
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProgrammable photoresponsive materials based on a single molecule via distinct topochemical reactions†
Xiao
Wei
,
Bao
Li
 ,
Zhiqiang
Yang
,
Ronglin
Zhong
,
Yufei
Wang
,
Yanan
Chen
,
Zeyang
Ding
,
Guangwen
Men
,
Zairan
Yang
,
Houyu
Zhang
,
Bing
Yang
,
Weiqing
Xu
and
Shimei
Jiang
*
,
Zhiqiang
Yang
,
Ronglin
Zhong
,
Yufei
Wang
,
Yanan
Chen
,
Zeyang
Ding
,
Guangwen
Men
,
Zairan
Yang
,
Houyu
Zhang
,
Bing
Yang
,
Weiqing
Xu
and
Shimei
Jiang
*
State Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, 2699 Qianjin Street, Changchun 130012, P. R. China. E-mail: smjiang@jlu.edu.cn
First published on 18th November 2021
Abstract
Engineering the preorganization of photoactive units remains a big challenge in solid-state photochemistry research. It is of not only theoretical importance in the construction of topochemical reactions but also technological significance in the fabrication of advanced materials. Here, a cyanostilbene derivative, (Z)-2-(3,5-bis(trifluoromethyl)phenyl)-3-(naphthalen-2-yl) acrylonitrile (BNA), was crystallized into two polymorphs under different conditions. The two crystals, BNA-α and BNA-β, have totally different intra-π-dimer and inter-π-dimer hierarchical architectures on the basis of a very simple monomer, which provides them with distinct reactivities, functions and photoresponsive properties. Firstly, two different types of solid-state [2 + 2] photocycloaddition reaction: (i) a typical olefin–olefin cycloaddition reaction within the symmetric π-dimers of BNA-α and (ii) an unusual olefin-aromatic ring cycloaddition reaction within the offset π-dimers of BNA-β have been observed, respectively. Secondly, the crystal of BNA-α can be bent to 90° without any fracture, exhibiting outstanding flexibility upon UV irradiation, while the reversible photocycloaddition/thermal cleavage process (below 100 °C) accompanied by unique fluorescence changes can be achieved in the crystal of BNA-β. Finally, micro-scale photoactuators and light-writable anti-counterfeiting materials have been successfully fabricated. This work paves a simple way to construct smart materials through a bottom-up way that is realized by manipulating hierarchical architectures in the solid state.
Introduction
Solid-state photoresponsive materials exhibit remarkable dynamic mechanical responses 1,2 or optical/electronic changes upon light irradiation,3,4 facilitating their applications in photoactuators,5 artificial muscle,6 smart switching7 and optical sensors.3 Photoactive molecules, such as azobenzenes,8 spiropyrans,9 diarylethenes10 and so on, are the building blocks to construct photoresponsive materials.11,12 However, achieving special functions in a confined environment and organized assemblies is not only determined by the molecular structure of photoactive molecules themselves, but also by the mode of molecular spatial arrangement.13–15 Organic supramolecular crystals with well-defined packing and intermolecular interaction are ideal systems for studying supramolecular organization and its control and operation.16–18 Therefore, how to provide organic supramolecular crystals with desirable photoresponsive behaviors attracts extensive attention in chemistry and materials science.Intermolecular [2 + 2] photocycloaddition reaction, especially photodimerization in the solid state, is highly susceptible to molecular spatial arrangement. It is expected to happen only when the two monomers in the reactive π-dimer are aligned as parallel as possible, and the proximity of them is within 4.2 Å.19–21 Such topochemical reactions have fascinating energy transfer, which is capable of converting light into chemical energy and kinetic energy rapidly and efficiently.18,22 On the one hand, the spatial movements of lattice atoms will generate local stress in surrounding π-dimers, enabling the deformation of crystals.23,24 For instance, Naumov and Vittal reported smart molecular crystals based on [2 + 2] photocycloaddition, achieving a variety of photomechanical dynamic behaviors such as bending, jumping, rolling, and photosalience.25–27 On the other hand, special fluorescence changes can be observed in photodimerization due to the breakage of the initial π-dimer structure.28,29 Recently, Tang et al. have demonstrated a significant fluorescence enhancement accompanied by [2 + 2] photoreactions in crystals.30,31 However, despite their charming potential in smart materials, the controllability of photomechanical effect as well as the reversibility of photoswitching remain unaddressed issues for the development of desirable crystal materials.
In this context, further fundamental investigation is required to reveal the underlying principles at work. In particular, the relationship between molecular/supramolecular structures and photoresponsive behaviors should be focused on.18,21 Therefore, constructing different π-dimer structures based on a single monomer and further topochemical reactions might be a solution to reveal the effect of molecular stacking modes on the reactivities, functions and photoresponsive behaviors. Here, a cyanostilbene derivative, (Z)-2-(3,5-bis(trifluoromethyl)phenyl)-3-(naphthalen-2-yl) acrylonitrile, has been synthesized as a basic photoactive building block (BNA in Scheme 1). The polymorphs of BNA were obtained under different conditions: colorless needle-like crystals (BNA-α) and green block crystals (BNA-β) (Fig. 1a and e). As expected, the two crystals have totally different intra-π-dimer (Fig. 1b and f) and inter-π-dimer (Fig. 1c and g) hierarchical architectures, which provides the crystals with distinct reactivities, functions and photoresponsive properties under UV irradiation (Scheme 1). Furthermore, controllable photoactuators and reversible fluorescence switching have been successfully achieved. This system exhibits a very rare instance of desired photoresponsive materials that are realized by utilizing different hierarchical architectures on the basis of a single molecule, which enables distinct topochemical [2 + 2] photocycloadditions.
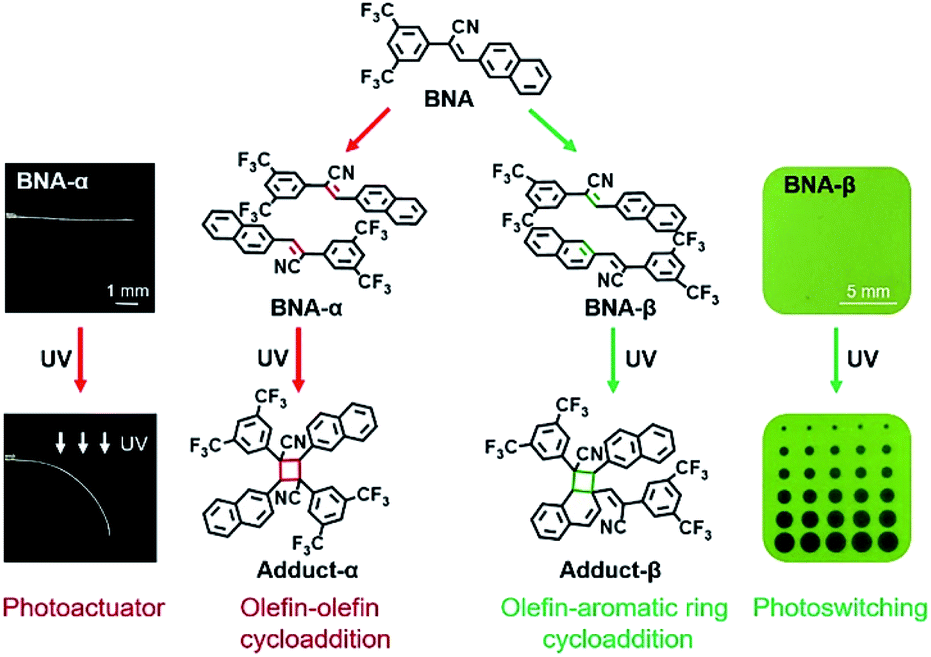 | ||
| Scheme 1 Programmable photoresponsive materials based on a single molecule BNA. Different preorganizations of BNA, BNA-α and BNA-β undergo different types of [2 + 2] photocycloaddition under UV irradiation, respectively. The crystal of BNA-α can be bent to 90° without any fracture upon UV irradiation, exhibiting outstanding potential as a photoactuator (the left of the figure), while the crystal of BNA-β has been fabricated as a light-writable smart material, in which clear patterns can be printed by UV irradiation through photomasks and erased by heating (the right of the figure). | ||
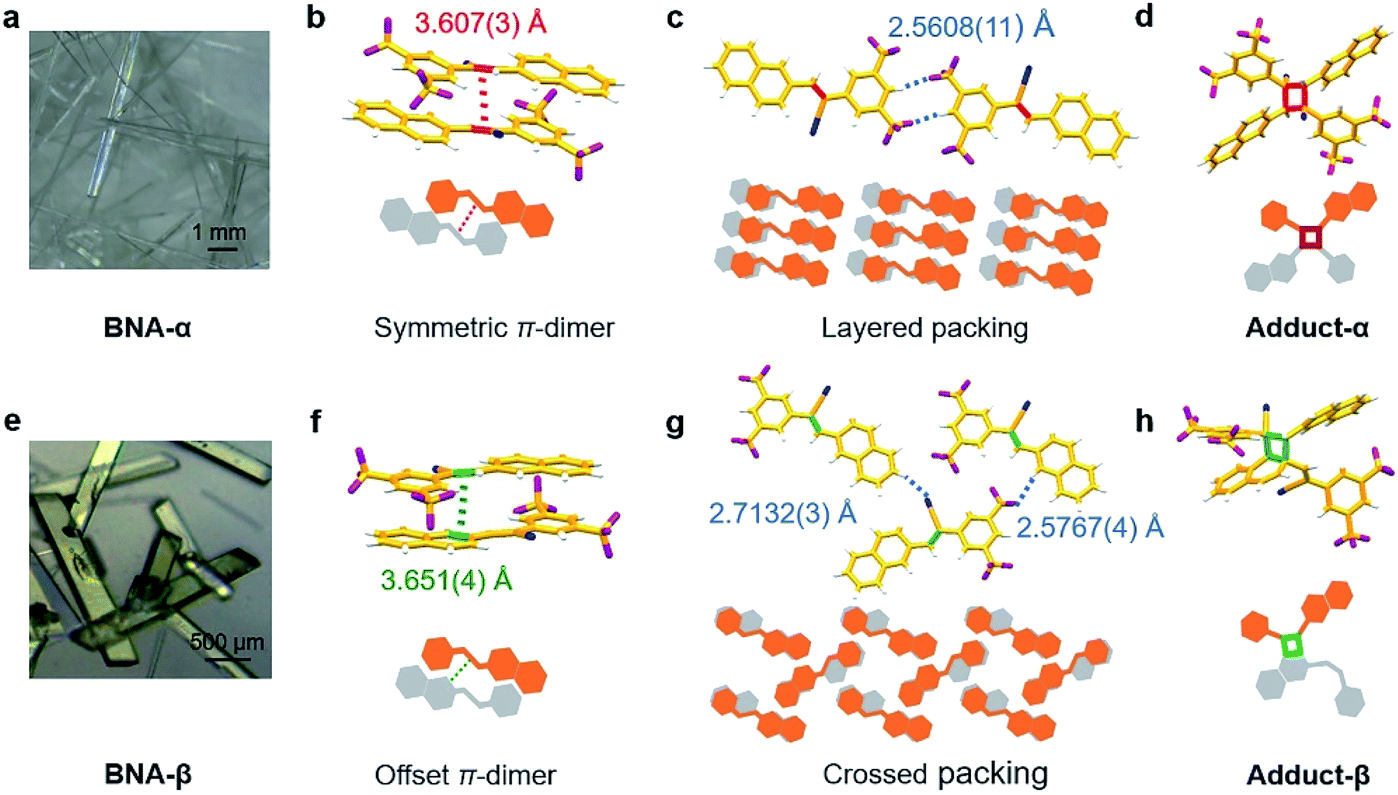 | ||
| Fig. 1 Molecular preorganization of BNA with different hierarchical architectures. (a) Crystals of BNA-α obtained from the ethanol/dichloromethane solution. (b) Symmetric π-dimer structure in BNA-α. (c) Layered packing of π-dimers in the direction of hydrogen bonding in BNA-α. (d) Single-crystal structure of Adduct-α. (e) Crystals of BNA-β obtained from the dichloromethane solution. (f) Offset π-dimer structure in BNA-β. (g) Crossed packing of π-dimers in the direction of hydrogen bonding in BNA-β. (h) Single-crystal structure of Adduct-β. | ||
Results and discussion
Molecular preorganization of BNA with different hierarchical architectures
BNA was prepared by Knoevenagel condensation between 2-(3,5-bis(trifluoromethyl) phenyl) acetonitrile and 2-naphthaldehyde (Scheme S1†). The product was confirmed by NMR spectroscopy (1H and 13C) and mass spectrometry (Fig. S1–S3†). Single crystals of BNA were obtained through the solvent evaporation method. Colorless needle-like crystals (BNA-α) were obtained from a mixed solvent of ethanol and dichloromethane (Fig. 1a and S4a†), while green block-like crystals (BNA-β) were obtained from dichloromethane (Fig. 1e and S4b†). Details of crystallization are provided in the ESI† The crystals of BNA-α and BNA-β were characterized by powder X-ray diffraction (PXRD). As shown in Fig. S5,† different diffraction peaks confirm that BNA-α and BNA-β are packed in different ways.To gain insight into the molecular packing in the two polymorphs, the single crystal structures of BNA-α and BNA-β were determined, respectively. The molecules of BNA-α crystallized in the triclinic crystal system and P-1 space group. More cell parameters and details are shown in Fig. S6 and Table S1†. In the structure of BNA-α, the BNA molecule shows a planar conformation due to its full conjugation structure (Fig. S8a†). The two adjacent monomers stack in an antiparallel and head-to-tail manner through π–π interactions.32,33 The olefin bonds are perfectly aligned (θ1 = 0.00(11)°, θ2 = 86.95(16)°, and θ3 = 78.39(14)°), and the distance between them is 3.607(3) Å, forming a symmetric π-dimer (Fig. 1b, S9, S10a and Table S3†). In addition, the C–H⋯F hydrogen bond (H⋯F: 2.5608(11) Å, C–H⋯F: 150.17(16)°) is formed between benzene and the CF3 group, causing the π-dimers to expand along the hydrogen bond and stack into a layered structure (Fig. 1c and S11a†).
The structure of BNA-β belongs to monoclinic space group P121/n1. The cell parameters and more details are shown in Fig. S7 and Table S2†. Although the pair of monomers in BNA-β also stacks in an antiparallel and head-to-tail manner, the olefin bonds in the π-dimer are dislocated. Surprisingly, the olefin bond and aromatic C–C bond of naphthalene are face to face, nearly parallel (θ1 = 16.33(12)°, θ2 = 100.29(18)°, and θ3 = 79.45(17)°). The distance between them is 3.651(4) Å, forming an offset π-dimer (Fig. 1f, Table S3 and Fig. S10b†). Due to the abundant intermolecular interactions provided by CF3 substitutions,34,35 another type of C–H⋯F hydrogen bond (H⋯F: 2.5767(4) Å, C–H⋯F: 159.12(18)°) is formed between naphthalene and CF3 groups (Fig. 1g). The C–H⋯F hydrogen bond and C–H⋯N hydrogen bond (H⋯N: 2.7132(3) Å, C–H⋯N: 150.58(19)°), as self-assembly driving forces are along different directions, make the offset π-dimers arrange in a crossed structure (Fig. 1g and S11b†). So, the two polymorphs have totally different intra-π-dimer and inter-π-dimer hierarchical architectures on the basis of a very simple monomer.
Programming two types of [2 + 2] photocycloaddition
According to the single crystal structure of BNA-α, the spatial packing of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond pairs in the symmetric π-dimer is parallel-aligned perfectly and the proximity of them is within 4.2 Å, which conforms to the rules of topological chemical reaction. Theoretical calculation also shows the obvious in-phase interaction between the lowest unoccupied molecular orbitals (LUMOs) of CC bond pairs (Fig. S12a†). Thus, the olefin–olefin [2 + 2] cycloaddition is expected to occur based on the preorganization of BNA-α, resulting in a cyclobutane product (Adduct-α, Scheme 1).19,20 For the crystals of BNA-β, although the CC bond and the aromatic C–C bonds in the offset π-dimer preorganized a little bit less parallelly, and the distance between them is slightly farther than that of CC bond pairs in BNA-α, the spatial packing of them still meets the topochemical rules. From the result of theoretical calculation, we can clearly see the overlap between the LUMOs of olefin and the aromatic ring (Fig. S12b†). Therefore, the offset π-dimer of BNA-β is expected to undergo an unusual olefin-aromatic ring cycloaddition reaction to yield a polycyclic product (Adduct-β, Scheme 1).36,37
C bond pairs in the symmetric π-dimer is parallel-aligned perfectly and the proximity of them is within 4.2 Å, which conforms to the rules of topological chemical reaction. Theoretical calculation also shows the obvious in-phase interaction between the lowest unoccupied molecular orbitals (LUMOs) of CC bond pairs (Fig. S12a†). Thus, the olefin–olefin [2 + 2] cycloaddition is expected to occur based on the preorganization of BNA-α, resulting in a cyclobutane product (Adduct-α, Scheme 1).19,20 For the crystals of BNA-β, although the CC bond and the aromatic C–C bonds in the offset π-dimer preorganized a little bit less parallelly, and the distance between them is slightly farther than that of CC bond pairs in BNA-α, the spatial packing of them still meets the topochemical rules. From the result of theoretical calculation, we can clearly see the overlap between the LUMOs of olefin and the aromatic ring (Fig. S12b†). Therefore, the offset π-dimer of BNA-β is expected to undergo an unusual olefin-aromatic ring cycloaddition reaction to yield a polycyclic product (Adduct-β, Scheme 1).36,37
1H NMR spectroscopy was used to monitor the course of the two reactions under UV irradiation. The microcrystalline powders of BNA-α and BNA-β irradiated for different durations were dissolved in DMSO-d6 for 1H NMR measurement, respectively. Compared with the 1H NMR spectrum of the sample before UV irradiation, a new signal at 6.50 ppm out of the range of aromatic protons appears in the irradiated BNA-α, corresponding to the proton in the newly formed cyclobutane ring (Fig. 2a).21 The integration area of this signal increased gradually with prolongation of the irradiation time. Correspondingly, the gradual enhancement of the proton signals at 5.71 ppm and 5.69 ppm in the irradiated BNA-β indicated the gradual increase of a polycyclic product (Fig. 2b). It has been found that almost 100% BNA-α was converted into the corresponding cyclobutane product (Adduct-α) within a very short exposure time of 5 min, indicative of a high efficiency of the reaction, while for BNA-β, a maximum photoconversion of 66% was achieved by irradiation for 30 min. The difference in reactivity might originate from the different spatial arrangements of the monomers. To further clarify the photodimerization products, we isolated and purified them from the photo-irradiated samples, respectively. The 1H NMR spectra, FT-IR analysis and importantly, the single crystal structures of products (Adduct-α and Adduct-β) demonstrate that the two types of [2 + 2] photodimerization have taken place (Fig. 1d and h and S13–S18, Tables S4 and S5†).
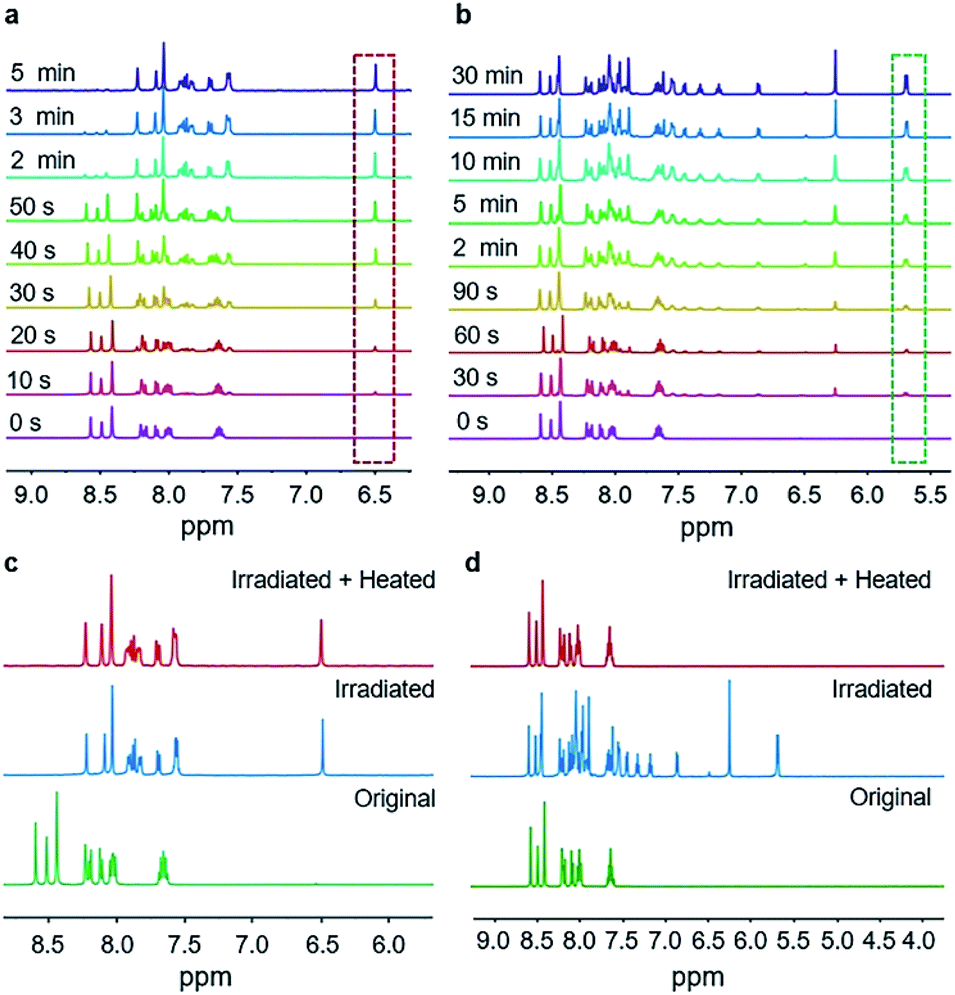 | ||
| Fig. 2 1H NMR spectra of BNA-α and BNA-β in different states. Photocycloaddition processes of (a) BNA-α and (b) BNA-β. Thermal stabilities of the irradiated (c) BNA-α and (d) BNA-β samples. | ||
It was surprising to find that the irradiated powders of BNA-α and BNA-β displayed different thermal stabilities. According to the results of differential scanning calorimetry (DSC), the irradiated BNA-α exhibits no peak before 180 °C (Fig. S19a†). At the same time, no change can be detected for the irradiated BNA-α after heating to 180 °C in the 1H NMR spectrum (Fig. 2c), which reveals that the cyclobutane product (Adduct-α) is very stable. In the case of irradiated BNA-β, before the melting endothermic peak of the monomer at 146 °C, there is an exothermic peak at 95 °C in the DSC curve (Fig. S19b†). Correspondingly, the proton signals of product Adduct-β at 5.71 ppm and 5.69 ppm completely disappeared after heating to 95 °C (Fig. 2d). This is consistent with the result of XRD patterns (Fig. S20†), and the diffraction peaks of the irradiated BNA-β after heating to 95 °C are the same as those of BNA-β without irradiation. These results indicate that the polycyclic ring (Adduct-β) can be thermally cleaved at 95 °C in the irradiated BNA-β.
The thermodynamic processes of the two irradiated samples were further analyzed by combining DSC curves, thermogravimetric analysis (TGA) curves and energy profiles (Fig. S21†). The irradiated product of Adduct-α with a lower energy demonstrates better stability. It undergoes a thermal cleavage and the corresponding monomers evaporate at a very high temperature (above 300 °C) with the absorption of energy (Fig. S21a and b†). In sharp contrast, the irradiated product of Adduct-β has higher energy due to the involvement of the aromatic ring. Adduct-β in irradiated microcrystal powders can be thermally cleaved to monomers at lower temperature with the release of energy (Fig. S21c and d†). This unique behavior in irradiated BNA-β mainly comes from the poor stability of the irradiated product, and the regain of aromaticity can be a driving force to enable the product to be thermally cleaved easier. In addition, the crystals of BNA-β possess a dense and crossed molecular arrangement. When the olefin-aromatic photocycloaddition reactions occur, the buildup of strain forces at the interfacial regions between reactant and product domains cannot be released in time, enabling geometrical constraints and easier thermal cleavage of product Adduct-β.38,39 From an application perspective, it is remarkable progress to find such a reversible photocycloaddition/thermal cleavage process that can be achieved under mild conditions, which will facilitate the development of smart materials based on [2 + 2] photocycloaddition.
Photomechanical effect and controllable photoactuators
The spatial movements of atoms during the [2 + 2] photocycloaddition reaction cause the deformation of crystals.25–27 Here, the single crystal of BNA-α was quickly bent when the UV light was applied (Fig. 3a). However, photoinduced cracks were detected for BNA-β (Fig. 4a). To understand the different photomechanical behaviors in depth, it is necessary to combine molecular packing with their crystal morphologies.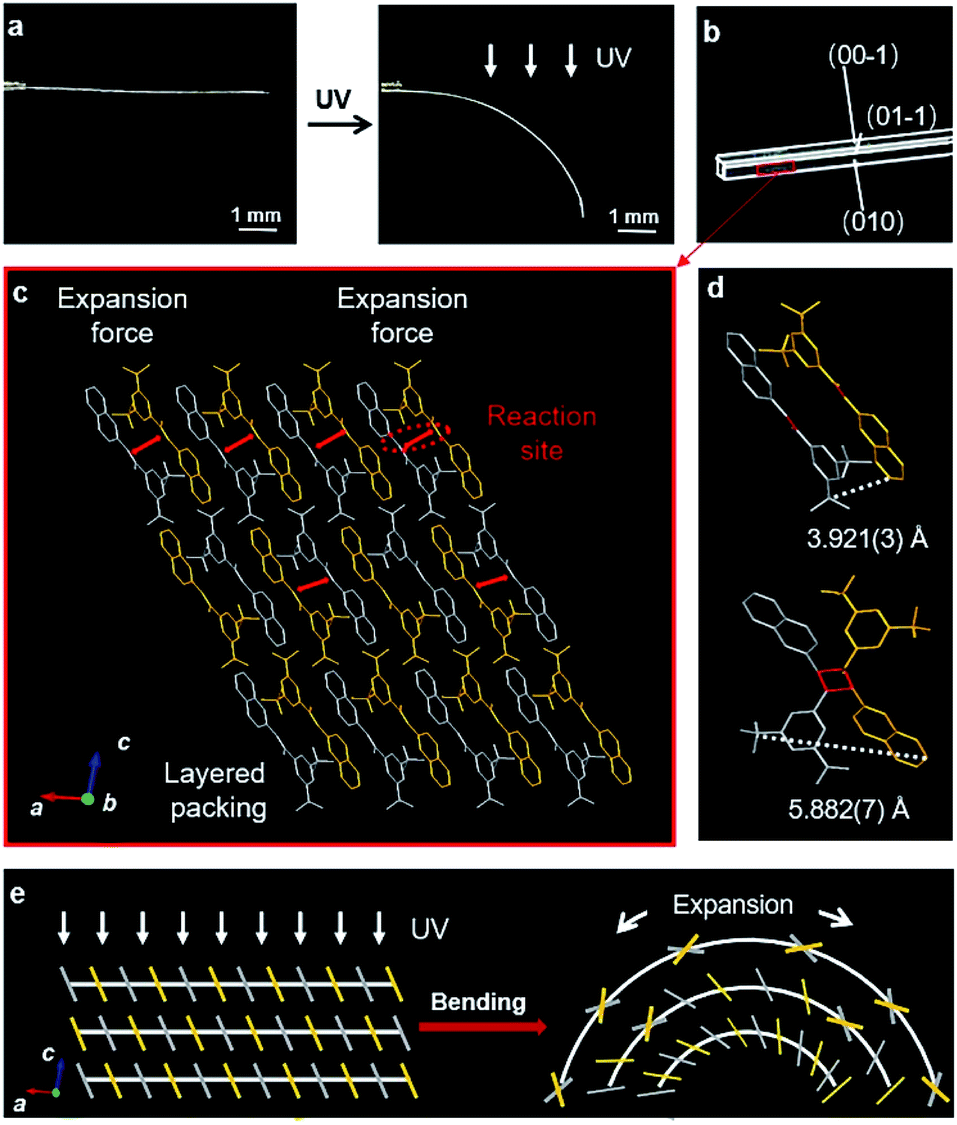 | ||
| Fig. 3 Photoinduced bending of the BNA-α crystal. (a) Optical micrographs of the BNA-α crystal before and after UV irradiation from top. (b) The crystal of BNA-α with face indices. (c) Molecular stack structure viewed from the side face (010) along the b-axis. (d) Adjacent molecular packing in the π-dimer and configuration of Adduct-α in the single crystal. (e) Schematic representation of the photoinduced bending process. When UV irradiation is perpendicular to the crystal downward, the upper π-dimers near the UV source exhibit more photodimerization than the lower π-dimers, and the stress generated by the gradient expansion in different layers leads to the bending of the crystal. | ||
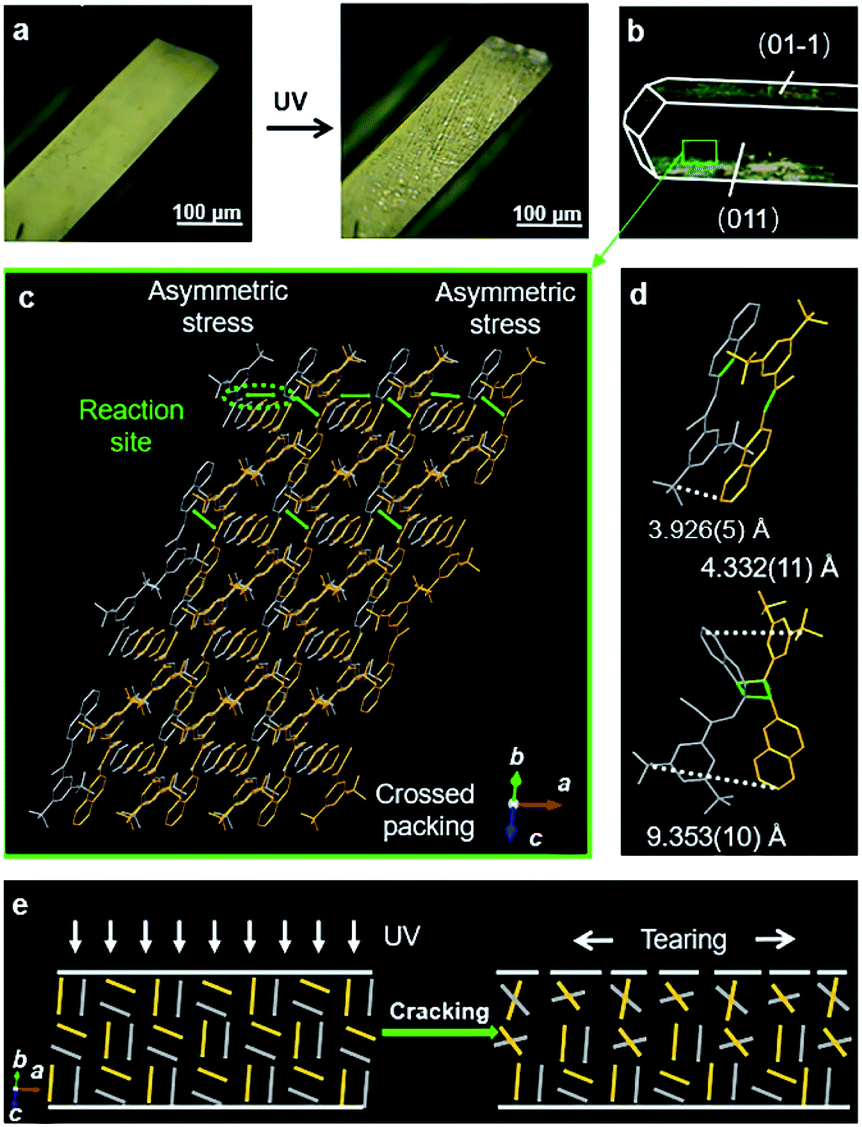 | ||
| Fig. 4 Photoinduced cracks of the BNA-β crystal. (a) Optical micrographs of the BNA-β crystal before and after UV irradiation. (b) The crystal of BNA-β with face indices. (c) Molecular stack structure viewed from the side face (011). (d) Adjacent molecular packing in the π-dimer and configuration of Adduct-β in the single crystal. (e) Schematic representation of the photoinduced cracking process. When UV irradiation is applied to the crystal, the photodimerization of the cross-stacked π-dimers generates internal anisotropic stress and results in the cracks of the crystal. | ||
The crystal of BNA-α with face indices demonstrates that the two pairs of side faces of the needle-like crystal are (010)/(0–10) and (001)/(00–1), and the length direction of the crystal is along the a axis (Fig. 3b). This is consistent with the crystal morphology predicted by Materials Studio (Fig. S22†). Viewed from the side face (010), the symmetric π-dimers are stacked along the a axis and finally form a layered structure by further stacking along the c axis (Fig. 3c). Upon UV irradiation, the crystal structure of the newly formed Adduct-α demonstrates that the atoms within the π-dimer have moved during photodimerization, as shown in Fig. 3d. The parallel benzene and naphthalene were forced away from the cyclobutane moiety, and the distance between the outermost C atoms changed from 3.921(3) Å to 5.882(7) Å, resulting in a symmetric expansion force on its neighboring π-dimers. The π–π stacking interactions within layers along with the hydrogen bonds between layers, as “structural buffers”, can respond to the force of expansion or contraction, enable the bending of the BNA-α crystal.40–42 To further clarify its mechanism, the schematic representation of the photoinduced bending process is illustrated based on the molecular packing in the (010) face (Fig. 3e). When UV irradiation is perpendicular to the (00–1) plane of the crystal downward, the irradiation intensity is diminished from top to bottom due to the gradient shielding effect of molecules. This will lead to more photodimerization of the upper π-dimers than those of the lower π-dimers. The photodimerization generates the symmetric expansion force along the π–π stacking direction parallel to the (00–1) face. The stress generated by the gradient expansion in different layers leads to the bending of the crystal away from the light source. Crystal mechanics tests also demonstrated that the crystal of BNA-α is remarkably flexible, capable of bending 180° by force, without any obvious fracture (Fig. S24a†). The three-point bending test and the nanoindentation technique were employed for the quantitative analysis of crystals (Fig. S25†). The results demonstrated that the bending of BNA-α crystals is elastic in nature, and its yield strain can reach 1.3% (Fig. S26a†). In addition, the results of single-crystal X-ray diffraction indicate that the bent crystals under UV light irradiation or external force remain crystalline (Fig. S27 and S28†).43
However, in the case of BNA-β, the crystal with face indices and crystal morphology simulation demonstrates that the two pairs of side faces of the block crystal are (011)/(0–1–1) and (01–1)/(0–11), respectively (Fig. 4b and S23†). Viewed from the (011) plane, the offset π-dimers stack along two different directions, forming a crossed structure (Fig. 4c). Before UV irradiation, the distance between the outermost C atoms is 3.926(5) Å in the offset π-dimer. The photocycloaddition between the olefin bond and naphthalene broke the π-dimer. The distance between the outermost C atoms of the newly formed Adduct-β increased to 4.332(11) Å and 9.353(10) Å, respectively (Fig. 4d). The asymmetric expansion of the cross-stacked π-dimers generates an internal anisotropic stress, resulting in the cracks of the crystal (Fig. 4e). The crystal mechanics tests also demonstrate a higher hardness and a smaller yield strain of BNA-β compared with BNA-α crystals, indicating the fragility of BNA-β crystals (Fig. S24b and S26†).
Different intra-π-dimer and inter-π-dimer hierarchical architectures of BNA-α and BNA-β provided the crystals with entirely different photomechanical responses. The crystals of BNA-α were employed to fabricate photoactuators owing to their excellent photo-induced manipulation. Firstly, the bending angle of the crystal depends on the irradiation time as shown in Fig. 5a. More photodimerization occurs along with the increase of irradiation time, leading to a larger bending angle of crystals (Fig. S29†). To quantitatively evaluate the controllable bending behavior, the response rate at varying thicknesses was determined by linear fitting of the temporal changes of curvature under irradiation (Fig. S30 and S31†). From the dependence of the response on the crystal thickness, the kinetics of bending depending on the crystal sizes was obtained (Fig. S32†).44 The thinner the crystal is, the faster it responds to light. Secondly, according to the mechanism of BNA-α deformation, the bending direction can be controlled by altering the direction of incident light. When the UV source irradiated from above, the crystal was bent downwards. The crystal recovered to flat and then was bent upwards under continuous UV light irradiation from the opposite direction (Fig. 5b and ESI Video 1†). Thirdly, the bending position of the crystal is regulated by the incident position of light. A wavy crystal was prepared successfully by controlling the position and direction of incident light (Fig. 5c). Hence, the crystal of BNA-α can be precisely controlled by regulating the irradiation time, direction and position of incident light. Based on this, light-controlled tweezers that could exactly grip micro-scale objects were designed. The tweezers were fabricated by fixing two crystals of BNA-α with different thicknesses on the tip of a capillary. As shown in Fig. 5d, the tweezers were moved near to an orange microcrystal through a 3-dimensional operation platform, and the faster bending of the thinner crystal shortened the distance between two crystals, successfully clamping the micro-scale object.
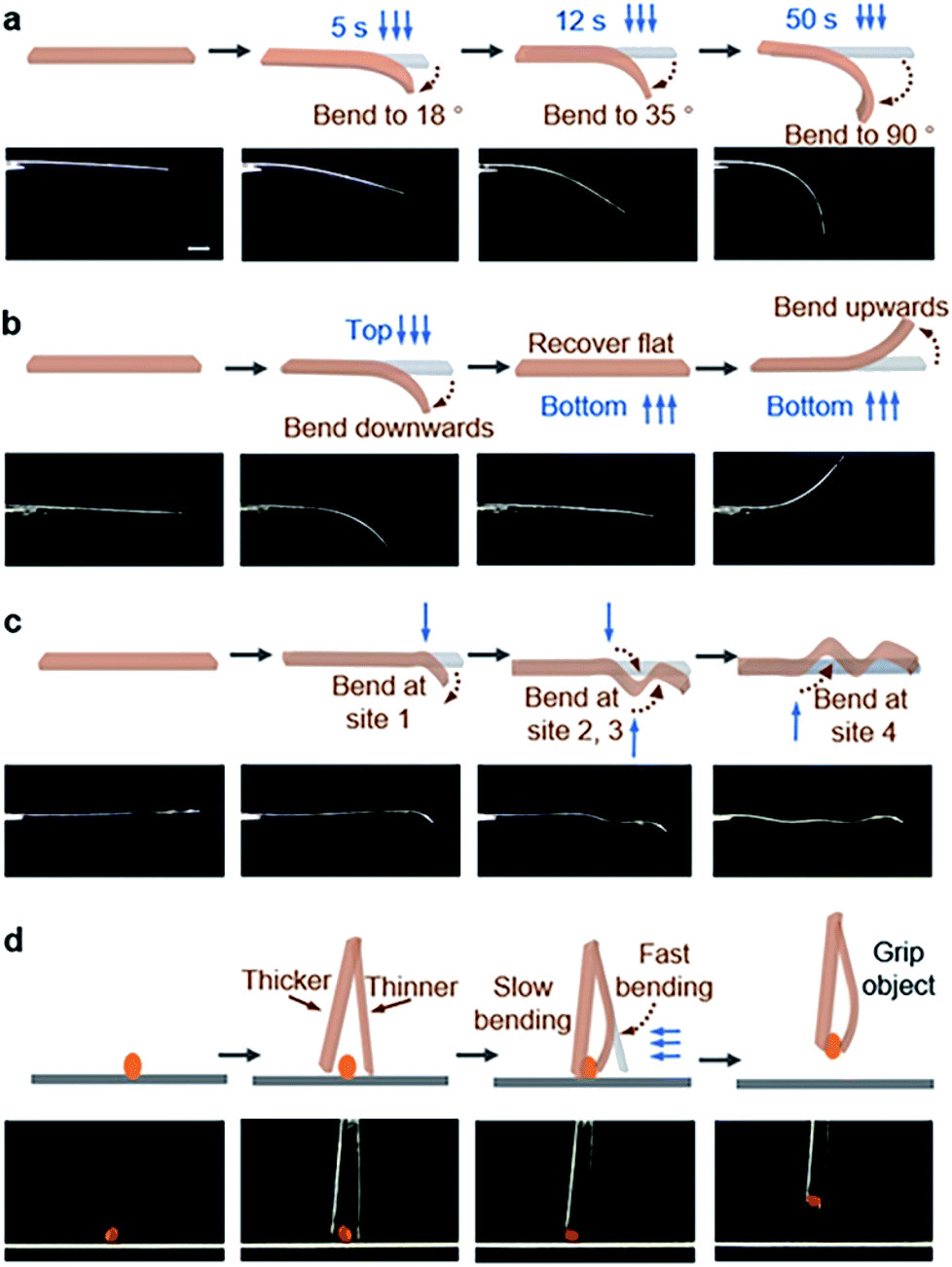 | ||
| Fig. 5 Photoactuators based on BNA-α crystals. Precisely controlling the movement of the crystal by regulating the irradiation conditions: (a) the bending angle of the crystal depending on irradiation time; (b) the bending direction of the crystal depending on the direction of light incidence; (c) the bending site of the crystal depending on the position of light incidence. (d) The tweezers consisting of two BNA-α crystals with different thicknesses can clamp a micro-scale object (the orange microcrystal) by applying UV irradiation. Scale bar is 1 mm. The blue arrows show the direction of UV irradiation, and crystals were bent away from the light source. | ||
Photoinduced fluorescence change and advanced photoswitching
Different spatial arrangements not only affect the photomechanical deformation, but also shift the emission color of the materials. The luminescence of cyanostilbene is sensitive to the molecular structure and spatial packing,45–49 and hence BNA-α and BNA-β exhibited unique fluorescence changes accompanied by [2 + 2] photodimerization. As shown in Fig. 6, BNA-α was non-fluorescent, while BNA-β emitted green fluorescence in its initial state due to their different molecular packings. Both of them began to exhibit blue emissions, and their intensities were gradually enhanced with increasing time of UV irradiation. To clarify these processes, UV-vis absorption and fluorescence spectra were utilized to monitor their courses. The powders of BNA-α and BNA-β exposed to UV light for different durations were dissolved in dichloromethane for UV-vis absorption measurement. Abs@334 nm, ascribed to the BNA monomer, decreased under UV irradiation, while new absorption bands appeared at 230 nm and at 228 nm, respectively (Fig. S33†). Such changes are related to the formation of the cyclobutane ring, suggesting the conversion from the BNA monomer to photocycloaddition products.29,50 Meanwhile, the intensities of the newly appeared blue emissions increased along with the increase of the photodimerization products (Fig. S34†). The Adduct-α and Adduct-β were separated and purified, respectively. As expected, the fluorescence peaks of these pure products were coincident with those of the aforementioned blue emissions, indicating that the photochromism resulted from the corresponding photodimerization reactions (Fig. S35 and S36†).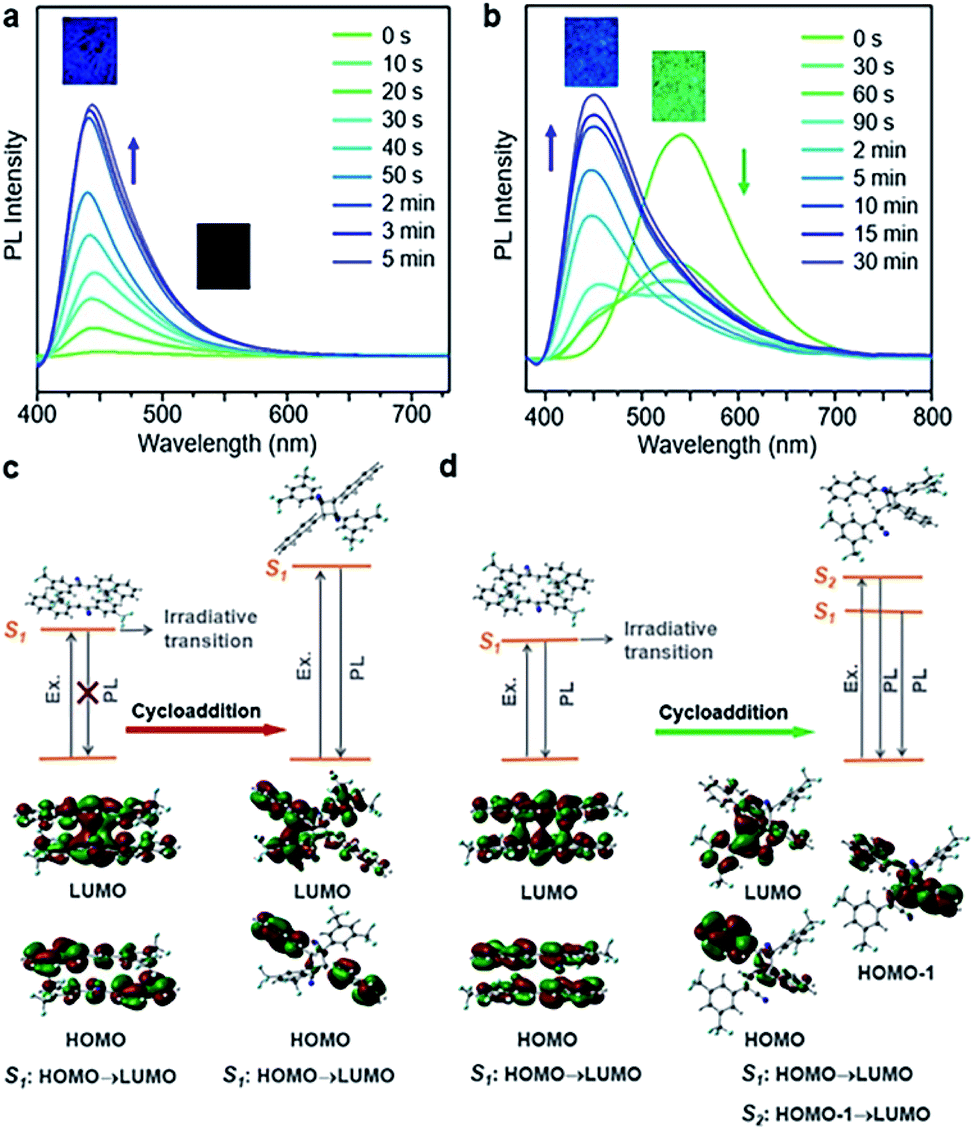 | ||
| Fig. 6 Photoinduced fluorescence change of BNA-α and BNA-β. Fluorescence spectra of (a) BNA-α and (b) BNA-β at different UV irradiation times. Calculation results of the processes of fluorescence changes in (c) BNA-α and (d) BNA-β. | ||
Theoretical calculation was further employed to investigate the processes of fluorescence changes. As shown in Fig. 6, the different in-phase interactions between the two molecules of the π-dimers in excited states reveal the different photodimerization reactions of BNA-α and BNA-β. According to previous reports, when the π-dimers are excited, there are two competing photophysical pathways available: emission transition and nonradiative transition.28 For BNA-α, the nonradiative transition is predominant in the symmetric π-dimer, resulting in no fluorescence initially, whereas BNA-β presents green fluorescence in its initial state, because the two pathways exist in the excited state based on the offset π-dimer. Upon exposure to UV light, for Adduct-α, obvious electronic delocalization of the lowest unoccupied molecular orbital (LUMO) indicates through-space conjugation between the stacked naphthyl ring (donor) and 2,4-bis(trifluoromethyl)phenyl group (acceptor).30,31 The charge transfer from the 2,4-bis(trifluoromethyl)phenyl group to the naphthyl ring results in the blue emission of Adduct-α (Fig. 6c). Similar to Adduct-α, Adduct-β also exhibits an obvious through-space conjugation character between the stacked naphthyl ring and 2,4-bis(trifluoromethyl)phenyl group (Fig. 6d). It is worth noting that the olefin-aromatic ring cycloaddition reaction yields an asymmetric Adduct-β. When the charge transfer is mainly carried out from the 2,4-bis(trifluoromethyl)phenyl group to the naphthyl ring that was not involved in the cycloaddition, the emission of Adduct-β is similar to that of Adduct-α, showing that blue fluorescence peaked at 453 nm, while the charge transfer mainly comes back to the naphthyl ring involved in the cycloaddition, and it emits green fluorescence at 514 nm (Fig. S37†). The fluorescence decay profiles of Adduct-β at 453 nm and 514 nm respectively related to the S2 state and S1 state showed different average fluorescence lifetimes, which further confirms a dual-state emission of Adduct-β (Fig. S38†).
BNA-β exhibits a unique fluorescence change as well as reversible photo-cycloaddition/thermal cleavage under UV irradiation and heating, which enables it to be an ideal candidate for advanced photoswitching. Here, BNA-β nanocrystals were attached to a backing with an adhesive to prepare a kind of rewritable fluorescent paper for UV light writing and printing (Fig. S39†). Consistent with the above-mentioned results, the fluorescence change of the paper depends on the irradiation time. The paper showed green fluorescence at the beginning (ON state), and its intensity rapidly weakened to the OFF state within 1 s under UV light irradiation (Fig. 7a and S40†). The OFF state of this paper recovered to the ON state after heating at 95 °C for 1 min. The fluorescence of this paper could circulate between ON and OFF states multiple times by alternate exposure to light and heat, which shows its fast response and good reversibility (Fig. 7b). Moreover, when we wrote with a UV laser pen, the paper exhibited a rapid fluorescence response and showed “CHEM” handwriting, which could be easily erased by heating (Fig. 7c and ESI Video 2†). The handwriting was hard to read under sunlight, whereas it appeared in a sharp contrast under the irradiation of the UV lamp, indicating that the paper can be applied to information encryption. In addition, it can also be used for anti-counterfeiting. Different patterns were printed on the fluorescence paper by UV light with different photomasks and were erased by heating (Fig. 7d and S41†). The patterns can always keep a high resolution and a high contrast after multiple print–erase cycles.
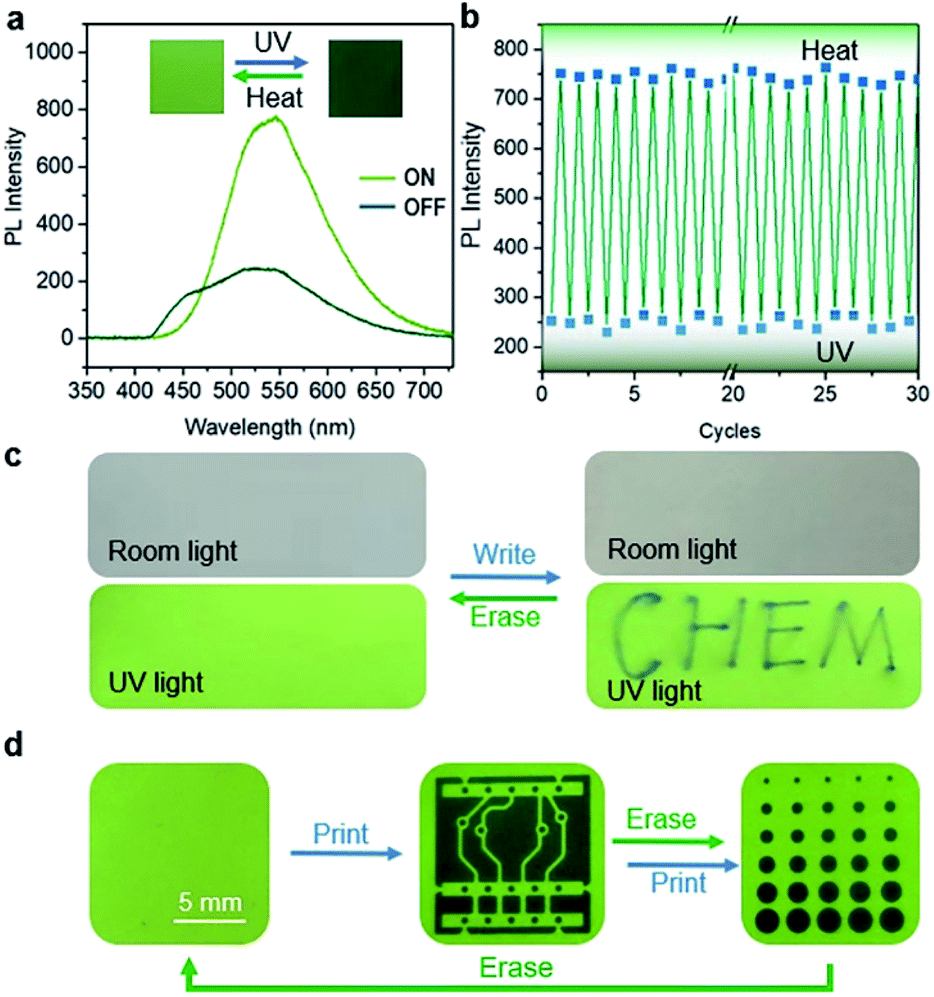 | ||
| Fig. 7 Photoswitching based on BNA-β nanocrystals. (a) Fluorescence spectra of the rewritable fluorescent paper in the ON and OFF state based on BNA-β nanocrystals. Inset: photographs of this paper in the ON and OFF state modulated by the UV–heat cycle. (b) Fluorescence intensities modulated by the multiple UV–heat cycles. (c) Optical and fluorescent photographs of the rewritable fluorescent paper with “CHEM” handwriting wrote with a UV laser pen and erased by heating. (d) Fluorescent photographs of the rewritable fluorescent paper with different patterns printed by UV irradiation through different photomasks and erased by heating. | ||
Conclusions
In summary, one single molecule bearing a cyanostilbene moiety could be crystallized into two polymorphs. As a consequence, two different types of solid-state [2 + 2] photocycloaddition reaction have been observed, respectively. It has been demonstrated that the hierarchical architectures, i.e. intra-π-dimer structure and inter-π-dimer structure, are crucial to topochemical reactions toward on-demand functions. For crystals fulfilling the demand of olefin–olefin [2 + 2] photocycloaddition (BNA-α), the symmetric π-dimers stack to form layered structures, which can act as “structural buffers” to drain the expansion force and provide the crystals with outstanding flexibility. However, for crystals fulfilling the demand of olefin-aromatic ring [2 + 2] photocycloaddition (BNA-β), the dense and crossed arrangement of the offset π-dimers as well as the involvement of aromatic ring enable easier thermal decomposition of the photochemical products. Based on their distinct photoresponsive behaviors, controllable photoactuators and reversible fluorescence switching have been successfully achieved. These results not only demonstrate the significant effect of hierarchical architectures in topochemical reactions but also open a new insight into further development of smart materials with finely tuned properties in a bottom-up way.Data availability
The data that support the findings of this study and crystallographic data are available in the ESI.†Author contributions
X. W. performed experiments. X. W. and S. J. wrote the manuscript. B. L. offered single crystal data. Z. Y, Y. C., H. Z. and B. Y. performed theoretical calculations. All authors contributed to the analysis and the interpretation of the results.Conflicts of interest
All the authors declare no competing financial interest.Acknowledgements
We thank Dr Shitong Zhang (Jilin University) for discussions relating to the theoretical calculations. X. W. is grateful to Songyuan Tao (Jilin University) and Dr Hanfei Gao (Ji Hua Laboratory) for useful discussions. This work was supported by the National Natural Science Foundation of China (52173167 and 21935005).Notes and references
- O. Sato, Nat. Chem., 2016, 8, 644–656 CrossRef CAS PubMed.
- M. Lahikainen, H. Zeng and A. Priimagi, Nat. Commun., 2018, 9, 4148 CrossRef PubMed.
- E. Orgiu and P. Samorì, Adv. Mater., 2014, 26, 1827–1845 CrossRef CAS PubMed.
- R. Pardo, M. Zayat and D. Levy, Chem. Soc. Rev., 2011, 40, 672–687 RSC.
- T. Muraoka, K. Kinbara and T. Aida, Nature, 2006, 440, 512–515 CrossRef CAS PubMed.
- M. K. McBride, A. M. Martinez, L. Cox, M. Alim, K. Childress, M. Beiswinger, M. Podgorski, B. T. Worrell, J. Killgore and C. N. Bowman, Sci. Adv., 2018, 4, eaat4634 CrossRef CAS PubMed.
- Y. Yao, H. Dong, F. Liu, T. P. Russell and W. Hu, Adv. Mater., 2017, 29, 201701251 Search PubMed.
- S. Crespi, N. A. Simeth and B. König, Nat. Rev. Chem., 2019, 3, 133–146 CrossRef CAS.
- L. Kortekaas and W. R. Browne, Chem. Soc. Rev., 2019, 48, 3406–3424 RSC.
- D. Kitagawa, H. Tsujioka, F. Tong, X. Dong, C. J. Bardeen and S. Kobatake, J. Am. Chem. Soc., 2018, 140, 4208–4212 CrossRef CAS PubMed.
- M. Irie, Chem. Rev., 2000, 100, 1683–1684 CrossRef CAS PubMed.
- S. Crespi, N. A. Simeth and B. König, Nat. Rev. Chem., 2019, 3, 133–146 CrossRef CAS.
- W. Peng, G. Zhang, J. Liu, S. Nie, Y. Wu, S. Deng, G. Fang, J. Zhou, J. Song, J. Qian, P. Pan, Q. Zhao and T. Xie, Adv. Funct. Mater., 2020, 30, 2000522 CrossRef CAS.
- F. Tong, W. Xu, M. Al–Haidar, D. Kitagawa, R. O. Al-Kaysi and C. J. Bardeen, Angew. Chem., Int. Ed., 2018, 57, 7080–7084 CrossRef CAS PubMed.
- Y. Gu, E. A. Alt, H. Wang, X. Li, A. P. Willard and J. A. Johnson, above Nature, 2018, 560, 65–69 CrossRef CAS PubMed.
- S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Nature, 2007, 446, 778–781 CrossRef CAS PubMed.
- R. Mohanrao, K. Hema and K. M. Sureshan, Nat. Commun., 2020, 11, 865 CrossRef CAS PubMed.
- P. Naumov, S. Chizhik, M. K. Panda, N. K. Nath and E. Boldyreva, Chem. Rev., 2015, 115, 12440–12490 CrossRef CAS PubMed.
- M. D. Cohen and G. M. J. Schmidt, Topochemistry.Part I. A survey.J.Chem.Soc., 1964, 1996–2000 CAS.
- G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647–678 CAS.
- V. Ramamurthy and J. Sivaguru, J. Chem. Rev., 2016, 116, 9914–9993 CrossRef CAS PubMed.
- D. J. Shields, D. P. Karothu, K. Sambath, R. Ranaweera, S. Schramm, A. Duncan, B. Duncan, J. A. Krause, A. D. Gudmundsdottir and P. Naumov, J. Am. Chem. Soc., 2020, 142, 18565–185675 CrossRef CAS PubMed.
- H. Wang, P. Chen, Z. Wu, J. Zhao, J. Sun and R. Lu, Angew. Chem., Int. Ed., 2017, 56, 9463–9467 CrossRef CAS PubMed.
- S. Li, B. Lu, X. Fang and D. Yan, Angew. Chem., Int. Ed., 2020, 59, 22812–22819 CrossRef.
- B. B. Rath and J. J. Vittal, J. Am. Chem. Soc., 2020, 142, 20117–20123 CrossRef CAS PubMed.
- P. Naumov, J. Kowalik, K. M. Solntsev, A. Baldridge, J.-S. Moon, C. Kranz and L. M. Tolbert, J. Am. Chem. Soc., 2010, 132, 5845–5857 CrossRef CAS PubMed.
- P. Naumov, S. Chizhik, P. Commins, et al.Bending, Jumping, and Self-Healing Crystals in Mechanically Responsive Materials for Soft Robotics, Wiley-VCH, Weinheim, 2020, pp. 105–138 Search PubMed.
- J. W. Chung, Y. You, H. S. Huh, B.-K. An, S.-J. Yoon, S. H. Kim, S. W. Lee and S. Y. Park, J. Am. Chem. Soc., 2009, 131, 8163–8172 CrossRef CAS PubMed.
- P. Wei, J. X. Zhang, Z. Zhao, Y. Chen, X. He, M. Chen, J. Gong, H. H. Sung, I. D. Williams, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2018, 140, 1966–1975 CrossRef CAS PubMed.
- J. Guo, J. Fan, X. Liu, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 8828–8832 CrossRef CAS PubMed.
- H. Wang, H. Xing, J. Gong, H. Zhang, J. Zhang, P. Wei, G. Yang, J. W. Y. Lam, R. Lu and B. Z. Tang, Mater. Horiz., 2020, 7, 1566–1572 RSC.
- S. Y. Yang, P. Naumov and S. Fukuzumi, J. Am. Chem. Soc., 2009, 131, 7247–7249 CrossRef CAS PubMed.
- G. W. Coates, A. R. Dunn, L. M. Henling, J. W. Ziller, E. B. Lobkovsky and R. H. Grubbs, J. Am. Chem. Soc., 1998, 120, 3641–3649 CrossRef CAS.
- S. Varghese, S. K. Park, S. Casado, R. Resel, R. Wannemacher, L. Lüer, S. Y. Park and J. Gierschner, Adv. Funct. Mater., 2016, 26, 2349–2356 CrossRef CAS.
- K. Reichenbächer, H. I. Süss and J. Hulliger, Chem. Soc. Rev., 2005, 34, 22–30 RSC.
- Y. Ito, S. Horie and Y. Shindo, Org. Lett., 2001, 3, 2411–2413 CrossRef CAS PubMed.
- R. Medishetty, Z. Bai, H. Yang, M. W. Wong and J. J. Vittal, Cryst. Growth Des., 2015, 15, 4055–4061 CrossRef CAS.
- T. Kim, L. Zhu, L. J. Mueller and C. J. Bardeen, J. Am. Chem. Soc., 2014, 136, 6617–6625 CrossRef CAS PubMed.
- L. Zhu, R. O. Al-Kaysi and C. J. Bardeen, J. Am. Chem. Soc., 2011, 133, 12569–12575 CrossRef CAS PubMed.
- S. Ghosh, M. K. Mishra, S. Ganguly and G. R. Desiraju, J. Am. Chem. Soc., 2015, 137, 9912–9921 CrossRef CAS PubMed.
- E. Ahmed, D. P. Karothu and P. Naumov, Angew. Chem., Int. Ed., 2018, 57, 8837–8846 CrossRef CAS PubMed.
- H. Liu, Z. Lu, Z. Zhang, Y. Wang and H. Zhang, Angew. Chem., Int. Ed., 2018, 57, 8448–8452 CrossRef CAS PubMed.
- P. Commins, D. P. Karothu and P. Naumov, Angew. Chem., Int. Ed., 2019, 58, 10052–10060 CrossRef CAS PubMed.
- J. M. Halabi, E. Ahmed, L. Catalano, D. P. Karothu, R. Rezgui and P. Naumov, J. Am. Chem. Soc., 2019, 141, 14966–14970 CrossRef CAS PubMed.
- Z. Ding, Y. Ma, H. Shang, H. Zhang and S. Jiang, Chem.–Eur. J., 2018, 25, 315–322 CrossRef PubMed.
- S. J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M.-G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS PubMed.
- Y. Xu, K. Wang, Y. Zhang, Z. Xie, B. Zou and Y. Ma, J. Mater. Chem. C, 2016, 4, 1257–1262 RSC.
- H. Shang, Z. Ding, Y. Shen, B. Yang, M. Liu and S. Jiang, Chem. Sci., 2020, 11, 2169–2174 RSC.
- D. Niu, Y. Jiang, L. Ji, G. Ouyang and M. Liu, Angew. Chem., Int. Ed., 2019, 58, 5946–5950 CrossRef CAS PubMed.
- X. Tang, Z. Huang, H. Chen, Y. Kang, J.-F. Xu and X. Zhang, Angew. Chem., Int. Ed., 2018, 57, 8545–8549 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, calculated growth morphology, mechanics tests, photomechanical and photochromism studies, theoretical calculation, photophysical property analysis, supplementary figures and NMR, DSC, PXRD and FT-IR spectra (PDF). Photomechanical and photochromism videos are shown in supplementary video 1 and supplementary video 2. CCDC The single crystal data of BNA-α, BNA-β, Adduct-α and Adduct-β have been deposited as CIF files in the Cambridge Crystallographic Data Center with publication numbers CCDC 2032920, 2113420, 2032927 and 2032930. Crystallographic data in CIF (supplementary CIF 1, CIF 2, CIF 3 and CIF 4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04053g |
| This journal is © The Royal Society of Chemistry 2021 |