
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComplexation and redox chemistry of neptunium, plutonium and americium with a hydroxylaminato ligand†
Jing
Su‡
 a,
Thibault
Cheisson
b,
Alex
McSkimming
b,
Conrad A. P.
Goodwin
c,
Ida M.
DiMucci
c,
Thomas
Albrecht-Schönzart
d,
Brian L.
Scott
e,
Enrique R.
Batista
*a,
Andrew J.
Gaunt
*c,
Stosh A.
Kozimor
*c,
Ping
Yang
*a and
Eric J.
Schelter
*b
a,
Thibault
Cheisson
b,
Alex
McSkimming
b,
Conrad A. P.
Goodwin
c,
Ida M.
DiMucci
c,
Thomas
Albrecht-Schönzart
d,
Brian L.
Scott
e,
Enrique R.
Batista
*a,
Andrew J.
Gaunt
*c,
Stosh A.
Kozimor
*c,
Ping
Yang
*a and
Eric J.
Schelter
*b
aTheoretical Division, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA. E-mail: erb@lanl.gov; pyang@lanl.gov
bP. Roy and Diana T. Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, 231 S 34th St., Philadelphia, Pennsylvania 19104, USA. E-mail: schelter@sas.upenn.edu
cChemistry Division, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA. E-mail: gaunt@lanl.gov; stosh@lanl.gov
dDepartment of Chemistry and Biochemistry, Florida State University, 95 Chieftan Way, Tallahassee, Florida 32306, USA
eMaterials and Physics Applications Division, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA
First published on 3rd September 2021
Abstract
There is significant interest in ligands that can stabilize actinide ions in oxidation states that can be exploited to chemically differentiate 5f and 4f elements. Applications range from developing large-scale actinide separation strategies for nuclear industry processing to carrying out analytical studies that support environmental monitoring and remediation efforts. Here, we report syntheses and characterization of Np(IV), Pu(IV) and Am(III) complexes with N-tert-butyl-N-(pyridin-2-yl)hydroxylaminato, [2-(tBuNO)py]−(interchangeable hereafter with [(tBuNO)py]−), a ligand which was previously found to impart remarkable stability to cerium in the +4 oxidation state. An[(tBuNO)py]4 (An = Pu, 1; Np, 2) have been synthesized, characterized by X-ray diffraction, X-ray absorption, 1H NMR and UV-vis-NIR spectroscopies, and cyclic voltammetry, along with computational modeling and analysis. In the case of Pu, oxidation of Pu(III) to Pu(IV) was observed upon complexation with the [(tBuNO)py]− ligand. The Pu complex 1 and Np complex 2 were also isolated directly from Pu(IV) and Np(IV) precursors. Electrochemical measurements indicate that a Pu(III) species can be accessed upon one-electron reduction of 1 with a large negative reduction potential (E1/2 = −2.26 V vs. Fc+/0). Applying oxidation potentials to 1 and 2 resulted in ligand-centered electron transfer reactions, which is different from the previously reported redox chemistry of UIV[(tBuNO)py]4 that revealed a stable U(V) product. Treatment of an anhydrous Am(III) precursor with the [(tBuNO)py]− ligand did not result in oxidation to Am(IV). Instead, the dimeric complex [AmIII(μ2-(tBuNO)py)((tBuNO)py)2]2 (3) was isolated. Complex 3 is a rare example of a structurally characterized non-aqueous Am-containing molecular complex prepared using inert atmosphere techniques. Predicted redox potentials from density functional theory calculations show a trivalent accessibility trend of U(III) < Np(III) < Pu(III) and that the higher oxidation states of actinides (i.e., +5 for Np and Pu and +4 for Am) are not stabilized by [2-(tBuNO)py]−, in good agreement with experimental observations.
Introduction
Developing a greater understanding of redox control for the actinide elements is an important aspect of devising separation strategies for used nuclear fuel and radioactive waste processing.1–10 Ligands with the capability to stabilize oxidation states higher than Am(III) is attractive as they offer a route to achieve challenging Ln(III)/An(III) separations.3,11–13 In lanthanide chemistry, oxidation of Ce(III) complexes to Ce(IV) are usually hampered by an inaccessible redox potential (large and positive) meaning many ligands do not form stable Ce(IV) complexes.14,15 However, the [2-(tBuNO)py]− hydroxylaminato ligand was recently shown to provide a −1.82 V stabilization of the +4 oxidation state of cerium, versus Fc/Fc+ in THF.16,17 It was also reported that [2-(tBuNO)py]− could stabilize the +5 oxidation state of uranium, as evidenced from electrochemical studies that demonstrated a one-electron oxidation of a U(IV)-hydroxylaminato complex to form a U(V) complex.17 Expanding understanding of the redox chemistry of hydroxylaminato/nitroxide ligands (and those with related bonding and electronic properties) to transuranium elements enhances fundamental understanding of f-element bonding and redox properties and may afford development of new classes of separations agents. The aim of this study was to determine how the strongly donating ability of [2-(tBuNO)py]−, which accounts for the stabilization afforded to Ce(IV), impacts oxidation state stability and complex formation with Np, Pu and Am. For context, the Ce(IV) to Ce(III) reduction potential of +1.7 V compares to the Am(IV) to Am(III) reduction potential of +2.3 V.16b Herein, we report our findings on reactions of the [(tBuNO)py]−/2-(tBuNO–H)py ligand with Pu(III), Pu(IV), Np(IV) and Am(III) precursors, that result in isolation of Pu(IV) (1), Np(IV) (2), and Am(III) (3) complexes, respectively. The products were characterized by a mixture of single-crystal X-ray diffraction, numerous spectroscopic techniques (including nuclear magnetic resonance (NMR), ultraviolet-visible-near infrared (UV-vis-NIR), X-ray absorption near edge spectroscopy (XANES), extended X-ray absorption fine structure (EXAFS)), cyclic voltammetry, and DFT and wave-function theory calculations. The results are discussed comparatively across the 5f series, and in analogy to Ce. These data are additionally discussed within the context of relative actinide redox stability and electronic structure trends. Quantifying redox trends into the transuranium series is useful because there are few reported cyclic voltammetry measurements on air- and moisture-sensitive Pu and Np compounds in organic solvents.18,19 Taken as a whole, this study provides a rare example of how using a common ligand, [2-(tBuNO)py]−, and chemical conditions can hold minor actinides (Am) in the +3 oxidation state, Pu and Np in the +4 oxidation state, and U in the +5 oxidation state. This knowledge supports contemporary efforts to (1) advance our abilities to control actinide redox chemistry and (2) design advanced separation processes specially tailored for solving 21st century problems in heavy element chemistry.Results/discussion
(a) Syntheses, single-crystal structural, and spectroscopic characterization
All syntheses with transuranium elements were conducted based on optimized reaction protocols developed first for Ce, Th, and U,16,17,20 and then scaled down and re-optimized with Ce salts on a 5 to 10 milligram scale. This strategy was employed to ensure efficient usage of transuranium stocks and maximize the probability of isolating reaction products on the small reaction scales typically associated with Np, Pu, and Am molecular research. We began with an initial reaction methodology designed to target a Pu(III) complex with the [2-(tBuNO)py]− hydroxylaminato ligand, in analogy to the previously reported Ce(III) dimer, [CeIII(μ2-(tBuNO)py)((tBuNO)py)2]2, that was obtained by treatment of Ce[N(SiMe3)2]3 with 2-(tBuNO–H)py.16 The analogous reaction of Pu[N(SiMe3)2]3, prepared in situ, with three equivalents of 2-(tBuNO–H)py resulted in an immediate color change to a very dark brown or black solution (Scheme 1). Upon standing at ambient temperature, very dark single-crystals formed, which were revealed by X-ray diffraction to have the formula Pu[(tBuNO)py]4 (1). The isolated crystalline yield of 1 after washing and drying was 33%. This result indicated that oxidation to Pu(IV) had occurred based on molecular formula and assuming ligand redox innocence. | ||
| Scheme 1 Synthesis of 1, PuIV[(tBuNO)py]4, via a Pu(III) precursor. | ||
Complex 1 is isostructural with CeIV[(tBuNO)py]4, which was prepared by oxidation of [CeIII(μ2-(tBuNO)py)((tBuNO)py)2]2. The X-ray structure of complex 1 was solved in the I2/c space group whereas the Ce analog was reported in the C2/c space group.16 The two space groups are interconvertible via a {1 0 1; 0 1 0; −1 0 0} matrix applied to the I2/c solution. We have chosen to report 1 in I2/c as this preserves the smallest β-angle, as is convention. The Pu(IV) center in 1 is eight coordinate and bound in a bidentate fashion to each of the four mono-anionic hydroxylaminato ligands through O and N atoms (Fig. 1). According to criteria from Haigh,21 the geometry around the Pu(IV) center is best described as distorted bicapped trigonal prismatic. The two crystallographically independent Pu–O distances in 1 are 2.227(3) and 2.235(3) Å compared to 2.2351(12) and 2.2361(12) Å for Ce–O in Ce[(tBuNO)py]4. The similarity in ionic radii of eight coordinate Pu(IV) and Ce(IV), 0.96 and 0.97 Å,22 respectively, is consistent with the observed similar metal–oxygen bond lengths, for which the average distances are statistically equivalent between Pu and Ce complexes when the standard deviation associated with the average values is taken into account. The independent Pu–N distances in 1 are 2.498(4) and 2.501(4) Å compared to 2.5399(15) and 2.5468(15) Å for Ce–N in Ce[(tBuNO)py]4. However, the average Pu–N distance of 2.500 Å is 0.043 Å shorter than the average Ce–N distance of 2.543 Å. For context with other structurally similar Pu(IV)/Ce(IV) N,O-donor complexes, the Pu–O vs. Ce–O and Pu–N vs. Ce–N distances in the Schiff-base coordination complexes ML2 (M = Pu, Ce; L = N,N′-bis[(4,4′-diethylamino)salicylidene]-1,2-phenylenediamine) were statistically equivalent and also very similar in comparison to the corresponding M–O and M–N hydroxylamide distances after taking standard deviations associated with average values into account.23
 | ||
| Fig. 1 Thermal ellipsoid plot (at the 50% probability level) of the solid-state structure of 1, PuIV[(tBuNO)py]4. Hydrogen atoms have been omitted for clarity, and tBu groups are shown as ball-and-stick depictions also for clarity. Atom labelling has been shown only for unique hetero atoms, and the Pu center. Symmetry operations: −X, +Y, 1/2 − Z. | ||
The exact mechanism responsible for obtaining a Pu(IV) complex is not known, but is likely driven by the strongly donating hydroxylaminato ligand conferring stability to the tetravalent oxidation state. Possible causes are disproportionation of Pu(III) to Pu(0) and 3 equiv. Pu(IV) or reaction with the solvent medium. In light of the observed ‘oxidation’ of Pu(III) to Pu(IV), the Np(IV) analog was targeted directly through use of NpCl4(DME)2 as the starting material, and employing the potassium salt of the hydroxylamide ligand, K[2-(tBuNO)py] to utilize a salt metathesis route as a means of ligand installation onto the Np(IV) metal center (Scheme 2). The reaction, conducted in THF, afforded an orange product, in 65% yield based upon a formula of Np[(tBuNO)py]4. Apparent crystals suitable for X-ray diffraction were grown on multiple occasions from repeat reactions, but issues stemming from insufficient diffraction intensity to twin components that could not be resolved and refined satisfactorily prevented full structural determination. However, it can be noted the inner-sphere connectivity around the Np(IV) metal center was consistent with N,O-bidentate coordination of four hydroxylaminato ligands, analogous to the Pu(IV) complex (see later section which provides EXAFS structural validation of this Np(IV) complex). We subsequently confirmed that complex 1 (Pu) could also be prepared rationally in the same manner as with Np(IV), by a salt metathesis reaction of PuCl4(DME)2 with four equivalents of K[2-(tBuNO)py] in THF (Scheme 2) and afforded a higher yield than the route from the Pu(III) precursor (88% vs. 33% – yield based on amount of Pu in the precursor prior to reaction with the ligand).
 | ||
| Scheme 2 Synthetic route to 1, PuIV[(tBuNO)py]4, and 2, NpIV[(tBuNO)py]4 from An(IV) precursors. | ||
The 1H NMR spectra of 1 and 2 (Fig. S28 and S29†), in benzene-d6, contained paramagnetically shifted resonances readily assignable to complexes with chemical formula An[(tBuNO)py]4. The tBu protons were observed at 0.60 ppm for 1 and 4.77 ppm for 2, respectively, while the four inequivalent pyridine-ring protons were observed for 1 at 10.11, 8.49, 7.19, 6.90 ppm and for 2 at 12.31, 7.01, 4.59, 2.95 ppm, respectively. The observed solution symmetry was consistent with the average C2v symmetry observed in the solid-state. Fingerprint UV-vis-NIR spectra (Fig. S30–S33†) in THF solution were also measured and exhibited a mixture of presumed 5f–5f and 5f–6d transitions along with metal/ligand charge transfer features in the higher energy region.
Moving beyond plutonium to elements with Z > 94 represents a significant step-up in the level of logistical and radiological challenge; limited quantities of the high specific-activity α-particle emitting 243Am isotope are available. Moreover, anhydrous, inert atmospheric, synthetic chemistry with americium is rare. Only in 2019 have the first reports been published containing full structural characterizations on air- and moisture-sensitive Am molecules prepared under inert atmosphere conditions. These were [AmCl(μ-Cl)2(THF)2]n and AmBr3(THF)4,24 followed by Am(C5Me4H)3.25 We had 5 milligrams of 243Am elemental content available to us for this study, taken from a stock of Am(III) prepared by dissolution of AmO2 in 6 M HCl. Although it was not known if the hydroxylaminato ligand would induce oxidation of Am(III) to Am(IV), for planning purposes it was predicted that the reaction may proceed analogously to cerium chemistry where a cerium(III) intermediate was isolated. Therefore, a synthetic protocol that was optimized with 5 mg of Ce(III) dissolved in 6 M HCl (leading to crystals of [CeIII(μ2-(tBuNO)py)((tBuNO)py)2]2) was adopted for the americium experiment. Evaporation of a pale-pink Am(III) 6 M HCl solution using a flow of argon gas afforded an off-white residue that was then introduced into a helium atmosphere glovebox. Addition of DME was followed by treatment with Me3Si–Cl (TMS–Cl) to scavenge residual water content, and the resulting off-white/pale-pink solid was subjected to washing and drying in vacuo (see Experimental section for full details). This putative ‘AmCl3(DME)x’ complex was used to generate putative ‘Am[N(SiMe3)2]3’ in situ, by treatment with K[N(SiMe3)2], based on the Ce scale-down. This procedure also echoed known uranium/plutonium silylamide chemistry. In the Am case, a distinct color change to peach-pink was noted, along with solubility of the peach colored product in hexanes, consistent with formation of a silylamide complex.26 Reaction of this silylamide intermediate with the hydroxylamine pro-ligand, 2-(tBuNO–H)py, caused an immediate color change to yellow-orange (Scheme 3). From this solution, amber-yellow single-crystals were obtained, by layering this Et2O solution of the Am complex with hexanes, and determined by X-ray diffraction to have formula [AmIII(μ2-(tBuNO)py)((tBuNO)py)2]2 (3), in direct analogy to the Ce(III) dimer previously reported.16,20 Indeed, compound 3 is isomorphous with the Ce(III) counterpart, crystallizing in the P21/n space group (Fig. 2). The two Am(III) cations are crystallographically identical due to the presence of an inversion center. The dimer consists of two N,O-bound terminal hydroxylaminato ligands for each Am(III) ion, with two bridging hydroxylaminato ligands. This bonding arrangement renders the Am(III) cation eight-coordinate through four O atoms and four N atoms. The geometry about the Am(III) cation is best described as a distorted triangular dodecahedron.21 For the two terminal hydroxylaminato ligands the Am–O distances are 2.286(3) and 2.307(3) Å, and the Am–N distances are 2.534(4) and 2.563(4) Å. These Am–O values are slightly shorter than the corresponding Ce–O distances in the Ce(III) analog of 2.306(2) and 2.332(2) Å, but the Am–N distances are significantly shorter compared to the Ce–N distances of 2.596(2) and 2.616(2) Å. For the bridging ligands, the Am–O(μ2-bridging) distances that directly link the two Am cations are 2.395(3) and 2.436(3) Å, compared to slightly longer values of 2.427(2) and 2.4624(18) Å in the Ce(III) isomorph. In the case of the N atoms in the bridging hydroxylaminato ligands, the N atoms are bound to only one Am cation (i.e. not a direct bridge) with the Am–N(py) distance being 2.634(4) Å and the Am–N(tBu) distance being 2.641(4) Å. Again, these metal–nitrogen distances are significantly shorter in 3 than in the Ce analog, with Ce–N(py) having a value of 2.707(2) Å and Ce–N(tBu) 2.687(2) Å. The comparison of bond distances between Ce(III) and Am(III) should be framed in the context of their reported eight-coordinate ionic radii of 1.143 and 1.09 Å, respectively,22 a difference of ∼0.05 Å. Therefore, it is likely that the shorter metal–ligand bond lengths in 3, compared to the Ce isomorph, are a result of the expected increase in ionic bond strength owing to the higher charge/size ratio of Am(III) vs. Ce(III). However, the differences in the metal–nitrogen bond distances could also be indicative of minor differences in covalent Am–N vs. Ce–N bonding character. For further context with other reported Am–N bonds for which similar Ce(III) molecules exist for comparison, the average Am–N distance in [Am(S2CNEt2)3(phen)] (phen = 1,10-phenanthroline) is 2.6031(8) Å,27 and the Ce–N distances in [Ce(S2CNMe2)3(dipphen)] (dipphen = 4,7-diphenyl-1,10-phenanthroline) are 2.665(10) Å and 2.664(11) Å,28 while in [Am{N(SePh2)2}3] the Am–N distance is 2.672(2) Å compared to 2.691(3) Å in [Ce{N(SePh2)2}3].29 Given the small quantity of 243Am available for this study, the low isolated yield of 3 and its very limited solubility, it was not possible to perform cyclic voltammetry on a solution of 3 or attempt chemical oxidation. However, we note that theoretical predictions (vide infra) suggest that a ligand oxidation event in 3 would occur before oxidation of Am(III) to Am(IV).
 | ||
| Scheme 3 Multi-step synthesis of 3, [AmIII(μ2-(tBuNO)py)((tBuNO)py)2]2, an Am(III) dimeric complex. | ||
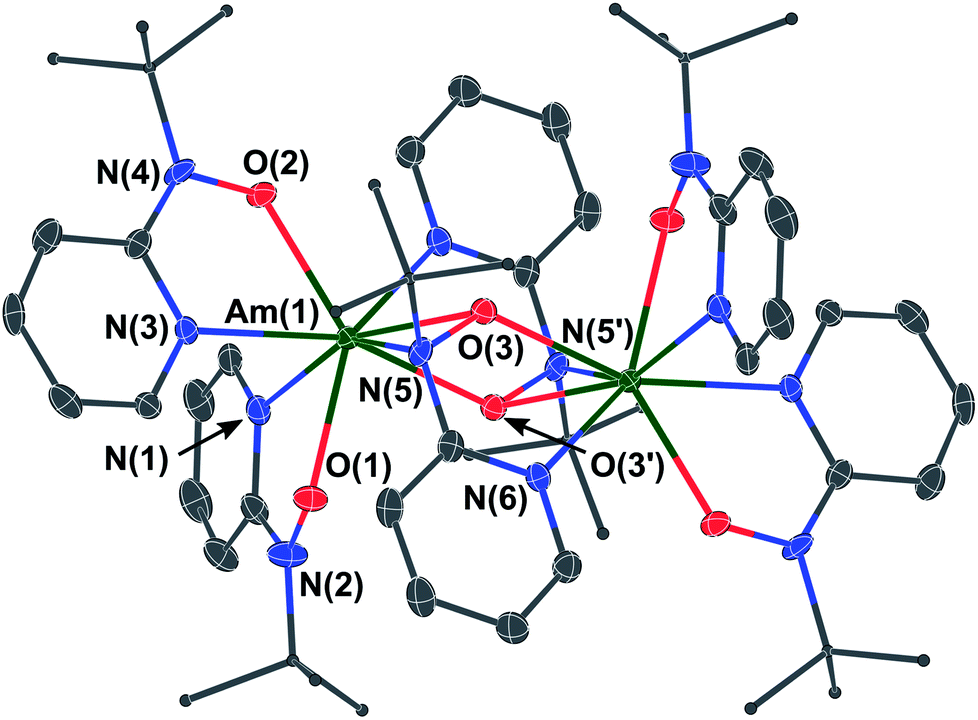 | ||
| Fig. 2 Thermal ellipsoid plot (at the 50% probability level) of the solid-state structure of 3, an Am(III) dimer. Hydrogen atoms have been omitted for clarity, and tBu groups are shown as ball-and-stick depictions also for clarity. Atom labelling has been shown only for unique hetero atoms and the symmetry-equivalent bridging N, O atoms, and the unique Am center. Symmetry operations: −X, 2 − Y, 2 − Z. | ||
(b) L3-edge X-ray absorption spectroscopy (XAS)
To provide support for metal oxidation state assignment in Pu[(tBuNO)py]4 (1) and Np[(tBuNO)py]4 (2), we turned to actinide L3-edge X-ray absorption near edge spectroscopy (XANES). Pu and Np L3-edge XANES measurements have been established for characterizing Pu and Np oxidation states,30 provided reasonable comparisons with appropriate standards are made. Here, the XANES data from 1 and 2 are evaluated within the context of a number of oxidation state standards that we also characterized, specifically PuIVCl4(DME)2,31 PuIIII3(NC5H5)4,26 an aqueous solution of PuVIO22+(aq),32a (PPh4)2NpVIO2Cl4,33 and NpIVCl4(DME)2.31 Note, the inflection point energies correlate with the effective nuclear charge for Pu and Np and typically increase by 1 to 2 eV with increasing oxidation states. In many ways, the background subtracted and normalized data from 1 and 2 are similar to the oxidation state standards (Fig. 3). Each spectrum shows a pronounced absorption peak loosely attributed to dipole allowed electronic excitations between core 2p and 6d orbitals, e.g., between the ground state 2p6 … 5fx 6d0 and excited state 2p5 … 5fx 6d1 electronic configurations. This absorption peak is superimposed on an absorption threshold that is attributed to ionization of the absorbing actinide cation. Closer inspection of the absorption peak energies and the line shapes of the post-edge tail revealed more subtle differences that are characteristic of Pu and Np oxidation state changes. The low +3 and +4 oxidation state standards have the lowest energy inflection points and the highest oxidation state AnVIO22+ (An = Np and Pu) standards have the highest energy inflection points (Table 2). The PuVIO22+ and NpVIO22+ standards also exhibit a defining characteristic. These spectra displayed a post-edge feature approximately +20 eV above the absorption edge. This post-edge peak has often been attributed to actinyl multiple scattering pathways.30f,34 For the Pu containing compounds, the spectra from 1 and the PuIVCl4(DME)4 +4 oxidation state standard are similar in energy. They are also bracketed by spectra from the two-oxidation state standard extremes (III and VI). For instance, the inflection point from 1 is 3.7 eV higher in energy than that from PuIIII3(NC5H5)4 and 0.9 eV lower than PuVIO22+. We remind the reader that inflection points from actinyl ions (like PuVIO22+) are often low in energy, slightly above those from +4 compounds.30h The post-edge line shape from compound 1 was also distinct from the PuVIO22+ standard, in that it did not contain multiple scattering features inherent to the linear O![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) PuO2+ dication. This comparison suggests that Pu in 1 is best described as being in the +4 oxidation state and is consistent with the EXAFS analyses described below. Similar observations are associated with Np. The inflection point from 2 is also 0.9 eV lower than the NpVIO22+ standard, 2 did not contain post-edge multiple scattering features associated with NpVIO22+ cations, and 2's inflection point was nearly equivalent to the Np(IV) standard. This comparison (as well as the structural metrics determined by EXAFS, vide infra) suggests that the Np oxidation state in 2 was also +4.
PuO2+ dication. This comparison suggests that Pu in 1 is best described as being in the +4 oxidation state and is consistent with the EXAFS analyses described below. Similar observations are associated with Np. The inflection point from 2 is also 0.9 eV lower than the NpVIO22+ standard, 2 did not contain post-edge multiple scattering features associated with NpVIO22+ cations, and 2's inflection point was nearly equivalent to the Np(IV) standard. This comparison (as well as the structural metrics determined by EXAFS, vide infra) suggests that the Np oxidation state in 2 was also +4.
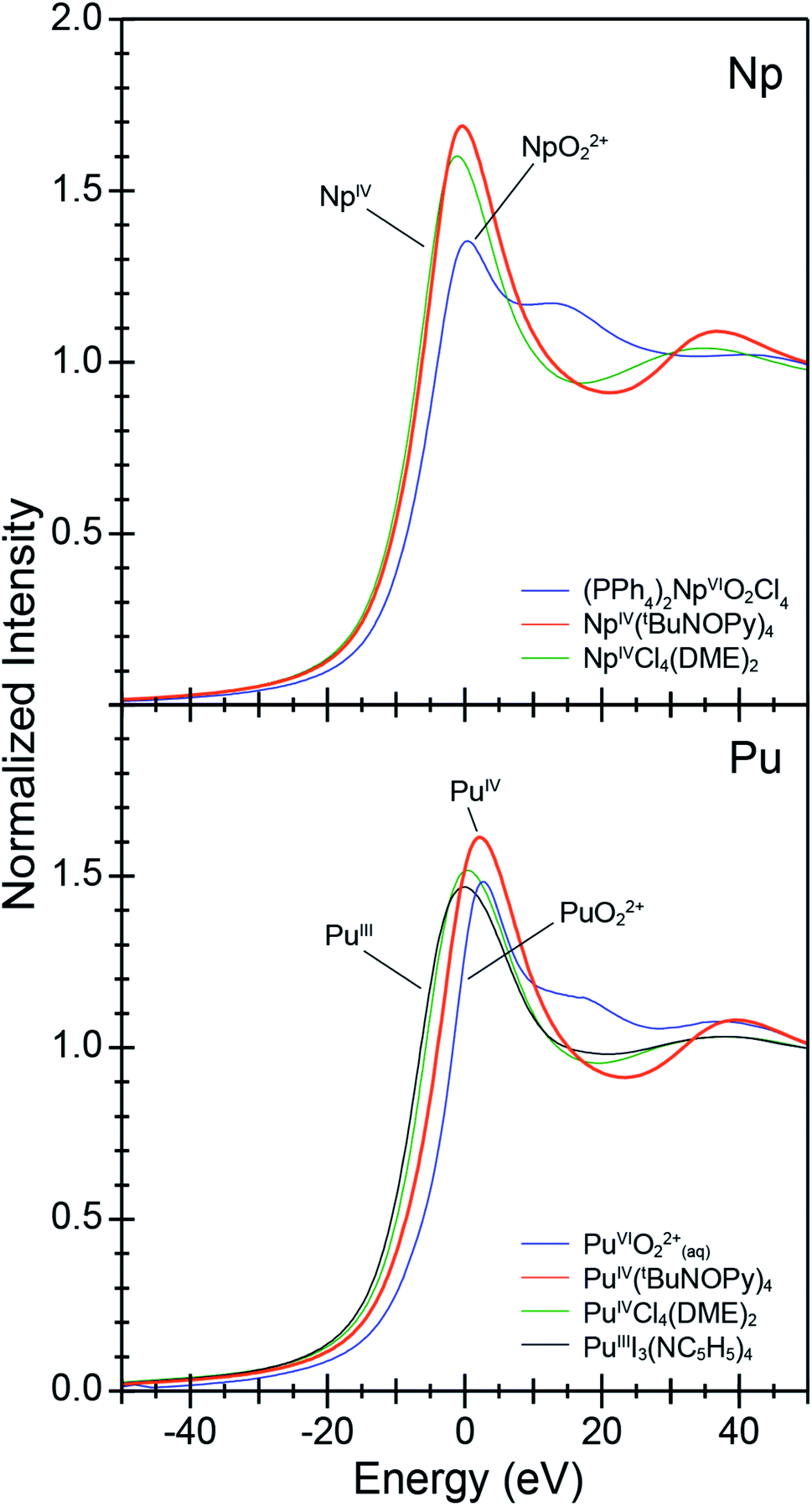 | ||
| Fig. 3 The actinide L3-edge XANES spectra from An[(tBuNO)py]4 [An = Pu (1), bottom; Np (2), top]. Data were compared with oxidation state references, (PPh4)2[NpVIO2Cl4], AnIVCl4(DME)2 (An = Np, Pu), PuIIII3(NC5H5)4, and an aqueous sample of PuVIO22+ dissolved in HNO3(aq) (1 M) that contained (NH4)2[Ce(NO3)6] to hold the Pu oxidation state at +6. | ||
| An–O | An–N(py) | An–N(tBu) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Calc. | XRD | EXAFS | Calc. | XRD | EXAFS | Calc. | XRD | EXAFS | |
| a py = N atom of pyridine ring in hydroxylaminato ligand; tBu = N atom connected to the tert-butyl group of the hydroxylaminato ligand b = bridging distance; distances are non-bridging unless denoted by (b). In monomeric 1 and 2 there are no An–N(tBu) or any bridging interactions. Complex 3 is a dimer, with each Am ion 8-coordinate containing terminal An–O and An–N(py) bonds as well as three types of bridging interactions: An–N(py), An–N(tBu) and An–O. | |||||||||
| Pu (1) | 2.296 | 2.231 | 2.239 | 2.546 | 2.500 | 2.496 | N/A | N/A | N/A |
| Np (2) | 2.245 | N/A | 2.245 | 2.576 | N/A | 2.528 | N/A | N/A | N/A |
| Am (3) | 2.329 | 2.297 | N/A | 2.544 | 2.549 | N/A | N/A | N/A | N/A |
| 2.430(b) | 2.416(b) | 2.673(b) | 2.634(b) | 2.805(b) | 2.641(b) | ||||
| Compound | Inflection point (eV) | Δ inflection point (analyte – AnVIO22+) (eV) |
|---|---|---|
| (PPh4)2[NpVIO2Cl4] | 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 615.7 615.7 |
−0.0 |
| NpIV[(tBuNO)py]4 (2) | 17614.8 |
−0.9 |
| NpIVCl4(DME)2 | 17613.9 |
−1.9 |
| PuVIO2(aq) (HNO3, 2 M) | 18063.6 |
−0.0 |
| PuIV[(tBuNO)py]4 (1) | 18062.0 |
−1.6 |
| PuIVCl4(DME)2 | 18059.3 |
−4.3 |
| PuIIII3(NC5H4)4 | 18058.3 |
−5.3 |
To better characterize the identity and structure of 2 (which evaded characterization by single-crystal X-ray diffraction), actinide L3-edge extended X-ray absorption fine structure experiments (EXAFS) were carried out on microcrystalline samples of 2 and single-crystals of 1. The low temperature (77 K) k3-weighted actinide L3-edge EXAFS spectra and Fourier transformed data from 1 and 2 are compared in Fig. 4–6. Notably, these spectra are all similar, both in terms of the oscillation frequencies and amplitudes observed in energy space and in terms of the peak height, width, and position in R-space. Superficially, these similarities suggest that the coordination environment around Pu in compound 1 is similar to that of Np in compound 2.
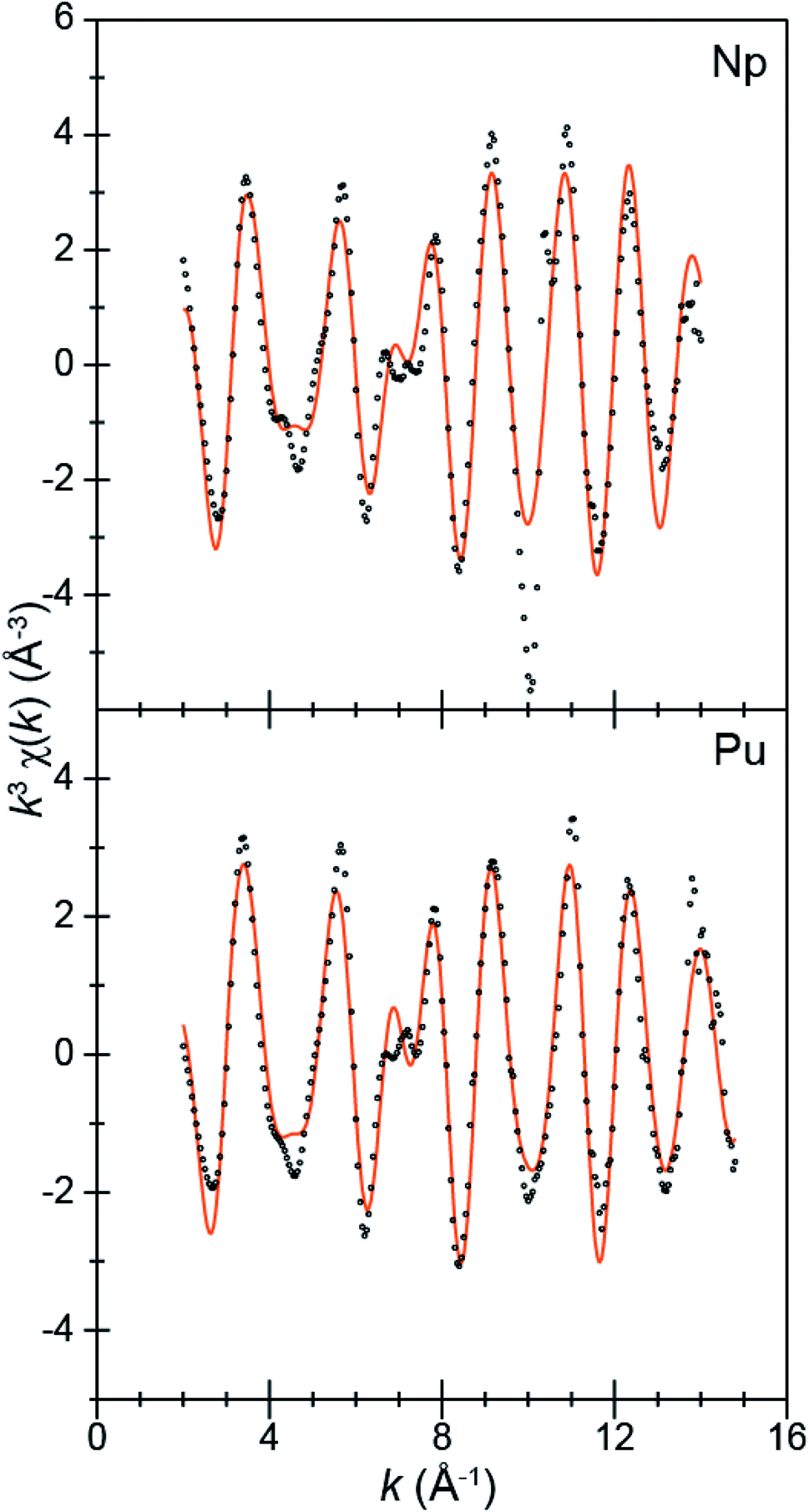 | ||
| Fig. 4 The EXAFS function k3χ(k) (Å) for solid samples of An[(tBuNO)py]4 [An = Pu, 1; bottom; Np, 2, top; Å; ○] and the fits for the data (orange traces). | ||
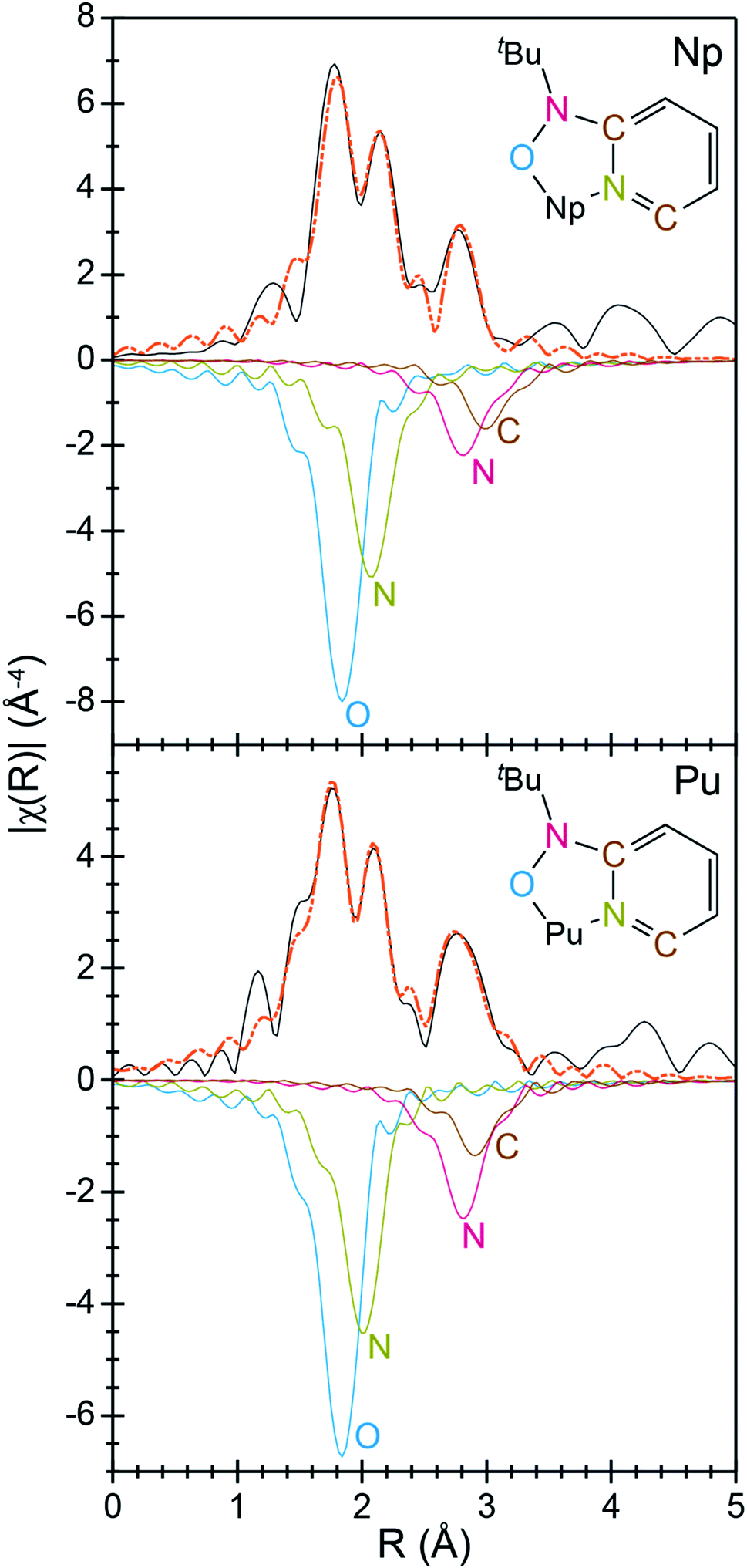 | ||
| Fig. 5 Interpretation of the Fourier transform of the k3-EXAFS spectra from An[(tBuNO)py]4 [An = Pu, 1; bottom; Np, 2, top; Å; black traces]. Fits are shown as orange dashed traces, while An–OO–NtBuNC5 (blue trace), An–NO–NtBuNC5 (red trace), An⋯NO–NtBuNC5 (olive trace), and An⋯CO–NtBuNC5 (brown trace) are shown as inverted functions. | ||
 | ||
| Fig. 6 Fourier transform of k3-EXAFS spectra from An[(tBuNO)Py]4 [An = Pu, 1; bottom; Np, 2, top; Å; black traces]. Fits for the data are shown as orange dashed traces. Also included are the real parts of the Fourier transform (olive traces) and the corresponding fit (blue dashed traces). | ||
A more quantitative evaluation involved shell-by-shell modeling of the data. These fits were constrained to maintain a reasonable number of free variables and avoid over-parameterization. The energy range over which usable data was obtained spanned from 2 to 15 Å−1. For compounds 1 and 2, fits were generated based on the single-crystal X-ray data of 1. Spectra were modeled with An–OO–NtBuNC5, An–NO–NtBuNC5, An⋯NO–NtBuNC5, and An⋯CO–NtBuNC5 scattering pathways. The An⋯CO–NtBuNC5 pathways involved only two ring carbon atoms that were para to the pyridine ring nitrogen. Because the structure for compound 1 was known, coordination numbers were fixed (based on the crystal structure) and the amplitude reduction factors (S02), the mean-squared displacement variables (σ2), and the actinide–atom distances were allowed to converge to reasonable values (Table 3). The obtained S02 parameter from 1 was then used in modeling data from compound 2. In this fit, S02 was fixed to be equivalent to that observed for 1, the mean-squared displacement variables (σ2) and the actinide–atom distances were allowed to converge to reasonable values. The coordination numbers were also floated with the following constraints. The oxygen coordination number was completely free to refine. Carbon and nitrogen parameters, however, were constrained based on the refined oxygen values. For example, the NO–NtBuNC5 and NO–NtBuNC5 parameters were set to be equal to OO–NtBuNC5, and the CO–NtBuNC5 value set to be two times the OO–NtBuNC5 coordination number. Structural metrics obtained from these data demonstrate that the structure of 2 is analogous to that for 1, revealing only minor differences between these two compounds.
| Compound | 1 | 2 |
|---|---|---|
| O coordination number | 4 (fixed) | 3.9(3) |
| N(1) coordination number | 4 (fixed) | 3.9 (fixed) |
| N(2) coordination number | 4 (fixed) | 3.9 (fixed) |
| C coordination number | 4 (fixed) | 3.9 (fixed) |
| σ 2 (O) | 0.004(1) | 0.002(1) |
| σ 2 (N1) | 0.004(1) | 0.003(2) |
| σ 2 (N2) | 0.003(2) | 0.001(4) |
| σ 2 (C) | 0.004(4) | 0.002(7) |
| An–OO–NtBuNC5 | 2.239(4) | 2.245(9) |
| An–NO–NtBuNC5 | 2.496(5) | 2.528(14) |
| An⋯NO–NtBuNC5 | 3.29(2) | 3.30(3) |
| An···CO–NtBuNC | 3.41(3) | 3.45(4) |
| S02 | 1.05(11) | 1.05 (fixed) |
| E 0 | −2.4(7) | 2.9(6) |
| Total variables used | 10 | 10 |
| Independent variables available for the fitting range | 16.44 | 17.04 |
Structural metrics obtained from compounds 1 and 2 by actinide L3-edge EXAFS are compared with the single-crystal X-ray data and the DFT calculations in Fig. 7. There is excellent agreement between all of the experimental and computational data. For instance, the EXAFS determined An–OO–NtBuNC5, An–NO–NtBuNC5, An⋯NO–NtBuNC5, and An⋯CO–NtBuNC5 distances are equivalent to those determined by single-crystal X-ray diffraction and to those optimized in the DFT calculation. These data are also compared with calculated structures for hypothetical compounds. These hypothetical compounds have structures that are similar to 1 and 2 and differ in that they contain Np and Pu cations in the +3 oxidation state (not the +4 oxidation state). Notice, calculated distances between the central actinide atom and the outer sphere N and C atoms, those associated with the pyridine ring (An⋯NO–NtBuNC5, and An⋯CO–NtBuNC5), are similar in the calculations for the +3 and +4 structures. There is also agreement between the single-crystal X-ray diffraction data and actinide L3-edge EXAFS measurements with these calculated values. This suggests that the actinide oxidation state does not impact the long-range An⋯NO–NtBuNC5, and An⋯CO–NtBuNC5 distances. In contrast, significant differences between the calculated An(IV) and An(III) structures are observed for the inner coordination sphere distances, those with direct An–OO–NtBuNC5 and An–NO–NtBuNC5 bonds. The An(III)–NO–NtBuNC5 calculated distances are approximately 0.05 Å longer and the An(III)–OO–NtBuNC5 calculated distances are approximately 0.1 Å longer than the observed distances and the AnIV calculations. The difference between the experimental and calculated An(III) distances, as well as the agreement between the experiment and calculated An(IV) distances, provide additional confidence that compound 1 has a similar structure to compound 2 and that the actinide oxidation states in compounds 1 and 2 are best described as +4.
 | ||
| Fig. 7 Comparison of interatomic distances for An[(tBuNO)py]4 (An = Pu, 1; Np, 2) determined by actinide L3-edge EXAFS, single-crystal X-ray diffraction (XRD), and density functional theory (DFT) with both An(III) and An(IV). | ||
(c) Electrochemical properties
To assess the redox properties of 1 and 2, cyclic voltammograms (CV) were acquired in THF solutions employing [nPr4N][BArF4] (BArF4 = [B(3,5-CF3-C6H3)4]) as the supporting electrolyte (0.1 M). For both 1 and 2, the CV data presented in Fig. 8 exhibits ligand-based oxidation events at −0.37 and −0.38 V vs. Fc+/0, respectively. These oxidation events are attributed to the one-electron oxidation of the [2-(tBuNO)py]1− ligand to its radical nitroxide form [2-(tBuNO)py]˙. This event was previously observed at −0.55 and −0.42 V vs. Fc+/0 for the isostructural Th(IV) and Ce(IV) complexes respectively.16,17 A positive correlation of the ligand-based oxidation potential with the decreasing ionic radii along the actinide series was observed in this data set. This result is in agreement with our observations on related hydroxylamine ligands for the lanthanide series where a similar correlation was drawn and which allowed for the design of a separation system based on these small differences of oxidation potentials and rates caused by the increasing Lewis acidity of the central cations.35 A second oxidation event can be observed and is attributed to the subsequent ligand-based oxidation of the hydroxylamine ligand to its oxoammonium form (NO˙/N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O+) at higher potential.20,36 On the reductive side of the CV of 2, small ligand-based return reduction events were observed, in analogy with the processes observed for the Th complex.17 On the other hand, the CV of 1 demonstrated a quasi-reversible redox event centered at E1/2 = −2.26 V attributed to a Pu(IV/III) couple (Ered value of −2.53 V). This event was similar to the Ce(IV/III) couple observed at E1/2 = −1.83 V vs. Fc+/0 and consistent with the qualitatively similar redox properties of these two cations as highlighted by other authors.37 Together, these data constitute an electrochemical series for the actinides Th, U, Np, Pu and the lanthanide Ce ion. Measurements were not possible on the Am complex due to the extremely low solubility of 3.
O+) at higher potential.20,36 On the reductive side of the CV of 2, small ligand-based return reduction events were observed, in analogy with the processes observed for the Th complex.17 On the other hand, the CV of 1 demonstrated a quasi-reversible redox event centered at E1/2 = −2.26 V attributed to a Pu(IV/III) couple (Ered value of −2.53 V). This event was similar to the Ce(IV/III) couple observed at E1/2 = −1.83 V vs. Fc+/0 and consistent with the qualitatively similar redox properties of these two cations as highlighted by other authors.37 Together, these data constitute an electrochemical series for the actinides Th, U, Np, Pu and the lanthanide Ce ion. Measurements were not possible on the Am complex due to the extremely low solubility of 3.
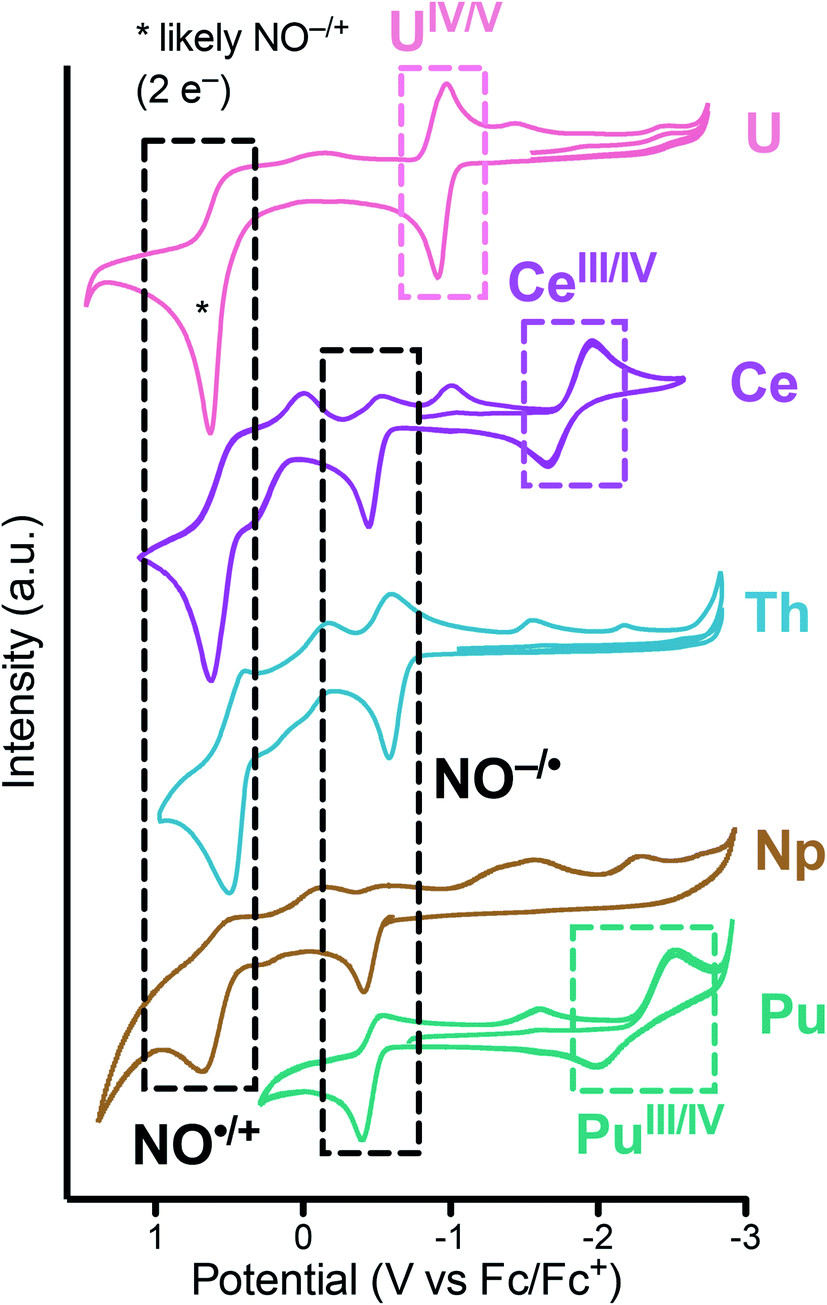 | ||
| Fig. 8 Cyclic voltammograms measured for An[(tBuNO)py]4 (An = Pu, 1; Np, 2) in THF (2 mM) with 0.1 M [nPr4N][BArF4] at 200 mV s−1 and compared with previous reports of M[(tBuNO)py]4 (M = Ce, Th, U). | ||
(d) Electronic structure and calculated redox properties
Density functional theory (DFT) and wave-function theory calculations, including spin-orbit coupling corrections, were carried out to gain insight into the electronic structures of the [2-(tBuNO)py] complexes with Pu(IV), Np(IV) and Am(III), 1–3, and their oxidation/reduction products. The computed average bond distances in An[(tBuNO)py]40/− (An = Pu, 1; Np, 2) as well as the experimental crystal structure and EXAFS data are summarized in Fig. 7 and Table S2.† The computed structure parameters are in reasonable agreement with the single-crystal X-ray diffraction and EXAFS data, with An–O and An–N bond distance deviation within 3% of experimental values, providing support for the theoretical modeling approach. The optimized average Am–O and Am–N bond lengths of Am(III) monomeric complex (Am[(tBuNO)py]4− theoretical only) and dimeric complex (AmIII(μ2-(tBuNO)py)((tBuNO)py)2]2) are displayed in Table S3† along with the experimental values for the dimer. The computed average terminal Am–O and bridging Am–O distances for the dimer are 2.329 and 2.430 Å, respectively, in good agreement with experimental values of 2.297 and 2.416 Å, respectively. The calculated monomeric structure gives an average terminal Am–O distance of 2.393 Å, longer than the above theoretical and experimental values of the dimeric structure. The computed dimeric complex has an average terminal Am–N distance of 2.544 Å, very similar to the average experimental value of 2.549 Å. In comparison to the dimeric complex, the calculated monomeric structure gives longer average terminal Am–O and Am–N distances, i.e., 2.393 Å and 2.590 Å, respectively. The N(tBu) coordination of bridging hydroxylamine ligands to Am in the optimized dimeric structure has an Am–N(tBu) distance of 2.805 Å, which significantly deviates from the experimental Am–N(tBu) value of 2.641 Å for the bridging hydroxylamine ligands. This deviation could possibly be due to the crystal packing effects in the crystal lattice that are not considered in the calculation. Detailed geometrical parameters for the above Np(IV), Pu(IV), and Am(III) complexes and their one-electron reduction/oxidation products are shown in Table S4† while selected experimental and computed bond distances are summarized in Table 1.The calculated and formal spin densities on the metal ions for An[(tBuNO)py]4−/0/+ are shown in Table 4, where the formal charge on the [(tBuNO)py] ligand was assumed to be −1. For An[(tBuNO)py]4− (An = Np, Pu, Am), all metal ions were unambiguously calculated as being in the +3 oxidation state. Upon one-electron oxidation, for An[(tBuNO)py]4 (An = Np, Pu) the deviations between calculated and formal spin densities are not remarkable, indicating metal-centered oxidation to the +4 metal oxidation state. In contrast, the calculated spin density on Am in Am[(tBuNO)py]4 differs from the formal by 0.93 and is very close to that in Am[(tBuNO)py]4−, indicating [(tBuNO)py]− centered oxidation. This result is consistent with the established high stability of Am(III). For the further one-electron oxidation of An[(tBuNO)py]4 (An = Np, Pu), the oxidation products An[(tBuNO)py]4+ show deviation of spin density larger than 1.0 between the calculated and formal one, also suggesting that the oxidation event is ligand centered. This is consistent with experimental observations of common stable An(IV) complexes and the relatively few examples of non-actinyl An(V) species for An = Np and Pu.38 It is worth mentioning that our previous report showed different oxidation chemistry for the U(IV) analogue, for which a metal-based oxidation product, UV[(tBuNO)py]4+, is assigned.17 The different oxidation behaviors across U(IV), Np(IV) and Pu(IV) are closely related with their electronic structures, as is discussed further below.
| An[(tBuNO)py]4− | An[(tBuNO)py]40 | An[(tBuNO)py]4+ | ||||
|---|---|---|---|---|---|---|
| Calc. | Formal | Calc. | Formal | Calc. | Formal | |
| Np | 3.96 | 4.0 | 3.13 | 3.0 | 3.02 | 2.0 |
| Pu | 5.05 | 5.0 | 4.39 | 4.0 | 4.51 | 3.0 |
| Am | 6.06 | 6.0 | 5.93 | 5.0 | — | — |
The calculated redox potentials of An[(tBuNO)py]40/− and An[(tBuNO)py]4+/0 couples are shown in Table 5 with the absolute values in Table S5.† The predicted potential (in THF vs. ferrocene) for the Pu(iv/iii) couple is −2.68 V in good agreement with the experimental Ered value of −2.53 V measured for 1 (E1/2 experimental value for 1 is −2.26 V). The calculated prediction for the NpIV/III couple is −3.29 V, beyond the electrochemical discharge limits of THF, and is consistent with the absence of any reduction process in the cyclic voltammogram of Np[(tBuNO)py]4. One electron oxidation potentials for An[(tBuNO)py]4 (An = Np, Pu) were calculated, affording values of −0.69 and −0.74 V, respectively, in reasonable agreement with the experimental results of −0.36 and −0.40 V for 2 and 1, respectively. The differences of ≤0.34 V are within typical errors for the methods applied and may be ascribed in part to insufficient consideration of solvation effects, lack of counter-ions, neglect of multiplet effects of the fn systems, and inadequate treatment of the strong electron correlation in the metal complexes with redox-active ligands.39–42 The predicted ligand-based oxidation potential for Am[(tBuNO)py]4− is −1.88 V, which would be within the experimentally observable window if the KAmIII[(tBuNO)py]4 complex could be obtained.
| An[(tBuNO)py]40/− | An[(tBuNO)py]4+/0 | |||
|---|---|---|---|---|
| Calc. | Expt. | Calc. | Expt. | |
| Np | −3.29 | −0.69 | −0.36 | |
| Pu | −2.68 | −2.53 | −0.74 | −0.40 |
| Am | −1.88 | |||
The frontier molecular orbital (MO) energy level diagram for NpIV[(tBuNO)py]4, PuIV[(tBuNO)py]4 and AmIII[(tBuNO)py]4− as well as UIV[(tBuNO)py]4 is shown in Fig. 9 with the envelope of singly occupied MOs (SOMOs) in Fig. S34.† For all of these complexes, the highest occupied MO (HOMO) is predominantly of ligand atomic orbital character and the SOMOs are of An 5f character. The HOMO is well above (≥0.76 eV) the SOMOs in Np(IV), Pu(IV), and Am(III) complexes, compared to being 0.25 eV lower than SOMOs in U(IV). This SOMO/HOMO energy separation trend results from the decreasing energy of An 5f orbitals as the actinide series is traversed from low to higher atomic number. The MO energy level ordering is consistent with our results, in which the electron is removed from the ligand upon one-electron oxidation of NpIV[(tBuNO)py]4, PuIV[(tBuNO)py]4 (both computed and experimental) and AmIII[(tBuNO)py]4− (computed only) complexes. These results are in contrast to electron removal from the metal, as is the case for UIV[(tBuNO)py]4 (see Table 3 in ref. 17). In comparison, the lowest unoccupied MO (LUMO) of Np(IV) and Pu(IV) complexes, and the LUMO+1 orbital of the U(IV) compound that is almost degenerate with LUMO, are mainly An 5f orbital in character with a small contribution from a ligand atomic orbital (Fig. 9 and S34†). This LUMO orbital picture is in accord with our observation that the electron is added to the metal upon one electron reduction of UIV[(tBuNO)py]4, NpIV[(tBuNO)py]4, and PuIV[(tBuNO)py]4 complexes. Furthermore, the decreasing orbital energy trend from the LUMO+1 of UIV[(tBuNO)py]4 (−0.94 eV) to the LUMO of NpIV[(tBuNO)py]4, and PuIV[(tBuNO)py]4 (−1.51 and −2.41 eV, respectively) corresponds to a less negative reduction potential from −3.79 (U) to −3.29 (Np) to −2.68 (Pu) eV for An(IV) + 1e– → An(III), which is consistent with the observed good correlation between the calculated LUMO energy and experimentally determined uranium(IV/III),43 uranium(V/IV),44 U(VI/V),45 and cerium(IV/III),46 redox potentials. In addition, appreciable mixing between An 5f and ligand atomic orbitals (mainly O 2p orbitals) is evident from the envelope of SOMOs of Np[(tBuNO)py]4, Pu[(tBuNO)py]4 and Am[(tBuNO)py]4− (see Fig. S34†), which is likely due to an effective energy match of the actinide metal 5f orbitals and ligand N,O-donor 2p orbitals. As the N 2p contribution to the MO mixing is much less than O 2p, the An–O bonding, with much shorter bond distances than An–N bonds by about 0.25–0.33 Å on average, is dominant in the metal–ligand bonding. Finally, the increase of metal–ligand orbital mixing in An[(tBuNO)py]4 complexes moving from An = U to Np and Pu is consistent with the observed trend in recently reported [AnIVCl6]2− complexes.47
 | ||
| Fig. 9 Frontier molecular orbital energy level diagram for UIV[(tBuNO)py]4, NpIV[(tBuNO)py]4, PuIV[(tBuNO)py]4 and AmIII[(tBuNO)py]4− complexes. For all complexes except UIV[(tBuNO)py]4, the envelope of the highest occupied MO (HOMO) and lowest unoccupied MO (LUMO) is given with contour values of 0.03 a.u. and the MO energy levels between the HOMO and singly occupied MOs (SOMOs) are not included. The Z direction is labeled with an arrow. The MOs of An 5f and ligand atomic orbital character are in red and black color, respectively. Only alpha orbital energies are shown. The orbital energies of the AmIII[(tBuNO)py]4− complex are shifted by −1.3 eV to facilitate visual presentation. | ||
Conclusions
Np(IV), Pu(IV) and Am(III) complexes with the [2-(tBuNO)py]− anionic ligand have been synthesized and structurally characterized. Spectroscopic (1H NMR, UV-vis-NIR, EXAFS, XANES) and cyclic voltammetry measurements were also performed on the Np and Pu complexes. Oxidation of a Pu(III) precursor was observed resulting in isolation and single-crystal structural determination of PuIV[(tBuNO)py]4 (1). Subsequent facile synthesis and isolation of 1 and Np[(tBuNO)py]4 (2) directly from Pu(IV) and Np(IV) precursors was achieved. Grounded through a range Pu and Np reference compound measurements, XANES data validated the +4 metal oxidation state assignment in 1 and 2. EXAFS measurements confirmed the structure of 2, which was otherwise not attainable by single-crystal X-ray diffraction structure determination. EXAFS measurements also provided M–O and M–N bond distances in 1 and 2. In contrast to the plutonium reactivity, complexation of an anhydrous Am(III) precursor with the hydroxylaminato ligand afforded the dimeric complex [AmIII(μ2-(tBuNO)py)((tBuNO)py)2]2 (3) as determined by single-crystal X-ray diffraction. Electrochemical measurements on non-aqueous molecular transuranic complexes in organic solvents are notably rare. Cyclic voltammetry on An[(tBuNO)py]4 complexes, in conjunction with DFT electronic structure calculations, revealed a metal-centered large negative reduction potential for Pu(IV) while the corresponding reduction event for the Np(IV) analogue was outside the observed electrochemical window. On the other hand, oxidation processes were observed for both the Np(IV) and Pu(IV) complexes, but are attributed to ligand-based oxidation events rather than an An(IV/V) couple. This ligand-based oxidation process for the Np(IV) and Pu(IV) complexes is different from our earlier finding of a facile metal-based oxidation and observation of U(IV) → U(V). Calculations on the model monomeric Am[(tBuNO)py]4− complex suggest that the redox-active [2-(tBuNO)py]− ligand cannot stabilize the +4 oxidation state of Am, in contrast to the previously observed remarkable stabilization of Ce(IV), and is consistent with isolation of 3, an Am(III) complex from reaction of [2-(tBuNO)py]− with an Am(III) precursor. This result stands in contrast to the analogous Pu(III) reaction from which only a Pu(IV) complex was isolated. The variable redox chemistry of the actinide and lanthanide hydroxylaminato complexes is closely associated with their electronic structures. Ultimately, studies such as these help to understand redox behaviors and trends across multiple transuranium metals with potential ligand candidates to develop various separation processes of importance to used nuclear fuel processing and waste remediation. In particular, demonstration that computational predictions correlate well to experimental redox observations lends great promise for rational, theoretical-driven, design of new ligands based on tuning electronic structure as a control mechanism for stabilizing different actinide oxidation states.Experimental
General
The primary transuranium isotopes utilized in the syntheses of the complexes were 237Np, 239Pu, and 243Am. Caution! These isotopes are high specific-activity α-particle emitters, along with associated γ-ray emissions, in addition to β-particle emission from daughters. These radiological hazards (and heavy metal chemical toxicity) pose serious health threats. Hence, all studies were conducted with appropriate controls for the safe handling and manipulation of radioactive materials, i.e. in a regulated radiological laboratory equipped with HEPA filtered hoods and continuous air monitors. All free-flowing solids that contained transuranium elements were handled in negative-pressure gloveboxes.Plutonium metal was obtained internally from Los Alamos National Laboratory. Neptunium oxide was purchased from the National Isotope Development Center, and americium oxide was supplied to Los Alamos from Florida State University as part of a research collaboration. All manipulations and syntheses described below were conducted with the rigorous exclusion of air and water using an MBraun Labmaster 130 helium atmosphere drybox equipped with stand-alone Vacuum Atmospheres Genesis oxygen and moisture removal systems, in addition to standard Schlenk and glovebox techniques under an argon atmosphere for non-radioactive ligand and reagent preparation at the University of Pennsylvania (see ESI† for small-scale lanthanide optimizations).
Solvents utilized for the transuranium chemistry were purchased in anhydrous grade and stored on a mixture of 3 and 4 Å molecular sieves and/or NaK alloy before use. Solvents were subjected to the sodium benzophenone ketyl test to ensure they held a deep purple color prior to use. Benzene-d6 and THF-d8 (Aldrich) were purchased in sealed glass ampules and stored over a mixture of 3 and 4 Å molecular sieves. AnCl4(DME)2 (An = Np, Pu), K(tBuNO)py, (tBuNO–H)py were prepared as previously described.16a,311H NMR spectra were recorded on a Bruker Avance 400 MHz spectrometer, at ambient temperature. 1H NMR spectra were referenced internally to solvent resonances. For radiological containment of transuranic solutions, NMR samples were placed inside 4 mm FEP tube liners inside of 5 mm borosilicate glass NMR tubes. The FEP liners were placed in vacuo overnight in the glovebox antechamber prior to use. Two plugs were used for each liner to aid in exclusion of air and moisture. Solution phase electronic absorption spectra were collected in oven dried screw-capped quartz cuvettes (loaded in a transuranic glovebox using parafilm to protect the exterior surface of the cuvette and cap from radioactive contamination) at ambient temperature using a Varian Cary 6000i UV-vis-NIR spectrophotometer. Cyclic voltammetry (CV) experiments were performed using a BioLogic SP-50 potentiostat, controlled with EC-Lab V11.18. All experiments were initially assessed at the open-circuit potential. The electrochemical cell consisted of a Pt-disc working electrode, a Pt wire counter-electrode and a Ag wire coated with AgCl as a pseudo-reference electrode. All potentials are referenced vs. the Fc0/+ couple measured as an internal standard.
Synthesis of 1, [PuIV((tBuNO)py)4]
UV-vis-NIR: 2.4 mg of 1 dissolved in 2.1910 g THF (nm): 672, 720 (sh, br), 766 (sh, br), 792 (sh), 837, 1049 (br), 1114 (br, sh). A more dilute solution was also prepared to observe maxima of ‘charge-transfer’ type peaks in the UV region by dissolving a 0.6 mg sample of 1 in 3.1578 g THF (nm): 369.
Synthesis of 2, [NpIV((tBuNO)py)4]
NpCl4(DME)2 (0.0243 g, 0.043 mmol) and K[(tBuNO)py] (0.0358 g, 0.175 mmol) were suspended in THF (2.5 mL) and stirred at ambient temperature to form an orange reaction mixture. After stirring overnight, volatiles were removed in vacuo and the product extracted into warm (∼40 °C) toluene (7 mL). The toluene solution was filtered through celite supported on a glass fiber plug to afford an orange-colored filtrate. The volume of the filtrate was reduced in vacuo to 2 mL (the point at which the solution began to appear turbid) and hexanes (2 mL) layered on top of the solution. After storage at −35 °C for one day, the pale yellow supernatant was pipetted away from the orange solid. The solid was washed with hexanes (5 mL) and dried in vacuo to afford an orange powder (0.0254 g, 65% yield). Crystals suitable for X-ray diffraction were grown from a toluene solution layered with hexanes at ambient temperature – unfortunately, the crystals exhibited too many twin domains to allow suitable refinement for publication. However, inner-sphere connectivity about the Np metal center was consistent with N,O-bidentate coordination of four nitroxide ligands.1H NMR (C6D6): δ 12.31 (br, 1H, pyH), 7.01 (dd, 1H, pyH) 4.77 (s, 9H, C(CH3)3), 4.59 (d, 1H, pyH), 2.95 (d, 1H, pyH).
UV-vis-NIR: 1.7 mg of 2 dissolved in 2.8542 g THF (nm): 559, 641 (br), 701 (br, sh), 726, 756, 844, 881 (br), 928, 958 (br, sh), 1005, 1174 (w), 1194. Further dilution to observe maxima of ‘charge-transfer’ type peaks in UV region was made by taking a 0.4318 g aliquot of the initial solution and adding 2.1472 g THF (nm): 374 (sh, br).
Synthesis of 3, [AmIII(μ-(tBuNO)py)((tBuNO)py)2]2
243AmO2 (0.0192 g, 0.070 mmol) was dissolved in 6 M HCl (1 mL) by heating at ∼50 °C for 10 min then stirring for 16 hours at ambient temperature inside an air-atmosphere negative pressure glovebox designed for handling of dispersible transuranium-containing powders. The resultant very pale yellow/pink (depending on the light) colored solution was transferred to another glass vial, the original vial washed with 6 M HCl, and the combined solution was determined to have a mass of 5.402 g (a 10 μL aliquot was added to 1 M HClO4 and the UV-vis-NIR spectrum confirmed that only the Am(III) oxidation state was present). An aliquot of this pale-pink stock solution containing 0.005 g (0.021 mmol) of Am metal content was blown to dryness over a stream of argon gas then placed in vacuo in the antechamber of a helium-atmosphere negative pressure glovebox for 16 hours. The off-white residue was brought inside the glovebox and DME added (2 mL), followed by stirring for 2 min, resulting in a pale- pink suspension. The suspension was heated for 20 min without any appreciable dissolution observed. After cooling to ambient temperature, TMS-Cl (1 mL) was added dropwise with stirring for 10 min, followed by stirring and heating to 50 °C for 2 hours. After cooling to ambient temperature, hexanes (2.5 mL) was added to the off-white/pale-pink suspension with stirring for 10 min, followed by placement in the glovebox freezer at −35 °C to allow solids to settle to the bottom of the vial. The supernatant was pipetted away and the solid stirred with hexanes (4 mL), allowed to settle again at −35 °C, the hexanes pipetted away and the solid dried in vacuo for 70 min. The solid was stirred with Et2O (1.5 mL) for 15 min, the mixture allowed to settle at −35 °C, the Et2O wash pipetted away and the solid dried in vacuo for 1 h. The solid was dissolved in THF (2 mL) to form a pale-pink solution. THF (3 mL) was added to K[N(SiMe3)2] (0.0123 g, 0.062 mmol) and the mixture added to the Am-containing solution and stirred for 16 hours. The cloudy mixture was pale peach-pink, a subtle but noticeable color change compared to before K[N(SiMe3)2] addition. Volatiles were removed in vacuo and the putative ‘Am[N(SiMe3)2]3’ complex was extracted into hexanes (6 mL) and filtered through a glass fiber circle inside a glass pipette. Volatiles were removed in vacuo from the filtrate to result in an oily residue, which was dissolved in freshly filtered Et2O (2 mL). The ligand (tBuNO–H)py (0.0103 g, 0.062 mmol) was dissolved in freshly filtered Et2O (1.5 mL). The Am-containing solution was filtered through a glass fiber plug directly into the ligand solution resulting in an instant color change to yellow-orange. After overnight storage at −35 °C, the solution volume was reduced to 1.5 mL by evaporation and layered with hexanes (1.5 mL). After 1 day, small amber-yellow colored crystals had deposited which were determined by single-crystal X-ray diffraction to be [AmIII(μ-(tBuNO)py)((tBuNO)py)2]2.X-ray crystallography
Crystals of 1 and 3 for single-crystal X-ray diffraction were mounted from Paratone-N oil inside 0.5 mm quartz capillaries (Charles Supper), which were sealed with capillary wax inside the glovebox and then coated in clear nail varnish (Hard as Nails™) outside the glovebox to provide shatter-resilience. The data were collected on a Bruker D8 diffractometer, with APEX II charge-coupled-device (CCD) detector, and Bruker Kryoflex liquid nitrogen low temperature device (100 K). The instrument was equipped with graphite monochromatized Mo Kα X-ray source (λ = 0.71073 Å), and a 0.5 mm monocapillary. Integration of data frames, including Lorentz-polarization corrections, and final cell parameter refinement were performed using SAINT+ software (version 6.45, 2003, Bruker AXS, Inc., Madison, Wisconsin 53719). The selected crystal of 1 had two twin components related by a 180° rotation; these were resolved and refined to a final contribution of 0.4671(9) for the reported component. The data were corrected for absorption using the TWINABS program for 1 (Sheldrick, G. M. (2012). TWINABS 2012/1. Bruker, Madison, Wisconsin, USA.) and the SADABS program for 3 (version 2.05, 2002, George Sheldrick, University of Göttingen, Germany). Decay of reflection intensity was monitored via analysis of redundant frames. The structure was solved using direct methods and difference Fourier techniques. All hydrogen atom positions were idealized and rode on the atom they were attached to. Structure solution, refinement, graphics, and creation of publication materials were performed using SHELXTL (version 6.10, 2001, Bruker AXS, Inc., Madison, Wisconsin 53719). See master CIF file for commentary regarding alerts and refinement details.Actinide L3-edge X-ray absorption spectroscopy (XAS)
The Pu and Np L3-edge X-ray absorption spectra were measured at the Stanford Synchrotron Radiation Lightsource (SSRL) under dedicated operating conditions (3.0 GeV, 5%, 500 mA) on beamline 11–2. At the time of these measurements, the beamline was equipped with a 26-pole and a 2.0 tesla wiggler. Using a liquid nitrogen-cooled double-crystal Si(220) (ϕ − 90°) monochromator, a single energy was selected from the incident white beam. The crystals were run fully tuned and higher harmonics were rejected by a Rh harmonic rejection mirror. Solid 1 and 2, as well as the (PPh4)2NpVIO2Cl4, AnIVCl4(DME)2 (An = Np, Pu), and PuIIII3(NC5H5)4 reference standards were prepared and loaded for XAS analysis in a negative pressure inert atmosphere glovebox (filled with helium or argon) in the following manner. The analyte was weighed out, such that the edge jump for the absorbing atom was calculated to be at 1 absorption length in transmission (∼10 mg of Np(IV) and Pu(IV) metal). The samples were diluted with boron nitride (BN; dried at 200 °C under vacuum, 10−3 torr, for 24 h) so that the total mass of the mixture was 80 mg. Samples were ground in a mortar and pestle for 2 min. The Np and Pu samples were loaded into Teflon tubes (7 mm OD) that were sealed with a Teflon Plug. The tubes were removed from the glovebox and were loaded into two nested aluminum sample holders equipped with Kapton windows. One set of windows was glued on, and one set was sealed with indium wire. The sample holder was shipped to SSRL. Upon arrival, the samples were opened at the beam-line and attached to the coldfinger of a liquid N2 cryostat, which was also attached to the SSRL's Beamline 11–2 rail, such that the samples were 90° to the incident radiation. The cryostat was evacuated (10−6 torr) and cooled with liquid N2.The plutonyl nitrate solution was prepared in a HEPA-filtered transuranium fume hood. An aliquot of the Pu(IV) stock solution that contained 0.7 mg of Pu in nitric acid (HNO3, 1 M), was synthesized as previously described.32 Then, the solution was contacted with excess of (NH4)2Ce(NO3)6. The CeIV converted PuIV to PuO22+ and the process was monitored by UV-visible spectroscopy. When the oxidation state adjustment was complete the solution was loaded into liquid sample holders. These holders consisted of a Teflon well (0.5 mL capacity) that was sealed with both a thin Teflon window and a Kapton window. These were secured to the sample holder using a metal frame that screwed to the Teflon well, clamping the windows in place. The top of the holder contained a hole, through which the sample was injected into the well. This hole was sealed with a Teflon plug that was held in place by a metal plate that was secured to the Teflon holder with two screws. This sealed “primary” holder was secured in a “secondary” container, which consisted of a metal box with a large Kapton window on one side. The Kapton window was secured by screwing a metal frame and Viton gasket onto the container. The back of the “secondary” was slotted (and the slots covered with Kapton). This “secondary” container maintained access to the X-ray beam used in the experiment (the beam could pass through the secondary container and into the downstream ion chambers) and provided an engineering control that guarded against accidental release of the highly radioactive samples. The engineering control provided by the “secondary” container was made more robust by nesting the “secondary” within a “tertiary” container. This third holder was essentially a larger version of the “secondary” container. The sample was mounted on SSRL's Beamline 11–2 at 90° to the incident radiation.
At the time of the measurements, the beamline 11–2 rail was best described as containing one chamber positioned before the sample holder, to monitor the incident radiation (I0, 10 cm). A second chamber was positioned after the sample holder, such that sample transmission (I1, 30 cm) could be evaluated against I0. A third chamber (I2, 30 cm) was positioned downstream from I1 so that a calibration foil could be measured in situ during the XAS experiments against I1. Samples were measured in transmission. All data were collected with vertical slits set at 1 mm and horizontal slits at 7 to 9 mm. All spectra were calibrated to a metal foil whose spectra were obtained in situ.
XAS data analysis
Individual scans for each sample were calibrated to the point at which the second derivative of the in situ calibration foil equaled zero. For Pu, data were calibrated to a Zr foil (17998.0 eV) and for Np a Y foil (17038.4 eV) was used. Solid-state spectra were obtained in duplicate and the solution data reproduced five times. Individual scans were merged to generate an average spectrum for each sample. Using the Athena software package,48 the spectral background was eliminated by (1st) fitting a line to the pre-edge region and (2nd) subtracting the line from the experimental data. Spectra were then normalized by fitting a second-order polynomial to the post-edge region of the spectrum and setting the edge jump at E0 to an intensity of 1.0. This procedure normalized the spectral intensities to a single Pu or Np atom. The EXAFS data were then analyzed by shell-by-shell fitting methods using IFEFFIT48 software and FEFF8 calculations.49 The spectra were k3-weighted and Fourier transformed prior to non-linear least squares curve fitting. For compound 1, atomic coordinates for the FEFF8 calculations were obtained from the single-crystal X-ray data. For compound 2, atomic coordinates for the FEFF8 calculations were obtained by substituting the Pu atom in the single-crystal X-ray data for Np.
Computational details
Kohn–Sham density functional theory (DFT)50,51 calculations were performed on the Np[(tBuNO)py]4+/0/−, Pu[(tBuNO)py]4+/0/−, Am[(tBuNO)py]40/−, and [AmIII(μ2-(tBuNO)py)((tBuNO)py)2]2 complexes. The generalized gradient approach with PBE52 exchange-correlation functional and the hybrid functional B3LYP53,54 implemented in the Gaussian 09.55 As PBE functional has been demonstrated to have good performance in reproducing experimental geometries of actinide complexes,56 it was employed in the geometry optimization and harmonic frequency calculations in the gas phase in this work. The calculated vibrational frequencies were positive confirming that the optimized structures were stationary points on the potential energy surface, and the corresponding thermochemical corrections to the free energy were obtained. For each molecule, a subsequent single point using the implicit CPCM solvation model57 with the Universal Force Field (UFF) radii58 and Solvent Excluding Surface (SES)59,60 was calculated at the DFT/B3LYP level to obtain more accurate electronic energy in the solvation environment. A relative permittivity of 7.4257 was assumed in the solvation calculations to simulate THF as the solvent experimentally used for all the cases. We applied the 6-311G* basis sets61,62 for nonmetal atoms, Stuttgart energy-consistent relativistic pseudopotentials ECP60MWB63,64 and the corresponding ECP60MWB-SEG basis for actinide atoms Np, Pu and Am. The spin–orbit coupling (SOC) corrections on the ground-state electronic energies of heavy-element compounds with open-shell electronic configurations at the metal centers, i.e., An[(tBuNO)py]4−/0 (An = Np, Pu) complexes, were obtained from active space self-consistent field (CASSCF)65 calculations using implemented in ORCA 4.0.1 quantum chemistry package.66 The spin–orbit coupling (SOC) and scalar relativistic (SR) effects are accounted for by means of quasi-degenerate perturbation theory (QDPT),67 using the second-order scalar relativistic Douglas–Kroll–Hess Hamiltonian formalism.68 The all electron scalar relativistic SARC basis sets69 SARC-TZVP for Np and Pu and DEF2-SVP basis sets70 for H, C, N and O were used in all the SO-CASSCF calculations unless stated otherwise. In addition, the AUTOAUX feature71 was used in order to automatically generate auxiliary basis sets for the resolution of identity approximation.72 To reduce the computational cost to a manageable degree, the CASSCF calculations were performed on the simplified complexes based on the DFT/PBE optimized geometries. The idea is to reduce the size of side groups in the ligand that are bulky and electronically unimportant to less-bulky and electronically similar ones. In this work, we replaced the original –C(CH3)3 substituent at the hydroxylamine nitrogen atom in the (tBuNO)py ligand with a –CH3 substituent with typical C–H bond lengths of 1.09 Å, and denote this simplified ligand as (tBuNO)py′. In the CASSCF calculations, the active orbitals contain all the 5f orbitals for An[(tBuNO)py′]4−/0 (An = Np, Pu) complexes, giving CAS(4e,7o) for Np[(tBuNO)py′]4− and Pu[(tBuNO)py′]40, and CAS(3e,7o) for Np[(tBuNO)py′]40, and CAS(5e,7o) for Pu[(tBuNO)py′]4−, respectively. The CASSCF calculations were performed on all the states arising from 5fn configuration for An[(tBuNO)py′]4−/0 (An = Np, Pu). The calculated SOC correction on the ground-state electronic energy is −1.097, −1.153, −1.271 and −1.452 eV for Np[(tBuNO)py′]4−, Np[(tBuNO)py′]40, Pu[(tBuNO)py′]4− and Pu[(tBuNO)py′]40, respectively. These SOC corrections were applied in the calculations of oxidation potentials of An(III) to An(IV) for Np and Pu, i.e., An[(tBuNO)py′]4− → An[(tBuNO)py′]40, as the oxidation events happen on the metal centers. For the other three oxidation reactions, where an electron is removed from ligands with no change in oxidation state of the metal centers, i.e., An[(tBuNO)py′]40 → An[(tBuNO)py′]4+ (An = Np, Pu) and Am[(tBuNO)py′]4− → Am[(tBuNO)py′]40, the SOC corrections to the oxidation potential were not included. The absolute reduction potential of −5.436 V for Fc+ → Fc0 and the calculation results of U[(tBuNO)py]4 complex are from our previous work.17Data availability
Crystallographic data for 2056537 and 2056538 has been deposited with the CCDC and can be obtained viahttp://www.ccdc.cam.ac.uk/data_request/cif.Author contributions
JS performed computational calculations. TC and AM prepared ligands samples and conducted lanthanide small-scale optimization reactions. CAPG performed electrochemistry measurements with analysis by TC and EJS. ID performed EXAFS/XANES data analysis and interpretation along with SAK, who collected that data. TEAS provided the americium. BLS was the crystallographer. ERB and PY interpreted the computational data along with JS. AJG performed the transuranium syntheses, except the route to 1 from a Pu(IV) precursor, which was conducted by CAPG. EJS, ERB, AJG, SAK and PY devised the project and were direct advisors to the students and postdoctoral researchers. JS and AJG wrote the first draft with extensive contributions and editing from all authors. AJG was the submitting author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Center for Actinide Science and Technology, an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences (BES), under Award Number DE-SC0016568 (AJG, JS, SAK, PY, ERB TEAS, EJS, TC, AM). CAPG was sponsored by a J. R. Oppenheimer Distinguished Postdoctoral Fellowship under the Laboratory Directed Research and Development program at Los Alamos National Laboratory (LANL-LDRD 20180703PRD1). Portions of the manuscript writing and editing were supported by U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences (BES), Heavy Element Chemistry program at LANL (DE-AC52-06NA25396; AJG).References
- A. Paulenova, Physical and chemical properties of actinides in Nuclear Fuel Reprocessing, in Advanced Separation Techniques for Nuclear Fuel Reprocessing and Radioactive Waste Treatment, Woodhead Publishing, Cambridge, 2011 Search PubMed.
- G. R. Choppin and M. P. Jensen, Actinides in Solution: Complexation and Kinetics, in The Chemistry of the Actinide and Transactinide Elements, ed. L. Morss, N. Edelstein and J. Fuger, Springer, The Netherlands, 2011 Search PubMed.
- W. H. Runde and B. J. Mincher, Chem. Rev., 2011, 111, 5723–5741 CrossRef CAS PubMed.
- K. L. Nash and J. C. Braley, Challenges for Actinide Separations in Advanced Nuclear Fuel Cycles, in Nuclear Energy and the Environment; ACS Symposium Series, American Chemical Society, Washington, DC, 2010, vol. 1046, pp. 19−38 Search PubMed.
- J. D. Burns and B. A. Moyer, Inorg. Chem., 2016, 55, 8913–8919 CrossRef CAS PubMed.
- J. Zhang, J. Nucl. Mater., 2014, 447, 271–284 CrossRef CAS.
- K. A. Venkatesan, C. J. Rao, K. Nagarajan and V. P. R. Rao, Int. J. Electrochem., 2012, 841456 Search PubMed.
- X. Sun, H. Luo and S. Dai, Chem. Rev., 2012, 112, 2100–2128 CrossRef CAS PubMed.
- E. J. O'Loughlin, M. I. Boyanov, D. A. Antonopoulos and K. M. Kemner, Redox processes affecting the speciation of technetium, uranium, neptunium, and plutonium in aquatic and terrestrial environments, in Aquatic Redox Chemistry, ed. P. G. Tratnyek, T. J. Grundl and S. B. Haderlein, American Chemical Society, Washington, DC, 2011, ACS Symposium Series, vol. 1071, pp. 477−517 Search PubMed.
- L. Newsome, K. Morris and J. R. LIoyd, Chem. Geol., 2014, 363, 164–184 CrossRef CAS.
- C. J. Dares, A. M. Lapides, B. J. Mincher and T. J. Meyer, Science, 2015, 350, 652–655 CrossRef CAS PubMed.
- M. N. Sokolova, A. M. Fedosseev, G. B. Andreev, N. A. Budantseva, A. B. Yusov and P. Moisy, Inorg. Chem., 2009, 48, 9185–9190 CrossRef CAS PubMed.
- (a) M. J. Lopez, M. V. Sheridan, J. R. McLachlan, T. S. Grimes and C. J. Dares, Chem. Commun., 2019, 55, 4035–4038 RSC; (b) K. McCann, B. J. Mincher, N. C. Schmitt and J. C. Braley, Ind. Eng. Chem. Res., 2017, 56, 6515–6519 CrossRef CAS; (c) K. McCann, D. M. Brigham, S. Morrison and J. C. Braley, Inorg. Chem., 2016, 55, 11971–11978 CrossRef CAS PubMed; (d) B. J. Mincher, N. C. Schmitt, B. K. Schuetz, T. C. Shehee and D. T. Hobbs, RSC Adv., 2015, 5, 27205–27210 RSC; (e) J. D. Bruns, M. Borkowski, A. Clearfield and D. T. Reed, Radiochim. Acta, 2012, 100, 901–906 CrossRef.
- N. A. Piro, J. R. Robinson, P. J. Walsh and E. J. Schelter, Coord. Chem. Rev., 2014, 260, 21–36 CrossRef CAS.
- N. T. Rice, J. Su, T. P. Gompa, D. R. Russo, J. Telser, L. Palatinusm, J. Bacsa, P. Yang, E. R. Batista and H. S. La Pierre, Homoleptic Imidophosphorane Stabilization of Tetravalent Cerium, Inorg. Chem., 2019, 58, 5289–5304 CrossRef CAS PubMed.
- (a) J. A. Bogart, A. J. Lewis, S. A. Medling, N. A. Piro, P. J. Carroll, C. H. Booth and E. J. Schelter, Inorg. Chem., 2013, 52, 11600–11607 CrossRef CAS PubMed; (b) S. Cotton, Lanthanide and Actinide Chemistry, Wiley, 2006 CrossRef.
- A. McSkimming, J. Su, T. Cheisson, M. R. Gau, P. J. Carroll, E. R. Batista, P. Yang and E. J. Schelter, Inorg. Chem., 2018, 57, 4387–4394 CrossRef CAS PubMed.
- P. L. Arnold, M. S. Dutkiewicz and O. Walter, Chem. Rev., 2017, 117, 11460–11475 CrossRef CAS PubMed.
- (a) D. C. Sonnenberger and J. Gaudiello, J. Less-Common Met., 1986, 126, 411–414 CrossRef CAS; (b) D. C. Sonnenberger and J. G. Gaudiello, Inorg. Chem., 1988, 27, 2747–2748 CrossRef CAS; (c) S. L. Staun, L. M. Stevens, D. E. Smiles, C. A. P. Goodwin, B. S. Billow, B. L. Scott, G. Wu, A. M. Tondreau, A. J. Gaunt and T. W. Hayton, Inorg. Chem., 2021, 60, 2740–2748 CrossRef CAS PubMed.
- J. A. Bogart, A. J. Lewis, M. A. Boreen, H. B. Lee, S. A. Medling, P. J. Carroll, C. H. Booth and E. J. Schelter, Inorg. Chem., 2015, 54(6), 2830–2837 CrossRef CAS PubMed.
- C. W. Haigh, Polyhedron, 1995, 14, 2871–2878 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 25, 925–945 CrossRef.
- B. E. Klamm, C. J. Windorff, M. L. Marsh, D. S. Meeker and T. E. Albrecht-Schmitt, Chem. Commun., 2018, 54, 8634–8636 RSC.
- S. S. Galley, J. M. Sperling, C. J. Windorff, M. Zeller, T. E. Albrecht-Schmitt and S. C. Bart, Organometallics, 2019, 38, 606–609 CrossRef CAS.
- C. A. P. Goodwin, J. Su, T. E. Albrecht-Schmitt, A. V. Blake, E. R. Batista, S. R. Daly, S. Dehnen, W. J. Evans, A. J. Gaunt, S. A. Kozimor, N. Lichtenberger, B. L. Scott and P. Yang, Angew. Chem., Int. Ed., 2019, 58, 11695–11699 CrossRef CAS PubMed.
- (a) L. R. Avens, S. G. Bott, D. L. Clark, A. P. Sattelberger, J. G. Watkin and B. D. Zwick, Inorg. Chem., 1994, 33, 2248–2256 CrossRef CAS; (b) A. J. Gaunt, A. E. Enriquez, S. D. Reilly, B. L. Scott and M. P. Neu, Inorg. Chem., 2008, 47, 26–28 CrossRef CAS PubMed.
- S. K. Cary, J. Su, S. S. Galley, T. E. Albrecht-Schmitt, E. R. Batista, M. G. Ferrier, S. A. Kozimor, V. Mocko, B. L. Scott, C. E. V. Alstine, F. D. White and P. Yang, Dalton Trans., 2018, 47, 14452–14461 RSC.
- P. B. Hitchcock, A. G. Hulkes, M. F. Lappert and Z. Li, Dalton Trans., 2004, 129–136 RSC.
- C. A. P. Goodwin, A. W. Schlimgen, T. E. Albrecht-Schönzart, E. R. Batista, A. J. Gaunt, M. T. Janicke, S. A. Kozimor, B. L. Scott, L. M. Stevens, F. D. White and P. Yang, Angew. Chem., Int. Ed., 2021, 60, 9459–9466 CrossRef CAS PubMed.
- (a) A. L. Smith, P. Martin, D. Prieur, A. C. Scheinost, P. E. Raison, A. K. Cheetham and R. J. M. Konings, Inorg. Chem., 2016, 55, 1569–1579 CrossRef CAS PubMed; (b) S. D. Conradson, I. Al Mahamid, D. L. Clark, N. J. Hess, E. A. Hudson, M. P. Neu, P. D. Palmer, W. H. Runde and C. D. Tait, Polyhedron, 1998, 17, 599–602 CrossRef CAS; (c) V. LoPresti, S. D. Conradson and D. L. Clark, J. Alloys Compd., 2007, 444–445, 540–543 CrossRef CAS; (d) K. P. Carter, K. F. Smith, T. Trantnjek, K. M. Shield, L. M. Moreau, J. A. Rees, C. H. Booth and R. J. Abergel, Chem.–Eur. J., 2020, 26, 2354–2359 CrossRef CAS PubMed; (e) D. Prieur, U. Carvajal-Nunez, T. Vitova and J. Somers, Eur. J. Inorg. Chem., 2013, 2013, 1518–1524 CrossRef CAS; (f) M. R. Antonio, C. W. Williams, J. A. Sullivan, S. Skanthakumar, Y.-J. Hu and L. Soderholm, Inorg. Chem., 2012, 51, 5274–5281 CrossRef CAS PubMed; (g) A. J. Nelson, W. K. Grant, J. A. Stanhold, W. J. Siekhaus, P. G. Allen and W. McClean, J. Vac. Sci. Technol., A, 2015, 33, 031401 CrossRef; (h) S. D. Conradson, K. D. Abney, B. D. Begg, E. D. Brady, D. L. Clark, C. Den Auwer, M. Ding, P. K. Dorhout, F. J. Espinosa-Faller, P. L. Gordon, R. G. Haire, N. J. Hess, R. F. Hess, D. W. Keogh, G. H. Lander, A. J. Lupinetti, L. A. Morales, M. P. Neu, P. D. Palmer, P. Paviet-Hartmann, S. D. Reilly, W. H. Runde, C. D. Tait, D. K. Veirs and F. Wastin, Inorg. Chem., 2004, 43, 116–131 CrossRef CAS PubMed.
- S. D. Reilly, J. L. Brown, B. L. Scott and A. J. Gaunt, Dalton Trans., 2014, 43, 1498–1501 RSC.
- (a) D. L. Clark, D. E. Hobart and C. Walther, Plutonium Handbook, American Nuclear Society, 2nd edn, 2019, ch. 46.8, vol. 6, p. 3212 Search PubMed; (b) S. K. Cary, K. S. Boland, J. N. Cross, S. A. Kozimor and B. L. Scott, Polyhedron, 2017, 126, 220–226 CrossRef CAS.
- D. D. Schnaars and R. E. Wilson, Inorg. Chem., 2018, 57, 3008–3016 CrossRef CAS PubMed.
- C. Den Auwer, E. Simoni, S. Conradson and C. Madic, Eur. J. Inorg. Chem., 2003, 2003, 3843–3859 CrossRef.
- H. Fang, B. E. Cole, Y. Qiao, J. A. Bogart, T. Cheisson, B. C. Manor, P. J. Carroll and E. J. Schelter, Angew. Chem., Int. Ed., 2017, 56, 13450–13454 CrossRef CAS PubMed.
- J. A. Bogart, H. B. Lee, M. A. Boreen, M. Jin and E. J. Schelter, J. Org. Chem., 2013, 78, 6344–6349 CrossRef CAS PubMed.
- M. R. Antonio and M.-H. Chiang, Inorg. Chem., 2008, 47, 8278–8285 CrossRef CAS PubMed.
- (a) A. J. Gaunt and M. P. Neu, C. R. Chim., 2010, 13, 821–831 CrossRef CAS; (b) M. B. Jones and A. J. Gaunt, Chem. Rev., 2013, 113, 1137–1198 CrossRef CAS PubMed; (c) P. L. Arnold, M. S. Dutkiewicz and O. Walter, Chem. Rev., 2017, 117, 11460–11475 CrossRef CAS PubMed; (d) M. Altmaier, X. Gaona and T. Fanghänel, Chem. Rev., 2013, 113, 901–943 CrossRef CAS PubMed.
- J. R. Levin, W. L. Dorfner, A. X. Dai, P. J. Carroll and E. J. Schelter, Inorg. Chem., 2016, 55, 12651–12659 CrossRef CAS PubMed.
- L. E. Roy, E. R. Batista and P. J. Hay, Inorg. Chem., 2008, 47, 9228–9237 CrossRef CAS PubMed.
- (a) A. E. V. Gorden and M. L. McKee, J. Phys. Chem. A, 2016, 120, 8169–8183 CrossRef CAS PubMed; (b) P. J. Hay, R. L. Martin and G. Schreckenbach, J. Phys. Chem. A, 2000, 104, 6259–6270 CrossRef CAS; (c) S. Tsushima, U. Wahlgren and I. Grenthe, J. Phys. Chem. A, 2006, 110, 9175–9182 CrossRef CAS PubMed; (d) H. M. Steele, D. Guillaumont and P. Moisy, J. Phys. Chem. A, 2013, 117, 4500–4505 CrossRef CAS PubMed.
- (a) A. W. Schlimgen and D. A. Mazziotti, J. Phys. Chem. A, 2017, 121, 9377–9384 CrossRef CAS PubMed; (b) A. W. Schlimgen, C. W. Heaps and D. A. Mazziotti, J. Phys. Chem. Lett., 2016, 7, 627–631 CrossRef CAS PubMed; (c) J. M. Montgomery and D. A. Mazziotti, J. Phys. Chem. A, 2018, 122, 4988–4996 CrossRef CAS PubMed.
- A. Elkechai, Y. Mani, A. Boucekkine and M. Ephritikhine, Inorg. Chem., 2012, 51, 6943–6952 CrossRef CAS PubMed.
- F. Kias, F. Talbi, A. Elkechai, A. Boucekkine, D. Hauchard, J.-C. Berthet and M. Ephritikhine, Organometallics, 2017, 36, 3841–3853 CrossRef CAS.
- A. Elkechai, F. Kias, F. Talbi and A. Boucekkine, J. Mol. Model., 2014, 20, 2294 CrossRef PubMed.
- Y.-X. Zhong, Y.-R. Guo and Q.-J. Pan, Chem. Phys. Lett., 2016, 646, 75–80 CrossRef CAS.
- J. Su, E. R. Batista, K. S. Boland, S. E. Bone, J. A. Bradley, S. K. Cary, D. L. Clark, S. D. Conradson, A. S. Ditter, N. Kaltsoyannis, J. M. Keith, A. Kerridge, S. A. Kozimor, M. W. Löble, R. L. Martin, S. G. Minasian, V. Mocko, H. S. La Pierre, G. T. Seidler, D. K. Shuh, M. P. Wilkerson, L. E. Wolfsberg and P. Yang, J. Am. Chem. Soc., 2018, 140, 17977–17984 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- (a) A. L. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7565–7576 CrossRef CAS; (b) J. J. Rehr and R. C. Albers, Rev. Mod. Phys., 2000, 72, 621–654 CrossRef CAS.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- J. P. Perdew, K. Burke and N. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision D.01, Gaussian Inc., Wallingford, CT, 2013 Search PubMed.
- (a) J. Su, C. J. Windorff, E. R. Batista, W. J. Evans, A. J. Gaunt, M. T. Janicke, S. A. Kozimor, B. L. Scott, D. H. Woen and P. Yang, J. Am. Chem. Soc., 2018, 140, 7425–7428 CrossRef CAS PubMed; (b) M. P. Kelley, J. Su, M. Urban, M. Luckey, E. R. Batista, P. Yang and J. C. Shafer, J. Am. Chem. Soc., 2017, 139, 9901–9908 CrossRef CAS PubMed; (c) B. Vlaisavljevich, P. Miro, C. J. Cramer, L. Gagliardi, I. Infante and S. T. Liddle, Chem.–Eur. J., 2011, 17, 8424–8433 CrossRef CAS PubMed; (d) D. Reta, F. Ortu, S. Randall, D. P. Mills, N. F. Chilton, R. E. P. Winpenny, L. Natrajan, B. Edwards and N. Kaltsoyannis, J. Organomet. Chem., 2018, 857, 58–74 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard III and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10039 CrossRef CAS.
- M. L. Connolly, J. Appl. Crystallogr., 1983, 16, 548–558 CrossRef CAS.
- M. L. Connolly, J. Mol. Graphics Modell., 1993, 11, 139–141 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- X. Cao, M. Dolg and H. Stoll, J. Chem. Phys., 2003, 118, 487–496 CrossRef CAS.
- X. Cao and M. Dolg, J. Mol. Struct.: THEOCHEM, 2004, 673, 203–209 CrossRef CAS.
- (a) B. O. Roos, P. R. Taylor and P. E. M. Siegbahn, Chem. Phys., 1980, 48, 157–173 CrossRef CAS; (b) P.-A. Malmqvist and B. Roos, Chem. Phys. Lett., 1989, 155, 189–194 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, J. Chem. Phys., 2005, 122, 034107 CrossRef PubMed.
- (a) T. Nakajima and K. Hirao, Chem. Rev., 2012, 112, 385–402 CrossRef CAS PubMed; (b) M. Reiher, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 139–149 CAS.
- D. A. Pantazis and F. Neese, J. Chem. Theory Comput., 2011, 7, 677–684 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef.
- G. L. Stoychev, A. A. Auer and F. Neese, J. Chem. Theory Comput., 2017, 13, 554–562 CrossRef CAS PubMed.
- F. Neese, J. Comput. Chem., 2003, 24, 1740–1747 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR, UV-vis-NIR, optimized bond distance tables, envelope of frontier valence orbitals, calculated redox potential table, and Cartesian coordinate table. CCDC 2056537 and 2056538. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03905a |
| ‡ Current address: School of Chemistry, Sichuan University, Chengdu 610064, China. |
| This journal is © The Royal Society of Chemistry 2021 |