol (Fig. S14†). To prepare sgRNA, we employed the in vitro transcription (IVT) procedure with the synthesized 117-mer DNA template (Fig. S15a†). First, the DNA template was synthesized by the fill-in PCR procedure. Two partly hybridized primers were used including (1) one containing the T7 promoter (TAATACGACTCACTATA) and sgRNA/tDNA target sequence, (2) and the other as the reverse primer. There was 20-nt length of these two complementary primers. The following thermal cycle was set in the PCR procedure: first, heating at 95 °C for 2 min; followed by 40 cycles of 95 °C for 20 s, 60 °C for 30 s, and 72 °C for 15 s; and at the end at 72 °C for 10 min. The 117-mer product was purified with a PCR Product Purification Kit (Sangon Biotech) and was employed as the template for the T7 RNA polymerase-mediated transcription reaction. T7 transcription was performed with the dsDNA template and T7 RNA polymerase for 6 h at 37 °C according to the protocol, followed by purification with a Spin Column RNA Cleanup & Concentration Kit (Sangon Biotech). The purified sgRNA was immediately employed for CRISPR/Cas9 cleavage or frozen at −80 °C. The primers, dsDNA template and sgRNA product were together characterized by polyacrylamide gel electrophoresis (PAGE) as shown in Fig. S15b.† Prior to cleavage, sgRNA was incubated to be folded as multiple tetraloops by heating to 95 °C for 5 min and then slowly cooling to 20 °C.EC-CRISPR detection of initial cleavage
Generally, six prepared [Ln]-dsDNA substrates were equivalently mixed to compose the element coding substrate. 300 nM sgRNA and Cas9 are pre-incubated in reaction buffer for 30 min, followed by introducing the 30 nM mixed [Ln]-dsDNA substrate. After 2 h incubation at 37 °C, the cleavage reaction is quenched by EDTA. Subsequently, 3 μL SA-MBs were added into the mixture to remove the biotin labeled residues and undigested substrates through 30 min incubation. Ultimately, after magnetic separation, the acidulated supernate was directly analyzed by ICPMS to acquire the intensity of 139La, 140Ce, 141Pr, 159Tb, 165Ho and 169Tm, while the remaining SA-MBs were acid digested with 20% HNO3 and then analyzed by ICPMS to read the 153Eu label intensity. The PCT process can only change the length of cleaved fragments, instead of the carrying element labels. Therefore, the initial cleavage pattern could be directly evaluated with no extra pre-treatment process. The acquired raw intensities of elements are shown in Fig. S17–S21† and ready to be further processed by following the protocol.Product processing and data analysis
In the PAGE control group, the separation of the dsDNA substrate and cleavage products employed denaturing PAGE (25% polyacrylamide, 8 M urea, 1× TBE). Gel was stained with 4S Red Plus Nucleic Acid Stain (Sangon Biotech) for 60 min and imaged by using a FluorChem M Imaging System. The cleavage efficiency quantification was calculated by using the formula as below.| Cleavage efficiency = 100 × (1 − a/a + b) |
Quantification in previous research was based on the electrophoresis band intensities, where a is the integrated intensity of the uncleaved substrate, and b stands for the sum of integrated intensities of all cleaved products ([site #1] + [site #2] + ⋯ + [site #6]). As for EC-CRISPR analysis, cleavage information can be obtained directly from element information. The concentration of each element could be quantified by using the calibration curve (Fig. S16†). Beyond 0.9999 R2 ensures the accuracy of concentration calculations. To minimize the interference, we would redetect the standard samples and refresh the calibration curves every time before sample detection. Besides, the 153Eu signal could be considered as an internal standard to verify the consistency of parallel samples. The concentration of Eu on MBs also indicates the uncleaved substrate. And the difference obtained after subtraction from the concentrations of adjacent element labels in the supernate indicates the relative intensity of the corresponding cleavage site (Fig. 1). Based on the correspondence, the cleavage efficiency formula can be revised as below.
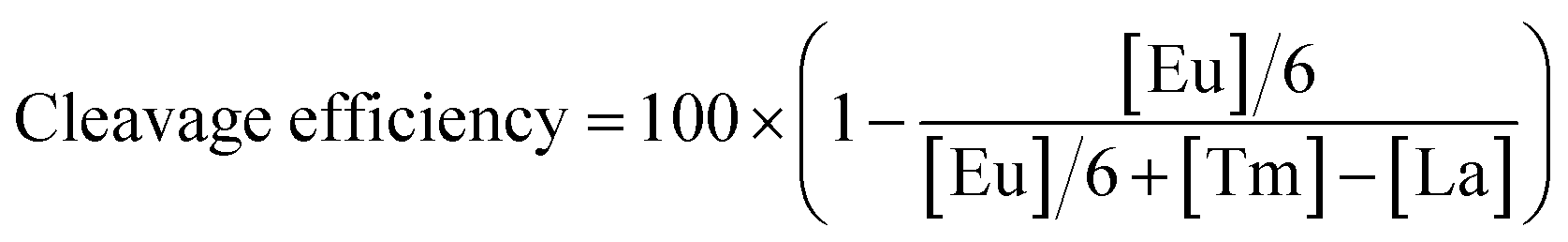
To evaluate the initial non-site #3 cleavage site selection, we defined the non-#3 ratio which is determined by the formula,
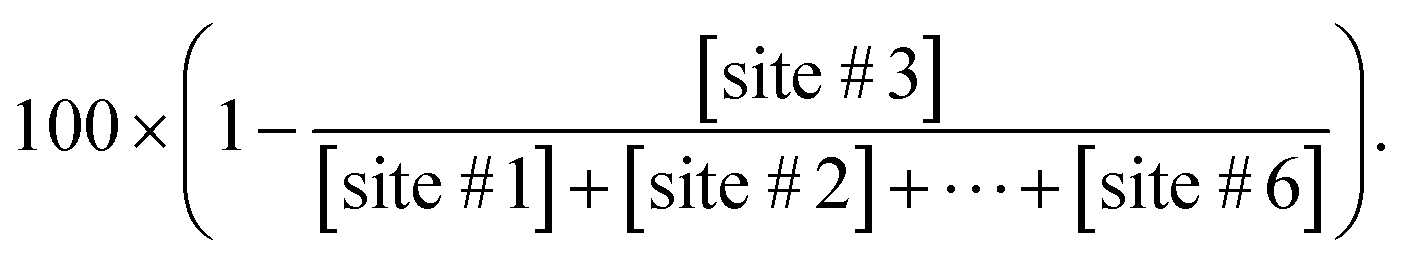
Integrated with the corresponding element label, we can rewrite the above formula as follows,
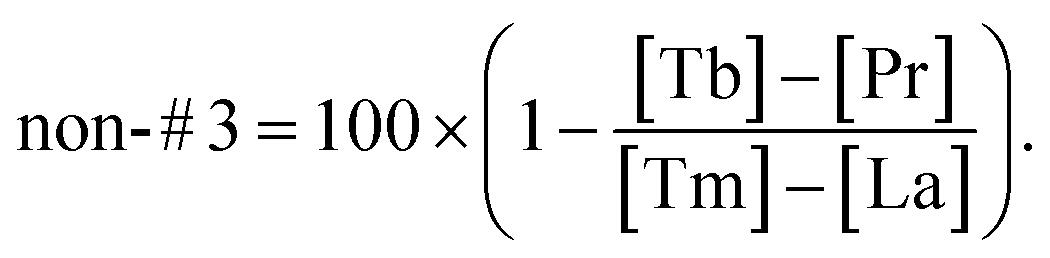
Kinetic behaviours of initial cleavage detection
For EC-CRISPR detection, similar to the general operations mentioned above, the cleavage reaction was quenched at successive time intervals (5–7200 s) with EDTA.For the electrophoresis assay, element label-free ntDNA was pre-hybridized with 3′-biotin-tDNA as the dsDNA substrate. 3 μM sgRNA and Cas9 were pre-incubated at 25 °C for 30 min to form the Cas9/sgRNA binary complex followed by introducing the 300 nM element label-free dsDNA substrate. All the above procedures were carried out in the Mg2+ free reaction buffer (100 mM HEPES, 625 mM KCl and 5 mM DTT, pH = 7.5). With 1 h incubation at 25 °C, 6 mM Mg2+ was rapidly introduced to trigger the cleavage at 37 °C. After the corresponding cleavage time, the reaction was quenched with 0.5 M EDTA. After heated above the melting temperature (Tm) and captured by SA-MBs, the 5′-terminal biotin labeled residues were removed. The remain ssDNA was analyzed by native polyacrylamide gel electrophoresis. For 25% hydrogel preparation, 2 mL of DEPC H2O, 25.0 mL of 30% Acr-Bis (30:1) gel solution, 3 mL of 10× TBE buffer (890 mM Tris, 890 mM boric acid, 20 mM EDTA, pH 8.3), 300 μL of 10% ammonium persulfate APS solution, and 30 μL N,N,N′,N′-tetramethylethylenediamine (TEMED) were rapidly mixed to trigger polymerization for 1 h at room temperature. Each sample was proportionally mixed with a loading buffer, and then loaded into the notches of the 25% gel. Electrophoresis was conducted at a constant 200 V voltage for about 2.5 h. After 2 h of staining with 4 s Red Plus, the gel was imaged by using a FluorChem M (ProteinSimple, USA).
Conclusions
By the proposed EC-CRISPR detection, we systematically studied the cleavage efficiency and site selection under different experimental conditions (temperature, Cas9 concentration), DNA targeting specificity and detailed kinetic behavior of initial cleavage. Despite EC-CRISPR being restrictive in the application of gene editing in cells, EC-CRISPR can provide a direct and accurate in vitro detection of the initial cleavage site, which avoids the interference of the degradation and has a higher sensitivity for elemental analysis. Thanks to this, several hidden facts have been revealed along with the conclusions consistent with previous studies. For site specificity, multiple mismatches will not only affect the efficiency of cleavage, but more than two base mismatches will also have a vast effect on the initial on-target cleavage ratio. In the further study of kinetic behavior, EC-CRISPR also captured the abnormal increase in the proportion of non-site #3 cleavage during the initial stage of the CRISPR reaction. The series of findings can provide support to accurately profile the initial cleavage, minimize the off-target and predict the final products. EC-CRISPR is assembled based on multiplex lanthanide labeling on the adjacent region of PAM. Therefore, EC-CRISPR can be used as a universal platform to evaluate other Cas nuclease systems with substrate specificity.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Jianyu Hu performed the experiments and the data analyses, and wrote the manuscript. Rui Liu contributed significantly to experimental guidance and manuscript revision. Jing Zhou helped perform the analysis with constructive discussions. Yi Lv contributed to the conception of the study.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The National Natural Science Foundation of China is appreciatively acknowledged (No. 22074098 & 22074096). This work is also supported by the Talents Program of Sichuan Province (No. 903), the Sichuan Science and Technology Program (19CXRC0047), and the Fundamental Research Funds for the Central Universities. We appreciate Dr Shuguang Yan and Dr Xiaobo Xie from the Analytical & Testing Center of Sichuan University for helpful technical assistance.References
- J. A. Doudna and E. Charpentier, Science, 2014, 346, 1258096 CrossRef PubMed.
- H. Yin, W. Xue, S. Chen, R. L. Bogorad, E. Benedetti, M. Grompe, V. Koteliansky, P. A. Sharp, T. Jacks and D. G. Anderson, Nat. Biotechnol., 2014, 32, 551–553 CrossRef CAS PubMed.
- W. Xue, S. D. Chen, H. Yin, T. Tammela, T. Papagiannakopoulos, N. S. Joshi, W. X. Cai, G. L. Yang, R. Bronson, D. G. Crowley, F. Zhang, D. G. Anderson, P. A. Sharp and T. Jacks, Nature, 2014, 514, 380–384 CrossRef CAS PubMed.
- M. Z. Alyami, S. K. Alsaiari, Y. Y. Li, S. S. Qutub, F. A. Aleisa, R. Sougrat, J. S. Merzaban and N. M. Khashab, J. Am. Chem. Soc., 2020, 142, 1715–1720 CrossRef CAS PubMed.
- R. T. S. C. Knight and J. A. Doudna, Angew. Chem., Int. Ed., 2018, 57, 4329–4337 CrossRef PubMed.
- K. C. Martin Jinek, I. Fonfara, M. Hauer, J. A. Doudna and E. Charpentier, Science, 2012, 337, 816–821 CrossRef PubMed.
- S. C. Knight, R. Tjian and J. A. Doudna, Angew. Chem., Int. Ed., 2018, 57, 4329–4337 CrossRef CAS PubMed.
- S. H. Sternberg, B. LaFrance, M. Kaplan and J. A. Doudna, Nature, 2015, 527, 110–113 CrossRef CAS PubMed.
- F. A. Ran, L. Cong, W. X. Yan, D. A. Scott, J. S. Gootenberg, A. J. Kriz, B. Zetsche, O. Shalem, X. B. Wu, K. S. Makarova, E. V. Koonin, P. A. Sharp and F. Zhang, Nature, 2015, 520, 186–U198 CrossRef CAS PubMed.
- P. D. Hsu, E. S. Lander and F. Zhang, Cell, 2014, 157, 1262–1278 CrossRef CAS PubMed.
- Z. Zuo and J. Liu, Sci. Rep., 2016, 5, 37584 CrossRef PubMed.
- J. S. C. Yavuz, S. Dagdas, S. H. Sternberg, J. A. Doudna and A. Yildiz, Sci. Adv., 2017, 3, eaao0027 CrossRef PubMed.
- K. Sung, J. Park, Y. Kim, N. K. Lee and S. K. Kim, J. Am. Chem. Soc., 2018, 140, 7778–7781 CrossRef CAS PubMed.
- A. T. Raper, A. A. Stephenson and Z. Suo, J. Am. Chem. Soc., 2018, 140, 2971–2984 CrossRef CAS PubMed.
- S. K. Jones Jr, J. A. Hawkins, N. V. Johnson, C. Jung, K. Hu, J. R. Rybarski, J. S. Chen, J. A. Doudna, W. H. Press and I. J. Finkelstein, Nat. Biotechnol., 2021, 39, 84–93 CrossRef CAS PubMed.
- A. A. Stephenson, A. T. Raper and Z. Suo, J. Am. Chem. Soc., 2018, 140, 3743–3750 CrossRef CAS PubMed.
- R. L. Frock, J. Hu, R. M. Meyers, Y.-J. Ho, E. Kii and F. W. Alt, Nat. Biotechnol., 2015, 33, 179–186 CrossRef CAS PubMed.
- W. X. Yan, R. Mirzazadeh, S. Garnerone, D. Scott, M. W. Schneider, T. Kallas, J. Custodio, E. Wernersson, Y. Li, L. Gao, Y. Federova, B. Zetsche, F. Zhang, M. Bienko and N. Crosetto, Nat. Commun., 2017, 8, 15058 CrossRef CAS PubMed.
- D. Kim, S. Bae, J. Park, E. Kim, S. Kim, H. R. Yu, J. Hwang, J.-I. Kim and J.-S. Kim, Nat. Methods, 2015, 12, 237–243 CrossRef CAS PubMed.
- B. Wienert, S. K. Wyman, C. D. Yeh, B. R. Conklin and J. E. Corn, Nat. Protoc., 2020, 15, 1775–1799 CrossRef CAS PubMed.
- U.-P. Guenther, L. E. Yandek, C. N. Niland, F. E. Campbell, D. Anderson, V. E. Anderson, M. E. Harris and E. Jankowsky, Nature, 2013, 502, 385–388 CrossRef CAS PubMed.
- S. Q. Tsai, Z. Zheng, N. T. Nguyen, M. Liebers, V. V. Topkar, V. Thapar, N. Wyvekens, C. Khayter, A. J. Iafrate, L. P. Le, M. J. Aryee and J. K. Joung, Nat. Biotechnol., 2015, 33, 187–197 CrossRef CAS PubMed.
- N. Crosetto, A. Mitra, M. J. Silva, M. Bienko, N. Dojer, Q. Wang, E. Karaca, R. Chiarle, M. Skrzypczak, K. Ginalski, P. Pasero, M. Rowicka and I. Dikic, Nat. Methods, 2013, 10, 361–365 CrossRef CAS PubMed.
- S. Q. Tsai, N. T. Nguyen, J. Malagon-Lopez, V. V. Topkar, M. J. Aryee and J. K. Joung, Nat. Methods, 2017, 14, 607–614 CrossRef CAS PubMed.
- R. Liu, S. Zhang, C. Wei, Z. Xing, S. Zhang and X. Zhang, Acc. Chem. Res., 2016, 49, 775–783 CrossRef CAS PubMed.
- X. Yan, Y. Luo, Z. Zhang, Z. Li, Q. Luo, L. Yang, B. Zhang, H. Chen, P. Bai and Q. Wang, Angew. Chem., Int. Ed., 2012, 51, 3358–3363 CrossRef CAS PubMed.
- J. Hu, D. Deng, R. Liu and Y. Lv, J. Anal. At. Spectrom., 2018, 33, 57–67 RSC.
- Z. Liu, X. Li, G. Xiao, B. Chen, M. He and B. Hu, TrAC, Trends Anal. Chem., 2017, 93, 78–101 CrossRef CAS.
- G. Han, S. Zhang, Z. Xing and X. Zhang, Angew. Chem., Int. Ed., 2013, 52, 1466–1471 CrossRef CAS PubMed.
- G. Han, M. H. Spitzer, S. C. Bendall, W. J. Fantl and G. P. Nolan, Nat. Protoc., 2018, 13, 2121–2148 CrossRef CAS PubMed.
- G. Gasiunas, R. Barrangou, P. Horvath and V. Siksnys, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E2579–E2586 CrossRef CAS PubMed.
- G. Xiang, X. Zhang, C. An, C. Cheng and H. Wang, J. Genet. Genomics, 2017, 44, 199–205 CrossRef PubMed.
- P. D. Hsu, D. A. Scott, J. A. Weinstein, F. A. Ran, S. Konermann, V. Agarwala, Y. Li, E. J. Fine, X. Wu, O. Shalem, T. J. Cradick, L. A. Marraffini, G. Bao and F. Zhang, Nat. Biotechnol., 2013, 31, 827–832 CrossRef CAS PubMed.
- Z. Hu, G. Sun, W. Jiang, F. Xu, Y. Zhang, M. Xia, X. Pan, Z. Xing, S. Zhang and X. Zhang, Anal. Chem., 2019, 91, 5980–5986 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Oligonucleotide sequence, ESI-MS and MALDI-TOF-MS results of the element tagging process, extra PAGE images and [Ln] raw concentration data. See DOI: 10.1039/d1sc03599a |
| This journal is © The Royal Society of Chemistry 2021 |