
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of structurally-defined polymeric glycosylated phosphoprenols as potential lipopolysaccharide biosynthetic probes†
Lei
Wang
a and
Todd L.
Lowary
 *abc
*abc
aDepartment of Chemistry, University of Alberta, Edmonton, AB T6G 2G2, Canada. E-mail: tlowary@ualberta.ca
bInstitute of Biological Chemistry, Academia Sinica, Academia Road, Section 2, #128, Nangang, Taipei, 11529, Taiwan
cInstitute of Biochemical Sciences, National Taiwan University, Section 4, #1, Roosevelt Road., Taipei 10617, Taiwan
First published on 13th August 2021
Abstract
The biosynthesis of lipopolysaccharide (LPS), a key immunomodulatory molecule produced by gram-negative bacteria, has been a topic of long-term interest. To date, the chemical probes used as tools to study LPS biosynthetic pathways have consisted primarily of small fragments of the larger structure (e.g., the O-chain repeating unit). While such compounds have helped to provide significant insight into many aspects of LPS assembly, understanding other aspects will require larger, more complex probes. For example, the molecular interactions between polymeric LPS biosynthetic intermediates and the proteins that transfer them across the inner and outer membrane remain largely unknown. We describe the synthesis of two lipid-linked polysaccharides, containing 11 and 27 monosaccharide residues, that are related to LPS O-chain biosynthesis in Escherichia coli O9a. This work has led not only to multi-milligram quantities of two biosynthetic probes, but also provided insights into challenges that must be overcome in the chemical synthesis of structurally-defined polysaccharides.
Introduction
Lipopolysaccharide (LPS), an essential component of the gram-negative bacterial outer membrane, is an important mediator of host–pathogen interactions.1 LPS has a tripartite structure consisting of lipid A, the core oligosaccharide, and the O-polysaccharide (O-PS).2 The first two of these components are semi-conserved across all species while the O-PS is highly variable from organism to organism. For example, more than 180 different Escherichia coli O-antigens are known.3,4 Structural differences in repeating units, chain length, and non-carbohydrate substituents (e.g., acetylation) make the O-PS one of the most diverse classes of naturally-occurring glycans.1Given its biological importance and structural diversity, understanding how LPS is biosynthesized has attracted significant attention.2,5,6 A common model organism to study this process is E. coli O9a.7–9 The structure of the LPS in this organism, with a focus on the O-PS, is shown in Fig. 1A. Key features are a ‘primer–adaptor’ trisaccharide containing two mannopyranose (Manp) and one N-acetylglucopyranosylamine (GlcpNAc) residues, which links the lipid A–core domain to the O-PS.10 Four Manp residues, in a mixture of α-(1→2) and α-(1→3)-linkages, comprise the repeating unit and each O-PS chain has 9–17 repeating units.10,11 The structure is terminated with a methyl phosphate group on O-3 of the non-reducing end Manp residue.7,12
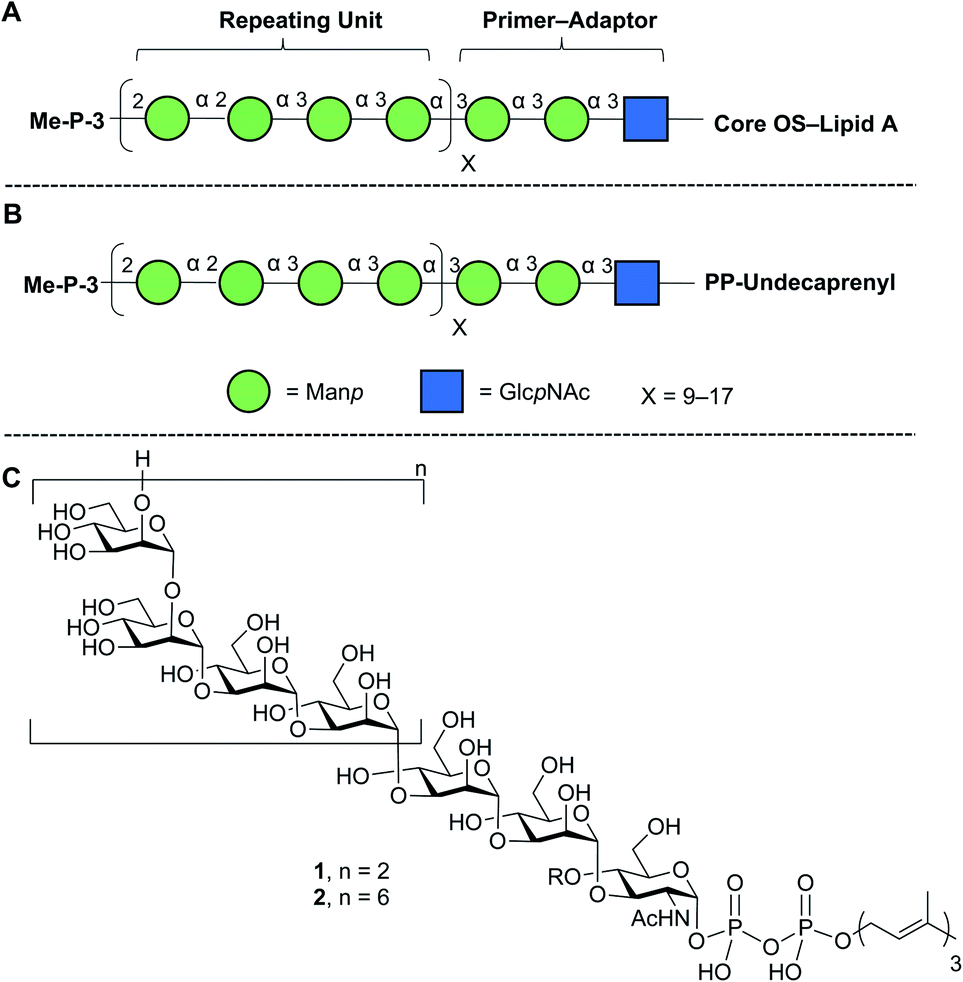 | ||
| Fig. 1 Structures of (A) E. coli O9a LPS; (B) its phosphopolyprenol biosynthetic precursor; (C) biosynthetic probes synthesized in this paper. The structures in A and B are drawn using the Consortium for Functional Glycomics symbolic nomenclature (green circles = Manp; blue squares = GlcpNAc).13. | ||
E. coli O9a LPS biosynthesis employs the ABC transporter-dependent pathway.2,10 This process involves the assembly of the full-length O-PS on an undecaprenol (C55) pyrophosphate carrier (Fig. 1B) on the cytoplasmic side of the inner membrane and then its transfer across the inner membrane to the periplasm by an ABC transporter. Ligation of the O-PS to the Lipid A–core domain occurs in the periplasm and then the entire structure is exported across the outer membrane. The assembly of the undecaprenol pyrophosphate polysaccharide intermediate (Fig. 1B) is achieved by the integrated action of four glycosyltransferase (GTs) that install the primer–adaptor and O-PS repeating units, followed by the action of a bifunctional kinase/methyltransferase (a protein named WbdD)12 that caps the reactive hydroxyl group on the terminal Manp. This capping process also signals, through an unknown mechanism, the transfer of the intermediate across the inner membrane by the ABC transporter.
Like other LPS biosynthetic investigations14–16 deciphering the assembly of the O9a O-PS has benefited greatly from the availability of small synthetic fragments of the larger molecule.7–9 These compounds have been used to characterise the activities of not only the GTs but also WbdD.7–9 However, questions remain about the specificity of WbdD and protein–substrate interactions in the ABC transporter that recognizes and transfers the large lipid-linked glycan across the inner membrane remain obscure. In general, despite impressive recent advances,17–19 the ‘flipping’ of large glycans across membranes remains poorly understood at the molecular level and it is likely that short O-PS fragments will not be effective probes of this process. In addition, the impact of chain length on the action of the biosynthetic GTs is unknown. Answering questions of this type, not only for O9a LPS, but also for other polysaccharides, will require access to larger glycan probes and the development of efficient strategies to assemble them.
In this paper, we describe an approach to synthesize two such probe molecules (Fig. 1C), containing either two (1) or six (2) tetrasaccharide E. coli O9a O-PS repeating units connected via the primer–adaptor trisaccharide to farnesyl pyrophosphate. These molecules thus contain 11 and 27 monosaccharide residues, respectively. Over the past several years, outstanding achievements have been made in the synthesis of structurally-defined polysaccharides.20–26 However, with some exceptions,24–26 most of the structures reported have either been homopolymers containing single glycosidic linkages or polymers with a disaccharide repeating unit. Less common have been syntheses of targets such as 1 and 2, the preparation of which are complicated both by the structure of a tetrasaccharide repeating unit and by the presence of the pyrophosphate and lipid moieties.
Results and discussion
Retrosynthetic analysis and strategy
Two possible synthetic routes to 1 and 2 are shown in Scheme 1A. One approach (Route 1) includes a trisaccharide primer–adaptor and a tetrasaccharide repeating unit, which could be obtained from five different protected Manp building blocks (3–7) and GlcpNAc derivative 8. The second approach (Route 2) involves a tetrasaccharide primer–adaptor, a tetrasaccharide repeating unit, and a trisaccharide cap, which can be assembled using one fewer building block than Route 1: compounds 4–8.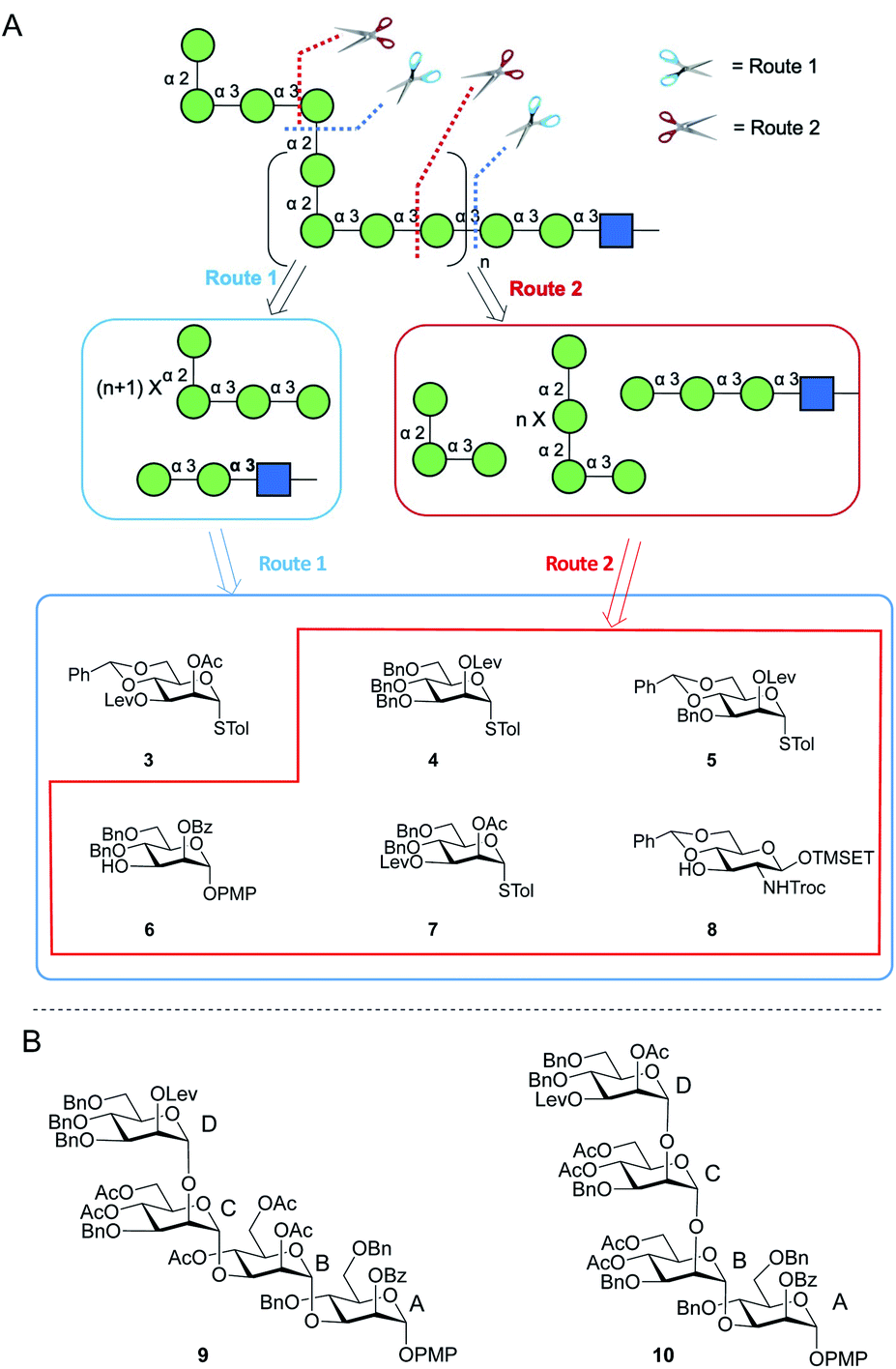 | ||
| Scheme 1 (A) Retrosynthesis of E. coli O9a O-PS fragments; (B) structures of tetrasaccharide repeating units 9 and 10. | ||
Regardless of the route employed, it was necessary to develop an approach to appropriate tetrasaccharide repeating unit building blocks and be able to prepare them efficiently in multi-gram scale. We selected two targets, p-methoxyphenyl (PMP) glycosides 9 and 10 (Scheme 1B), that could be used in Route 1 or Route 2, respectively.
Protecting groups greatly influence glycosylation reactivity, glycosylation stereoselectivity and deprotection efficiency.27,28 These factors must be balanced with the complexity of the routes needed to assemble (usually monosaccharide) building blocks. Thus, great care was taken in choosing them in this investigation. In particular, an overall goal was to limit the number of benzyl (Bn) ethers as we envisioned their removal on the large target compounds could be challenging.22 That said, we did select benzyl ethers to protect the majority of the hydroxyl groups on mannose residues A and D in both 9 and 10 to increase reactivities in glycosylation reactions. A benzoyl (Bz) ester was used to protect the C-2 hydroxyl group on residue A to control the selectivity of the α-glycosylation. As the temporary protecting group in residue D, a levulinate (Lev) ester was selected, given the orthogonality of this group to others used in the synthesis. Finally, we chose acetate (Ac) esters to protect the majority of the hydroxyl groups on residues B and C as we expected that they would be easier to remove than Bn ethers. In addition, given their location on the internal portion of the building block, they would not be expected to greatly affect glycosylation reactivity. In cases where hydroxyl groups on residues B and C are benzylated, this was done to simplify the preparation of the monosaccharide precursors. The synthesis of 9 and 10 is discussed below. The preparation of the monosaccharide building blocks needed to synthesize these tetrasaccharides is described in the ESI (Scheme S2†).
Synthesis of two repeating units and comparison of Routes 1 and 2
To compare the two routes shown in Scheme 1A, we prepared tetrasaccharide 16 and 21 (Scheme 2), which can be converted to the key building blocks 9 and 10, respectively, by manipulation of the protecting groups. The synthesis of tetrasaccharide 16 (Scheme 2A), started with the coupling of glycosyl acceptor 6 with thioglycoside donor 3 mediated by NIS (1.3 equiv.) and AgOTf (0.1 equiv.). The product was produced in poor (45%) yield, mainly due to the formation of the 1,2-othoester 12 (25%). To improve the yield, a larger amount of AgOTf (0.35 equiv.) was used in the reaction. Under these more acidic conditions, disaccharide 11 was isolated in 65% yield. Chemoselective removal of the levulinate group using hydrazine acetate gave acceptor 13 in 92% yield. Subsequent NIS/AgOTf-promoted glycosylation of 13 with 5 gave a 67% yield of trisaccharide 14. Removal of the levulinate ester provided 15, which was then glycosylated with 4 leading to the formation of tetrasaccharide 16 in 62% yield over the two steps. The synthesis of tetrasaccharide 21, which was needed for Route 2, also involved an alternating series of NIS/AgOTf-promoted glycosylations and levulinate protecting group removals with hydrazine acetate (Scheme 2B). In all glycosylations leading to mannosidic linkages described in this paper, the stereochemistry of the newly formed glycosidic linkage was determined by measuring its 1JC-1,H-1. These values were in the range of 171–177 Hz, as expected for an α-linkage.29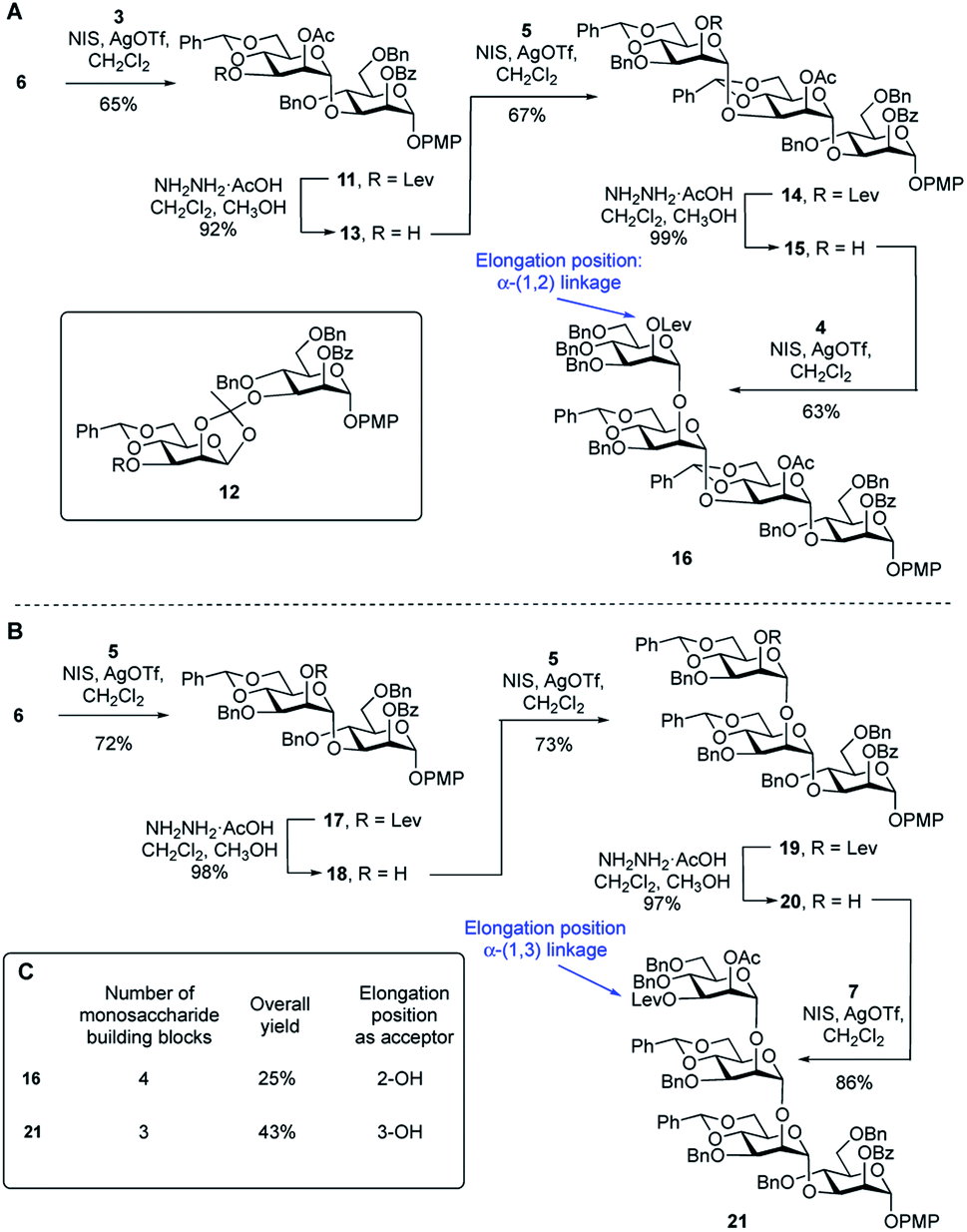 | ||
| Scheme 2 (A) Synthesis of tetrasaccharide repeating unit 16 (Route 1); (B) synthesis of tetrasaccharide repeating unit 21 (Route 2); (C) comparison of Routes 1 and 2, leading to 16 and 21, respectively. | ||
A comparison of the synthesis of 16 and 21 is provided in Scheme 2C. Four monosaccharide building blocks (3–6) are needed for the synthesis of 16. In contrast, only three building blocks (5–7) are needed to prepare 21. In addition, compared with the overall yield for the synthesis of 16 (25%), the overall yield for the synthesis of repeating unit 21 was much higher: 43%. We then considered the relative glycosylation reactivities that could be expected in both routes in the reactions leading to longer oligomers of the repeating units. In Route 1, chain extension would involve the formation of an α-(1→2) glycosidic linkage. In Route 2, this process would require generation of an α-(1→3)-linkage. In a previous study30 the relative reactivity between the C-2 and C-3 hydroxyl groups on mannose was investigated. Using a mannose acceptor with unprotected C-2 and C-3 hydroxyl groups, the α-(1→3)-disaccharide was isolated in 80% yield and no α-(1→2)-disaccharide was observed. This suggests that the equatorial C-3 hydroxyl group is more reactive than the axial C-2 hydroxyl group. After these considerations, we decided to focus on Route 2 to assemble the targets. Although this ‘frame-shift’ approach may appear more complicated, requiring three oligosaccharide building blocks compared to only two for Route 1, the former has three advantages: (1) fewer monosaccharide building blocks are needed for the preparation of 21 compared to 16; (2) there is a higher yield in the synthesis of 21 compared to 16 and (3) the key chain extension process will involve reactions at the more reactive C-3 hydroxyl group.
Synthesis of tetrasaccharide primer–adaptor and trisaccharide cap domains needed for Route 2
To synthesize the targets via Route 2, a protected primer–adaptor tetrasaccharide intermediate was needed (Scheme 3A). The preparation of this compound started by the coupling of glycosyl acceptor 8 (Scheme S1, ESI†) to mannose thioglycoside donor 7 mediated by NIS and AgOTf, which gave disaccharide 22 in 85% yield. The benzylidene acetal in 22 was then hydrolyzed and the resulting diol was acetylated leading to, in 94% overall yield, the formation of 23. From this disaccharide, the levulinate ester was removed using hydrazine acetate, providing 24 (98% yield), which was then subjected to glycosylation with 7 affording a 91% yield of trisaccharide 25. Another cycle of levulinate deprotection and NIS/AgOTf promoted glycosylation with thioglycoside 7 provided the tetrasaccharide 27, which was then treated with hydrazine acetate to afford the primer–adaptor tetrasaccharide intermediate 28 in 73% yield over the three steps.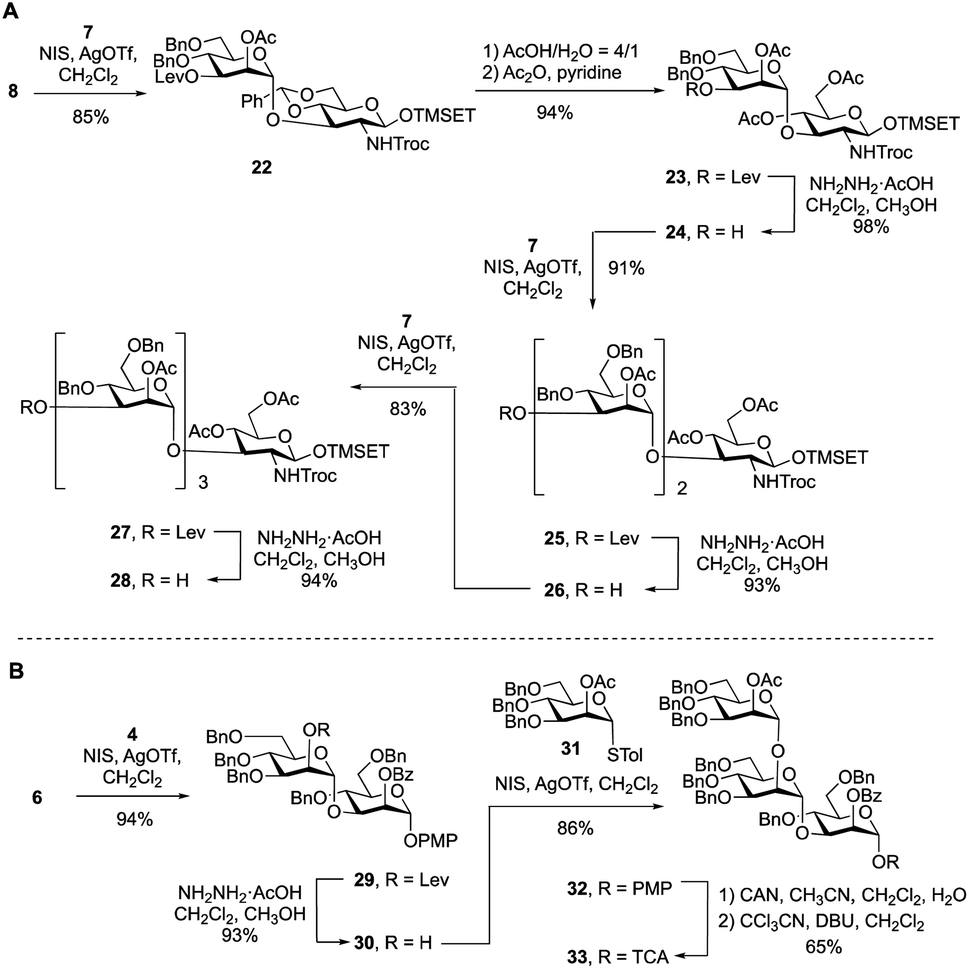 | ||
| Scheme 3 (A) Synthesis of primer–adaptor acceptor building block 28; (B) synthesis of trisaccharide cap donor 33. | ||
The final intermediate needed for the targets was the trisaccharide cap at the nonreducing end (Scheme 3B). This building block was accessed by coupling of glycosyl acceptor 6 and glycosyl donor 4 using NIS/AgOTf-promoted glycosylation and subsequent levulinate deprotection to produce disaccharide alcohol 30 in 87% yield over the two steps. Subsequent glycosylation of 30 with thioglycoside 31 (see Scheme S2 in the ESI† for its synthesis) proceeded in 86% yield producing the target trisaccharide 32. Finally, a two-step functional group transformation sequence, from PMP glycoside to glycosyl trichloroacetimidate (TCA), led to the activated donor 33 in 65% yield over the two steps. This was achieved by ceric ammonium nitrate-mediated cleavage of the PMP group and then treatment of the resulting alcohol with tricholoracetonitrile and DBU.
Exploration of 4 + 4 glycosylation conditions and synthesis of undecasaccharide 1
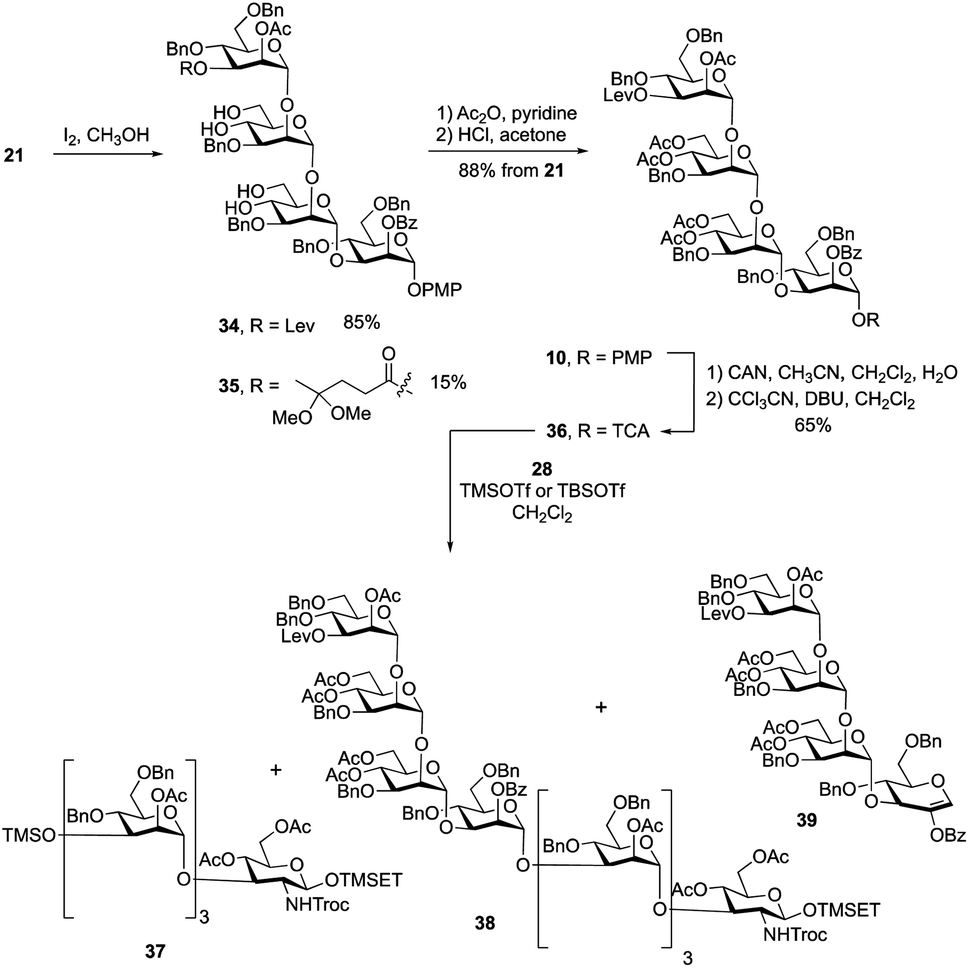
Having obtained the primer–adaptor and cap domains, we moved forward on assembling the first target, undecasaccharide 1. Before doing this, we chose to exchange the benzylidene acetals on residues B and C in 21 with acetate esters to simplify the final deprotection steps (Scheme 4). We initially used 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetic acid–water at 80 °C to hydrolyze the two acetals. Unfortunately, the yield for this reaction was only ∼60%. Successful acetal cleavage could, however, be achieved by reaction with iodine in methanol at reflux.31 The 1H NMR spectrum and mass spectrometric data of the crude reaction mixture showed that, in addition to the desired product 34, there was ∼15% of 35, formed by conversion of the ketone in the levulinate ester to a ketal. Tetrasaccharides 34 and 35 were inseparable; therefore, the mixture was treated with acetic anhydride and pyridine to give the corresponding acetylated products, which were then dissolved in a 2% solution of HCl in acetone. This led to hydrolysis of the ketal affording the levulinoyl-protected tetrasaccharide 10 in 88% yield over three steps. Conversion of 10 into O-trichloroacetimidate 36 was carried out under the standard conditions described above giving the activated donor in 65% yield.
:1 acetic acid–water at 80 °C to hydrolyze the two acetals. Unfortunately, the yield for this reaction was only ∼60%. Successful acetal cleavage could, however, be achieved by reaction with iodine in methanol at reflux.31 The 1H NMR spectrum and mass spectrometric data of the crude reaction mixture showed that, in addition to the desired product 34, there was ∼15% of 35, formed by conversion of the ketone in the levulinate ester to a ketal. Tetrasaccharides 34 and 35 were inseparable; therefore, the mixture was treated with acetic anhydride and pyridine to give the corresponding acetylated products, which were then dissolved in a 2% solution of HCl in acetone. This led to hydrolysis of the ketal affording the levulinoyl-protected tetrasaccharide 10 in 88% yield over three steps. Conversion of 10 into O-trichloroacetimidate 36 was carried out under the standard conditions described above giving the activated donor in 65% yield.
 | ||
| Scheme 4 Synthesis of repeating unit 10 and exploration of the 4 + 4 glycosylation between 28 and 36. | ||
With the primer–adaptor tetrasaccharide acceptor 28 and repeating unit donor 36 in hand, their coupling was studied (Table 1). When 0.4 equiv. of TMSOTf was used as promoter (Entry 1), two side products – TMS ether 37 (Scheme 4, 35%) and glycal 39 (30%) – were obtained and the yield for desired product, 38, was modest (52%). Side products 37 and 39 come from acceptor 28 and donor 36, respectively. We concluded that under these conditions, the hydroxyl group on 28 reacted with the TMSOTf promotor to form 37, which does not undergo glycosylation. As a result, some of trichloroacetimidate 36 has no substrate to glycosylate and it undergoes elimination to give 39. To minimize the formation of by-products, we reduced the amount of TMSOTf to 0.2 equiv. (Entry 2). The yield for 38 was improved to 65%; however, 37 and 39 were still produced in 30% and 15% yields, respectively. This suggests that the silylation of 28 is rapid and that using a more sterically-demanding Lewis acid might result in less of these two side products being produced. Indeed, when using 0.4 equiv. of TBSOTf (Entry 3), the yield of the desired product 38 was greatly improved, to 86%, and no 37 or 39 could be isolated. Although the newly formed H-1 resonance could not be identified in the one-dimensional 1H NMR spectrum due to overlap, all of the 1JC-1, H-1 values could be measured from the 1H-coupled HSQC spectrum. Given this finding, we used TBSOTf as the promotor for all trichloroacetimidate glycosylations carried out during the synthesis of the targets (see below).
| Entry | 1 | 2 | 3 |
|---|---|---|---|
| Donor 36 | 1.2 eq. | 1.0 eq. | 1.0 eq. |
| Acceptor 28 | 1.0 eq. | 1.2 eq. | 1.2 eq. |
| Activator/eq. | TMSOTf/0.4 | TMSOTf/0.2 | TBSOTf/0.4 |
| TMS ether 37 | 35% | 30% | — |
| Glycal 39 | 30% | 15% | — |
| Octasaccharide 38 | 52% | 65% | 86% |
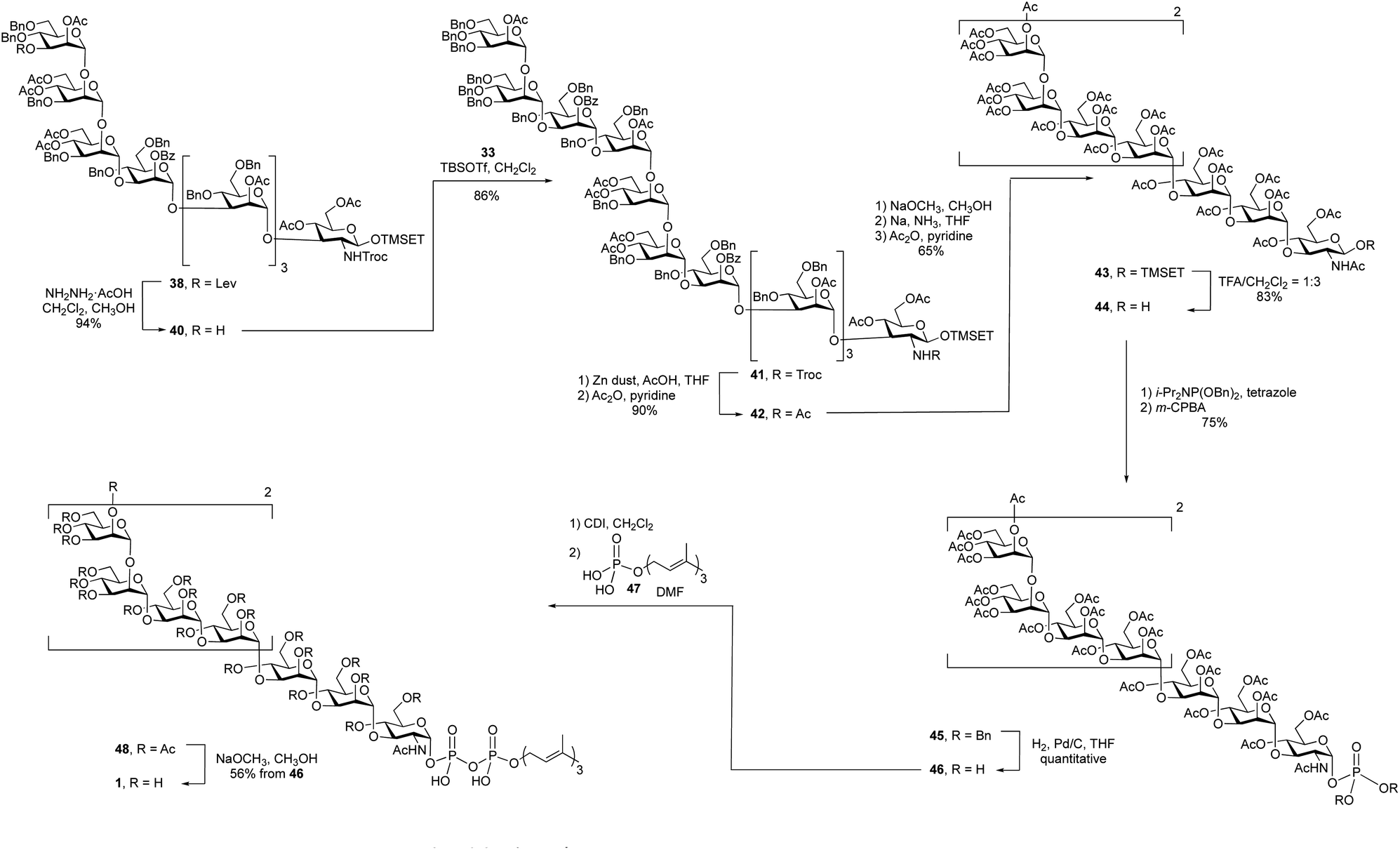
With an approach for the construction of octasaccharide 38 in place, the focus shifted to the final glycosylation with the cap moiety and elaboration to the target (Scheme 5). Thus, cleavage of the levulinate protecting group in 38 with hydrazine acetate gave octasaccharide alcohol 40 (94% yield), which was subsequently reacted with trichloroacetimidate 33 to afford undecasaccharide 41. This 3 + 8 glycosylation provided 41 in 86% yield.
 | ||
| Scheme 5 Synthesis of undecasaccharide lipid pyrophosphate 1. | ||
After all of the monosaccharide residues were in place, the focus became changing the functional group on the nitrogen atom, introduction of the phosphate and lipid moiety and the final deprotection. The Troc group in 41 was removed in 86% yield via reductive elimination, which employed activated zinc in AcOH/THF, to afford a crude product with a free amine. A common problem of this reaction is the formation of the dichloroethoxy-carbamate by-product,32 which was minimized by using freshly activated zinc dust. After N-acetylation using acetic anhydride and pyridine, the Troc protecting group was converted to an acetyl group to give undecasaccharide 42 in 90% yield over the two steps.
It was next necessary to remove the benzyl ether protecting groups and replace them with acetate esters. This would simplify the deprotection at the end of the synthesis to a single ester cleavage step with an easy to remove by-product (methyl acetate). It can be difficult to remove large numbers of benzyl groups in large oligosaccharides using hydrogenolysis;22 therefore, we chose to use Birch reduction. The Birch reduction is a strongly basic reaction and, as such, esters are readily cleaved under these conditions. It was then, in principle, possible to remove all of the protecting groups in a single step before replacing them with acetate esters, which was required before the pyrophosphate coupling reaction. However, after exploring this reaction, we found it more convenient to first cleave the acyl groups, purify the molecule and execute the dissolving metal reduction. This approach has been used for the synthesis of other molecules.33,34 and in our hands this strategy greatly simplified the purification of the product after removal of the benzyl ethers. Thus, undecasaccharide 42 was treated with sodium methoxide to remove all of the acetyl and benzoyl groups. This intermediate was purified and then subjected to Birch conditions giving a fully deprotected oligosaccharide, which was then acetylated to afford 43 in 65% yield over the three steps.
The final steps in the synthesis involved the introduction of the lipid phosphate moiety. To do this, the anomeric TMSET protecting group was removed by treatment of 43 with 25% trifluoroacetic acid (TFA) in dichloromethane to give 44 in 83% yield. This hemiacetal was treated with dibenzyl N,N-diisopropyl phosphoramidite and tetrazole to afford a phosphite intermediate, which was oxidized with m-CPBA providing a 75% overall yield of glycosyl phosphate 45. Hydrogenolysis of the benzyl groups on the phosphate gave a glycosyl phosphate intermediate, 46, that was coupled to farnesyl phosphate (47)35 using a carbonyldiimidazole (CDI)-mediated phosphoesterification.36,37 Such coupling reactions are typically low yielding,38 but through careful optimization of the conditions, including the use of a large excess of farnesyl phosphate and long (seven-day) reaction times, reasonable yields could be obtained. The product, 48, was then deacetylated with catalytic sodium methoxide in methanol affording the farnesyl pyrophosphate-linked oligosaccharide 1 in 56% yield from 45 over the three steps.
The high-resolution electrospray mass spectrum of undecasaccharide 1 showed a molecular ion with two negative charges (M–2H)−2 at m/z = 1101.8628, which agrees with calculated exact mass. The identity 1 was further confirmed by NMR spectroscopy (Fig. S1 and S2†).
Assembly of eicosaheptasaccharide 2
After the synthesis of the undecasaccharide 1 was secure, we moved to the preparation of 2, an eicosaheptasaccharide containing 27 sugar residues. Although the approach detailed above could be used to make larger oligosaccharides, we considered that using tetrasaccharide imidate 36 would be inefficient as it would allow chain extension only by one repeating unit in each glycosylation. We therefore decided to synthesize an octasaccharide donor (a dimer of 36) to facilitate chain extension. To do this (Scheme 6), a tetrasaccharide acceptor (49) was obtained in 93% yield by removal of the levulinate group in 10. Next, trichloroacetimidate donor 36 was used to glycosylate 49 promoted by TBSOTf to afford the desired octasaccharide 50 in 88% yield. As was observed in the synthesis of 38, spectral overlap prevented the identification of 1H signal arising from the nascent glycosidic bond in the 1H NMR spectrum of 50. However, all of the 1JC-1,H-1 values could be measured from the 1H-coupled HSQC spectrum, which confirmed the α-stereochemistry of the eight mannose residues in 50. Conversion of 50 to the trichloroacetimidate donor 51 was achieved by selective cleavage of the PMP group and subsequent reaction of the resulting hemiacetal with tricholoracetonitrile in the presence of DBU (65% yield over two steps).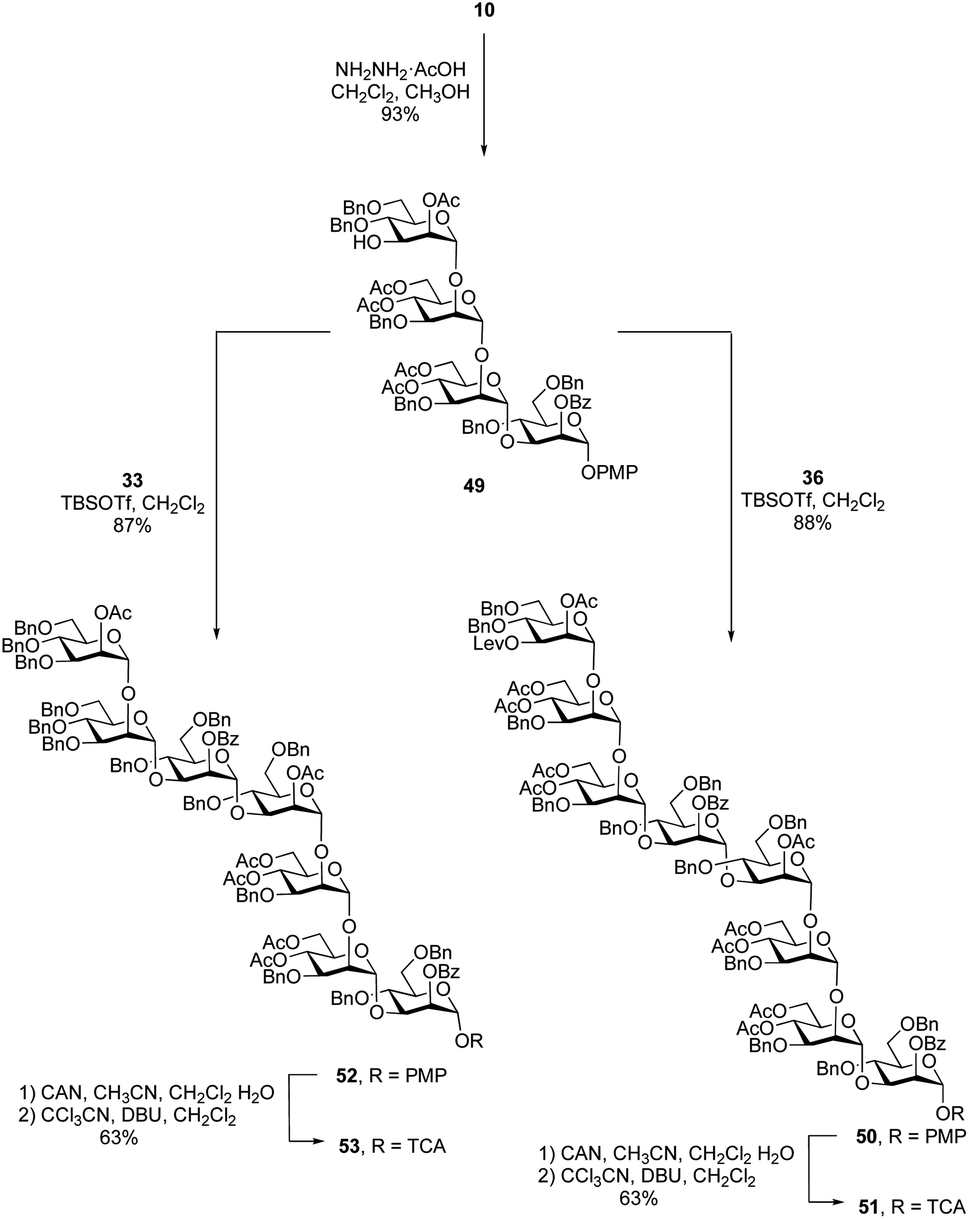 | ||
| Scheme 6 Synthesis of octasaccharide 51 and heptasaccharide 53 donors. | ||
Starting from primer–adaptor tetrasaccharide 28 and octasaccharide donor 51, the 20-mer could be synthesized following a 4 + 8 + 8 glycosylation sequence. To obtain the desired 27-mer, it was necessary to synthesize a heptasaccharide donor (53, Scheme 6), analogous to the trisaccharide ‘cap’ used for the synthesis of 1. Glycosylation of 49 with imidate 33 in presence of TBSOTf led to the desired heptasaccharide 52 in 87% yield. Conversion of 52 into heptasaccharide donor 53 was achieved in 63% overall yield using the same method used to synthesize 51.
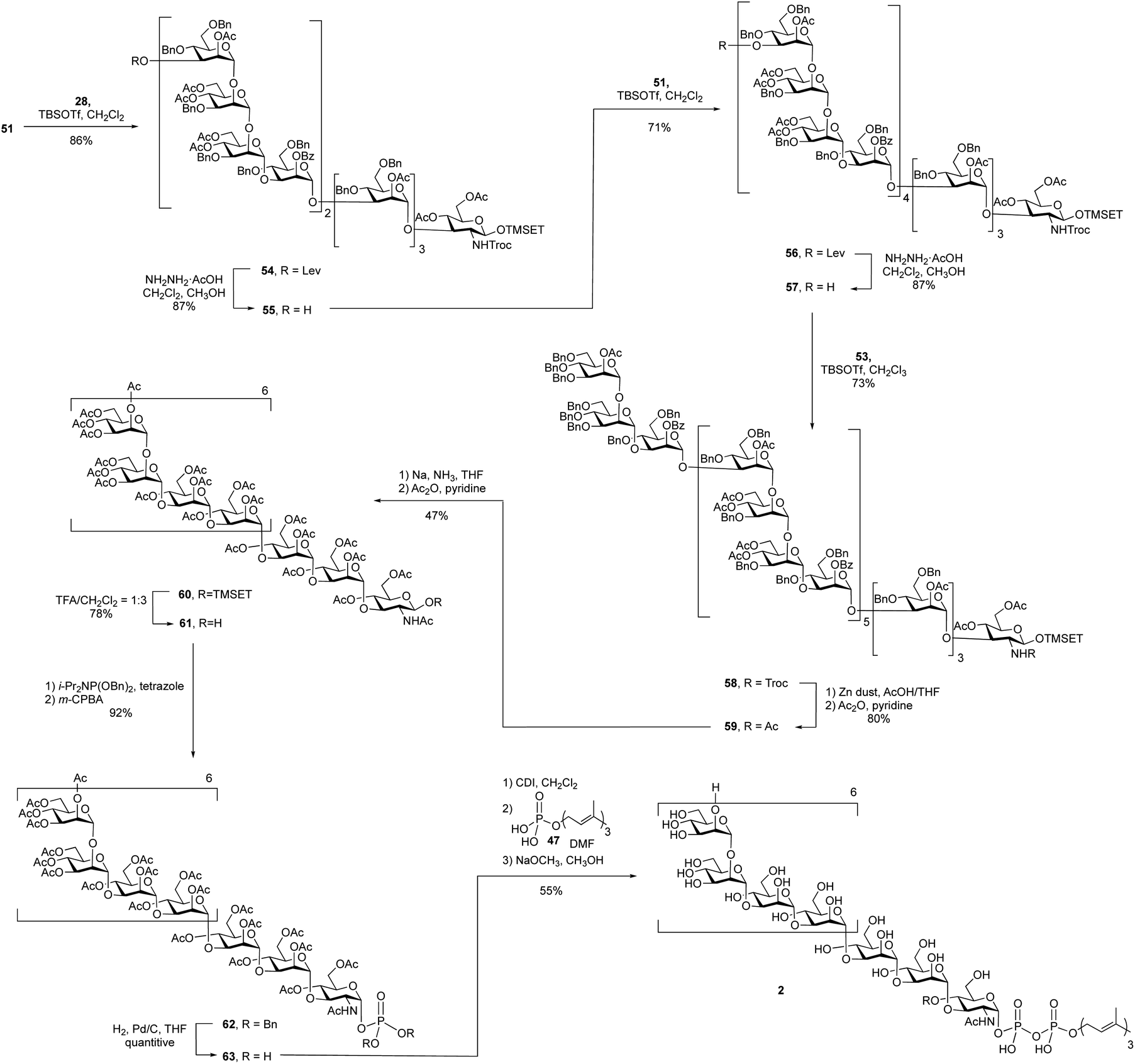
A 4 + 8 + 8 + 7 reaction sequence was used to assemble the polysaccharide domain of the eicosaheptasaccharide (Scheme 7). The initial 8 + 4 glycosylation between octasaccharide 51 and tetrasaccharide 28 using TBSOTf proceeded in 86% yield. The subsequent deprotection of the levulinate ester on the product dodecasaccharide 54 using hydrazine hydrate at room temperature, conditions that had worked well on the smaller oligosaccharides, was surprisingly slow. Following this reaction by TLC was also complicated by the fact that the starting material and product were inseparable. In the 1H NMR spectrum of dodecasaccharide 54, the resonance for H-3 of the terminal mannose residue (the hydrogen adjacent to the levulinate ester) appears at 5.37 ppm as a doublet of doublets (3J = 9.5, 3.5 Hz). This signal could be easily identified in the crude 1H NMR spectrum of the mixture and thus NMR spectrometry was used to follow the reaction. After four hours at room temperature, the spectrum showed that only 15% of the levulinate group was removed. To achieve full deprotection, a rotary evaporator was used to concentrate the reaction mixture and then the flask was kept rotating for 30 min at 40 °C. Under these conditions, the crude 1H NMR spectrum showed complete disappearance of the peak at 5.37 ppm, suggesting 100% conversion; the yield of 55 was 87% after purification. The reason for the low reactivity is unclear. We postulate that the molecule adopts a three-dimensional structure that hinders either the formation of the hydrazone intermediate of the levulinolyl ketone moiety, or its subsequent intramolecular cyclization that releases the alcohol. While, in principle, this could be ascertained by TLC analysis of the reaction mixture, this was not possible given the size of the molecules, which rendered their chromatographic mobilities very similar.
 | ||
| Scheme 7 Synthesis of eicosaheptasaccharide 2. | ||
To further extend dodecasaccharide 55, a TBSOTf-mediated 8 + 12 glycosylation using octasaccharide donor 51 was executed, which provided eicosasaccharide 56 in 71% yield. The levulinate ester group on 56 was removed by using the same method for deprotection as that used on the dodecasaccharide to generate, in 87% yield, 57. Finally, TBSOTf-promoted glycosylation using heptasaccharide 53 as the donor and 57 as the acceptor, generated the desired protected eicosaheptasaccharide 58 in 73% yield. The identity of 53 was supported by its MALDI-TOF mass spectrum, which showed a molecular ion of the sodium adduct at m/z = 10,578, consistent with the calculated mass for this molecule.
With the polysaccharide core of the molecule assembled, the next step was the exchange of the Troc group on 58 for an acetate to produce 59. This was carried out in two steps and in 80% yield upon treatment with zinc in acetic acid and then acetic anhydride and pyridine. Deprotection by Birch reduction to remove all of the benzyl ethers was then investigated. Initially, consistent with what had been done in the synthesis of 1, all of the acetate and benzoate groups in 59 were removed using sodium methoxide in methanol to afford a partially deprotected product. However, this deacylated product was insoluble in THF, the solvent used for the Birch reduction. Therefore, it was necessary to use the fully protected molecule 59 in this step. Application of the same conditions used for the undecasaccharide led to a mixture of two products: the desired product and one or more side products in which the 2-(trimethylsilyl)ethyl (TMSEt) group appeared to have been lost. Based on the 1H NMR spectrum of the crude reaction mixture, the ratio between the desired and undesired product(s) was 1:1.5 (integration of the methyl signal of the NHAc group). The exact structure of the side product could not be determined by NMR spectroscopy and mass spectroscopy of the crude mixtures showed a number of species with molecular weights lower than that of the desired product. In addition to dramatically lowering the yield, the product and side products were impossible to separate. Fortunately, it was discovered that decreasing the reaction temperature to −80 °C and shortening the reaction time to 1.5 h resulted in only a trace amount of the side products being produced. Under these conditions, the two-step yield for the Birch reduction and acetyl protection was improved to 47%.
Removal of the TMSEt group using TFA in dichloromethane gave oligosaccharide 61 in 78% yield. Following the same phosphorylation reaction described in the undecasaccharide synthesis, phosphate 62 was obtained in 92% yield. After removal of the benzyl groups on phosphate 62 by hydrogenolysis, coupling between the resulting glycosyl 1-phosphate 63 and farnesyl phosphate, mediated by CDI, led to the formation of protected glycosyl phospholipid, which was then deacetylated using sodium methoxide in a mixture dichloromethane in methanol. The desired product 2 was obtained in 55% yield over three steps.
The high-resolution electrospray mass spectrum of 2 showed a molecular ion with three negative charges (M–3H)−3 at m/z = 1599.1826, consistent with the exact mass of the molecule. Considering the size of 1 and 2, we envisioned that their NMR spectra would not be very informative, but that was not the case. Both 1H and 13C NMR spectra (Fig. S1 and S2†) provide strong support for the structure of the compounds. For example, in the 1H NMR of 2 (Fig. S1†), the peak at 5.50 ppm (dd, 1H, J = 7.0, 3.0 Hz) is from the GlcpNAc H-1. The small 2JP,H-1 (GlcpNAc) coupling constant (J = 3.0 Hz) indicates the connection between pyrophosphate and GlcpNAc residue. Three peaks at 5.46, 5.22 and 5.20 confirm the presence of farnesyl residue. Four peaks at 5.38, 5.31, 5.13 and 5.05 ppm are from repeating unit Manp residue C H-1, Manp residue D H-1, Manp residue B H-1 and Manp residue A H-1, respectively. Adjacent to the resonance for residue A is another smaller peak that we ascribe to the residue attached to the primer–adaptor trisaccharide. The 13C NMR spectrum of 2 (Fig. S2†) has a resonance at 95.6, split into a doublet (2JP,C-1 = 6.5 Hz) corresponding to C-1 of the GlcpNAc residue thus further confirming the GlcpNAc-PP linkage. The peak at 103.0 belongs to C-1 of the Manp C and Manp D residues, the peak at 101.6 ppm is from C-1 of Manp A and Manp B residues. The repeating unit peaks in 1H and 13C NMR are in agreement with that of the natural O-PS reported in literature.39,40
Conclusion
In summary, we report the first chemical synthesis of large lipid pyrophosphate-linked LPS O-PS intermediates (1 and 2). An important design feature was a ‘frame-shift’ strategy, in which the molecule was assembled not by using building blocks corresponding to natural repeating unit, but instead one where disconnections were made between different residues. This allowed for a reduction in the number of monosaccharide building blocks required and better yields of the glycosylations throughout the synthesis. This non-conventional strategy should thus be considered in future when designing routes to large glycans. Other key features of the route were the preparation three building blocks – repeating unit donor 36, tetrasaccharide primer–adapter acceptor 28 and trisaccharide cap donor 33 – via an iterative cycle of NIS/AgOTf-promoted glycosylations and hydrazine acetate-mediated levulinate ester cleavages. In addition, a TBSOTf-promoted glycosylation method was developed for glycosylations between oligosaccharide acceptors and trichloroacetimidate donors generated from the oligosaccharide building blocks.Following a 4 + 4 + 3 strategy, the protected undecasaccharide 41 was assembled in glycosylation yields between 82% and 88%. A 4 + 8 + 8 + 7 strategy was employed to synthesize the protected eicosaheptasaccharide 58 in good to excellent yields (70–86%). After nine additional steps, including protecting group manipulation, phosphorylation, coupling with farnesyl phosphate and final deprotection, we produced 13 mg and 7 mg quantities of 1 and 2, respectively. While we employed farnesol as the polyprenol in these targets, the use of longer polyprenols should be straightforward.41
In addition to providing access to valuable probe molecules that are currently being used in biosynthetic investigations, the strategy developed here can be extended to prepare even larger fragments of this O-PS. Moreover, the study provides insights into the challenges faced when assembling structurally-defined polysaccharides and solutions to circumvent them. In particular, it was necessary to overcome not only the formation of unproductive side products in glycosylation reactions (conversion of 36 into 38), but also difficulties in removing protecting groups either selectively (synthesis of 55), or in bulk (the Birch reduction of 59). Indeed, this work suggests that the efficiency of the glycosylations is not dramatically affected when carried out molecules of increasing size, something that has been previously reported for other couplings of large mannose-containing oligosaccharides23 and furanose-containing oligosaccharides.24,25 On the other hand, issues such as low reactivity and more mundane problems such as poor solubility, similar chromatographic mobilities, and spectral overlap, complicated the analysis and deprotection of large intermediates. These latter issues underscore the importance of considering these factors when designing synthetic routes to structurally-defined polysaccharides. Such problems have been previously encountered22,42 and suggest that the development of new protecting groups that can be removed in quantitative yield, and creative methods for reaction monitoring and execution, are needed additions to the arsenal of methods for synthetic polysaccharide chemistry. Such advances, in addition to improved methods for glycoside bond synthesis, would allow more straightforward and efficient access to structurally-defined complex polysaccharides.
Author contributions
TLL conceived the project and supervised WL who carried out all of the experimental work. TLL and WL jointly wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Alberta Glycomics Centre and the Natural Sciences and Engineering Research Council of Canada. LW thanks Alberta Innovates Technology Futures for a studentship award. We thank Dr Larry S. Lico for helpful comments on the manuscript.Notes and references
- B. W. Simpson and M. S. Trent, Nat. Rev. Microbiol., 2019, 17, 403–416 CrossRef CAS PubMed.
- C. Whitfield and M. S. Trent, Annu. Rev. Biochem., 2014, 83, 99–128 CrossRef CAS PubMed.
- R. Stenutz, A. Weintraub and G. R. Widmalm, FEMS Microbiol. Rev., 2006, 30, 382–403 CrossRef CAS PubMed.
- B. Liu, A. Furevi, A. V. Perepelov, X. Guo, H. Cao, Q. Wang, P. R. Reeves, Y. A. Knirel, L. Wang and G. Widmalm, FEMS Microbiol. Rev., 2020, 44, 655–683 CrossRef CAS PubMed.
- J. M. Cote and E. A. Taylor, Int. J. Mol. Sci., 2017, 18, 2256 CrossRef PubMed.
- P. Sperandeo, A. M. Martorana and A. Polissi, in Bacterial Cell Walls and Membranes, ed. A. Kuhn, Springer International Publishing, Cham, 2019, pp. 9–37 Search PubMed.
- B. R. Clarke, M. R. Richards, L. K. Greenfield, D. Hou, T. L. Lowary and C. Whitfield, J. Biol. Chem., 2011, 286, 41391–41401 CrossRef CAS PubMed.
- L. K. Greenfield, M. R. Richards, J. Li, W. W. Wakarchuk, T. L. Lowary and C. Whitfield, J. Biol. Chem., 2012, 287, 35078–35091 CrossRef CAS.
- S. D. Liston, B. R. Clarke, L. K. Greenfield, M. R. Richards, T. L. Lowary and C. Whitfield, J. Biol. Chem., 2015, 290, 1075–1085 CrossRef CAS PubMed.
- L. K. Greenfield and C. Whitfield, Carbohydr. Res., 2012, 356, 12–24 CrossRef CAS PubMed.
- J. D. King, S. Berry, B. R. Clarke, R. J. Morris and C. Whitfield, Proc. Natl. Acad. Sci. U.S.A., 2014, 111, 6407–6412 CrossRef CAS PubMed.
- G. Hagelueken, H. Huang, B. R. Clarke, T. Lebl, C. Whitfield and J. H. Naismith, Mol. Micribiol., 2012, 86, 730–742 CrossRef CAS PubMed.
- A. Varki, R. D. Cummings, M. Aebi, N. H. Packer, P. H. Seeberger, J. D. Esko, P. Stanley, G. Hart, A. Darvill, T. Kinoshita, J. J. Prestegard, R. L. Schnaar, H. H. Freeze, J. D. Marth, C. R. Bertozzi, M. E. Etzler, M. Frank, J. F. G. Vliegenthart, T. Lütteke, S. Perez, E. Bolton, P. Rudd, J. Paulson, M. Kanehisa, P. Toukach, K. F. Aoki-Kinoshita, A. Dell, H. Narimatsu, W. York, N. Taniguchi and S. Kornfeld, Glycobiology, 2015, 25, 1323–1324 CrossRef CAS PubMed.
- C. Xu, B. Liu, B. Hu, Y. Han, L. Feng, J. S. Allingham, W. A. Szarek, L. Wang and I. Brockhausen, J. Bacteriol., 2011, 193, 449–459 CrossRef CAS PubMed.
- R. Woodward, W. Yi, L. Li, G. Zhao, H. Eguchi, P. R. Sridhar, H. Guo, J. K. Song, E. Motari, L. Cai, P. Kelleher, X. Liu, W. Han, W. Zhang, Y. Ding, M. Li and P. G. Wang, Nat. Chem. Biol., 2010, 6, 418–423 CrossRef CAS PubMed.
- B. R. Clarke, O. G. Ovchinnikova, R. P. Sweeney, E. R. Kamski-Hennekam, R. Gitalis, E. Mallette, S. D. Kelly, T. L. Lowary, M. S. Kimber and C. Whitfield, Nat. Chem. Biol., 2020, 16, 450–457 CrossRef CAS PubMed.
- C. Whitfield, D. M. Williams and S. D. Kelly, J. Biol. Chem., 2020, 295, 10593–10609 CrossRef CAS PubMed.
- Y. Bi, E. Mann, C. Whitfield and J. Zimmer, Nature, 2018, 553, 361–365 CrossRef CAS PubMed.
- R. P. Sweeney and T. L. Lowary, Curr. Opin. Chem. Biol., 2019, 53, 37–43 CrossRef CAS PubMed.
- A. Ishiwata and Y. Ito, J. Am. Chem. Soc., 2011, 133, 2275–2291 CrossRef CAS PubMed.
- T. L. Lowary, Curr. Opin. Chem. Biol., 2013, 17, 990–996 CrossRef CAS PubMed.
- Q. Zhu, Z. Shen, F. Chiodo, S. Nicolardi, A. Molinaro, A. Silipo and B. Yu, Nat. Commun., 2020, 11, 4142 CrossRef CAS PubMed.
- A. A. Joseph, A. Pardo-Vargas and P. H. Seeberger, J. Am. Chem. Soc., 2020, 142, 8561–8564 CrossRef CAS PubMed.
- S. Pasari, S. Manmode, G. Walke and S. Hotha, Chem. Eur. J., 2018, 24, 1128–1139 CrossRef CAS PubMed.
- Y. Wu, D.-C. Xiong, S.-C. Chen, Y.-S. Wang and X.-S. Ye, Nat. Commun., 2017, 8, 14851 CrossRef CAS PubMed.
- Z. Hu, A. F. Bongat White and L. A. Mulard, Chem.–Asian J., 2017, 12, 419–439 CrossRef CAS PubMed.
- J. Guo and X.-S. Ye, Molecules, 2010, 15, 7235–7265 CrossRef CAS PubMed.
- B. Ghosh and S. S. Kulkarni, Chem.–Asian J., 2020, 15, 450–462 CrossRef CAS PubMed.
- K. Bock and C. Pedersen, J. Chem. Soc., Perkin Trans., 1974, 2, 293–297 RSC.
- J. Kalikanda and Z. Li, Tetrahedron Lett., 2010, 51, 1550–1553 CrossRef CAS.
- W. A. Szarek, A. Zamojski, K. N. Tiwari and E. R. Ison, Tetrahedron Lett., 1986, 27, 3827–3830 CrossRef CAS.
- M. F. Semmelhack and G. E. Heinsohn, J. Am. Chem. Soc., 1972, 94, 5139–5140 CrossRef CAS.
- P. Nagorny, B. Fasching, X. Li, G. Chen, B. Aussedat and S. J. Danishefsky, J. Am. Chem. Soc., 2009, 131, 5792–5799 CrossRef CAS PubMed.
- P. Wang, J. Zhu, Y. Yuan and S. J. Danishefsky, J. Am. Chem. Soc., 2009, 131, 16669–16671 CrossRef CAS PubMed.
- L. M. Lira, D. Vasilev, R. A. Pilli and L. A. Wessjohann, Tetrahedron Lett., 2013, 54, 1690–1692 CrossRef CAS.
- F. Cramer, H. Schaller and H. A. Staab, Chem. Ber., 1961, 94, 1612–1621 CrossRef CAS.
- H. Schaller, H. A. Staab and F. Cramer, Chem. Ber., 1961, 94, 1621–1633 CrossRef CAS.
- Z. Xu, Bioorg. Med. Chem. Lett., 2015, 25, 3777–3783 CrossRef CAS PubMed.
- L. A. S. Parolis, H. parolis and G. G. S. Dutton, Carbohydr. Res., 1986, 155, 272 CrossRef CAS PubMed.
- E. Vinogradov, E. Frirdich, L. L. MacLean, M. B. Perry, B. O. Petersen, J. Ø. Duus and C. Whitfield, J. Biol. Chem., 2002, 277, 25070–25081 CrossRef CAS PubMed.
- X. Xue, R. B. Zheng, A. Koizumi, L. Han, J. S. Klassen and T. L. Lowary, Org. Biomol. Chem., 2018, 16, 1939–1957 RSC.
- M. Delbianco, A. Kononov, A. Poveda, Y. Yu, T. Diercks, J. Jiménez-Barbero and P. H. Seeberger, J. Am. Chem. Soc., 2018, 140, 5421–5426 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectroscopic data. See DOI: 10.1039/d1sc03852d |
| This journal is © The Royal Society of Chemistry 2021 |