
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox and catalase-like activities of four widely used carbon monoxide releasing molecules (CO-RMs)†
Zhengnan
Yuan
,
Xiaoxiao
Yang
 and
Binghe
Wang
*
and
Binghe
Wang
*
Department of Chemistry and Center for Diagnostics and Therapeutics, Georgia State University, Atlanta, Georgia 30303, USA. E-mail: wang@gsu.edu
First published on 13th September 2021
Abstract
The pathophysiological roles of the endogenous signaling molecule, carbon monoxide (CO), have been extensively studied and validated in cell culture and animal models. Further, evidence supporting the therapeutic effects of CO in various human diseases has been mounting over the last two decades. Along this line, there has been intensive interest in developing various delivery forms including CO gas, CO in solution, metal–carbonyl complexes widely known as CO-releasing molecules (CO-RMs), and organic CO prodrugs. Among them, two ruthenium-based carbonyl complexes, CORM-2 and -3, occupy a very special place because they have been used in over 500 published studies. One of the mechanisms for CO's actions is known to be through attenuation of oxidative stress and regulation of production of reactive oxygen species (ROS). For this reason, it is important that CO delivery forms do not have intrinsic chemical redox properties. Herein, we describe our findings of catalase-like activities of CORM-2 and -3 in a CO-independent fashion, leading to the rapid degradation of hydrogen peroxide (H2O2) in PBS buffer (pH = 7.4) and in cell culture media. Further, we have found that CORM-2 and CORM-3 possess potent radical scavenging abilities. We have also studied two other widely used CO donors: CORM-401 and CORM-A1. Both showed chemical reactivity with ROS, but to a lesser degree than CORM-2 and -3. Because of the central role of ROS in some of the proposed mechanisms of actions for CO biology, the discovery of intrinsic chemical redox properties for these CO-RMs means that additional attention in designing proper controls is needed in future biological experiments using these CO-RMs for their CO-donating functions. Further, much more work is needed to understand the true implications of the chemical reactivity of these CO-RMs in cell-culture and animal-model studies of CO biology.
Introduction
As a small endogenous signaling molecule, carbon monoxide (CO) is produced in all mammals mostly through heme oxygenase-mediated heme degradation.1,2 The commonly accepted physiological functions of CO include cytoprotection and effects on immune responses, mitochondrial functions, bioenergetics, and the circadian clock. The therapeutic effects from exogenous delivery of CO have also been extensively studied in various disease models such as cancer,3,4 colitis,5 organ injury,6–9 and systematic inflammation.10 Along with the work in examining the therapeutic effects of CO, there has been intense interest in developing various delivery forms of CO including CO in solution, metal–carbonyl complexes as CO-releasing molecules (CO-RMs), and organic CO prodrugs.11,12 Among all the delivery forms of CO,13–16 CORM-2,17 CORM-3,18 CORM-401 (manganese-based)19 and CORM-A1 (boron-based)20 are among the most widely used CO donors in a large number of reported studies (Fig. 1). A search of Pubmed in August 2021 yielded 538 entries using “CORM-2 or CORM-3,” 76 using CORM-A1, and 22 using CORM-401. CORM-2 and CORM-3 are ruthenium carbonyl complexes, which were initially found to quantitatively transfer CO to myoglobin in the presence of dithionites (Fig. 1).21 However, under near-physiological conditions, later studies suggested limited CO release from CORM-2 and -3.21,22 Instead, CO2 was found to be the major product due to the water–gas shift reaction, which is fundamentally a redox reaction.22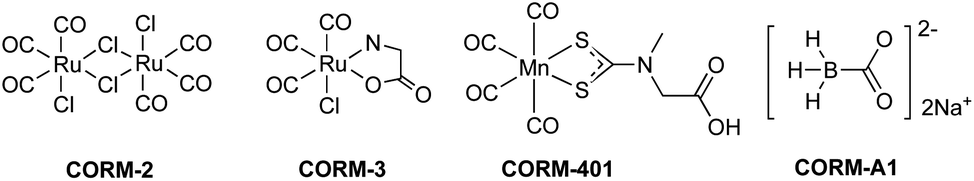 | ||
| Fig. 1 Chemical structures of representative CO-RMs: CORM-2, CORM-3, CORM-A1 and CORM-401. | ||
As for CO's molecular mechanism(s) of action in biology, it is widely accepted that redox signaling in general plays a critical role.23 In this context, CO has been reported to affect the production of reactive oxygen species (ROS) such as H2O2, the level of hypoxia-inducible factor-1a (HIF-1a), mitochondrial biogenesis, cellular bioenergetics, NADPH oxidase, and other redox-related events.12 For example, CO was reported to bind to cytochrome c oxidase, causing the inhibition of the mitochondrial respiratory chain to generate ROS.24,25 Studies also suggested the dependence of the anti-inflammatory effects of CO on ROS production in macrophages.26 In astrocytes, CO exposure was found to exert cytoprotective effects against ROS-induced apoptosis.27 At the organ level, there have been reports supporting one mechanism of CO as binding to the heme moiety on cytochromes P450 (CYP), which prevents CYP degradation from releasing free heme as a source of catalytic iron to generate oxidative stress.28 As surrogates of CO, CORM-2 and CORM-3 have been applied to examining CO's effects on ROS production and other redox-related processes in various cellular and animal models.29–31 Hydrogen peroxide (H2O2) is one of the major components of ROS in living organisms, and is primarily generated during cellular metabolism.32–34 The production of H2O2 can also be triggered by different stimuli and molecules, such as growth factors,35 cytokines36 and small drug molecules.37 Numerous studies have indicated the significant functions of H2O2 in cellular apoptosis and proliferation,38,39 inflammation,40 and cancer development41 among others. For cytoprotection studies, H2O2 has been widely used to induce oxidative stress in cell-culture models in CO-related studies. Overall, the central role of redox signaling has been widely accepted in CO research.
In our own work of studying CO's therapeutic roles using organic prodrugs,42 we were in need of using some of the most widely used CO-RMs as controls. In doing so, we have found catalase-like activities for CORM-2 and -3, leading to rapid degradation of H2O2, and the ability of these two CO-RMs to scavenge radicals. For comparison, the effects of CORM-401 (ref. 19) and CORM-A1 (ref. 20) have also been evaluated under near-physiological conditions. We have found that CORM-401 and CORM-A1 have the ability to react with H2O2 and to scavenge radicals as well. Given the central roles of redox signaling in CO's mechanism(s) of actions, our findings suggest the need to consider the redox properties of these CO-RMs in designing future biological experiments. It is imperative to consider the need for proper control experiments for deconvoluting the effects of these CO-RMs possibly arising from both their chemical reactivities and CO-donating ability. Below, we describe our findings and their implications.
Results and discussion
For examining the effect of these CO-RMs on the stability of peroxide, we incubated 5 mM of H2O2 with CORM-2 (20 μM) in PBS buffer (pH = 7.4) and monitored the concentration of H2O2 over time by using a literature titration method (see ESI† for details).43 About 90% of H2O2 decomposed within 8 min, suggesting a catalytic role of CORM-2 in H2O2 degradation because of the large excess of H2O2 relative to CORM-2 and a high turnover number of the reaction (Fig. 2). There have been reported methods of using UV by monitoring the absorbance at 230 nm and commercially available test strips for determining peroxide concentrations.44 We used both methods to confirm H2O2 degradation upon incubation with CORM-2. Results from all the methods showed rapid decomposition of H2O2 in the presence of CORM-2 (Fig. S1†). Under the same conditions, we used catalase (2–5 U) as a positive control and observed the reduction of H2O2 concentration to the same level after 9 min of incubation (Fig. 2). Ru(DMSO)4Cl2 (Ru complex A) has been reported to catalyze H2O2 disproportionation.45 Thus, Ru complex A was used as a secondary positive control (Fig. 2). We observed an initial induction period for ruthenium-based complexes (CORM-2 and ruthenium complex A), but not with catalase (Fig. 2). The results are consistent with a proposed mechanism from a previous report,45 indicating an initial conversion of the ruthenium(II) complexes into “activated” intermediates, which catalyze H2O2 decomposition (Fig. S2†). After the activation step, the initial decomposition rates were found to be about 3 × 10−5 M s−1, 9 × 10−6 M s−1 and 1 × 10−5 M s−1 for CORM-2 (turnover rate: 1.5 s−1), the ruthenium complex A and catalase, respectively. Along with H2O2 disproportionation, oxygen generation was also measured using a GC system (TCD detector, ESI†). Specifically, in a head space vial, 4 ml of H2O2 PBS solution (5 mM or 10 mM) was incubated with CORM-2 (20 μM) for one hour before GC detection. For a comparison group, 230 μl (∼0.01 mmol) or 450 μl (∼0.02 mmol) of pure oxygen was added into a headspace vial and incubated under the same conditions. GC analysis showed generation of similar amounts of oxygen between the H2O2–CORM-2 reaction (5 mM or 10 mM) and the corresponding oxygen-spiked groups (0.01 mmol or 0.02 mmol) (Table S1†). Such results are in agreement with the production of 1 molecule of O2 from two molecules of H2O2 through disproportionation. Together with the observations from H2O2 titration studies, the results further confirm the catalytic activity of CORM-2 in H2O2 disproportionation.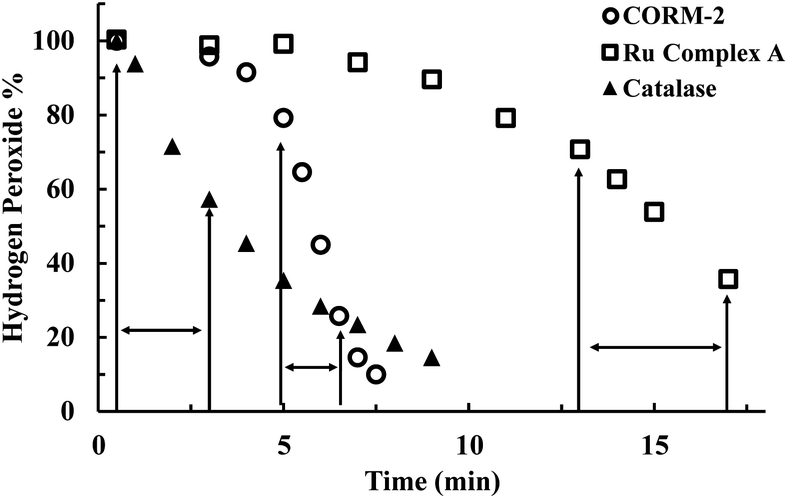 | ||
| Fig. 2 Time-dependent changes of H2O2 concentration (expressed as a percentage of the initial concentration: 5 mM) in the presence of CORM-2 (20 μM), Ru complex A (Ru(DMSO)4Cl2, 20 μM) and catalase (2–5 units per ml) in PBS (pH = 7.4, 0.01 M) at room temperature. The concentration of H2O2 was monitored by using a titration method.43 (The arrows label the period used to compare the initial rates among experiments using different catalysts). | ||
To further evaluate the reaction kinetics, we also studied H2O2 decomposition in the presence of different concentrations of CORM-2. Fig. 3A shows the dose-dependent effects of CORM-2 on H2O2 concentration. Considering the commonly used H2O2 concentrations in cytoprotection studies, CORM-2 was also incubated with lower concentrations (0.4 to 0.8 mM) of H2O2 at 37 °C. In the presence of 20 μM of CORM-2, H2O2 solutions at 0.4 mM, 0.6 mM and 0.8 mM were almost completely consumed after 20 to 45 min of incubation (Fig. S3†). By decreasing the concentration of H2O2 from 0.8 to 0.4 mM, the initial induction period was prolonged by around 2-fold (Fig. S3†). In earlier mechanistic studies of the catalase-like activity of Ru(II) complexes, Bienvenüe suggested the first step to be H2O2-mediated oxidation to generate a ruthenium–oxo complex as the “activated” species for catalysis (Fig. S2†).45 This proposed mechanism is consistent with the prolonged induction period associated with the lowering of the initial H2O2 concentration.
 | ||
| Fig. 3 CORM-2 catalyzes H2O2 degradation. The concentration of H2O2 was monitored by using a titration method.43 (A) Decrease of the concentration of H2O2 (expressed as a percentage of the initial concentration: 5 mM) over time upon incubation with various concentrations of CORM-2 in PBS (pH = 7.4, 0.01 M) at room temperature; (B) reinjection of H2O2 during the CORM-2 (20 μM)-induced H2O2 degradation in PBS (pH = 7.4, 0.01 M) at room temperature (n = 3). | ||
As additional efforts to probe the proposed induction period, H2O2 was reinjected at two time points after the consumption of H2O2 in solution in the initial preparation (Fig. 3B). After both reinjections, the concentration of H2O2 immediately started to drop without an induction period, indicating the presence of the “catalytic form” before each reinjection. This observation is also consistent with findings using other ruthenium complexes,45 suggesting the general applicability of the previously proposed mechanism in explaining the catalase-like activity of CORM-2.
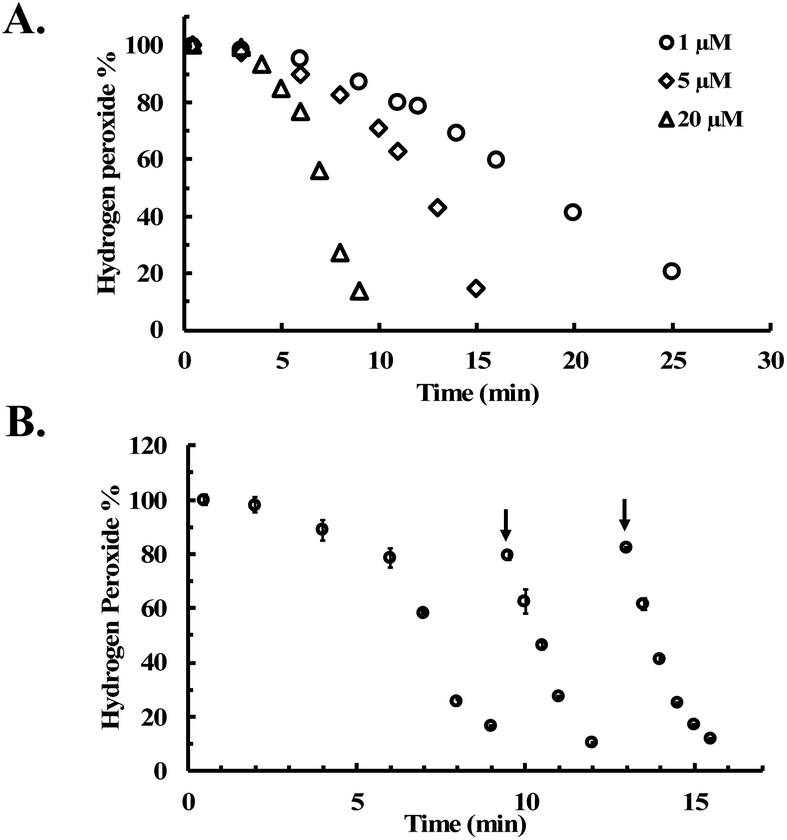
CORM-2 is insoluble in water. As a result, DMSO is widely used as a solvent to prepare stock solutions of CORM-2. However, CORM-2 is known to undergo ligand exchange with DMSO replacing the CO ligand, leading to a mixture of ruthenium complexes.22 Because of this, DMSO stock solutions of CORM-2 are often freshly prepared. The commonly used negative control compound is what is referred to as inactive CORM or iCORM-2, prepared by treatment of CORM-2 with DMSO for ≥18 h.30 The resulting mixture is unable to transfer CO to myoglobin. In this case, we also evaluated the activity of CORM-2 (stock solution freshly prepared with DMSO) and iCORM-2 for the ability to degrade H2O2. Incubation with both species (20 μM) led to the consumption of 5 mM H2O2 within 8 min, suggesting the ability of iCORM-2 to catalyze H2O2 degradation (Fig. S4†). CORM-3 is another ruthenium-based CO-RM. Due to its good water solubility, CORM-3 has been widely used as a CO surrogate in cell-culture and animal-model studies. Therefore, we also evaluated the catalytic activity of CORM-3. In the presence of CORM-3 (5–20 μM), 5 mM of H2O2 was almost completely consumed within 10 min of incubation in PBS at 37 °C (Fig. 4), suggesting similar catalase-like activity for CORM-3. Additionally, concentration-dependent effect of H2O2 on the initial lag period was also observed with CORM-3. Increasing the initial concentration of H2O2 from 0.8 mM to 8 mM significantly shortened the induction period (Fig. S5†), suggesting a similar mechanistic pathway involving H2O2-mediated oxidation of the ruthenium core as the initial step, leading to the formation of a catalytically active form. Along the same line, the catalytic activity of iCORM-3 was also assessed. In the presence of 5 μM and 20 μM of iCORM-3, 5 mM H2O2 was completely consumed within 6 min and 15 min, respectively (Fig. S6†). The difference in reactivity among CORM-2, CORM-3, and their iCORMs is likely due to the effect of coordination chemistry.22,46 As a control, we also incubated an organic CO prodrug (CO-111)47 and its release byproduct (CP-111) with H2O2 under the same conditions, and we did not observe similar catalase-like activity to CORM-2 and CORM-3 (Fig. S7†).
 | ||
| Fig. 4 Time-dependent change in H2O2 concentration (expressed as a percentage of the initial concentration: 5 mM) upon incubation with various concentrations of CORM-3 in PBS (pH = 7.4, 0.01 M) at 37 °C. The concentration of H2O2 was monitored by using a titration method.43 | ||
Besides H2O2, free radical species are also important ROS in affecting cellular signaling and metabolism. For this reason, we were interested in examining whether CORM-2 and -3 possess radical scavenging abilities. ABTS (2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) decolorization assay was used for the assessment.48,49 Stock solutions of ABTS radical were prepared according to literature procedures (see ESI† for details).49,50 Upon incubation with ruthenium-based CO-RMs, the absorbance at 734 nm was monitored to indicate the concentration of a stable ABTS radical. CORM-2 (2 to 6 μM) caused significant decreases of ABTS radical concentrations in a dose-dependent manner (Fig. 5A). At 6 μM, CORM-2 caused an almost complete consumption of the ABTS radical in solution after 6 min of incubation (Fig. 5A). As a reference antioxidant, Trolox was used as a positive control for the ABTS assay. A linear relationship was established between Trolox concentration and the rate of decrease in UV absorption at 734 nm (Fig. S8†). According to this standard curve, 2 and 4 μM of CORM-2 possess equivalent radical scavenging ability to 4 and 12 μM of Trolox, respectively. Such results suggest a strong radical scavenging capacity of CORM-2. We then wondered if iCORM-2 also exhibits similar activity. For this, various concentrations of iCORM-2 were incubated with ABTS radical solution under the same conditions as for CORM-2. A dose-dependent scavenging effect from iCORM-2 was found within the concentration range of 2 to 10 μM (Fig. S9A†). However, the radical scavenging ability of iCORM-2 seems to be weaker than that of CORM-2. Specifically, 6 and 10 μM of iCORM-2 showed similar radical scavenging ability to that of 2 and 7 μM of Trolox, respectively, against the ABTS radical. Considering the potent scavenging effect of CORM-2 against the ABTS radical, we took a further step to see if such activity is catalytic. As shown in Fig. 5B, after the initial consumption of 50 μM ABTS radical by 5 μM CORM-2, the ABTS radical was sequentially reinjected at two time points. A similar fast degradation of the ABTS radical was observed after both reinjections, showing the catalytic activity of CORM-2. Trolox is known to be a non-catalytic reductant, and each molecule can donate two electrons to reduce the ABTS radical in ethanol. As a result, incubation with 15 μM Trolox led to a quick drop of ABTS radical concentration by 30 μM (Fig. S9B†). After the consumption of 2 equivalents of ABTS, the ability of Trolox to scavenge additional portions of the ABTS radical was lost (Fig. S9B†). Such results suggest the stoichiometric nature of the reaction between Trolox and the ABTS radical. Under the same conditions, reinjection studies also showed the catalytic activity of iCORM-2 to degrade the ABTS radical (Fig. S9C†). In terms of a possible mechanism, Tennyson suggested the involvement of alcohols (solvent) as a reducing agent in the catalytic reduction of the ABTS radical by a ruthenium complex.51 To probe if CORM-2 follows a similar mechanistic pathway, we performed the ABTS-radical scavenging experiments with CORM-2 in acetate buffer containing various ratios (20%, 50% and 80%) of ethanol. It was found that lowering the ratio of ethanol also decreased the ABTS degradation rate, indicating the participation of ethanol in scavenging the ABTS radical (Fig. S10†). Along the same line, CORM-3 and iCORM-3 were also assessed for their catalytic activity to scavenge the ABTS radical. Incubation with both species resulted in a substantial decrease in ABTS concentrations (Fig. S11†). Interestingly, reinjection studies suggest iCORM-3 to be more potent in catalyzing the degradation of the ABTS radical than CORM-3 (Fig. S11†). Taken together, these results indicate the catalytic radical scavenging ability in vitro for both the ruthenium-based CO-RMs and their corresponding iCORMs.
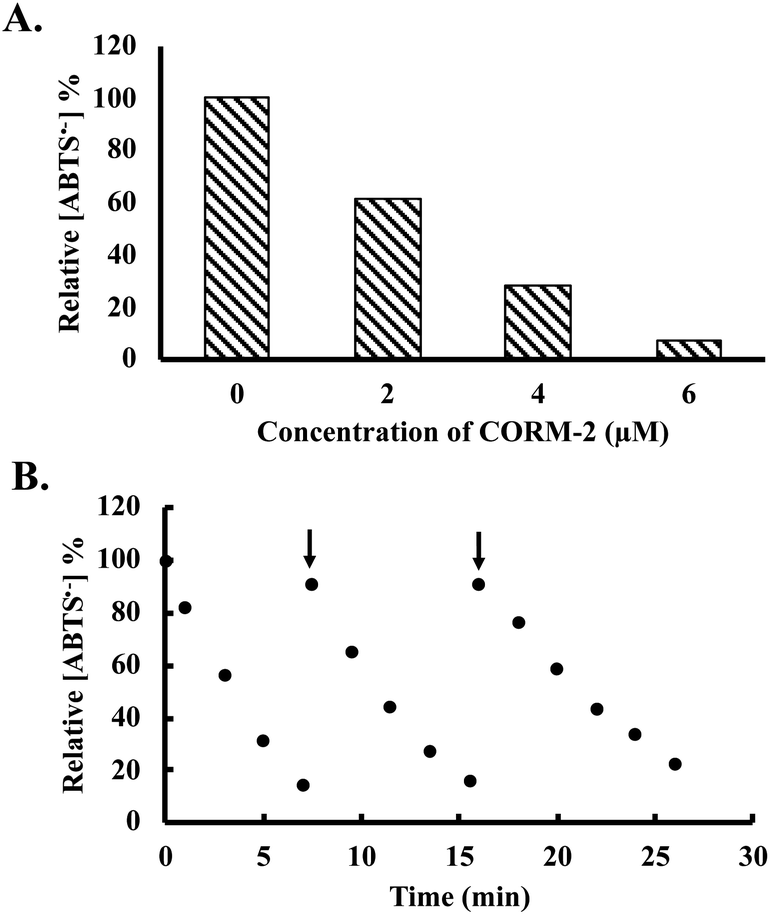 | ||
| Fig. 5 Decrease of ABTS radical concentration (expressed as a percentage of the initial concentration (50 μM), determined using absorbance measured at 734 nm (ref. 49)) upon incubation with (A) 0, 2, 4 and 6 μM of CORM-2 for 6 min; (B) reinjection of the ABTS radical after the initiation of CORM-2 (5 μM)-induced consumption of the ABTS radical in ethanol containing 20% acetate buffer (20 mM, pH 4.5, v/v). | ||
For comparison with ruthenium-based CO-RMs, we further conducted evaluation of a manganese (Mn)- and a boron (B)-based CO-RM (CORM-401 and CORM-A1, Fig. 1). We found that both were able to reduce H2O2 concentrations in PBS (Fig. 6 and S12†). For example, in the presence of 40 to 100 μM of CORM-401, the concentration of H2O2 decreased by 10% to 30% after 1 h of incubation (initial concentration: 300 μM; Fig. 6). Such results are more in line with stoichiometric reactions than for CORM-401 to be catalytic. However, because we did not study the end species of this complex mixture, we cannot say for sure whether the reaction is catalytic or stoichiometric. Further, prolonged incubation (6 h and 16 h) led to a higher degree of decomposition. After 16 h of incubation, 100 μM of CORM-401 further decreased the H2O2 concentration by another 15% as compared to the data point at 1 h, suggesting the possibility of a more complex reaction than a simple 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric reaction (Fig. 6). As compared to CORM-401, CORM-A1 showed a weaker ability to decompose H2O2. Incubation with 40 to 100 μM of CORM-A1 caused a decrease of H2O2 (300 μM) in PBS by 6% to 20% after 1–16 h (Fig. S12†). Such numbers are less than 1:1 stoichiometry and are consistent with CORM-A1 being a reagent capable of reacting with H2O2, instead of being a catalyst.
:1 stoichiometric reaction (Fig. 6). As compared to CORM-401, CORM-A1 showed a weaker ability to decompose H2O2. Incubation with 40 to 100 μM of CORM-A1 caused a decrease of H2O2 (300 μM) in PBS by 6% to 20% after 1–16 h (Fig. S12†). Such numbers are less than 1:1 stoichiometry and are consistent with CORM-A1 being a reagent capable of reacting with H2O2, instead of being a catalyst.
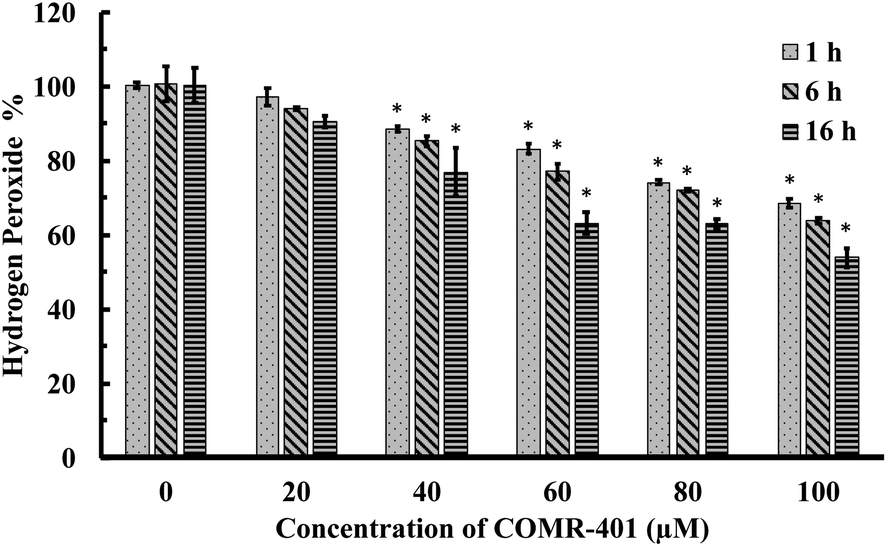 | ||
| Fig. 6 Time-dependent decrease of H2O2 concentration (expressed as a percentage of the initial concentration: 300 μM) upon incubations with CORM-401 in PBS (pH = 7.4, 0.01 M) at 37 °C. The concentration of H2O2 was monitored by using a titration method.43 Values are means ± SD, n = 3. Data with “*” indicates statistically significant differences between CORM-401 samples and the negative control as determined by the t-test. *P < 0.01 versus the control group. | ||
Next, we also assessed the radical scavenging abilities of CORM-A1 and CORM-401 by using the ABTS decolorization assay. In comparison to CORM-2 and -3, CORM-A1 showed weaker reactivity with the ABTS radical at the same concentration (Fig. S13A†). Specifically, 200 μM of CORM-A1 showed equivalent radical scavenging ability to that of 4 μM of Trolox. In contrast, a comparable scavenging ability of 10 μM CORM-401 was seen when compared with that of Trolox at the same concentration (Fig. S13B†). Further, reinjection experiments (ABTS radical) were performed upon incubation with CORM-A1 (100 μM) and CORM-401 (10 μM). Results suggest a non-catalytic nature of the ability of CORM-A1 and -401 to scavenge the ABTS radical with a 20:1 (CORM-A1:ABTS radical) and a 1:3 (CORM-401:ABTS radical) stoichiometry, respectively (Fig. S13C and D†). Additionally, another stable free radical, DPPH (1,1-diphenyl-2-picrylhydrazyl),52 was used to examine the antioxidative activity of CORM-401 and CORM-A1. Decolorization of DPPH was observed upon incubation with CORM-401 (12.5 to 50 μM) and CORM-A1 (25 to 100 μM) for 1 h (Fig. 7A and S14†). Under the same conditions, Trolox was also incubated with DPPH as a positive control (Fig. 7B). Such results further confirm the radical scavenging ability of CORM-401 and -A1.
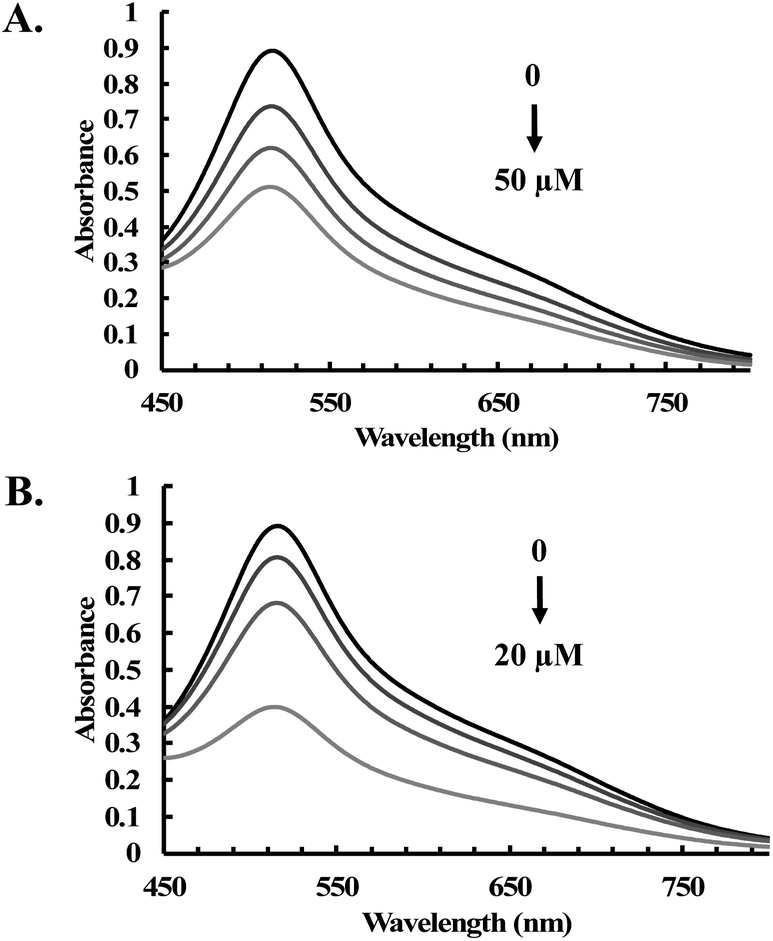 | ||
| Fig. 7 UV-vis spectra of DPPH (200 μM) upon treatment with various concentrations of (A) CORM-401 (12.5, 25 and 50 μM); and (B) Trolox (0, 5, 10 and 20 μM) in methanol for 1 h at room temperature. | ||
The observed reactivity of CORM-401 and CORM-A1 with H2O2 and the free radical is not entirely unexpected. The propensity for Mn(II) to be oxidized and thus serve as a reducing agent has been widely known;53,54 and CORM-401 is a Mn(II) complex. Further, certain Mn(II) complexes have been reported to possess superoxide dismutase-like properties.55 CORM-A1, on the other hand, is a complex of BH3, which is a textbook reducing agent in organic chemistry, capable of reacting with peroxide.
Considering the widespread application of CORM-2 and -3 in studying CO biology, we conducted additional experiments to examine their reactions with H2O2 in biological milieus. In doing so, we incubated 100 μM of H2O2 with different concentrations of CORM-2 or CORM-3 in the cell culture medium (DMEM with or without 10% FBS) for 1 h. Then, concentrations of H2O2 in the culture medium were determined by a widely used H2O2 probe, namely, PF-1 (Fig. S15†).56 Both CORM-2 and CORM-3 induced significant decreases in H2O2 concentration in cell culture medium as reflected by the fluorescence intensity from PF-1 (Fig. 8). Specifically, upon addition of 100 μM of CORM-2 or CORM-3, H2O2 concentration decreased by about 90% and 80% after 1 h of incubation from an initial concentration of 100 μM. iCORM-2 and iCORM-3 were also incubated with H2O2 under the same conditions. Results from iCORM-2 and iCORM-3 treated groups indicate their ability to cause H2O2 degradation as well (Fig. S16†). Interestingly, iCORM-2 seems to be more potent (compared with CORM-2) in consuming H2O2 in cell culture medium. In the presence of 50 μM of iCORM-2, H2O2 (100 μM) was found to be undetectable after 1 h incubation (Fig. S16A†). Together with the H2O2 decomposition study in PBS buffer, these results strongly support the notion that ruthenium-based CORM-2 and -3 and iCORM-2 and -3 are potent antioxidants in vitro. It should be noted that the H2O2 concentration in the cell culture assay was lower than that in PBS. Such concentration difference affects both induction period and reaction time profiles.
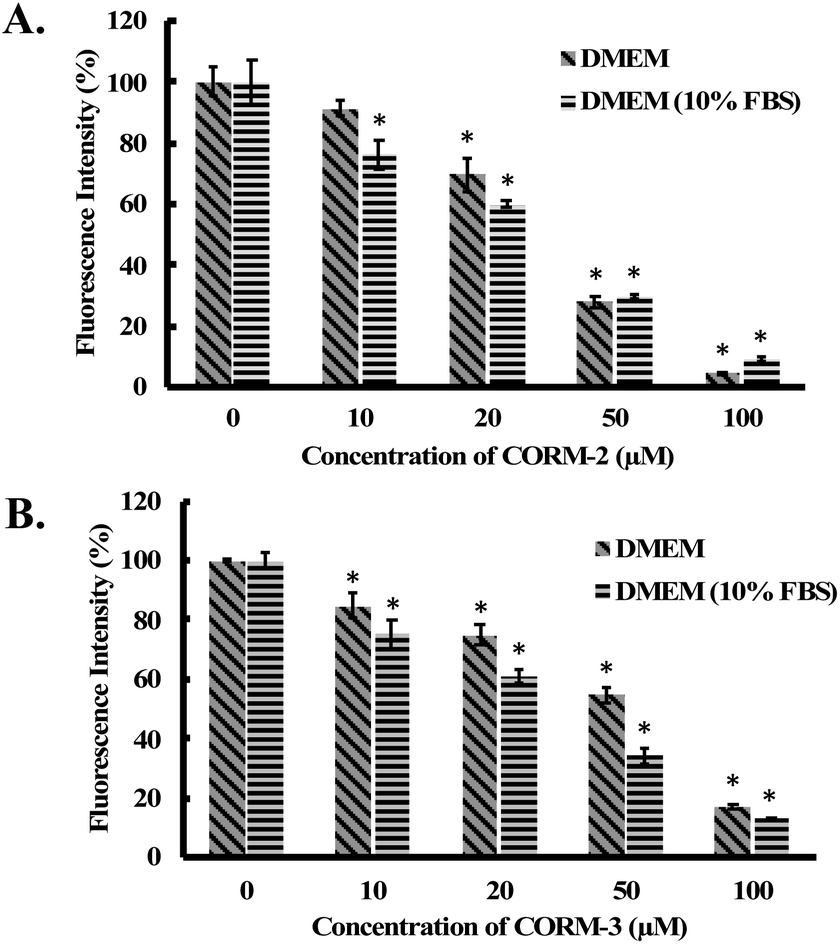 | ||
| Fig. 8 Fluorescence detection of H2O2 in cell culture medium by PF-1.56 100 μM of H2O2 solutions were incubated with (A) CORM-2 and (B) CORM-3 in DMEM or DMEM (10% FBS) for 1 h at 37 °C. Then H2O2 concentration was measured using PF-1 (ref. 56) (see ESI† for details). The concentrations of H2O2 are expressed as a percentage of the fluorescence intensity from the vehicle group. Values are means ± SD, n = 3. Data with “*” indicate statistically significant differences between CORM-2 and -3 samples and the respective negative control as determined by the t-test. *P < 0.01 versus the control group. | ||
Conclusions
Because of the critical roles of redox signaling in the mechanism(s) of action of carbon monoxide as an endogenous signaling molecule and the telltale signs of the redox properties of some metal-based CO-RMs, we undertook this extensive study to examine their chemical reactivities, especially in the context of the stability of ROS. Such information will be critically important for assessing the CO-dependent biological functions when such CO-RMs are used as CO donors. In this present work, we report for the first time the catalase-like activity of ruthenium-based CO-RMs, CORM-2 and CORM-3, leading to the rapid degradation of H2O2 under near-physiological conditions. Further, we have also observed the potent ability of CORM-2 and -3 to catalytically scavenge free radicals when ABTS was used as an example and Trolox was used as a positive control. It is important to note that the corresponding iCORMs also showed antioxidant activity, but to varying degrees. As a comparison, another two widely used CO-RMs, CORM-A1 (boron-based) and CORM-401 (manganese-based), were also examined. It was found that both CORM-401 and CORM-A1 were reactive toward H2O2 and ABTS, however, most likely in a non-catalytic manner. These results are summarized in Table 1.| CO-RM species | Roles in H2O2 degradation | Roles in ATBS radical degradation |
|---|---|---|
|
a Stoichiometric ratio of CORM:ROS.
|
||
| CORM-2 | Catalytic | Catalytic |
| CORM-3 | Catalytic | Catalytic |
| iCORM-2 | Catalytic | Catalytic |
| iCORM-3 | Catalytic | Catalytic |
| CORM-A1 | Non-catalytic (3:1)a |
Non-catalytic (20:1)a |
| CORM-401 | Non-catalytic (1:1)a |
Non-catalytic (1:3)a |
In the context of the chemical reactivity of ruthenium-based CO-RMs, it is important to discuss related reports to put our studies in a proper context. Several studies suggested that some previously reported biological effects from CORM-2 and -3 cannot be attributed to their CO-donating effects. For example, the antimicrobial effects from CORM-2 and CORM-3 are likely from the accumulation of ruthenium species in cells and their chemical reactivity toward functional groups on proteins.21,57 CORM-3 was found to directly interact with proteins including lysozyme, fibronectin and p62.58–60 CORM-2 was reported to form Ru adducts with histidine residues on proteins to modulate the activities of potassium ion channels,61 snake venoms54 and bee venom phospholipase.62 In aqueous solutions, cysteine and glutathione can bind to CORM-3 with low micromolar Kd.21 Recently, we reported the ability of CORM-2 and -3 to directly consume GSSG and nitrites in PBS buffer.63 CORM-2 and -3 also possess the ability to reduce arylnitro and N-oxide compounds.64
As to the implications of the varying antioxidant ability of these CO-RMs and iCO-RMs and their chemical reactivity with proteins and other molecules in the context of using them as CO donors in studying CO-dependent biological effects, one can only assess specifically in each biological experiment. Further, experiments described in this study were all conducted in solution only. Therefore, there is an information gap among chemical behaviors in solution, in cell culture, and in animal models, which we anticipate a higher level of complexes both in terms of chemical reactions and biological consequences. As a result, we do not feel comfortable drawing general conclusions on the biological implications of our findings. Neither do we have sufficient information to assess the implications of these chemical reactions of CORM-2, CORM-3, CORM-401, and CORM-A1 in the interpretation of the results from various studies in the literature. We leave these issues to experts in the respective fields to assess. Further, there is much more to do to truly understand how the chemical reactivity of these CO-RMs affects (or not) the studies of CO-dependent effects when they are used as CO donors. However, one thing is clear: there is a convoluted picture of the interlays among the CO-donating ability, the anti-oxidation effects, and the chemical reactivities with proteins and other molecules when these CO-RMs are used in studying the intrinsic effects of CO in biology. In future studies, carefully designed control experiments are needed for the deconvolution of a potentially very complex picture when CORM-2 CORM-3, CORM-401, or CORM-A1 is used as a CO surrogate for studying the intrinsic effects of CO in vitro and in vivo.
Data availability
Supporting data for this article have been uploaded as part of the ESI material.†Author contributions
ZY and XY designed and conducted all the experiments, and analyzed the data. ZY drafted the manuscript. BW provided overall guidance throughout the process including experimental design, data analysis, and manuscript preparation. BW also revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the National Institutes of Health (R01DK119202), the Georgia Research Alliance Eminent Scholar fund, and internal resources at Georgia State University.Notes and references
- L. Wu and R. Wang, Pharmacol. Rev., 2005, 57, 585–630 CrossRef CAS PubMed
.
- R. F. Coburn and N. Eng, J. Med., 1970, 282, 207–209 CAS
- L. Shao, Y. Y. Gu, C. H. Jiang, C. Y. Liu, L. P. Lv, J. N. Liu and Y. Zou, Eur. Rev. Med. Pharmacol. Sci., 2018, 22, 1948–1957 CAS
- C. Lv, Q. Su, J. Fang and H. Yin, Biochem. Biophys. Res. Commun., 2019, 520, 320–326 CrossRef CAS PubMed
- C. Steiger, K. Uchiyama, T. Takagi, K. Mizushima, Y. Higashimura, M. Gutmann, C. Hermann, S. Botov, H.-G. Schmalz, Y. Naito and L. Meinel, J. Control. Release, 2016, 239, 128–136 CrossRef CAS PubMed
- X. Yang, M. de Caestecker, L. E. Otterbein and B. Wang, Med. Res. Rev., 2019, 40, 1147–1177 CrossRef PubMed
- Y. E. Yoon, K. S. Lee, Y. J. Lee, H. H. Lee and W. K. Han, Transplant. Proc., 2017, 49, 1175–1182 CrossRef CAS PubMed
- D. Bakalarz, M. Surmiak, X. Yang, D. Wójcik, E. Korbut, Z. Śliwowski, G. Ginter, G. Buszewicz, T. Brzozowski, J. Cieszkowski, U. Głowacka, K. Magierowska, Z. Pan, B. Wang and M. Magierowski, Acta Pharm. Sin. B, 2020, 11, 456–475 CrossRef PubMed
- Y. Tayem, T. R. Johnson, B. E. Mann, C. J. Green and R. Motterlini, Am. J. Physiol. Ren. Physiol., 2006, 290, F789–F794 CrossRef CAS PubMed
- X. Ji, Z. Pan, C. Li, T. Kang, L. K. C. De La Cruz, L. Yang, Z. Yuan, B. Ke and B. Wang, J. Med. Chem., 2019, 62, 3163–3168 CrossRef CAS PubMed
- C. C. Romão, W. A. Blättler, J. D. Seixas and G. J. Bernardes, Chem. Soc. Rev., 2012, 41, 3571–3583 RSC
- R. Motterlini and L. E. Otterbein, Nat. Rev. Drug Discovery, 2010, 9, 728–743 CrossRef CAS PubMed
- X. Ji, C. Zhou, K. Ji, R. E. Aghoghovbia, Z. Pan, V. Chittavong, B. Ke and B. Wang, Angew. Chem., Int. Ed., 2016, 55, 15846–15851 CrossRef CAS PubMed
- M. Popova, T. Soboleva, S. Ayad, A. D. Benninghoff and L. M. Berreau, J. Am. Chem. Soc., 2018, 140, 9721–9729 CrossRef CAS PubMed
- P. Peng, C. Wang, Z. Shi, V. K. Johns, L. Ma, J. Oyer, A. Copik, R. Igarashi and Y. Liao, Org. Biomol. Chem., 2013, 11, 6671–6674 RSC
- X. Ji and B. Wang, Acc. Chem. Res., 2018, 51, 1377–1385 CrossRef CAS PubMed
- R. Motterlini, E. Clark James, R. Foresti, P. Sarathchandra, E. Mann Brian and J. Green Colin, Circ. Res., 2002, 90, e17–e24 CrossRef CAS PubMed
- E. Clark James, P. Naughton, S. Shurey, J. Green Colin, R. Johnson Tony, E. Mann Brian, R. Foresti and R. Motterlini, Circ. Res., 2003, 93, e2–e8 CAS
- S. H. Crook, B. E. Mann, A. J. H. M. Meijer, H. Adams, P. Sawle, D. Scapens and R. Motterlini, Dalton Trans., 2011, 40, 4230–4235 RSC
- R. Motterlini, P. Sawle, S. Bains, J. Hammad, R. Alberto, R. Foresti and C. J. Green, FASEB J., 2005, 19, 1–24 CrossRef PubMed
- H. M. Southam, T. W. Smith, R. L. Lyon, C. Liao, C. R. Trevitt, L. A. Middlemiss, F. L. Cox, J. A. Chapman, S. F. El-Khamisy, M. Hippler, M. P. Williamson, P. J. F. Henderson and R. K. Poole, Redox Biol., 2018, 18, 114–123 CrossRef CAS PubMed
- J. D. Seixas, M. F. A. Santos, A. Mukhopadhyay, A. C. Coelho, P. M. Reis, L. F. Veiros, A. R. Marques, N. Penacho, A. M. L. Gonçalves, M. J. Romão, G. J. L. Bernardes, T. Santos-Silva and C. C. Romão, Dalton Trans., 2015, 44, 5058–5075 RSC
- C. A. Piantadosi, Free Radic. Biol. Med., 2008, 45, 562–569 CrossRef CAS PubMed
- Q. H. Gibson and C. Greenwood, Biochem. J., 1963, 86, 541–554 CrossRef CAS PubMed
- Ò. Miró, J. Casademont, A. Barrientos, Á. Urbano-Márquez and F. Cardellach, Pharmacol. Toxicol., 1998, 82, 199–202 CrossRef PubMed
- B. S. Zuckerbraun, B. Y. Chin, M. Bilban, J. de Costa d'Avila, J. Rao, T. R. Billiar and L. E. Otterbein, FASEB J., 2007, 21, 1099–1106 CrossRef CAS PubMed
- A. S. Almeida, C. S. F. Queiroga, M. F. Q. Sousa, P. M. Alves and H. L. A. Vieira, J. Biol. Chem., 2012, 287, 10761–10770 CrossRef CAS PubMed
- A. Nakao, G. Faleo, H. Shimizu, K. Nakahira, J. Kohmoto, R. Sugimoto, A. M. K. Choi, K. R. McCurry, T. Takahashi and N. Murase, Kidney Int., 2008, 74, 1009–1016 CrossRef CAS PubMed
- M. Juszczak, M. Kluska, D. Wysokiński and K. Woźniak, Sci. Rep., 2020, 10, 12200 CrossRef CAS PubMed
- D. Babu, G. Leclercq, R. Motterlini and R. Lefebvre, Front. Pharmacol., 2017, 8, 31 Search PubMed
- E.-Y. Choi, S.-H. Choe, J.-Y. Hyeon, J.-I. Choi, I. S. Choi and S.-J. Kim, Eur. J. Pharmacol., 2015, 764, 22–29 CrossRef CAS PubMed
- E. A. Veal, A. M. Day and B. A. Morgan, Mol. Cell, 2007, 26, 1–14 CrossRef CAS PubMed
- S. Neill, R. Desikan and J. Hancock, Curr. Opin. Plant Biol., 2002, 5, 388–395 CrossRef CAS PubMed
- B. Halliwell, M. V. Clement and L. H. Long, FEBS Lett., 2000, 486, 10–13 CrossRef CAS PubMed
- Y. S. Bae, S. W. Kang, M. S. Seo, I. C. Baines, E. Tekle, P. B. Chock and S. G. Rhee, J. Biol. Chem., 1997, 272, 217–221 CrossRef CAS
- T. Böhler, J. Waiser, H. Hepburn, J. Gaedeke, C. Lehmann, P. Hambach, K. Budde and H.-H. Neumayer, Cytokine, 2000, 12, 986–991 CrossRef PubMed
- H. Mizutani, S. Tada-Oikawa, Y. Hiraku, M. Kojima and S. Kawanishi, Life Sci., 2005, 76, 1439–1453 CrossRef CAS PubMed
- M. B. Hampton and S. Orrenius, FEBS Lett., 1997, 414, 552–556 CrossRef CAS PubMed
- S. Sigaud, P. Evelson and B. González-Flecha, Antioxid. Redox Signal., 2004, 7, 6–13 CrossRef PubMed
- Z. Ungvari, Z. Orosz, N. Labinskyy, A. Rivera, Z. Xiangmin, K. Smith and A. Csiszar, Am. J. Physiol.: Heart Circ. Physiol., 2007, 293, H37–H47 CrossRef CAS PubMed
- M. López-Lázaro, Cancer Lett., 2007, 252, 1–8 CrossRef PubMed
- X. Yang, W. Lu, C. P. Hopper, B. Ke and B. Wang, Acta Pharm. Sin. B., 2021, 11, 1434–1445 CrossRef PubMed
- C. D. Fernando and P. Soysa, MethodsX, 2015, 2, 283–291 CrossRef PubMed
- R. J. Ruch, S.-j. Cheng and J. E. Klaunig, Carcinogenesis, 1989, 10, 1003–1008 CrossRef CAS PubMed
- S. Choua, P. Pacheco, C. Coquelet and E. Bienvenüe, J. Inorg. Biochem., 1997, 65, 79–85 CrossRef CAS
- T. R. Johnson, B. E. Mann, I. P. Teasdale, H. Adams, R. Foresti, C. J. Green and R. Motterlini, Dalton Trans., 2007, 1500–1508, 10.1039/B613629J
- Z. Pan, V. Chittavong, W. Li, J. Zhang, K. Ji, M. Zhu, X. Ji and B. Wang, Chem.–Eur. J., 2017, 23, 9838–9845 CrossRef CAS PubMed
- R. L. Prior, X. Wu and K. Schaich, J. Agric. Food Chem., 2005, 53, 4290–4302 CrossRef CAS PubMed
- R. Re, N. Pellegrini, A. Proteggente, A. Pannala, M. Yang and C. Rice-Evans, Free Radic. Biol. Med., 1999, 26, 1231–1237 CrossRef CAS PubMed
- M. Ozgen, R. N. Reese, A. Z. Tulio, J. C. Scheerens and A. R. Miller, J. Agric. Food Chem., 2006, 54, 1151–1157 CrossRef CAS PubMed
- Y. Htet and A. G. Tennyson, Chem. Sci., 2016, 7, 4052–4058 RSC
- W. Brand-Williams, M. E. Cuvelier and C. Berset, LWT - Food Sci. Technol., 1995, 28, 25–30 CrossRef CAS
- M. Sjödin, J. Gätjens, L. C. Tabares, P. Thuéry, V. L. Pecoraro and S. Un, Inorg. Chem., 2008, 47, 2897–2908 CrossRef PubMed
- M. M. Morrison and D. T. Sawyer, Inorg. Chem., 1978, 17, 333–337 CrossRef CAS
- O. Iranzo, Bioorg. Chem., 2011, 39, 73–87 CrossRef CAS PubMed
- M. C. Y. Chang, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2004, 126, 15392–15393 CrossRef CAS PubMed
- H. M. Southam, M. P. Williamson, J. A. Chapman, R. L. Lyon, C. R. Trevitt, P. J. F. Henderson and R. K. Poole, Antioxidants, 2021, 10, 915 CrossRef CAS PubMed
- T. Santos-Silva, A. Mukhopadhyay, J. D. Seixas, G. J. Bernardes, C. C. Romão and M. J. Romão, J. Am. Chem. Soc., 2011, 133, 1192–1195 CrossRef CAS PubMed
- T. Aki, K. Unuma, K. Noritake, N. Hirayama, T. Funakoshi and K. Uemura, PloS One, 2019, 14, e0210474 CrossRef CAS PubMed
- T. Aki, K. Unuma, K. Noritake, H. Kurahashi, T. Funakoshi and K. Uemura, Toxicol. in Vitro, 2018, 50, 201–209 CrossRef CAS PubMed
- G. Gessner, N. Sahoo, S. M. Swain, G. Hirth, R. Schönherr, R. Mede, M. Westerhausen, H. H. Brewitz, P. Heimer, D. Imhof, T. Hoshi and S. H. Heinemann, Eur. J. Pharmacol., 2017, 815, 33–41 CrossRef CAS PubMed
- V. G. Nielsen, J. Thromb. Thrombolysis, 2020, 49, 100–107 CrossRef CAS PubMed
- Z. Yuan, X. Yang, Y. Ye, R. Tripathi and B. Wang, Anal. Chem., 2021, 93, 5317–5326 CrossRef CAS PubMed
- Z. Yuan, X. Yang, L. K. C. De La Cruz and B. Wang, Chem. Commun., 2020, 56, 2190–2193 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03832j |
| This journal is © The Royal Society of Chemistry 2021 |