
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom crystallographic data to the solution structure of photoreceptors: the case of the AppA BLUF domain†
Shaima
Hashem
 ,
Veronica
Macaluso
,
Michele
Nottoli
,
Filippo
Lipparini
,
Lorenzo
Cupellini
* and
Benedetta
Mennucci
*
,
Veronica
Macaluso
,
Michele
Nottoli
,
Filippo
Lipparini
,
Lorenzo
Cupellini
* and
Benedetta
Mennucci
*
Dipartimento di Chimica e Chimica Industriale, Università di Pisa, Via G. Moruzzi 13, 56124 Pisa, Italy. E-mail: lorenzo.cupellini@unipi.it; benedetta.mennucci@unipi.it
First published on 9th September 2021
Abstract
Photoreceptor proteins bind a chromophore, which, upon light absorption, modifies its geometry or its interactions with the protein, finally inducing the structural change needed to switch the protein from an inactive to an active or signaling state. In the Blue Light-Using Flavin (BLUF) family of photoreceptors, the chromophore is a flavin and the changes have been connected with a rearrangement of the hydrogen bond network around it on the basis of spectroscopic changes measured for the dark-to-light conversion. However, the exact conformational change triggered by the photoexcitation is still elusive mainly because a clear consensus on the identity not only of the light activated state but also of the dark one has not been achieved. Here, we present an integrated investigation that combines microsecond MD simulations starting from the two conflicting crystal structures available for the AppA BLUF domain with calculations of NMR, IR and UV-Vis spectra using a polarizable QM/MM approach. Thanks to such a combined analysis of the three different spectroscopic responses, a robust characterization of the structure of the dark state in solution is given together with the uncovering of important flaws of the most popular molecular mechanisms present in the literature for the dark-to-light activation.
1 Introduction
The availability of high-resolution crystallographic data has revolutionized our understanding of proteins.1 However, a crystal structure gives a single static representation, which is often not sufficient to explain how the protein really works. Moreover, in some cases, different structures can be solved for the same protein, thus making the structure–function prediction even more difficult. Such potential flaws become especially dangerous for proteins whose function is activated by a structural change. This is the case of photoreceptors, light-sensitive machines that regulate processes like growth, circadian rhythms and photomovement in bacteria, plants, and even some animals. Photoreceptor proteins bind a chromophore which, upon light absorption, undergoes a change in its internal geometry or in its interactions with some close protein residues. These local changes then propagate into the whole protein, finally inducing a conformational change, which switches the protein from an inactive, resting state to an active, signaling one.2–4At variance with other proteins, what is compelling in photoreceptors is a detailed knowledge of the chromophore local environment, namely the types of residues, their 3D arrangement, the network of interactions among them, and their dynamics. Unfortunately, all this information cannot be easily and unequivocally obtained from the crystal structure. The latter in fact gives one single snapshot, which is not necessarily representative of the structures sampled by the system in solution. Moreover, relevant interactions can be missing or, conversely, artificial ones can be present due to the unavoidable limitations in the crystallographic resolution at the scale of atomic interactions. A possible strategy to overcome these problems is to supplement the crystallographic data with molecular dynamics (MD) simulations of the protein within its biological environment.7 However, for the MD results to be really useful, the conformational space should be properly sampled to avoid biases arising from the limitations of the initial crystal structures. An effective test to validate the completeness of the MD simulations is to integrate them with a quantum mechanical (QM) description, to predict spectroscopic properties that can be compared with experiments. This strategy requires the introduction of a multiscale approach, where the QM description is combined with classical models such as the Molecular Mechanics (MM) force fields. In the resulting QM/MM model, three main aspects determine the quality of the description: the choice of the QM subsystem, the selected QM level and the coupling between the QM and the MM subsystems.8 In the case of photoreceptors, the first aspect is straightforward, as the QM description will necessarily include the chromophore and possibly a few close residues. Moreover, the dimensions of the chromophores present in photoreceptors and the need for a large sample size necessitate the use of relatively inexpensive QM levels, such as Density Functional Theory (DFT), and its time-dependent (TDDFT) extension for simulating UV-Vis spectra. As regards the third aspect, the common strategy is to use an electrostatic embedding QM/MM formulation where the QM system “sees” the MM atoms as fixed point charges (or fixed multipoles). A more complete formulation of the QM–MM interactions is represented by a polarizable embedding, which explicitly accounts for mutual polarization between the QM and MM subsystems. The integration of polarizable QM/MM descriptions with extensive MD simulations has proven particularly effective in simulating spectra and, more generally, light-induced processes in complex biological systems.9
Here, this strategy is applied to a member of the Blue Light-Using Flavin (BLUF) family of photoreceptors present in bacteria. These proteins non-covalently bind a flavin chromophore, which upon absorption of blue light, initiates the cascade of structural changes leading to the active state.10–15 These changes have been connected with a rearrangement of the hydrogen bond network around the flavin on the basis of two spectroscopic signatures: a redshift in both the maximum of the UV-visible absorption spectrum and in the carbonyl infrared (IR) frequency of flavin.11,16 However, the exact conformational change triggered by the photoexcitation is still elusive. Strikingly, one of the reasons that prevents a complete and definitive characterization of the light-activation mechanism is the lack of a clear consensus on the identity not only of the light activated state of the protein but also of the initial (dark adapted) one. This problem is enhanced in AppA, a BLUF protein that controls photosynthesis in the gene expression of the purple bacterium Rhodobacter sphaeroides.17 Two different crystallographic structures have been resolved for its resting state, which differ both in the composition and the arrangement of the protein residues of the flavin binding pocket (see Fig. 1).
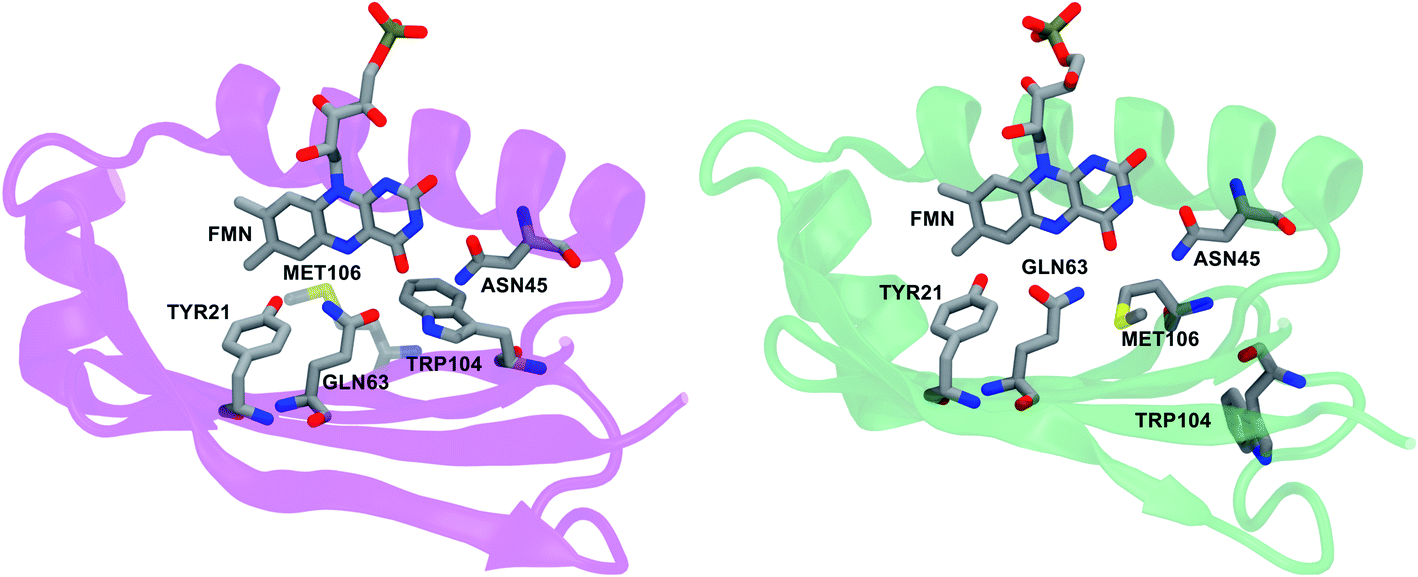 | ||
| Fig. 1 Flavin chromophore and the residues of the binding pocket in the two available crystal structures of AppA: Trpin5 (left, PDB ID: 1YRX) and Metin6 (right, PDB ID: 2IYG). | ||
One structure shows a tryptophan (Trp104) in the binding pocket, and it is called Trpin,5 while in the second, more recent structure, a methionine (Met106) replaces Trp104 in the binding pocket. This structure is denoted as Metin.6 Moreover, according to the Trpin structure, the Gln63 carbonyl group is oriented toward Trp104, while the Metin structure suggests that the glutamine side chain is oriented with its carbonyl oxygen near the OH group of the tyrosine residue (Tyr21) and its amide nitrogen close to the flavin O4. These differences have significantly complicated the analysis due to the crucial role that all these residues have been shown to play in the photoactivation. For example, the rotation (“flipping”) and tautomerization of Gln63 have both been suggested as possible changes in the dark-to-light transformation5,15,18–25 and mutagenesis studies have shown the importance of Trp104 in the AppA activity.11,26–28
In the last fifteen years many computational studies have been performed to solve this controversy and identify the structures of both dark and light states of AppA.22,23,25,29–34 However, the investigations have often been based on crystal structures only or they have used excitation energies only. Even in the case of more extensive investigations, such as those combining the simulation of excitation energies and IR frequencies, a detailed analysis of the conformational space of the two alternative structures was not performed because the MD simulations were limited to ten to few hundreds of nanoseconds. Here, we present an integrated investigation that combines 20 µs MD simulations starting from the two available structures of AppA with calculations of nuclear magnetic resonance (NMR), IR and UV-Vis spectra using a polarizable QM/MM approach. Moreover, the classical MD dynamics are here supplemented by polarizable QM/MM dynamics to investigate the IR frequencies beyond the standard harmonic approximation. By combining the outputs of the different simulations, a robust characterization of the structure of the dark state in solution is given together with the uncovering of important flaws of the most popular molecular mechanisms present in the literature for the dark-to-light activation.
2 Methods
2.1 Molecular dynamics simulations
The initial structures of AppA were extracted from the Protein Data Bank (PDB), describing Trpin (PDB ID: 1YRX)5 and Metin (PDB ID: 2IYG)6 conformations. In both structures the flavin chromophore is in the mononucleotide form (FMN). In addition to the main differences between the two structures reported in the Introduction and shown in Fig. 1, we recall that in the Metin structure, Cys20 is substituted by a serine.6We applied our calculations on the monomer AppA instead of the naturally found dimer, so that our results can be easily compared to the previous computational studies.32 Additionally, it was found that the dimer interface significantly differs among the available crystal structures of AppA.5,6 We thus considered chain A, using residues 17 to 121, in order to simulate the same sequence length for both structures. Hydrogen atoms were added using the LeAP module of AmberTools to refine the crystal structures. Protonation of the protein was set at pH 7 using the H++ web server.35 His residues were predicted to have pKA between 5.8 and 6.5, except for His85 in the Metin structure, which had a pKA of around 7. To avoid introducing differences in the protonation of the two structures, we considered all His residues in their standard protonation state. The positions of the hydrogen atoms were optimized using the Amber ff14SB force field.36
MD simulations were carried out using Amber18.37,38 Each system was soaked in a truncated octahedral box of TIP3P water molecules, ensuring a minimum 30 Å separation between periodic images of the protein. The system was neutralized with Na+ counterions. Furthermore, NaCl was added to achieve an ionic strength of 0.4 M. The protein was described using the Amber ff14SB force field,36 together with the general AMBER force field (GAFF);39 the parameters for the flavin mononucleotide molecule were obtained from a previous study.40 Periodic boundary conditions were applied. The Particle Mesh Ewald (PME) method was used to treat electrostatic interactions, using a cut-off of 10 Å. In all simulations, an integration time step of 2 fs was employed together with the SHAKE algorithm to constrain all the bonds involving hydrogen atoms. The Langevin thermostat was used to control the temperature. Each system was minimized through 1000 steps of steepest descent followed by 1000 steps of conjugate gradient. A 50 ps NVT simulation was run increasing the temperature from 0 to 50 K, followed by a 250 ps NPT simulation letting the system heat up to 300 K. Positional restraints were applied on heavy atoms during the heating using a force constant of 4 kcal mol−1 Å−2. An additional 750 ps NPT simulation was then run to allow the box to equilibrate at 300 K. NPT production simulations were performed for 2.5 µs for both Trpin and Metin structures.
We initially performed two MD replicas of 2.5 µs for each structure (MD1 and MD2). As one of the Trpin simulations manifested some instabilities (see below), we performed two additional control simulations (MD3 and MD4) with an extended equilibration for the loops in order to ensure the robustness of our findings. For the Metin simulations, we also performed two control simulations reverting the C20S mutation back to the original cysteine present in the AppA sequence (referred to in the following as “Metin–S20C” simulations. The total simulated time adds up to 20 µs (8 × 2.5 µs). A list of all simulations is reported in Table S1 in the ESI.†
2.2 QM/MM optimizations
Snapshots were extracted from the MD trajectories for each structure. These snapshots were taken from 2 µs to 2.5 µs of the production run every 5 ns, yielding a total of 200 snapshots for each structure. All the snapshots were refined by ONIOM(QM:MM) optimizations.41 In these optimizations, only the FMN molecule was allowed to move, while the protein and solvent molecules were kept frozen. Only the isoalloxazine ring, including the C1′ atom (Fig. 4a), was treated at the QM level of theory (B3LYP-D3/6-31G(d)),42 while the ribityl tail was treated at the MM level along with the rest of the system. All atoms within 30 Å of the isoalloxazine ring, except the Na+ and Cl− ions, were included in the MM part for the optimizations. The MM part was described with the same force field36,40 as in the MD simulations.2.3 Excited-state calculations
The 200 selected snapshots were used for excited-state calculations performed using a polarizable QM/MM model43,44 (from now on, QM/MMPol), which is based on the induced point dipole formulation and describes the MM part as a set of point charges and isotropic polarizabilities from the pol12 AL Amber force field.45,46 The QM part consisted of the isoalloxazine ring of the FMN, including the C1′ atom, and was treated at the ωB97X-D/6-31+G(d) level. The protein, ions, and water molecules within 40 Å of the chromophore were treated at the MMPol level. Although the long-range corrected ωB97X-D gives a systematic blue shift with respect to the experiment (∼0.3 eV), we chose this functional in order to avoid artificial mixing of the first excited state given by standard hybrid functionals such as B3LYP, as done in our previous work on another flavoprotein.47The homogeneous line shape of the flavin excitation was computed in the second-order cumulant expansion formalism.48 In this formalism the vibronic couplings with all the normal modes are encoded in the spectral density, namely
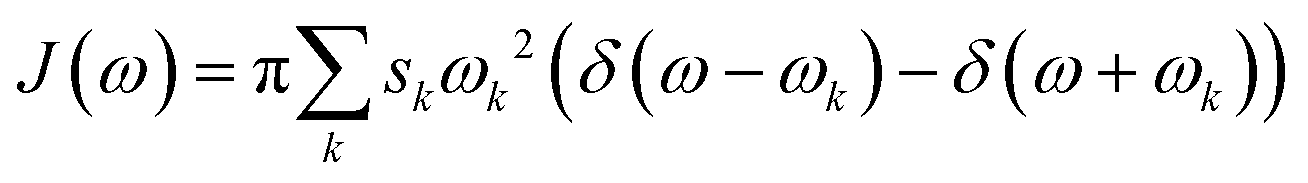 | (1) |
2.4 Chemical shift calculations
For all of the optimized structures, we computed the chemical shifts for all protons of the FMN ring and the four most tightly interacting protein side chains, as defined in our previous work.49 The environment effect on chemical shifts was treated via the same QM/MMPol approach used for the excited state calculations, where this time the QM part comprised the isoalloxazine ring and the side chains of the hydrogen-bonded residues (Gln63, Ty21 and Asn45) plus Trp104 and Met106, respectively, for Trpin and Metin structures. The QM part was described at the B3LYP/6-311+G(d,p) level, which was shown to well reproduce the much more expensive MP2/6-311G(d,p) calculations.49 The chemical shifts were averaged on either the Trpin or the Metin frames for comparison with experiments. Confidence intervals for the mean value were estimated by dividing the frames into contiguous blocks of 10–15 frames, and then using the block average to estimate error bars with the bootstrap method. This allows a more reliable error estimation that is not affected by correlations between consecutive frames.2.5 Polarizable QM/MM MD simulations and IR frequencies
Polarizable QM/MM MD simulations were performed starting from 10 frames extracted, respectively, from the Trpin and Metin simulations. For each starting frame, a 10 ps QM/MM MD was performed in the NVT ensemble with open boundary conditions. The QM/MMPol simulations were carried out using an interface50–52 between the molecular dynamics package Tinker53,54 and the development version of Gaussian.44,55 For these simulations, the QM part consisted of the FMN isoalloxazine ring, which was treated at the ωB97X-D/6-31G(d) level. The MM part consisted of all atoms within 30 Å of the QM part, and was described with the AMOEBA force field.56,57 Further details on the preparation of the QM/MMPol MD simulations are given in the ESI.†Vibrational frequencies were extracted from the last 5 ps of the QM/MM MD simulations. Power spectra were computed by Fourier transforming the autocorrelation function of bond lengths and bond angles, as well as their linear combinations. Since the normal modes are delocalized on the flavin ring, different modes contribute to the power spectrum of a single coordinate, and the power spectra show peaks at various frequencies. In order to better separate normal-mode frequencies, we resorted to a signal-processing technique called second-order blind identification (SOBI).58 Briefly, SOBI disentangles signals at different frequencies by performing a joint diagonalization of time-lagged autocorrelation matrices at various lag times. These autocorrelation matrices are here built on a basis of internal coordinates comprising all bond lengths and those bond angles that involve hydrogen atoms. The resulting linear combinations of internal coordinates showed power spectra that are well localized in frequency. More details on the frequency extraction are given in the ESI.†
3 Results
3.1 MD of the Trpin and Metin structures
As detailed in the Methods section, various MD trajectories were run starting from both crystal structures (PDB IDs: 1YRX5 and 2IYG6), referred to from now on as Trpin and Metin, respectively.We first examined the evolution of the backbone root mean square deviation (RMSD) of the protein core of Trpin and Metin to their respective crystal structures, excluding the loop region (residues 95–103) and the last few residues of the C-terminal which are expected to be highly disordered.26 Two example RMSD plots are shown in Fig. 2a. The Metin simulation is generally more stable along the whole trajectory compared to the Trpin one. This is confirmed by the distribution of RMSD values (Fig. S1†), which are shifted to larger values for all Trpin simulations, including the control Trpin simulations with a modified equilibration protocol (MD3 and MD4). In contrast, the Metin–S20C simulations are more dynamic and reach higher RMSD values.
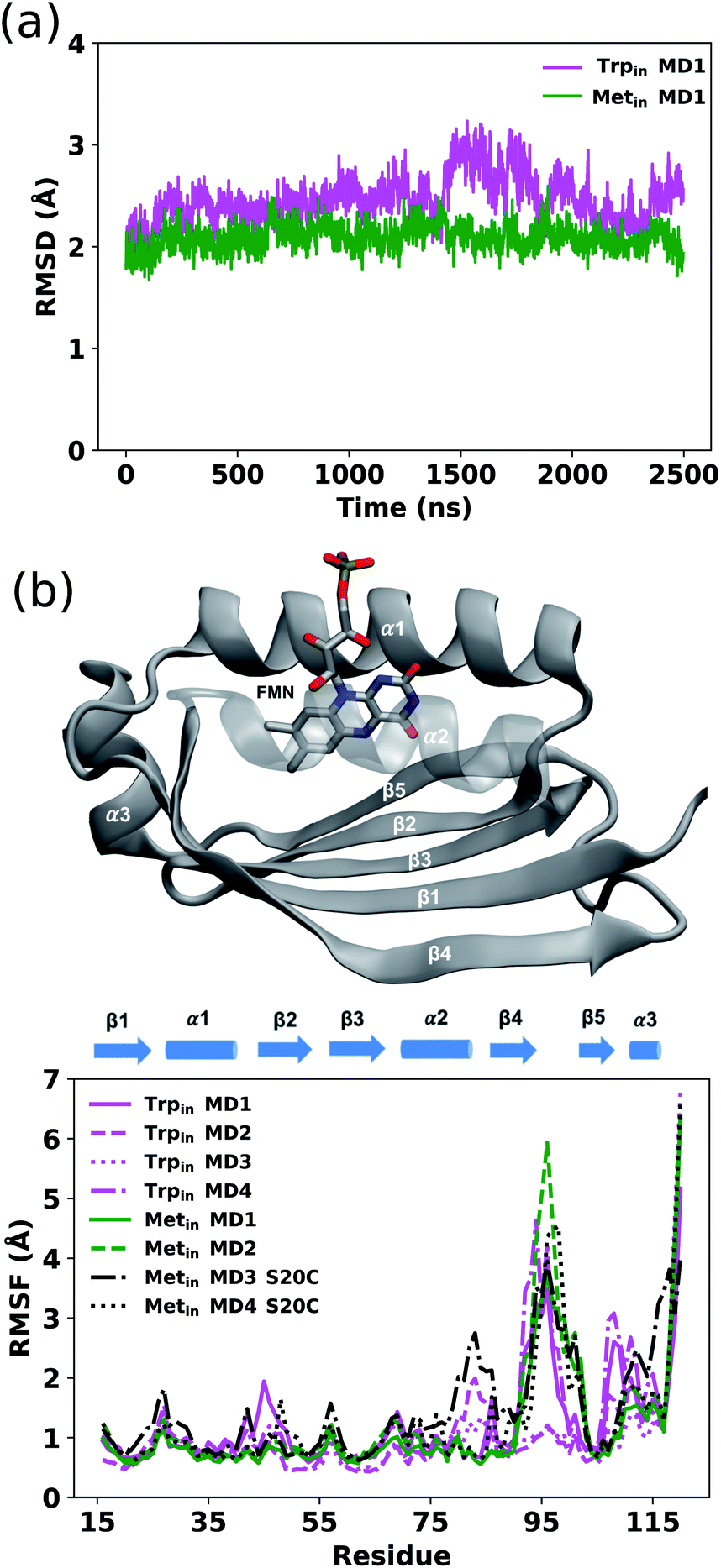 | ||
| Fig. 2 (a) Backbone RMSD during the Trpin (magenta) and Metin (green) MD simulations. (b) Profiles of RMSF values for Trpin (magenta), Metin (green) and Metin–S20C (black) MD simulations (lower panel). All replicas are reported. RMSF values were calculated on Cα atom coordinates of snapshots extracted from the production trajectory every 100 ps. The structure of Holo-AppA binding FMN, showing the different subdomains, is also reported (upper panel). | ||
We evaluated the overall dynamics of both structures as the root mean square fluctuation (RMSF) on the Cα atoms (Fig. 2b). In addition to the flexible regions of the loop and the C-terminal, all Trpin simulations exhibit large fluctuations especially in the α1 helix, β5-strand and C-terminal helix (α3 helix) compared to the Metin ones. Only the two Metin–S20C simulations display large motions similar to the Trpin ones, or even larger. These results suggest that the C20S mutation performed in ref. 6 has a stabilizing effect on the Metin conformation of AppA.
In all Trpin simulations, we observed relevant changes in the flavin binding pocket. The side chain of Gln63 adopts a conformation analogous to the Metin structure, and opposite to the crystal conformation depicted in Fig. 1. The hydrogen bonding pattern thus changes, and becomes more similar to the Metin structure (see Fig. 3a). This new conformation of Gln63 can be quantified by the distance distributions shown in Fig. 3b. In both Trpin and Metin simulations, the oxygen of Gln63 remains close to the C6 atom of the flavin, at a distance comparable to the Metin structure. The same conclusion can be reached by looking at the difference between FMN(O4)–Gln63(O) and FMN(O4)–Gln63(N) distances (dOO − dON) in Fig. 3c, which shows that the Gln63 amide nitrogen is closer to the flavin O4 atom than the amide oxygen. With this conformational change, Gln63 establishes an interaction with Tyr21 similar to that observed in Metin, with Gln63(O) and Tyr21(OH) approaching a H-bonding distance (Fig. 3d). However, this interaction is more dynamic than in the Metin structure, resulting in broader distributions. Control Trpin replicas performed after equilibration of the mobile loops (MD3 and MD4) all showed the same characteristics (Fig. S2†), with the Gln63 side chain oriented as in the Metin structure. These results strongly suggest that the orientation of Gln63 proposed by Anderson et al.5 (Fig. 1) is not stable in the µs time scale. However, while rotation of Gln63 was observed already at the beginning of Trpin simulations, the conformation shown in Fig. 3a only stabilized after 200–900 ns (see Fig. S5 in the ESI†), depending on the replica. This fact underlines the importance of long-scale MD simulations to correctly identify the most stable conformation of the flavin binding pocket.
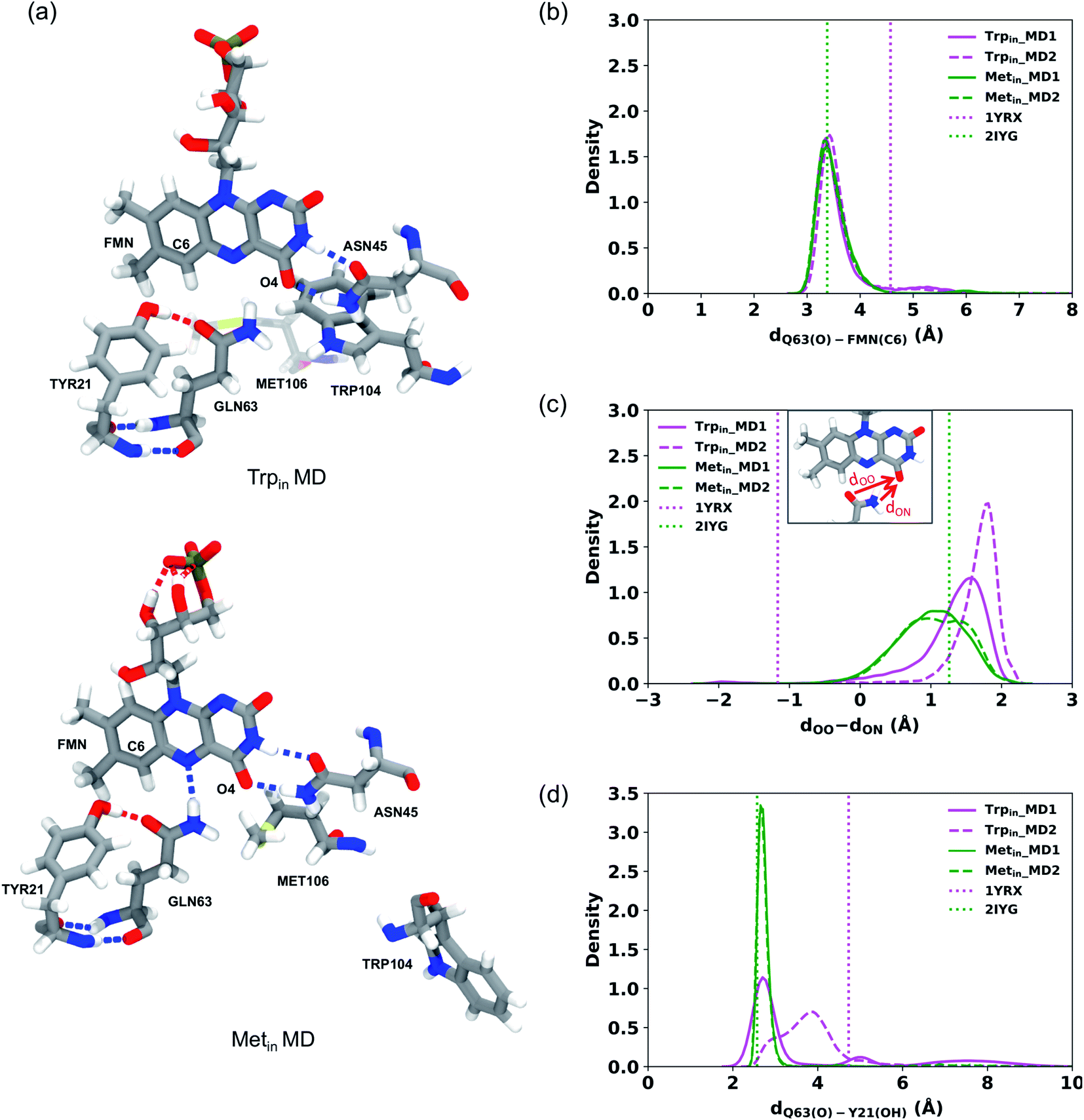 | ||
| Fig. 3 (a) FMN binding pocket in a representative snapshot of the Trpin (left) and Metin (right) MD simulations. (b–d) Distributions of the distances detecting Gln63 motion during the Trpin (magenta) and Metin (green) MD simulations. (b) Distribution of the distance between the oxygen of Gln63 and C6 of FMN. (c) Distribution of the difference between the FMN(O4)–Gln63(O) distance denoted as dOO and FMN(O4)–Gln63(N) distance denoted as dON. (d) Distribution of the distance between the oxygen of Gln63 and the hydroxyl group of Tyr21. The distances obtained from the Trpin (1YRX) (magenta) and Metin (2IYG) (green) crystal structures are represented as dotted lines. | ||
To better identify and understand the structural changes occurring in the binding pocket, hydrogen bond analysis was performed among the main key residues surrounding the flavin and between these residues and the flavin. These hydrogen bonds were previously proposed to have an important role in the AppA photocycle.59,60 The probability of occurrence of these hydrogen bonds according to the time of the simulations is reported in Table S2.† The hydrogen bond between Gln63(O) and Tyr21(OH) was observed during all the simulations with higher probability in the Metin simulations (with an average of 85%) than the Trpin ones (with an average of 55%). In contrast, the Gln63(NH)⋯Tyr21(O) hydrogen bond has negligible occupancy in the Trpin simulations (5%), and it was never observed in the Metin simulations.
In the Trpin simulations, the hydrogen bonds between Asn45 and FMN are temporarily lost in two of the four replicas that were performed (Fig. S3†), and are only recovered after ∼2 µs. This loss of interaction mirrors a change in the tertiary structure of the protein, as seen from the increased RMSD to the crystal structure (Fig. 2b). Asn45(NH) binds to FMN(O4) with average probabilities of 76% and 67% for Trpin and Metin simulations, respectively. In contrast, the hydrogen bond between Asn45(O) and FMN(N3H) is retained all along the Metin trajectories (90%) (Fig. S4†) while it is more frequently lost along the Trpin trajectories (63%) (Fig. S3†).
Notwithstanding the conformational change of Gln63, and the consequent loss of the Gln63⋯Trp104 hydrogen bond, we do not observe substantial changes in the Trp104 position during our simulations. The flavin binding pocket thus remains stable in the Trpin conformation. Analogously, in the Metin simulation, Met106 retains its position inside the binding pocket for all the simulations.
From this analysis, we conclude that both Trpin and Metin conformations are local free-energy minima, i.e. metastable states in the microsecond time scale and any conformational rearrangement between these two structures requires a significantly longer time.23
3.2 NMR chemical shifts
The structures obtained from the MD simulations have been used for the QM/MMPol calculation of chemical shifts. For this investigation all the snapshots extracted from the MD simulations were refined by geometry optimizations of the flavin molecule within the frozen environment made of the protein and solvent molecules (see the Methods section for details). To compare with experiments, we present the data relative to the protons of the isoalloxazine ring and those of the common surrounding residues in the flavin binding pocket, i.e. Tyr21, Asn45, and Gln63 (see Fig. 4a).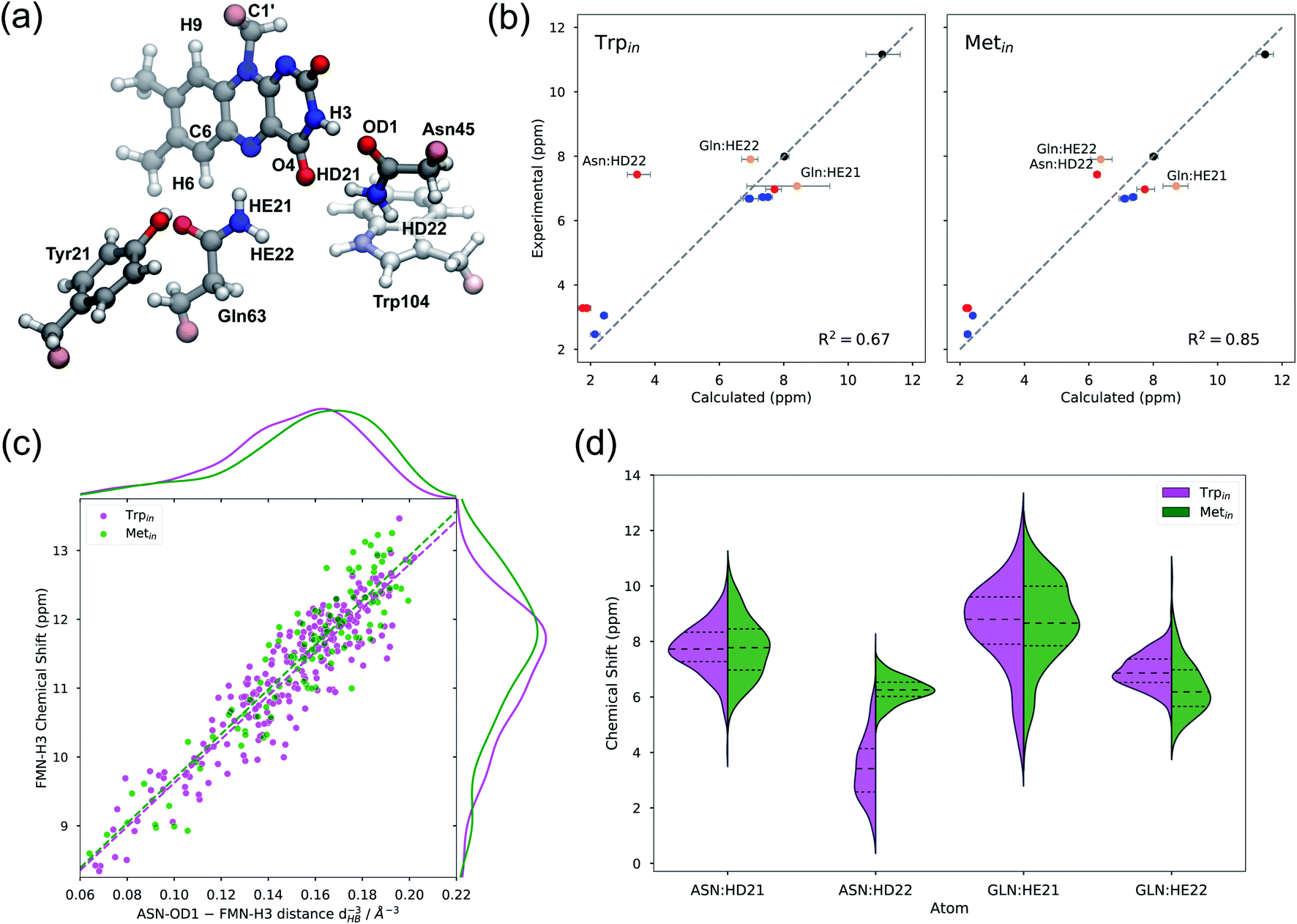 | ||
| Fig. 4 (a) Representation of the QM system considered for the QM/MMPol calculations, from a representative frame of the Trpin simulation. In the Metin structure, Met106 is included in place of Trp104. Link atoms are shown in pink color. (b) Comparison between calculated and experimental chemical shifts of Trpin (left) and Metin (right), including the protons of FMN (black), Tyr21 (blue), Asn45 (red), and Gln63 (yellow). Points with the largest deviations are labeled. Error bars represent bootstrapped 95% confidence intervals. (c) Dependence of the H3 chemical shift on the inverse third power of the H-bond distance (dHB−3), between the Asn45 OD1 atom and the FMN H3 atom. The dashed line represents the fit δ = δ0 + a·dHB−3, where a different slope a is allowed for Trpin (magenta) and Metin (green). The marginal distributions for dHB−3 (top) and for the chemical shift (right) are calculated with Gaussian kernel density estimation. (d) Distribution of amide proton chemical shifts of Asn45 and Gln63 for both Trpin (magenta) and Metin (green) structures. | ||
Fig. 4b shows the calculated Trpin/Metin chemical shifts compared to experiments.59 The agreement is generally good for both structures, and in all cases calculations reproduce the highly de-shielded H3 proton. However, the coefficient of determination (R2) is better for the Metin structure than for the Trpin structure. The largest discrepancy with respect to experiments is found for the amide HD22 of Asn45 for the Trpin simulations (Fig. 4b, left): the calculated chemical shift of this proton which is close to the aromatic ring of Trp104 is in fact much lower than the measured one. In contrast, on the Metin structure, our calculations predict a chemical shift much closer to the experiment, and similar to the corresponding Gln63 amide proton.
To gain a more detailed understanding of these findings, in Fig. 4d we report the distributions of amide chemical shifts calculated for Asn45 and Gln63 residues. The plots show similarities between Metin and Trpin structures, except for the aforementioned HD22 atom of Asn45, which has a completely different distribution in the two structures. In Trpin, the HD22 chemical shift has a broader distribution, centered at lower chemical shift values.
We hypothesize that Trp104 might be directly responsible for the unusual shielding of the Asn45 HD22 proton, owing to the shielding cone generated by the aromatic indole ring of the tryptophan. In fact, in the Trpin structure, the Asn45 amide group lies above the center of the pyrrolic ring of Trp104 (Fig. 4a), while in the Metin structure Trp104 is far from the pocket. In order to assess the direct role of the tryptophan, we recomputed the chemical shifts on the Trpin frames after moving Trp104 to the MM part. Comparing the two calculations (Fig. S5a†), it becomes clear that Trp104 has a direct, quantum mechanical, shielding effect on HD22 in most of the frames, while the other amide protons are only marginally affected. As a consequence, the shielding effect of Trp104 modifies the HD22 chemical shift distribution both in terms of position and broadening (Fig. S5b†). Notably, the anomalous shielding of the Asn45 amide proton was also previously found in the calculations on the Trpin crystal structure 1YRX, at both B3LYP and MP2 levels of theory,49 indicating that this result is affected neither by our sampling of the Trpin structure, nor by the level of theory used in this work.
Protons involved in hydrogen bonding interactions, or close to polar residues, are the most sensitive to changes in the local side-chain arrangement. Among these, flavin H3 was shown to be sensitive to the dark-to-light transition of AppA, giving the largest chemical shift change (+∼0.6 ppm).20 Our calculations predict very similar H3 chemical shifts (within error) in Metin and Trpin structures, thus suggesting caution in connecting the dark-to-light change (or vice versa) to an exchange between tryptophan and methionine inside the binding pocket.
In order to quantify the sensitivity of the H3 chemical shift to the H-bond pattern, we sought a relationship with the H-bonding distance to Asn45(OD1) (dHB). Our results show that the H-bond effect on this chemical shift is proportional to the inverse third power of dHB (Fig. 4c), and increases to 11–12 ppm when the Asn45 oxygen is at hydrogen bonding distance. Notably, the same trend can be observed for both Metin and Trpin structures, with a minimal difference in the slope. The distribution of dHB−3 (Fig. 4c, top) is slightly different for the two structures, with Metin showing closer H-bonding interactions, and is reflected in the distribution of chemical shifts (Fig. 4c, right). However, the difference between the two means (∼0.3 ppm) is smaller than the statistical error, and it is too small to explain the observed ∼0.6 ppm dark-to-light change.
Looking at the relationship in Fig. 4c, it is clear that structures with a tight hydrogen bond (dHB < 1.85 Å) give rise to H3 chemical shifts at around 12 ppm, i.e. compatible with the light-induced state of AppA.20 We can thus hypothesize that the light-induced AppA state will be characterized by a strong hydrogen bond with smaller fluctuations compared to what is observed in our MDs.
3.3 IR and UV-Vis signatures
As reported in the Introduction, the two spectroscopic signatures used to characterize the structural changes between the dark (inactive) and the light-activated forms of AppA are the redshifts observed in the flavin absorption maxima (∼10 nm, i.e. ∼0.07 eV) and in the IR frequency of its C4![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O4 stretching mode (20 cm−1).10,61 It thus becomes interesting to investigate if the differences previously found in the chemical shifts of the Metin and Trpin configurations have any connection with changes in the absorption maxima and the carbonyl frequencies which can finally be used to explain the molecular origin of the dark-to-light signatures.
O4 stretching mode (20 cm−1).10,61 It thus becomes interesting to investigate if the differences previously found in the chemical shifts of the Metin and Trpin configurations have any connection with changes in the absorption maxima and the carbonyl frequencies which can finally be used to explain the molecular origin of the dark-to-light signatures.
To simulate the IR frequencies of the carbonyl stretching modes, we randomly extracted a subset of ten Trpin and ten Metin configurations and used them in combination with normal-mode harmonic calculations. The obtained values however showed an unexpected problem: for almost all the configurations the C2O2 stretching frequency is blue-shifted with respect to the C4O4 one, which is in contrast to experiments.27,62 We note that in the isolated flavin, the same QM calculations indicate a C2O2 frequency slightly redshifted with respect to the other carbonyl. Therefore, the most probable reason for the observed wrong behavior is the way the harmonic calculations account for the environment. Here, in fact, the frequency differences are due to the different local environments of the two carbonyl groups both in terms of the number and the types of protein residues surrounding them, their orientations and the eventual presence of water molecules at hydrogen-bonding distances with the two carbonyl oxygens. To investigate this issue, we checked possible correlations between the frequencies of the two modes and the distances with potential hydrogen bonding donors. From the analysis reported in the previous sections, the most interesting candidate for O4 is Gln63. The results of this investigation are shown in Fig. S7 of the ESI† where we report a correlation between the C4O4 frequency and the O4–Gln63(H) distance in the different Trpin and Metin optimized structures. As can be seen, a clear correlation is present for Metin, for which a shorter distance corresponds to a smaller frequency as expected by the destabilization induced by the H-bond on the carbonyl double bond. In contrast, the Trpin structures do not show any clear correlation with such a distance, even if the sampled H-bond distances are similar to those found in Metin. Moving to the analysis of C2O2, the only possible H-bond donor is represented by water molecules. In all the selected frames, only a few water molecules are present at H-bond distances but also in those cases the C2O2 frequency is higher than the one localized on the other carbonyl.
This analysis clearly shows the limitations of harmonic calculations associated with optimized structures in the presence of an environment that can establish very dynamic H-bond interactions. To overcome these important limitations, we therefore resorted to a dynamical approach based on polarizable QM/MM MDs. We used the same 10 + 10 configurations used for the harmonic calculations and we ran 10 ps QM/MM MDs starting from each of these structures (see the Methods section for details).
The CO mode frequencies were computed from the power spectra of linear combinations of internal coordinates. These linear combinations were determined by the SOBI signal-processing analysis (see the Methods) and allowed to obtain power spectra well localized in frequency. As seen in Fig. 5a, these frequencies are now in the same order as the experiments, with the C4O4 frequency being ∼20 cm−1 higher than that of C2O2, indicating a more realistic description of environment effects.
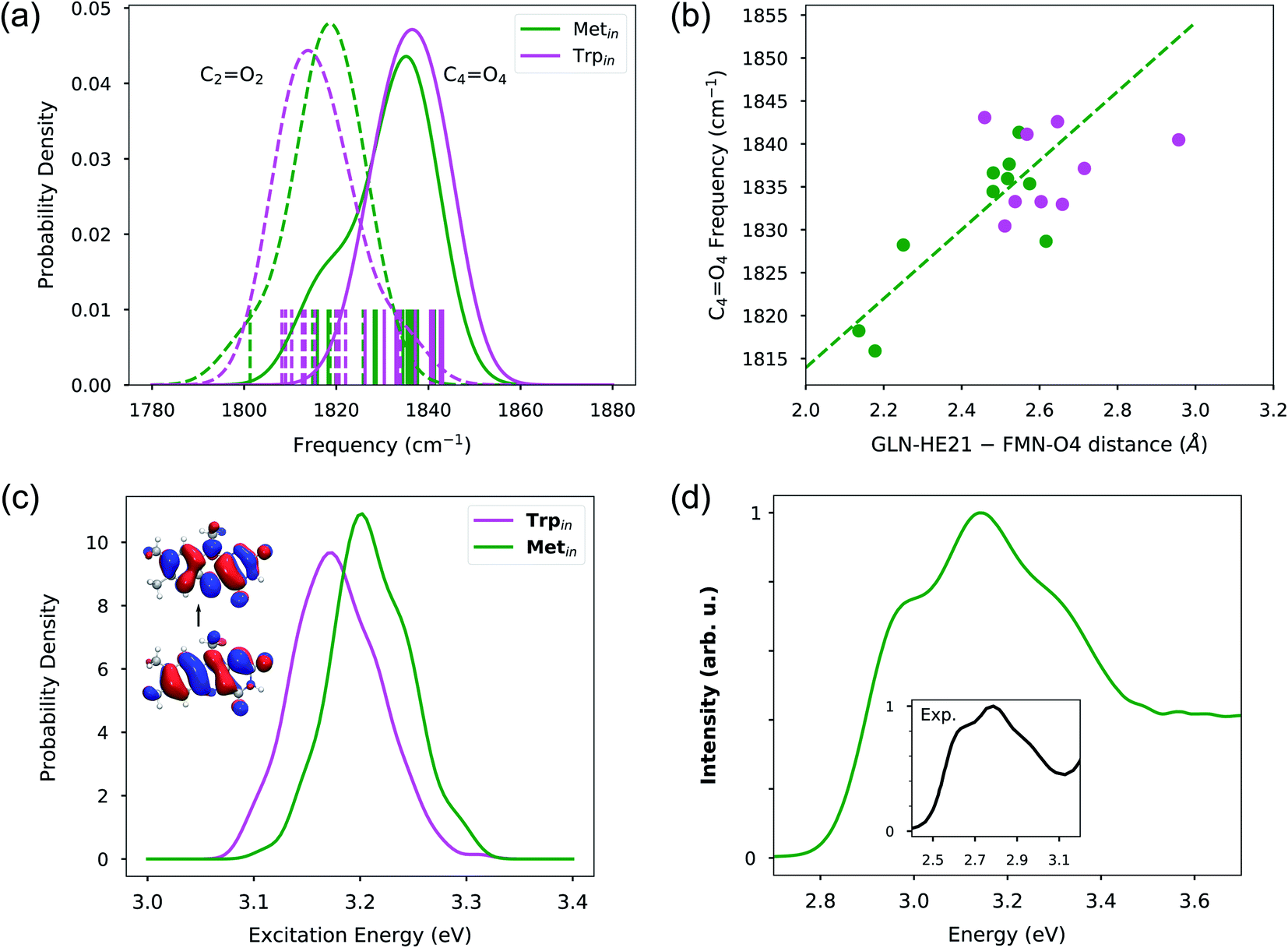 | ||
| Fig. 5 (a) Distribution of CO frequencies extracted from 10 Trpin and 10 Metin MDs. Individual frequencies are shown as vertical bars. The dotted and continuous lines represent the frequencies for C2O2 and C4O4 for Trpin (magenta) and Metin (green) simulations, respectively. (b) Correlation between extracted C4O4 frequencies and hydrogen bond distance from the Gln63 amino hydrogen to the FMN O4 atom. (c) Distribution of excitation energies for Trpin and Metin configurations. The inset shows the electron–hole NTOs. (d) Absorption spectra calculated on Metin configurations. The inset shows the experimental absorption spectrum in black.61 The spectral intensities were normalized in order to have a maximum of 1. | ||
In contrast to what is observed in the harmonic calculations, in the MD trajectories, the C2O2 group is always hydrogen bonded to at least one water molecule, and because of that it is difficult to assess the effect of such interaction on the frequencies. However, the C4O4 experiences different situations within our trajectory samples. As before, here we have investigated the relationship between the C4O4 frequency and the hydrogen bond with Gln63, through a correlation plot with the O4–Gln(H) distance (see Fig. 5b). From the plot, it is evident that in the Metin configurations the hydrogen bond well explains the value of the carbonyl frequency, and a clear correlation with the Gln(H)–O4 distance is found. Moreover, we see that the distances explored in the QM/MM dynamics trajectories correspond to a variability of the order of 30 cm−1 in frequencies, i.e. even more than what is seen experimentally in the dark-to-light transition. This suggests that such a signature alone cannot be used to safely distinguish between potential dark or light-activated forms. On the other hand, in Trpin, the range of explored of Gln(H)–O4 distances is much smaller and mostly concentrated in the large-value side of the plot. This indicates that the hydrogen bond is generally looser than in Metin. This behaviour can be traced back to the interaction of Gln63 with Tyr21. Indeed, in the Metin QM/MM MDs, Tyr21 always presents a tight interaction with the amide oxygen of Gln63. This interaction is much looser in the Trpin configurations (Fig. S8 in the ESI†). In some Trpin trajectories, Tyr21 is very far from Gln63 but even in those trajectories where a hydrogen bond is possible, the interaction is much looser than in the Metin QM/MM MDs. Even though the QM/MM MDs necessarily represent a limited sampling, they are completely in line with the classical MD results of Fig. 3c, which show a tighter interaction between Gln63 and Tyr21 for the Metin simulations.
To complete the investigation of the dark-to-light spectroscopic signatures, we calculated the excitation energies of Metin and Trpin configurations using the computational protocol described in the Methods section. The results, reported in Fig. 5c in terms of distributions for the two sets of structures, show a rather unexpected behavior. As can be seen from the Natural Transition Orbitals (NTOs) depicted in the inset of Fig. 5c, the π–π* excitation involves a large part of the molecule but still a weak transfer of electron density is visible between the two carbonyls. It is thus expected that H-bonds on those groups will induce a red-shift. Here, however, what we observe is that the Trpin configurations which have shown much looser H-bond interactions at O4 present an excitation energy distribution which is red-shifted by ∼0.03 eV with respect to Metin. To better understand this finding, we have isolated the effects that the protein has on the geometry of flavin, and indirectly on its excitation energy. We have thus recalculated the flavin excitation energies removing all the MMPol sites but still keeping the geometry as obtained in the original configurations. This analysis, which has been repeated for both Metin and Trpin configurations, clearly shows that indeed the observed red-shift of Trpin is due to the different geometrical constraints imposed on the flavin by the different local environments.
This result indicates that a very careful analysis has to be done when using calculated shifts on excitation energies to search for the correct dark and light structures, and that simulations of multiple spectroscopies have to be combined together in order to reach a physically sound picture. Here, in fact, the structures that have shown the worst agreement with experiments for both NMR and IR data, if incorrectly used as candidates for the dark state, would introduce an artificial red-shift in the absorption energy, which could invalidate the entire analysis.
Finally, the Metin structures have been used to calculate the lineshape using the approach described in the Methods section. The resulting spectrum is reported in Fig. 5d where it is also compared with the experimental one. Our calculations well reproduce the partially resolved vibronic structure, presenting a main 0 → 1 peak and two shoulders corresponding to the fundamental and 0 → 2 transitions. The vibronic lineshape arises from the coupling of the excitation with several flavin ring modes; analysis of the Huang–Rhys factors (see the Methods section) shows that both CC and C–C ring modes have significant vibronic coupling. Comparison with our previous work,47 where a slightly different lineshape was obtained for flavin absorption in mutant LOV2 flavoproteins, suggests that the lineshape can be tuned by the interaction with the protein.
4 Discussion
The microsecond scale simulations performed in this work have provided a new picture of the AppA structure in solution. Most strikingly, our results strongly indicate that the conformation adopted by Gln63 is essentially the same for Trpin and Metin binding pockets (Fig. 3). In fact, irrespective of the position of Trp104, Gln63 is preferentially oriented with the oxygen away from the flavin O4 atom. As a consequence, the hydrogen bond pattern in the AppA pocket is very different from what was inferred by Anderson et al.5 Gln63 acts as a H-bond acceptor towards the hydroxyl group of Tyr21, in the Trpin as in the Metin structure, while the Gln63(O)⋯Trp104(NH) H-bond is lost. It should also be noted that, in Trpin simulations, Gln63 only stabilized in the final conformation after at least 200 ns of simulation, suggesting that the nanosecond MDs employed in previous studies29,32,33 are not sufficient to equilibrate the Trpin structure.Importantly, all four Trpin replicas converged within 900 ns to a structure where Gln63 is rotated opposite to the X-ray structure. After 1 µs, all replicas show a stable conformation of Gln63 (see Fig. S5†), and the X-ray conformation is only marginally populated. Yet, our MDs show occasional rotation of Gln63, suggesting that Gln rotation is a fast, low-barrier motion that adapts to the relaxation of the protein towards an equilibrium conformation.
Our results also pose a challenge to some model mechanisms used to explain the dark-to-light conversion in AppA,20,23 which were based on the Trpin crystal structure.5 In fact, a structure with the Gln63 oxygen oriented towards Trp104 would be highly unstable, and would relax in less than a µs towards the opposite Gln63 orientation. Such a structure is obviously incompatible with either the dark-adapted or the light-induced state. It is important to stress that X-ray crystallography cannot distinguish the two orientations of Gln63, and in the crystal structure of Trpin (PDB: 1YRX) the orientation with the amide oxygen towards Trp104 was chosen only on the basis of a possible hydrogen bond with the pyrrolic NH group.5 However, our results demonstrate that this hydrogen bond is not stable, and in turn suggest that the Gln63 orientation present in the Trpin crystal is an artifact. Notably, it was also argued that the orientation chosen by Anderson et al. does not fit well the X-ray electron density.63
In contrast to Gln63, Met106 and Trp104 are both stable when they are inside the flavin binding pocket. Therefore, both Trpin and Metin are possible candidates for the dark-adapted structure of AppA, or, similarly, for the light-induced state. Our results also confirm that Trp104 is stable inside the pocket even when the Gln63 amide nitrogen is close to the Trp-NH group, in contrast with previous speculations.34 Our MDs suggest that the Metin structure is more stable than the Trpin one, especially in the binding pocket, which is more compact and exhibits stronger hydrogen bonds. However, when the mutation of Metin is reverted back to the WT analog, Metin–S20C, our simulations show larger instabilities and larger fluctuations in the core of the protein, similar to the original Trpin simulations. These results suggest that the C20S mutation can actively stabilize the Metin structure, possibly at the expense of Trpin. It would therefore be important to have an experimental demonstration of the Metin structure stability that does not rely on amino acid mutations.
The integration of MD analysis with an investigation of NMR chemical shifts has revealed further important aspects. According to our results, the Trpin and Metin structures give very similar chemical shifts for the flavin protons. The H3 proton, which is a reporter of the light-induced state,20 did not present statistically significant chemical shift differences between Metin and Trpin. Only one amide proton of Asn45 gave strikingly different results for Metin and Trpin. In particular, the chemical shift calculated for the Trpin structure is much lower than that from the experiment. We traced back this unusual chemical shift to a strong shielding effect of the aromatic Trp104 ring, which is located close to Asn45 in the Trpin structure. Our NMR results indicate that the Trpin structure is not compatible with the experiments, thus challenging the position and orientation of Trp104 in the Trpin structure. A different orientation of tryptophan was detected inside the binding pocket of another protein of the BLUF family (Slr1694).64 In this structure, the indole ring of the tryptophan is flipped with its nitrogen atom pointing away from the pocket. A detailed computational study by Hammes-Schiffer and co-workers65 examined the stability and the effect of the latter conformation of the tryptophan on the energy of Slr1694 protein and compared them to those of the conformation adopted in AppA by changing the orientation of the tryptophan manually in Slr1694. The authors concluded that the conformation of the tryptophan pointing away from the pocket is thermodynamically more stable than the conformation found in AppA. Our NMR simulations seem to suggest that this finding can also be valid for AppA.
Finally, Trpin and Metin configurations have been tested within the context of the two spectroscopic signatures used to characterize the dark-to-light conversion, the redshifts in the frequency of the C4O4 stretching mode and in the UV-Vis absorption maximum. Both analyses show much weaker H-bonding interactions within the binding pocket for the structures resulting from the Trpin trajectories, which do not allow identification of any significant correlation between the investigated spectroscopic properties and specific arrangement of the residues around the flavin. Instead, a more predictable picture comes out for Metin configurations, for which we found a clear correlation between the frequency of the C4O4 stretching mode and the O4–Gln63(H) distance. The analysis of the vibrational frequencies, however, has also shown that the dynamics and the relative weakness of the intermolecular interactions within the binding pocket require us to go beyond a harmonic model to achieve a realistic description. Finally, our results have shown that the analysis of excitation energies is very delicate, as their values depend more strongly on small distortions of the flavin geometry rather than on the direct effects of nearby residues on the relative energies of the electronic states.
Several studies have suggested that dark-to-light spectral differences could be explained by keto–enol tautomerization of Gln63, possibly also involving a flipping of the side chain.22,30,63,66 This mechanism could explain the spectral differences between the dark and light states of AppA22 or Slr1694,67 better than the Trpin/Metin differences. Our simulations, however, suggest that both the spectral features and the orientation of Gln63 are insensitive to whether Trp104 or Met106 is present inside the pocket. This result should be taken into account when considering the tautomerization mechanism of BLUF photoactivation.
5 Conclusions
In this work, we have combined microsecond molecular dynamics with multiscale calculations of very different spectroscopies to elucidate the structure of the dark state of the AppA BLUF photoreceptor in solution and to investigate some of the most popular molecular mechanisms present in the literature for the dark-to-light activation. Only the combination of NMR, IR and UV-Vis simulated spectroscopies has allowed a robust structural characterization of the dark state showing, for example, that the Gln63 side chain presents a unique and definite preferential orientation, irrespective of whether Trp104 is inside or outside the flavin-binding pocket, and that Trp104 should present another conformation inside the pocket. These findings finally challenge the popular model of AppA BLUF activation based on Gln flipping and the Met106/Trp104 substitution inside the binding pocket, and show that the dark-to-light transition cannot be explained by these structural changes.This study clearly represents a first step towards the complete characterization of AppA and its light-dependent function as an explicit description of the excited state processes is still lacking. Important results have been recently presented by Hammes-Schiffer and coworkers for a different BLUF photoreceptor (Slr1694),67 for which a complex light-induced mechanism involving a forward and backward proton-coupled electron transfer process connected to the formation of the Gln tautomer has been proposed. It is now very interesting to check if the same mechanism applies to AppA; simulations in this direction are now in progress in our group using the solvated structures identified in the present work.
Data availability
Raw data of chemical shifts, excitation energies, CO frequencies calculated on QM/MM MDs, and selected distances are available on Zenodo at https://zenodo.org/record/5507569.
Author contributions
B. M., L. C. and S. H. designed the study. B. M. acquired funding for the work. S. H. ran the MD simulations, performed analyses, and ran QM/MMPol calculations. L. C. oversaw the QM calculations and analyzed the NMR data. M. N. and F. L. developed the code for QM/AMOEBA MDs. V. M. and S. H. ran and analyzed the QM/AMOEBA MDs. L. C. developed the code for SOBI analysis of IR spectra. L. C., S. H. and B. M. wrote the first version of the manuscript. All authors contributed to the final version.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge funding by the European Research Council, under the grant ERC-AdG-786714 (LIFETimeS).References
- J. M. Thornton, A. E. Todd, D. Milburn, N. Borkakoti and C. A. Orengo, Nat. Struct. Biol., 2000, 7(l), 991–994 CrossRef CAS PubMed.
- M. A. van der Horst and K. J. Hellingwerf, Acc. Chem. Res., 2004, 37, 13–20 CrossRef CAS.
- A. Möglich, X. Yang, R. A. Ayers and K. Moffat, Annu. Rev. Plant Biol., 2010, 61, 21–47 CrossRef.
- T. Kottke, A. Xie, D. S. Larsen and W. D. Hoff, Annu. Rev. Biophys., 2018, 47, 291–313 CrossRef CAS.
- S. Anderson, V. Dragnea, S. Masuda, J. Ybe, K. Moffat and C. Bauer, Biochemistry, 2005, 44, 7998–8005 CrossRef CAS PubMed.
- A. Jung, J. Reinstein, T. Domratcheva, R. L. Shoeman and I. Schlichting, J. Mol. Biol., 2006, 362, 717–732 CrossRef CAS PubMed.
- E. Brini, C. Simmerling and K. Dill, Science, 2020, 370, eaaz3041 CrossRef CAS PubMed.
- H. M. Senn and W. Thiel, Angew. Chem., Int. Ed., 2009, 48, 1198–1229 CrossRef CAS PubMed.
- M. Bondanza, M. Nottoli, L. Cupellini, F. Lipparini and B. Mennucci, Phys. Chem. Chem. Phys., 2020, 22, 14433–14448 RSC.
- S. Masuda, K. Hasegawa, A. Ishii and T. aki Ono, Biochemistry, 2004, 43, 5304–5313 CrossRef CAS.
- S. Masuda, K. Hasegawa and T. A. Ono, Plant Cell Physiol., 2005, 46, 1894–1901 CrossRef CAS PubMed.
- M. Unno, R. Sano, S. Masuda, T. aki Ono and S. Yamauchi, J. Phys. Chem. B, 2005, 109, 12620–12626 CrossRef CAS.
- A. L. Stelling, K. L. Ronayne, J. Nappa, P. J. Tonge and S. R. Meech, J. Am. Chem. Soc., 2007, 129, 15556–15564 CrossRef CAS PubMed.
- S. Y. Park and J. R. Tame, Biophys. Rev., 2017, 9, 169–176 CrossRef CAS PubMed.
- T. Fujisawa and S. Masuda, Biophys. Rev., 2018, 10, 327–337 CrossRef CAS PubMed.
- T. Iwata, T. Nagai, S. Ito, S. Osoegawa, M. Iseki, M. Watanabe, M. Unno, S. Kitagawa and H. Kandori, J. Am. Chem. Soc., 2018, 140, 11982–11991 CrossRef CAS.
- S. Masuda and C. E. Bauer, Cell, 2002, 110, 613–623 CrossRef CAS.
- M. Unno, S. Masuda, T.-A. Ono and S. Yamauchi, J. Am. Chem. Soc., 2006, 128, 5638–5639 CrossRef CAS.
- S. Masuda, Y. Tomida, H. Ohta and K. ichiro Takamiya, J. Mol. Biol., 2007, 368, 1223–1230 CrossRef CAS PubMed.
- J. S. Grinstead, M. Avila-Perez, K. J. Hellingwerf, R. Boelens and R. Kaptein, J. Am. Chem. Soc., 2006, 128, 15066–15067 CrossRef CAS.
- M. Gauden, S. Yeremenko, W. Laan, I. H. Van Stokkum, J. A. Ihalainen, R. Van Grondelle, K. J. Hellingwerf and J. T. Kennis, Biochemistry, 2005, 44, 3653–3662 CrossRef CAS PubMed.
- F. Collette, T. Renger and M. Schmidt am Busch, J. Phys. Chem. B, 2014, 118, 11109–11119 CrossRef CAS PubMed.
- P. Goyal and S. Hammes-Schiffer, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1480–1485 CrossRef CAS.
- M. G. Khrenova, A. V. Nemukhin and T. Domratcheva, J. Phys. Chem. B, 2013, 117, 2369–2377 CrossRef CAS PubMed.
- T. Domratcheva, E. Hartmann, I. Schlichting and T. Kottke, Sci. Rep., 2016, 6, 1–14 CrossRef.
- V. Dragnea, A. I. Arunkumar, H. Yuan, D. P. Giedroc and C. E. Bauer, Biochemistry, 2009, 48, 9969–9979 CrossRef CAS PubMed.
- R. Brust, A. Lukacs, A. Haigney, K. Addison, A. Gil, M. Towrie, I. P. Clark, G. M. Greetham, P. J. Tonge and S. R. Meech, J. Am. Chem. Soc., 2013, 135, 16168–16174 CrossRef CAS PubMed.
- K. Karadi, S. M. Kapetanaki, K. Raics, I. Pecsi, R. Kapronczai, Z. Fekete, J. N. Iuliano, J. T. Collado, A. A. Gil, J. Orban, M. Nyitrai, G. M. Greetham, M. H. Vos, P. J. Tonge, S. R. Meech and A. Lukacs, Sci. Rep., 2020, 10, 1–15 CrossRef.
- K. Obanayama, H. Kobayashi, K. Fukushima and M. Sakurai, Photochem. Photobiol., 2008, 84, 1003–1010 CrossRef CAS.
- M. G. Khrenova, A. V. Nemukhin, B. L. Grigorenko, A. I. Krylov and T. M. Domratcheva, J. Chem. Theory Comput., 2010, 6, 2293–2302 CrossRef CAS.
- Y.-W. Hsiao, J. P. Götze and W. Thiel, J. Phys. Chem. B, 2012, 116, 8064–8073 CrossRef CAS.
- J. P. Götze, C. Greco, R. Mitrić, V. Bonačić-Koutecký and P. Saalfrank, J. Comput. Chem., 2012, 33, 2233–2242 CrossRef PubMed.
- K. Meier, W. Thiel and W. F. Van Gunsteren, J. Comput. Chem., 2012, 33, 363–378 CrossRef CAS.
- T. Mathes and J. P. Götze, Front. Mol. Biosci., 2015, 2, 1–14 Search PubMed.
- R. Anandakrishnan, B. Aguilar and A. V. Onufriev, Nucleic Acids Res., 2012, 40, W537–W541 CrossRef CAS.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS.
- D. A. Case, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, V. W. D. Cruzeiro, T. A. Darden, R. Duke, D. Ghoreishi, M. K. Gilson, H. Gohlke, A. W. Goetz, D. Greene, R. Harris, N. Homeyer, S. Izadi, A. Kovalenko, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D. J. Mermelstein, K. M. Merz, Y. Miao, G. Monard, C. Nguyen, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, J. Smith, R. Salomon-Ferrer, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, L. Xiao, D. M. York and P. A. Kollman, AMBER 2018, University of California, San Francisco, 2018 Search PubMed.
- T.-S. Lee, D. S. Cerutti, D. Mermelstein, C. Lin, S. LeGrand, T. J. Giese, A. Roitberg, D. A. Case, R. C. Walker and D. M. York, J. Chem. Inf. Model., 2018, 58, 2043–2050 CrossRef CAS PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- C. Schneider and J. Suhnel, Biopolymers, 1999, 50, 287–302 CrossRef CAS.
- L. W. Chung, W. M. C. Sameera, R. Ramozzi, A. J. Page, M. Hatanaka, G. P. Petrova, T. V. Harris, X. Li, Z. Ke, F. Liu, H.-B. Li, L. Ding and K. Morokuma, Chem. Rev., 2015, 115, 5678–5796 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- C. Curutchet, A. Muñoz Losa, S. Monti, J. Kongsted, G. D. Scholes and B. Mennucci, J. Chem. Theory Comput., 2009, 5, 1838–1848 CrossRef CAS PubMed.
- F. Lipparini, J. Chem. Theory Comput., 2019, 15, 4312–4317 CrossRef CAS.
- J. Wang, P. Cieplak, J. Li, T. Hou, R. Luo and Y. Duan, J. Phys. Chem. B, 2011, 115, 3091–3099 CrossRef CAS PubMed.
- J. Wang, P. Cieplak, J. Li, J. Wang, Q. Cai, M. Hsieh, H. Lei, R. Luo and Y. Duan, J. Phys. Chem. B, 2011, 115, 3100–3111 CrossRef CAS PubMed.
- F. Cardoso Ramos, L. Cupellini and B. Mennucci, J. Phys. Chem. B, 2021, 125, 1768–1777 CrossRef CAS PubMed.
- S. Mukamel, Principles of Nonlinear Optical Spectroscopy, Oxford University Press, New York, 1995 Search PubMed.
- S. Hashem, L. Cupellini, F. Lipparini and B. Mennucci, Mol. Phys., 2020, 118, e1771449 CrossRef.
- D. Loco, L. Lagardère, S. Caprasecca, F. Lipparini, B. Mennucci and J.-P. Piquemal, J. Chem. Theory Comput., 2017, 13, 4025–4033 CrossRef CAS.
- M. Nottoli, B. Mennucci and F. Lipparini, Phys. Chem. Chem. Phys., 2020, 22, 19532–19541 RSC.
- M. Nottoli, M. Bondanza, F. Lipparini and B. Mennucci, J. Chem. Phys., 2021, 154, 184107 CrossRef CAS PubMed.
- J. A. Rackers, Z. Wang, C. Lu, M. L. Laury, L. Lagardère, M. J. Schnieders, J.-P. Piquemal, P. Ren and J. W. Ponder, J. Chem. Theory Comput., 2018, 14, 5273–5289 CrossRef CAS PubMed.
- L. Lagardère, L.-H. Jolly, F. Lipparini, F. Aviat, B. Stamm, Z. F. Jing, M. Harger, H. Torabifard, G. A. Cisneros, M. J. Schnieders, N. Gresh, Y. Maday, P. Y. Ren, J. W. Ponder and J.-P. Piquemal, Chem. Sci., 2018, 9, 956–972 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, A. V. Marenich, M. Caricato, J. Bloino, B. G. Janesko, J. Zheng, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian Development Version, Revision J.13, Gaussian, Inc., Wallingford CT, 2020 Search PubMed.
- P. Ren and J. W. Ponder, J. Phys. Chem. B, 2003, 107, 5933–5947 CrossRef CAS.
- J. W. Ponder, C. Wu, P. Ren, V. S. Pande, J. D. Chodera, M. J. Schnieders, I. Haque, D. L. Mobley, D. S. Lambrecht, R. A. DiStasio, M. Head-Gordon, G. N. I. Clark, M. E. Johnson and T. Head-Gordon, J. Phys. Chem. B, 2010, 114, 2549–2564 CrossRef CAS PubMed.
- A. Belouchrani, K. Abed-Meraim, J.-F. Cardoso and E. Moulines, IEEE Trans. Signal Process., 1997, 45, 434–444 CrossRef.
- J. S. Grinstead, S. T. D. Hsu, W. Laan, A. M. Bonvin, K. J. Hellingwerf, R. Boelens and R. Kaptein, Chembiochem, 2006, 7, 187–193 CrossRef CAS PubMed.
- Q. Wu, W. H. Ko and K. H. Gardner, Biochemistry, 2008, 47, 10271–10280 CrossRef CAS PubMed.
- B. J. Kraft, S. Masuda, J. Kikuchi, V. Dragnea, G. Tollin, J. M. Zaleski and C. E. Bauer, Biochemistry, 2003, 42, 6726–6734 CrossRef CAS PubMed.
- A. Haigney, A. Lukacs, R. Brust, R.-K. Zhao, M. Towrie, G. M. Greetham, I. Clark, B. Illarionov, A. Bacher, R.-R. Kim, M. Fischer, S. R. Meech and P. J. Tonge, J. Phys. Chem. B, 2012, 116, 10722–10729 CrossRef CAS PubMed.
- A. Udvarhelyi and T. Domratcheva, J. Phys. Chem. B, 2013, 117, 2888–2897 CrossRef CAS PubMed.
- H. Yuan, S. Anderson, S. Masuda, V. Dragnea, K. Moffat and C. Bauer, Biochemistry, 2006, 45, 12687–12694 CrossRef CAS PubMed.
- J. J. Goings, C. R. Reinhardt and S. Hammes-Schiffer, J. Am. Chem. Soc., 2018, 140, 15241–15251 CrossRef CAS PubMed.
- T. Domratcheva, B. Grigorenko, I. Schlichting and A. Nemukhin, Biophys. J., 2008, 94, 3872–3879 CrossRef CAS PubMed.
- J. J. Goings, P. Li, Q. Zhu and S. Hammes-Schiffer, Proc. Natl. Acad. Sci., 2020, 117, 26626–26632 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03000k |
| This journal is © The Royal Society of Chemistry 2021 |