
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of degradable and chemically recyclable polymers using 4,4-disubstituted five-membered cyclic ketene hemiacetal ester (CKHE) monomers†
Xin Yi
Oh
 ,
Yicen
Ge
and
Atsushi
Goto
*
,
Yicen
Ge
and
Atsushi
Goto
*
Division of Chemistry & Biological Chemistry, School of Physical & Mathematical Science, Nanyang Technological University, 21 Nanyang Link, Singapore 637371, Singapore. E-mail: agoto@ntu.edu.sg
First published on 28th September 2021
Abstract
Novel degradable and chemically recyclable polymers were synthesized using five-membered cyclic ketene hemiacetal ester (CKHE) monomers. The studied monomers were 4,4-dimethyl-2-methylene-1,3-dioxolan-5-one (DMDL) and 5-methyl-2-methylene-5-phenyl-1,3-dioxolan-4-one (PhDL). The two monomers were synthesized in high yields (80–90%), which is an attractive feature. DMDL afforded its homopolymer with a relatively high molecular weight (Mn >100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, where Mn is the number-average molecular weight). DMDL and PhDL were copolymerized with various families of vinyl monomers, i.e., methacrylates, acrylates, styrene, acrylonitrile, vinyl pyrrolidinone, and acrylamide, and various functional methacrylates and acrylate. Such a wide scope of the accessible polymers is highly useful for material design. The obtained homopolymers and random copolymers of DMDL degraded in basic conditions (in the presence of a hydroxide or an amine) at relatively mild temperatures (room temperature to 65 °C). The degradation of the DMDL homopolymer generated 2-hydroxyisobutyric acid (HIBA). The generated HIBA was recovered and used as an ingredient to re-synthesize DMDL monomer, and this monomer was further used to re-synthesize the DMDL polymer, demonstrating the chemical recycling of the DMDL polymer. Such degradability and chemical recyclability of the DMDL polymer may contribute to the circular materials economy.
000, where Mn is the number-average molecular weight). DMDL and PhDL were copolymerized with various families of vinyl monomers, i.e., methacrylates, acrylates, styrene, acrylonitrile, vinyl pyrrolidinone, and acrylamide, and various functional methacrylates and acrylate. Such a wide scope of the accessible polymers is highly useful for material design. The obtained homopolymers and random copolymers of DMDL degraded in basic conditions (in the presence of a hydroxide or an amine) at relatively mild temperatures (room temperature to 65 °C). The degradation of the DMDL homopolymer generated 2-hydroxyisobutyric acid (HIBA). The generated HIBA was recovered and used as an ingredient to re-synthesize DMDL monomer, and this monomer was further used to re-synthesize the DMDL polymer, demonstrating the chemical recycling of the DMDL polymer. Such degradability and chemical recyclability of the DMDL polymer may contribute to the circular materials economy.
Introduction
Degradable polymers find numerous applications in, e.g., drug delivery, packaging, and agriculture fields.1–9 In the field of radical polymerization, degradable polymers are prepared via radical ring-opening polymerization of cyclic ketene acetals (CKAs),10–14 for example, gaining significant attention. Examples of CKA are 2-methylene-1,3-dioxepane (MDO),15–23 5,6-benzo-2-methylene-1,3-dioxepane (BMDO),24–26 and 2-methylene-4-phenyl-1,3-dioxolane (MPDL)27–30 (Scheme 1). CKAs are homo-polymerized and co-polymerized with other vinyl monomers, affording degradable ester linkages in the polymer backbones. Mechanistically (Scheme 1), the radical addition to the carbon–carbon double bond of CKA generates a ring-retained radical. The subsequent intramolecular fragmentation (ring opening) generates a ring-opened radical and provides a degradable ester linkage in the backbone. MDO and BMDO are seven-membered ring, and the ring strain would promote the ring opening. BMDO and MPDL produce benzylic ring-opened radicals, and the stabilization of the radical by the phenyl group would promote the ring opening. The obtained homopolymers and copolymers have been exploited for applications in, e.g., packaging, agrochemical, personal care, and biomedical applications.10–13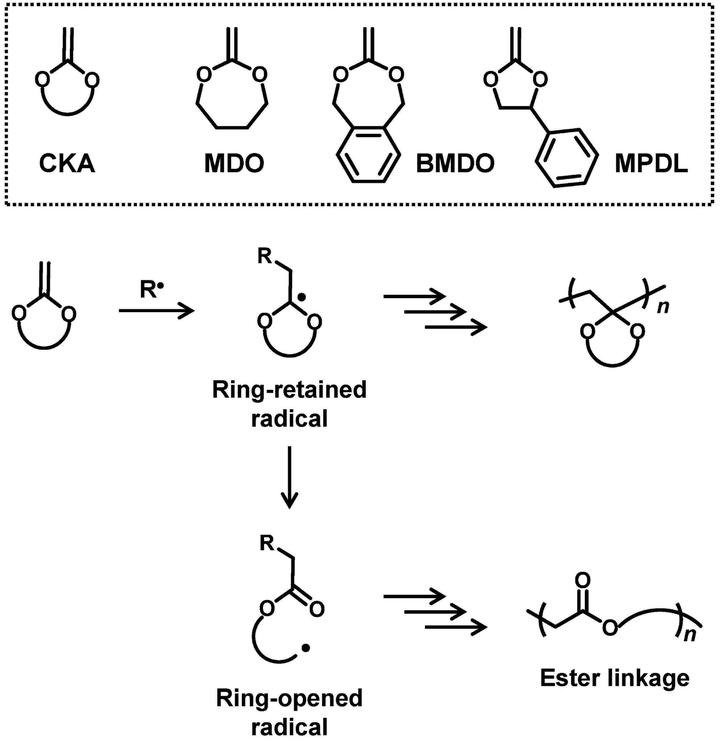 | ||
| Scheme 1 CKA monomers and their possible polymerization mechanisms. | ||
In the present work, we are motivated to use cyclic ketene hemiacetal esters (CKHEs) (Scheme 2) as alternatives to CKAs. A CKHE bears a carbonyl group, while a CKA does not. We choose 4,4-disubstituted five-membered CKHEs, i.e., 4,4-dimethyl-2-methylene-1,3-dioxolan-5-one (DMDL) and 5-methyl-2-methylene-5-phenyl-1,3-dioxolan-4-one (PhDL) (Scheme 2). If their ring opening occurs (Scheme 2), a relatively stable polymethacrylate-like radical (a tertiary radical with an ester group and two alkyl groups) is generated. Hence, we hypothesize that the polymerizations of these monomers would prefer ring opening and provide polymers with anhydride degradable linkages in the backbone, which is our initial motivation, although this hypothesis is found to be incorrect as described below.
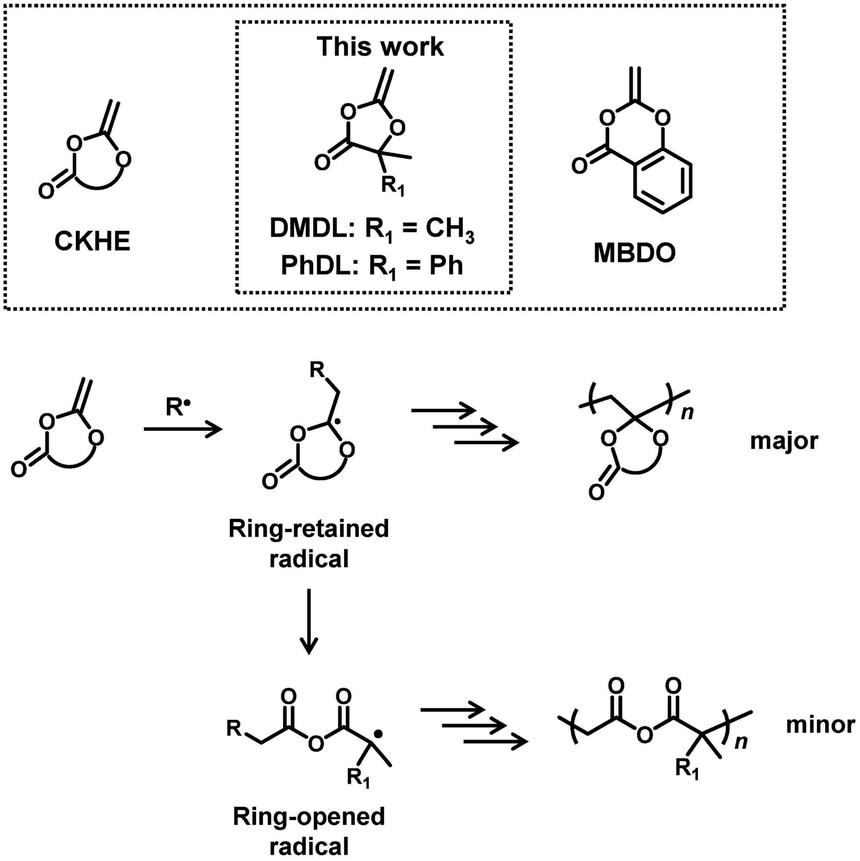 | ||
| Scheme 2 CKHE monomers and their possible polymerization mechanisms. | ||
To the best of our knowledge, there is one report of radical polymerization of a CKHE. The studied CKHE was a six-membered CKHE, i.e., 2-methylene-4H-benzo[d][1,3]dioxin-4-one (MBDO (Scheme 2)).31 MBDO yielded polymers with ring-retained structures, possibly because its ring-opened radical is a phenyl radical, which is unlikely to generate. The homopolymer of MBDO was insoluble in most of common organic solvents. MBDO was copolymerized with vinyl acetate (VAc), yielding soluble random copolymers. Interestingly, the obtained polymers degraded via acid-assisted hydrolysis. After 5 days of the hydrolysis, only small molecules were detected. The authors suggested the backbone (main chain) scission of the polymers. The nuclear magnetic resonance (NMR) analysis showed that the polymers degraded to acetic acid and salicylic acid, which are raw ingredients of MBDO. Therefore, the authors suggested that the polymers are potentially recyclable, although the recycling was not demonstrated experimentally. These results are interesting findings.
In the present paper, we report the use of DMDL and PhDL as CKHEs to synthesize their homopolymers and random copolymers with several co-monomers. DMDL and PhDL are 4,4-disubstituted five-membered CKHEs bearing two methyl groups (DMDL) or a phenyl group and a methyl group (PhDL) at the 4-position. Unlike the previously reported MBDO, DMDL was able to generate homopolymers soluble in common organic solvents. DMDL and PhDL were also able to be copolymerized with a range of co-monomers. Mechanistically, contrary to our initial hypothesis, DMDL and PhDL generated polymers with ring-retained structures (Scheme 2) like MBDO, meaning that the activation energy for generating the ring-opened radical is still high for DMDL and PhDL. Nevertheless, despite the ring-retained structure, the PDMDL homopolymers and copolymers degraded via the backbone scission in basic conditions, where PDMDL is poly(4,4-dimethyl-2-methylene-1,3-dioxolan-5-one). Importantly, NaOH-assisted degradation of PDMDL homopolymers generated 2-hydroxyisobutyric acid (HIBA), which is a raw ingredient of DMDL (Scheme 3). We recovered HIBA to synthesize the DMDL monomer and PDMDL, demonstrating the recycling of PDMDL (Scheme 3). In this recycling, four of the six carbons of DMDL are recycled via HIBA (four carbons). Thus, the polymers serve as (partly) recyclable polymers. Markedly, the degradation of our polymers does not generate mere CO2 but generates their raw ingredient. This type of polymers are classified to chemically recyclable polymers that can degrade into their raw ingredients or useful oligomers to build other new polymeric materials.2 Chemically recyclable polymers are attractive in sustainability and may provide more economic values compared with polymers that can degrade into mere CO2.
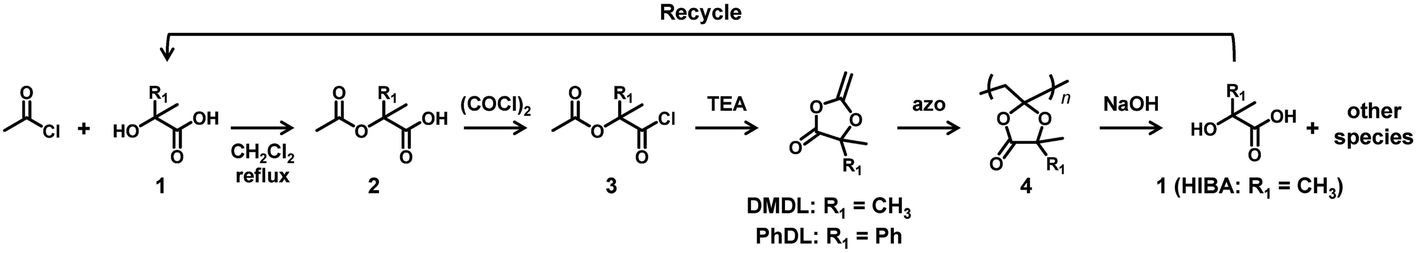 | ||
| Scheme 3 Synthesis of monomer, polymerization, hydroxide-assisted degradation of polymer, and recycle of HIBA. | ||
Results and discussion
Synthesis of DMDL and PhDL
DMDL was previously synthesized in the field of synthetic organic chemistry32 but has not been utilized in polymerization. According to the literature,32 we synthesized DMDL via an intramolecular cyclization of commercially available compound 3 (R1 = CH3 in Scheme 3) (1 equiv.) using triethylamine (TEA) (1.2 equiv.) in dichloromethane under reflux for 5 h. The conversion determined with 1H NMR was nearly 100%. There was no report for the synthesis of PhDL. We synthesized PhDL from commercially available compound 1 (R1 = Ph in Scheme 3) by referring to the synthesis of a similar compound.33,34 The compound 1 (1 equiv.) was reacted with acetyl chloride (2.8 equiv.) in dichloromethane under reflux for 2 h, giving compound 2 (R1 = Ph in Scheme 3) in a 96% yield after purification (recrystallization). The purified compound 2 (1 equiv.) was subsequently reacted with oxalyl chloride (1.1 equiv.) in a mixture of dichloromethane (99%) and dimethyl formamide (1%) at room temperature overnight, giving compound 3 (R1 = Ph in Scheme 3) in a 99% conversion (as determined with 1H NMR). The crude compound 3 (1 equiv.) was used for its intramolecular cyclization in the presence of TEA (4.7 equiv.) in dichloromethane under reflux for 2 h, yielding PhDL (Fig. S1 and S2 in ESI†). Both DMDL and PhDL were purified by distillation, and we obtained pure DMDL and PhDL in 80% and 90% yields (based on the amount of compound 3), respectively. The high yields of these two monomers are attractive features in practical use.In a synthetic point of view, the 4-position of compound 3 must be di-substituted. If the 4-position is not di-substituted (but mono-substituted or non-substituted), the compound 3 is unstable, spontaneously generating an undesired compound (a ketene).35 This requirement is also another reason why we studied 4,4-disubstituted DMDL and PhDL. The stability of the DMDL monomer (8.9 wt%) was studied in DMSO-d6 (89.3 wt) under moisture (1.8 wt% water) at room temperature. DMDL decomposed to form compound 2 (Scheme 3) via hydrolysis (Fig. S3 in ESI†). The extent of the DMDL hydrolysis was 5%, 11%, and 64% for 0.5 h, 12 h, and 4 days, respectively (ESI).
Homopolymerizations of DMDL and PhDL
We conducted a homopolymerization of DMDL (100 equiv., 50 wt%) using 2,2′-azoisobutyronitrile (AIBN) (1 equiv.) as a radical initiator in toluene (50 wt%) at 70 °C for 12 h (Table 1, entry 1). The monomer conversion reached 82% (as determined with 1H NMR), yielding a PDMDL with Mn = 10300 and Đ (= Mw/Mn) = 1.61, where Mn and Mw are the number- and weight-average molecular weights, respectively, and Đ is the dispersity. The Mn and Đ values are not absolute values but PMMA-calibrated GPC values, where PMMA is poly(methyl methacrylate). The obtained PDMDL was soluble in organic solvents such as chloroform, tetrahydrofuran, and dimethylsulfoxide. Fig. 1a and b show the 1H NMR spectra (CDCl3) of DMDL (monomer) and PDMDL (polymer) (after purification via reprecipitation in hexane/diethyl ether (1/1 (v/v)) (non-solvent)), respectively. The vinyl protons (a) of DMDL appearing at 3.62 and 3.70 ppm were converted to the backbone methylene protons (a′) of PDMDL appearing at 2.17–3.25 ppm. For PDMDL (Fig. 1b), the integration ratio of methylene protons (a′) and dimethyl protons (b′) was nearly 1 (a′) to 3 (b′), agreeing with the PDMDL structure.
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Co-monomer (M) | Initiator | Solventa | [M]0/[DMDL]0/[initiator]0 (equiv.) | T (°C) | t (h) | Conv (M/DMDL)b (%/%) | M n | Đ | F DMDL (%) |
| a Dilution with 50 wt% solvent for entries 1 and 3–17. EC is ethylene carbonate. DMF is N,N-dimethylformamide. b Monomer conversion obtained with 1H NMR. c PMMA-calibrated THF-GPC values for entries 1, 3–5, 7, and 12–17 and PMMA-calibrated DMF-GPC values for entries 2, 6, and 8–11. d F DMDL value determined by the 1H NMR analysis of the purified polymer. In the parenthesis, FDMDL value calculated from the monomer conversions of DMDL and co-monomer determined using 1H NMR is given. e Monomer conversion of DMDL was unable to determine because the DMDL monomer peak overlapped with the co-monomer peak in the 1H NMR spectrum. | ||||||||||
| 1 | None | AIBN | Toluene | 0/100/1 | 70 | 12 | NA/82 | 10300 |
1.61 | 100 (100) |
| 2 | None | AIBN | Bulk | 0/100/0.5 | 70 | 1.5 | NA/79 | 104000 |
2.04 | 100 (100) |
| 3 | MMA | AIBN | Toluene | 50/50/1 | 70 | 7 | 99/23 | 19000 |
1.78 | 18 (19) |
| 4 | BA | AIBN | Toluene | 50/50/1 | 70 | 1 | 98/56 | 130000 |
1.53 | 37 (36) |
| 5 | St | AIBN | Toluene | 50/50/2 | 80 | 12 | 79/NAe | 5100 | 1.46 | 8 (NA) |
| 6 | AN | AIBN | EC | 50/50/1 | 70 | 1 | 80/44 | 77000 |
1.54 | 34 (35) |
| 7 | VAc | AIBN | Toluene | 50/50/1 | 70 | 12 | 84/48 | 11000 |
1.60 | 35 (36) |
| 8 | NVP | AIBN | DMF | 50/50/1 | 70 | 2 | 96/28 | 29000 |
1.81 | 19 (23) |
| 9 | DMA | AIBN | Toluene | 50/50/1 | 70 | 1 | 100/39 | 91000 |
1.95 | 28 (29) |
| 10 | PEGMA | V65 | Toluene | 50/50/1 | 60 | 4 | 92/NAe | 39000 |
2.22 | 17 (NA) |
| 11 | DMAEMA | AIBN | Toluene | 50/50/1 | 70 | 4 | 98/20 | 27000 |
1.62 | 17 (17) |
| 12 | BMA | AIBN | Toluene | 50/50/1 | 70 | 5 | 93/11 | 46000 |
1.54 | 10 (11) |
| 13 | LMA | AIBN | Toluene | 50/50/1 | 70 | 3 | 85/15 | 42000 |
1.76 | 16 (15) |
| 14 | BzMA | AIBN | Toluene | 50/50/1 | 70 | 3 | 92/26 | 39000 |
1.76 | 19 (22) |
| 15 | EHMA | AIBN | Toluene | 50/50/1 | 70 | 2 | 97/18 | 38000 |
1.95 | 13 (16) |
| 16 | SMA | AIBN | Toluene | 50/50/1 | 70 | 2 | 77/29 | 86000 |
1.96 | 28 (27) |
| 17 | MEA | AIBN | Toluene | 50/50/1 | 70 | 1 | 100/54 | 82000 |
2.00 | 32 (35) |
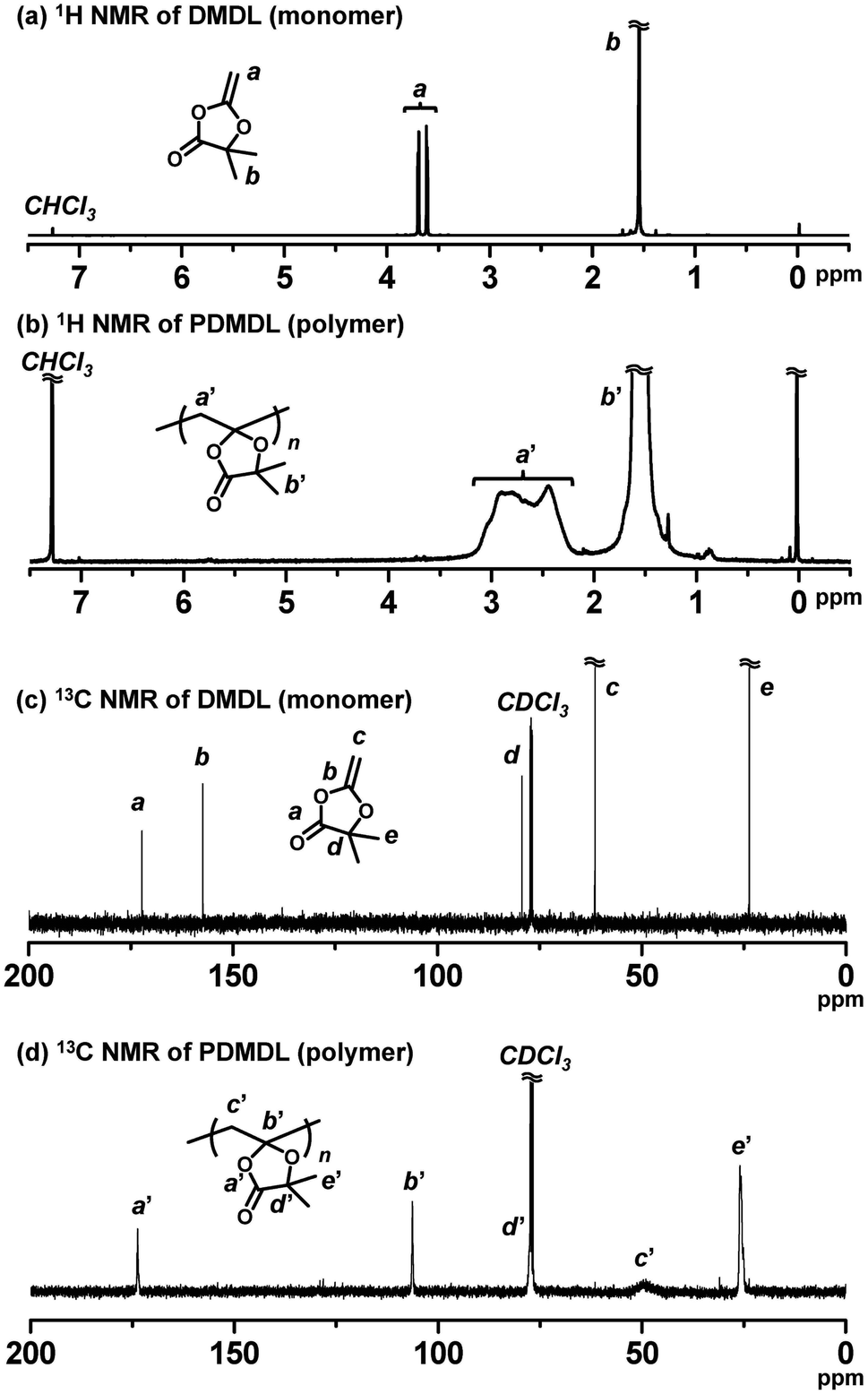 | ||
| Fig. 1 1H NMR spectra of (a) DMDL and (b) PDMDL. 13C NMR spectra of (c) DMDL and (d) PDMDL. The synthesis of PDMDL is given in Table 1 (entry 1). 400 MHz for 1H NMR (100 MHz for 13C NMR) in CDCl3. | ||
As mentioned, there are two possible polymerization pathways, i.e., the ring-retaining and ring-opening pathways (Scheme 2). Fig. 1c and d show the 13C NMR spectra (CDCl3) of DMDL and PDMDL, respectively. Through the polymerization, the vinyl carbon (b) of DMDL (Fig. 1c) appearing at 157 ppm can be converted to the backbone quaternary carbon (b′) via the ring-retaining pathway (Fig. 1d) or to a backbone anhydride carbonyl carbon via the ring-opening pathway (Scheme 2). The 13C NMR spectrum of PDMDL (Fig. 1d) shows a clear signal for the characteristic backbone quaternary carbon (b′) at 106 ppm, meaning the occurrence of the ring-retaining pathway. The signal of the ester carbonyl carbon (a) of DMDL appearing at 173 ppm (Fig. 1c) was only slightly sifted to 174 ppm ((a′) in Fig. 1d) in PDMDL, supporting the ring retention. If the ring-opened structure was also formed, an additional signal for the anhydride carbonyl carbon should have appeared, as anhydride carbonyl carbons generally appear at 160–170 ppm.36 However, we observed only one signal (for the ester carbonyl carbon for the ring-retained structure) but did not observe other signals in the region at 160–180 ppm, where any carbonyl carbons are to appear. The result means that the obtained PDMDL predominantly contained the ring-retained monomer units and that the ring-opened monomer units were minor units if any present. A possible reason why the ring-opened radical was hardly generated (Scheme 2) is the presence of the trigonal carbonyl carbon in CKHE (DMDL). The trigonal carbonyl carbon can make the CKHE ring more planar than the CKA ring (without the trigonal carbonyl carbon), and the planner conformation may enhance the stabilization of the ring-retained radical.37
Instead of the solution polymerization in toluene (Table 1, entry 1) (described above), we carried out a bulk polymerization of DMDL (100 equiv.) using AIBN (0.5 equiv.) at 70 °C for 1.5 h (Table 1, entry 2). The absolute monomer concentration increased, and the viscosity of the polymerization solution increased, suppressing radical–radical termination by the so-called gel effect and yielding a higher molecular weight PDMDL with Mn = 104000 and Đ = 2.04 at a 79% monomer conversion.
Despite the higher molecular weight, the polymer was still soluble in organic solvents. We measured the glass transition temperature (Tg), crystallization temperature (Tc), melting temperature (Tm), and 50%-decomposition temperature (Td) of this polymer (PDMDL) using differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). The Tg, Tc, Tm, and Td values were 41 °C, 117 °C, 186 °C, and 269 °C, respectively (Fig. S4 in ESI†).
The homopolymerization of PhDL (using AIBN) did not proceed at 70 °C. At an elevated temperature of 120 °C, the homopolymerization of PhDL (using t-butyl peroxybenzoate (TBPB)) slowly proceeded but yielded only an oligomer with Mn <1000 (Table 2, entry 1) (Fig. S5 in ESI†). In PhDL, one of the two methyl groups of DMDL is replaced by a phenyl group (Scheme 2). The steric hindrance of the bulky phenyl group might hinder the successive connection of the ring-retained monomer units. Another possibility is that the phenyl group of PhDL stabilizes the ring-opened radical and facilitates the formation of the ring-opened radical but that the radical might be too stable to polymerize. Also, the ring-opened radical might generate polymers with anhydride linkages but the anhydride linkages might be cleaved during the polymerization at a high temperature of 120 °C. These are not definitive but possible explanations. Due to the low molecular weight, the obtained oligomer was difficult to unequivocally analyze, and the exact explanation is not clear at this moment.
| Entry | Co-monomer (M) | Initiatora | Solventb | [M]0/[PhDL]0/[initiator]0 (mM) | T (°C) | t (h) | Conv (M/PhDL)c (%/%) | M n | Đ | F PhDL (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| a TBPB is t-butyl peroxybenzoate. b Dilution with 50 wt% solvent. c Monomer conversion obtained with 1H NMR. d PMMA-calibrated THF-GPC values. e F PhDL value determined by the 1H NMR analysis of the purified polymer. In the parenthesis, FPhDL value calculated from the monomer conversion (monomer consumption) determined using 1H NMR is given. | ||||||||||
| 1 | None | TBPB | Toluene | 0/100/1 | 120 | 12 | NA/37 | <1000 | NA | 100 (100) |
| 2 | MMA | AIBN | Toluene | 50/50/1 | 70 | 7 | 93/15 | 15000 |
1.83 | 11 (14) |
| 3 | MMA | TBPB | Toluene | 50/50/1 | 120 | 5 | 94/33 | 21000 |
1.45 | 24 (26) |
| 4 | BA | AIBN | Toluene | 50/50/1 | 70 | 3 | 96/39 | 47000 |
2.08 | 26 (29) |
| 5 | BA | TBPB | Toluene | 50/50/1 | 120 | 3.5 | 96/48 | 13000 |
2.63 | 31 (33) |
Random copolymerizations of DMDL and PhDL with various co-monomers
We carried out a random copolymerization of DMDL (50 mol%) with methyl methacrylate (MMA) (50 mol%) in toluene at 70 °C (Table 1, entry 3). Fig. 2a shows the plots of the conversions of MMA (square) and DMDL (circle) vs. time. MMA was consumed faster than DMDL. After 7 h, the conversions of MMA and DMDL were 99% and 23%, respectively, yielding a random copolymer of MMA and DMDL with Mn = 19000 and Đ = 1.78. The fraction (FDMDL) of DMDL in the copolymer was 18%. The 1H and 13C NMR analysis showed that the polymer contained both MMA and DMDL units and the DMDL units were incorporated in the ring-retained form (Fig. S6 and S7 in ESI†). The monomer reactivity ratio was determined by the Fineman–Ross method38 (Fig. S8–S12 in ESI†) to be rMMA = 4.60 and rDMDL ≈ 0 (Table 3, entry 1). The result means that the MMA terminal radical reacts with both MMA and DMDL and 4.60 times more reacts with MMA than DMDL and that the DMDL terminal radical virtually exclusively reacts with MMA.
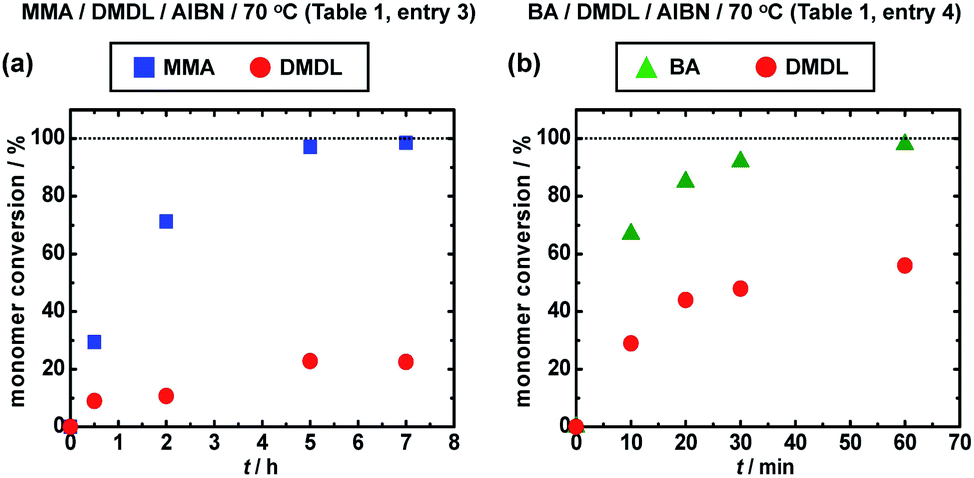 | ||
| Fig. 2 Plots of monomer conversion vs. time for (a) the MMA/DMDL/AIBN (50/50/1) system (70 °C) (Table 1, entry 3) and (b) the BA/DMDL/AIBN (50/50/1) system (70 °C) (Table 1, entry 4). The solvent was toluene (50 wt%) for both systems. The symbols are indicated in the figure. | ||
We studied n-butyl acrylate (BA) as another family of monomer. We carried out a random copolymerization of DMDL (50 mol%) with BA (50 mol%) in toluene at 70 °C (Table 1, entry 4). Fig. 2b shows the plots of the conversions of BA (triangle) and DMDL (circle) vs. time. Similar to MMA, the co-monomer BA was consumed faster than DMDL. After 1 h, the conversions of BA and DMDL were 98% and 56%, respectively, yielding a random copolymer of BA and DMDL with Mn = 130000 and Đ = 1.53 (Fig. S13 in ESI†). The monomer reactivity ratios were rBA = 0.83 and rDMDL ≈ 0.03 (a range of 0–0.10 with an experimental error) (Table 3, entry 2). These values means that the BA/DMDL system (rBA = 0.83 (Table 3, entry 2)) has a more alternating tendency than the MMA/DMDL system (rMMA = 4.60 (Table 3, entry 1)) but that the DMDL terminal radical still virtually exclusively reacts with BA over DMDL (rDMDL ≈ 0.03 (Table 3, entry 2)).
We also studied styrene (St), acrylonitrile (AN), and vinyl acetate (VAc) as other families of monomer (Table 1, entries 5–7). We carried out random copolymerizations of DMDL (50 mol%) with the co-monomer (50 mol%) in toluene (for St and VAc) or ethylene carbonate (for AN), yielding polymers with Mn = 5100–77000 and Đ = 1.46–1.60 at FDMDL = 8–35% (Fig. S14–S16 in ESI†). For the St and AN systems, the monomer reactivity ratios were rSt = 1.08 and rDMDL ≈ 0.01 (0–0.10 with an experimental error) (Table 3, entry 3) and rAN = 0.28 and rDMDL ≈ 0.04 (0–0.10 with an experimental error) (Table 3, entry 4). The rSt and rAN values mean that the St terminal radical reacts nearly equally with DMDL and St, whereas the AN terminal radical reacts 3.6 (= 1/0.28) times more with DMDL than AN. The nearly zero rDMDL values mean that the DMDL terminal radical predominantly reacts with St and AN over DMDL. The VAc system had rVAc = 1.65 and rDMDL = 0.29 (Table 3, entry 5). The rDMDL value is not nearly zero in the VAc system (Table 3, entry 5), in contrast to the mentioned four systems (Table 3, entries 1–4). The result means that the DMDL terminal radical reacts with VAc slowly and hence can react with DMDL to some extent in the VAc system.
In the literatures, random copolymerizations of CKAs and vinyl co-monomers also showed similar tendency, i.e., slower consumption of CKAs than the co-monomers. The rco-monomer values are typically 2–8 and the rCKA values are 0.01–1 (Table S1 in ESI†).29,39–43 For MBDO (previously reported CKHE monomer) (Scheme 2), its random copolymerization with VAc had rVAc = 0.587 and rMBDO = 1.51 (60 °C), showing that MBDO has higher reactivity than VAc.31
Thermal properties of the obtained DMDL copolymers (Table 1, entries 3 and 5–7) with MMA, St, AN, and VAc were studied (Fig. S17–S20 in ESI†). The Tg values of the copolymers with MMA (104 (±5) °C), St (90 (±5) °C), and AN (76 (±5) °C) were slightly lower than those of homopolymers of MMA (ca. 105 °C),44 St (ca. 90–100 °C),44 and AN (ca. 95 °C),44 while the Tg value of the copolymer with VAc (38 (33–46) °C) was slightly higher than that of a homopolymer of VAc (ca. 30 °C),44 because the Tg value of a homopolymer of DMDL is 41 °C.
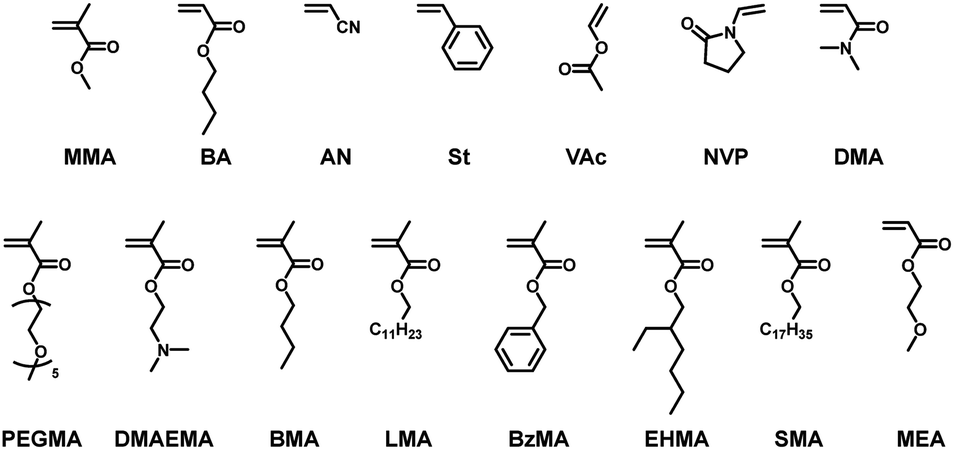
We further expanded the co-monomer scope to N-vinylpyrrolidone (NVP) and N,N-dimethylacrylamide (DMA) as other families of monomer (Table 1, entries 8 and 9) and functional methacrylates with poly(ethylene glycol) methyl ether (PEGMA), dimethylamino (DMAEMA), butyl (BMA), lauryl (LMA), benzyl (BzMA), ethylhexyl (EHMA), and stearyl (SMA) groups (Table 1, entries 10–16) as well as a functional acrylate with a methoxyethyl group (MEA) (Table 1, entry 17), yielding co-polymers with Mn = 27000–91000 and Đ = 1.54–2.22 at FDMDL = 10–32% (Fig. S21–S30 in ESI†). Such a wide range of amenable monomer families and functional monomers, including hydrophobic (BMA, LMA, BzMA, EHMA, SMA, and MEA), hydrophilic (NVP, DMA, PEGMA, and DMAEMA), and biocompatible (PEGMA and MEA) monomers, are attractive for polymer design by DMDL.
While the homopolymerization of PhDL generated only an oligomer (Table 2, entry1), PhDL was able to be copolymerized with a methacrylate and an acrylate (Table 2, entries 2–5). We carried out a random copolymerization of PhDL (50 mol%) with MMA (50 mol%) in toluene at 70 °C (Table 2, entry 2), which is the same condition as that in the DMDL/MMA system (Table 1, entry 3) except using PhDL instead of DMDL. After 7 h, the conversions of MMA and PhDL were 93% and 15%, respectively, yielding a random copolymer of MMA and PhDL with Mn = 15000 and Đ = 1.83 (Fig. S31 in ESI†). The conversion of PhDL (15%) was smaller than that of DMDL (23%) at similar conversions of MMA (93–99%). In order to increase the conversion of PhDL, we elevated the polymerization temperature from 70 °C to 120 °C (Table 2, entry 3). After 5 h, the conversions of MMA and PhDL were 94% and 33%, respectively, yielding a random copolymer with Mn = 21000 and Đ = 1.45. The conversion of PhDL increased by 18% (from 15% to 33%) by elevating the temperature from 70 °C to 120 °C at the similar (93–94%) conversions of MMA.
We also conducted a random copolymerization of PhDL (50 mol%) with BA (50 mol%) in toluene at 70 °C (Table 2, entry 4). After 3 h, the conversions of BA and PhDL were 96% and 39%, respectively, yielding a random copolymer with Mn = 47000 and Đ = 2.08 (Fig. S32 in ESI†). At a higher temperature of 120 °C, the conversions of BA and PhDL were 96% and 48%, respectively (Table 2, entry 5), showing an increase in the conversion of PhDL by 9% by elevating the temperature from 70 °C to 120 °C at the same (96%) conversion of BA.
Thus, DMDL and PhDL were able to be copolymerized with a range of vinyl monomers, offering a wide scope of accessible polymers. This feature is in sharp contrast to the limited co-monomer scope of the previously reported CKHE monomer, i.e., MBDO, which was copolymerized only with VAc.
Hydroxide-assisted degradation of PDMDL
We studied the degradation of PDMDL. We dissolved a PDMDL homopolymer (Mn = 104000 and Đ = 2.04 (Table 1, entry 2)) (0.064 g, 2.4 wt%, 1 equiv. of the DMDL monomer unit) in DMSO-d6 (2.08 g, 77.2 wt%), which was a colorless transparent solution. To this solution, a D2O solution of 1 M NaOH (0.55 g, 20.4 wt%, 1 equiv. of NaOH) was added at room temperature. The mixture became slightly turbid, because PDMDL chains became globular due to the presence of water (D2O), but the mixture soon turned reddish brown and transparent upon stirring at room temperature. PDMDL seemed to degrade to small molecules. The reaction mixture was subsequently analyzed using GPC and NMR within 5 h after the reaction started.
In the GPC chromatogram (DMF eluent) (Fig. 3a), no peak was observed in the polymer region (molecular weight >2000), suggesting that PDMDL degraded. (The peak at the molecular weight <2000 observed in Fig. 3a would be ascribed to generated oligomers and/or possible clusters of lithium bromide (LiBr) used in the DMF eluent.) The 1H (Fig. 3b) and 13C (Fig. 3c) NMR spectra also show no polymer (PDMDL) signals (no signal at 2.15–3.25 ppm for the backbone CH2 protons in the 1H NMR spectrum and no signal at 174 ppm for the ester carbon of the DMDL unit in the polymer in the 13C NMR spectrum) (the 1H and 13C NMR spectra of (non-degraded) PDMDL (2.4 wt%) in the studied DMSO-d6/D2O (77.2/20.4 wt%) mixed solvent are given in ESI (Fig. S33 and S34†)). The only species clearly detected was HIBA (Scheme 3) whose CH3 protons appeared at 1.13 ppm (Fig. 3b) and whose CH3 carbons, quaternary carbon, and carbonyl carbon appeared at 28, 73, and 181 ppm, respectively (Fig. 3c).
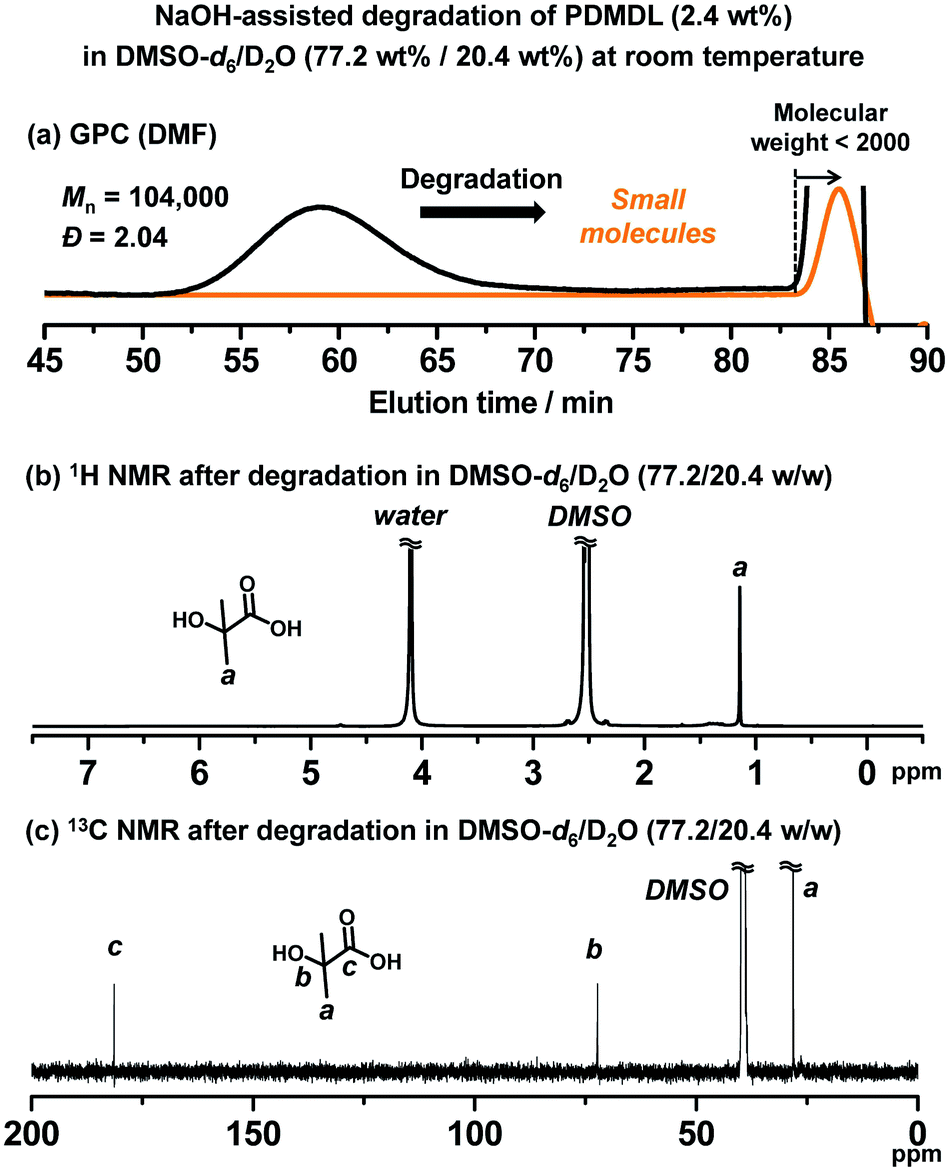 | ||
| Fig. 3 Degradation of PDMDL (Mn = 104000, Đ = 2.04, 2.4 wt%, 1 equiv. of the DMDL monomer unit) using 1 equiv. of NaOH in a mixture of D2O (20.4 wt%) and DMSO-d6 (77.2 wt%) at room temperature within 5 h. (a) GPC chromatogram before (black line) and after (orange line) degradation. (b) 1H NMR and (c) 13C NMR spectra of the reaction mixture after degradation (400 MHz for 1H NMR (100 MHz for 13C NMR) in DMSO-d6/D2O). | ||
Scheme 4 shows a possible degradation mechanism of PDMDL in the presence of OH−. It should be emphasized that this scheme is a possible mechanism but is not a definitive one at this moment. Firstly, OH− attacks the carbonyl carbon of the hemiacetal ester, generating an alkoxide anion (Scheme 4, compound 5). A subsequent elimination can result in either (a) the side-chain cleavage or (b) the main-chain cleavage. The side-chain cleavage can generate a ketone in the backbone chain (compound 6) and HIBA. If the side-chain cleavage is significant, a polyketone can be generated. The main-chain cleavage can generate a carboxylic acid chain-end PDMDL (compound 8) and a carboxylate chain-end PDMDL (compound 9). These two compounds can further generate another form of carboxylic acid chain-end PDMDL (compound 10) and a ketone chain-end PDMDL (compound 11) as well as two molecules of HIBA. If the main-chain cleavage is significant, the polymers (compounds 8–11) will further undergo the main-chain cleavage and degrade into small molecules. A main-chain cleavage mechanism was previously proposed for the degradation of the MBDO polymer, for which the mechanism was studied in an acidic condition.31
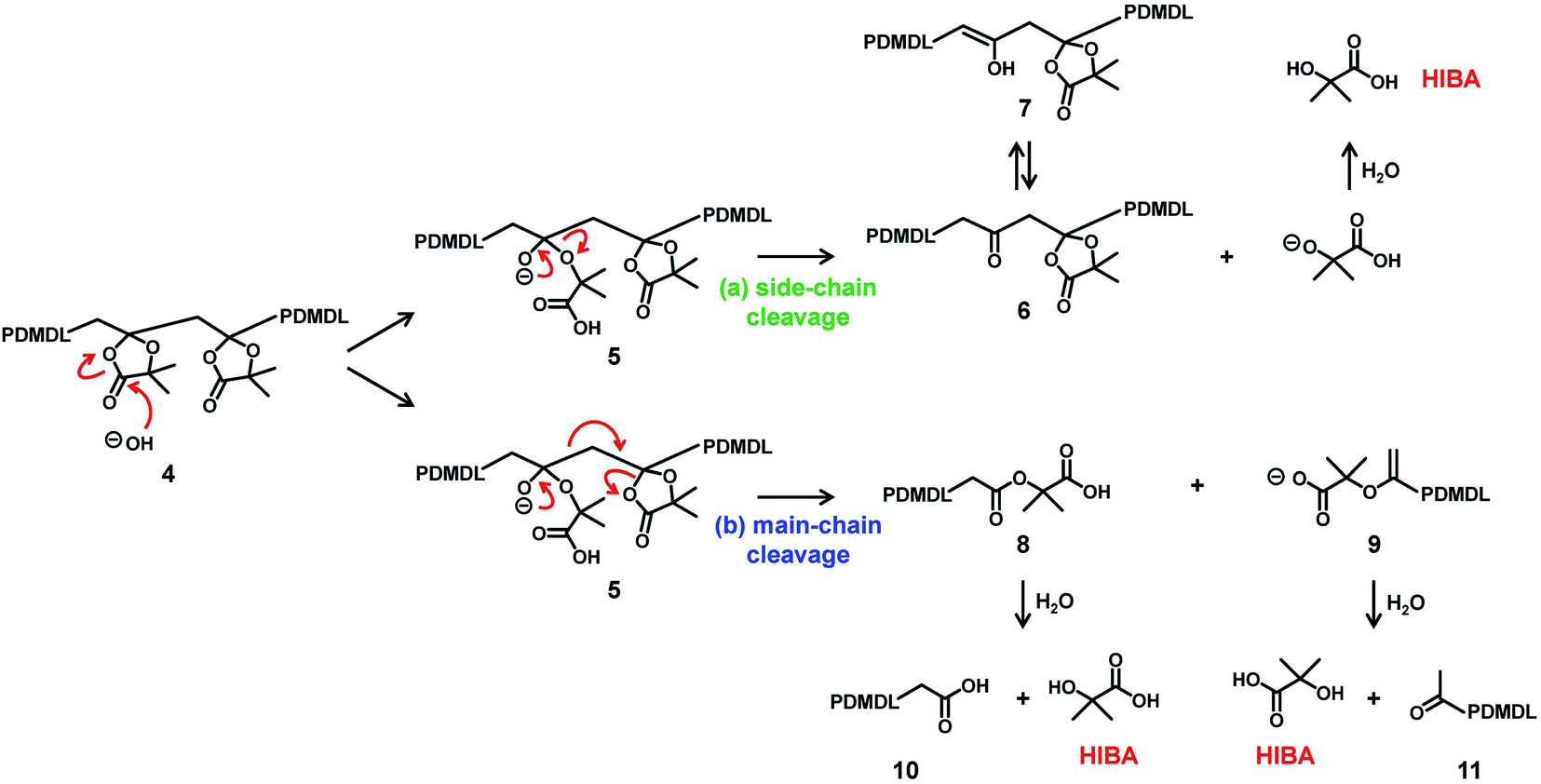 | ||
| Scheme 4 Possible mechanisms of hydroxide-assisted degradation of PDMDL. | ||
No polymer peak observed in the GPC chromatogram (Fig. 3a) suggests that the main-cleavage operated significantly in the present system so that the polymer chains could degrade into small molecules or oligomers. The side-chain cleavage is not excluded at this moment and might also operate to some extent. HIBA is a product in both pathways. The observation of HIBA in the NMR analysis (Fig. 3b and c) supports the proposed degradation scheme (either or both of the two pathways). However, the 1H and 13C NMR analyses detected no other species, and the reason is unclear at the moment. When the main-chain cleavage operates, small molecules or oligomers with carboxylic acid (compounds 8 and 10), carboxylate (compound 9), and/or ketone (compound 11) can be generated. However, the 13C NMR analysis (Fig. 3c) detected no other C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbons than that of HIBA. When the side-chain cleavage operates, a polyketone (compound 6) or its isomer, i.e., a polyenol (compound 7) can be generated, and the intramolecular cyclization of polyketone can also generate aromatic species.45 However, the 13C NMR analysis (Fig. 3c) detected no ketone (>190 ppm), no enol (90–100 ppm), or no aromatic species (110–140 ppm). Small molecules that were possibly generated might be volatile and evaporate during the degradation. Thus, we also conducted the same experiment in a valved (sealed) NMR tube (Fig. S35 in ESI†). However, again, only HIBA was detected. A possible reason for no detection of other species (compounds 6–11) is the solubility of the product. To check this possibility, we studied the degradation of PDMDL in a more hydrophobic solvent (a mixture of THF-d8 and D2O instead of a mixture of DMSO-d6 and D2O), as described in detail in ESI. The 1H NMR and 13C NMR spectra of the reaction mixture (Fig. S36 in ESI†) again showed only HIBA even in this more hydrophobic solvent. Thus, the reason for no detection of other species is not clear at this moment, and hence the proposed degradation scheme (Scheme 4) is viewed as a tentative mechanism.
O carbons than that of HIBA. When the side-chain cleavage operates, a polyketone (compound 6) or its isomer, i.e., a polyenol (compound 7) can be generated, and the intramolecular cyclization of polyketone can also generate aromatic species.45 However, the 13C NMR analysis (Fig. 3c) detected no ketone (>190 ppm), no enol (90–100 ppm), or no aromatic species (110–140 ppm). Small molecules that were possibly generated might be volatile and evaporate during the degradation. Thus, we also conducted the same experiment in a valved (sealed) NMR tube (Fig. S35 in ESI†). However, again, only HIBA was detected. A possible reason for no detection of other species (compounds 6–11) is the solubility of the product. To check this possibility, we studied the degradation of PDMDL in a more hydrophobic solvent (a mixture of THF-d8 and D2O instead of a mixture of DMSO-d6 and D2O), as described in detail in ESI. The 1H NMR and 13C NMR spectra of the reaction mixture (Fig. S36 in ESI†) again showed only HIBA even in this more hydrophobic solvent. Thus, the reason for no detection of other species is not clear at this moment, and hence the proposed degradation scheme (Scheme 4) is viewed as a tentative mechanism.
Nevertheless, as an experimental fact, PDMDL clearly degraded in the presence of NaOH. The degradation occurred within 5 h at room temperature or markedly fast (within 30 min) at 50 °C, which would be an attractive feature of PDMDL.
Acid-assisted degradation of PDMDL
We also studied the degradation of PDMDL in an acidic condition, using the same condition as previously reported condition for the acid-assisted degradation of the MBDO polymer.31 We dissolved a PDMDL (Mn = 106000 and Đ = 1.88) (0.064 g, 2.4 wt%, 1 equiv. of the DMDL monomer unit) in DMSO (2.08 g, 78.7 wt%), which was a colorless transparent solution. To this solution, an aqueous 1 M HCl solution (0.5 g, 18.9 wt%, 1 equiv. of HCl) was added, and the mixture became turbid. The mixture was heated at 80 °C for 24 h, and the generated precipitate was analyzed with GPC (Fig. S37 in ESI†). The precipitate was found to be unreacted PDMDL, and its Mn (= 100000) and Đ (= 2.00) values were close to its original values (Mn = 106000 and Đ = 1.88). The amount of the precipitate (unreacted PDMDL) was 0.057 g, which is 89% of the original amount (0.064 g). Thus, PDMDL did not degrade in the same condition as the MBDO polymer did, mainly because of the low solubility of PDMDL in the studied acidic condition.
Thus, we used a more hydrophobic solvent (a mixture of DMSO-d6 (79.4 wt%) and H2O (1.4 wt%)) to dissolve PDMDL (10.2 wt%) and used an organic acid, i.e., trifluoroacetic acid (9 wt%), that is soluble in this solvent. We heated the mixture at 80 °C for 3 days and observed a decrease in the Mn value by 25%, showing that PDMDL slowly degraded in this acidic condition (Fig. S38 in ESI†). We observed HIBA in this acid-assisted degradation, as in the NaOH-assisted degradation. Furthermore, acetone was observed in the acid-assisted degradation (Fig. S38 in ESI†), which may suggest a chain-end degradation of 11 in the acidic condition (Scheme S1 in ESI†), although this mechanism is a tentative mechanism. While the acid-assisted degradation of the MBDO polymer generated acetic acid,31 we did not observe acetic acid in the degradation of PDMDL. The degradation mechanism might be different for the MBDO polymer and PDMDL.
Amine-assisted degradation of PDMDL
The NaOH-assisted degradation was too fast to monitor over time. Hence, we used a milder base, i.e., an amine to probe the degradation of PDMDL. The studied amine was a primary amine, i.e., pentylamine (R–NH2). We dissolve a PDMDL (Mn = 39000 and Đ = 1.42) (0.064 g, 4.0 wt%, 1 equiv. of the DMDL monomer unit) and pentylamine (0.065 g, 4.0 wt%, 1.5 equiv.) in CD3CN (1.48 g, 92.0 wt%), which was a colorless transparent solution. The solution was heated at 65 °C. Fig. 4a and b show the GPC chromatograms and the plot of Mnvs. the reaction time, respectively. The original Mn (= 39000) value of PDMDL started to decay (Mn = 33000) right after the addition of the amine and became approximately a half (Mn = 20000) after the heating for 4 h, approximately a quarter (Mn = 10000) for 8 h, and approximately 1/10 (Mn = 3000) for 24 h. The polymer degraded to small molecules or oligomers (Mn <2000) for 60 h.
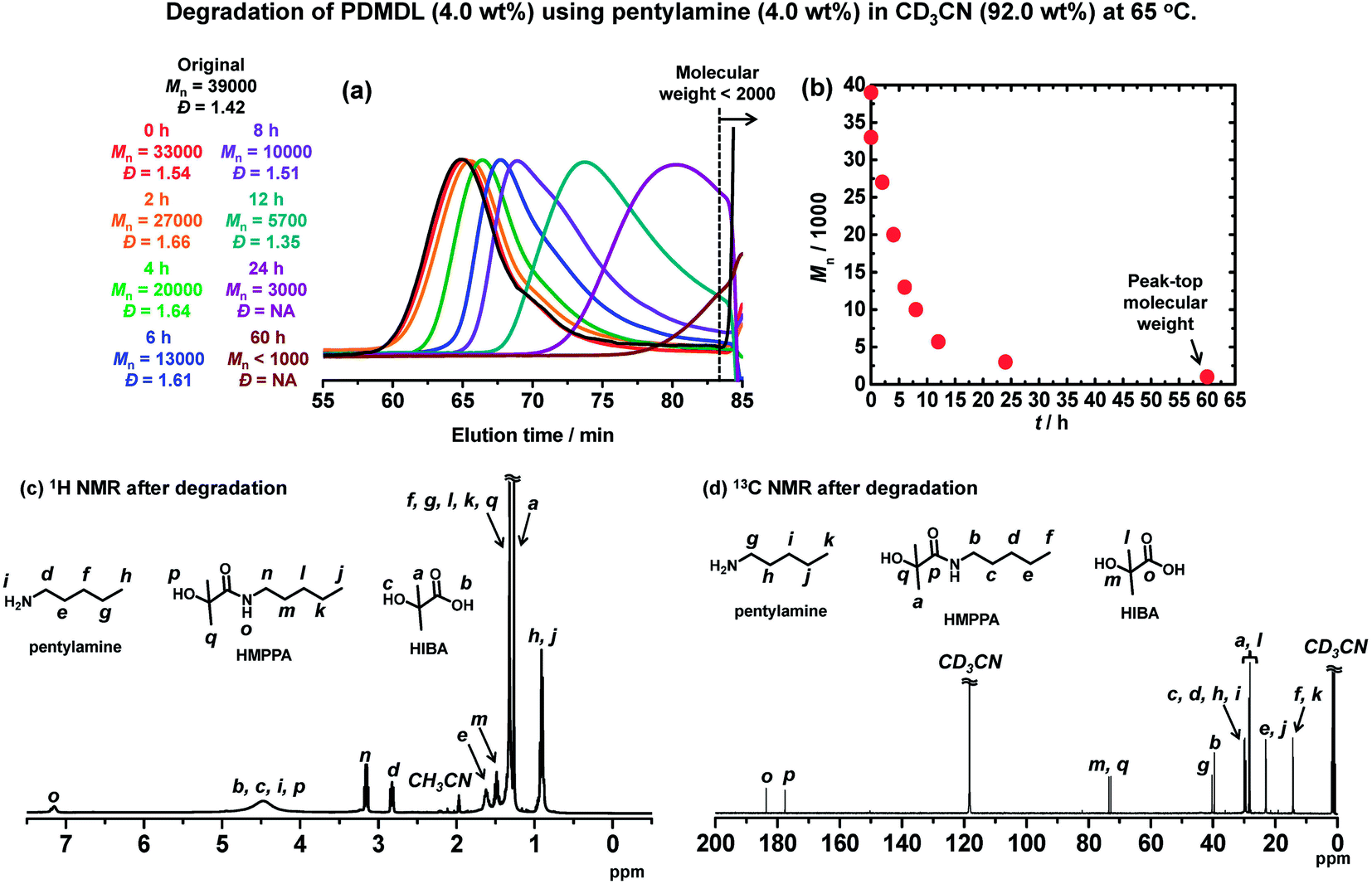 | ||
| Fig. 4 Degradation of PDMDL (Mn = 39000, Đ = 1.42, 4.0 wt%, 1 equiv. of the DMLD unit) using pentylamine (4.0 wt%, 1.5 equiv.) in CD3CN (92.0 wt%) at 65 °C. (a) GPC chromatograms and (b) plot of Mnvs. degradation time. The 0 h sample in (a) is a sample right after the addition of pentylamine. (c) 1H NMR and (d) 13C NMR spectra of the reaction mixture after degradation for 60 h (400 MHz for 1H NMR (100 MHz for 13C NMR) in CD3CN). | ||
The 1H (Fig. 4c) and 13C (Fig. 4d) NMR spectra of the reaction mixture for 60 h show no polymer (PDMDL) signals (no signal at 2.38–3.13 ppm for the backbone CH2 protons in the 1H NMR spectrum or no signal at 174 ppm for the ester carbon of the DMDL unit in the polymer in the 13C NMR spectrum) (the 1H and 13C NMR spectra of (non-degraded) PDMDL in the studied CD3CN are given in Fig. S39 and S40 in ESI†). A possible mechanism is given in Scheme S2 in ESI.† We observed 2-hydroxy-2-methyl-N-pentylpropanamide (HMPPA) (product), HIBA (product), and pentylamine (remaining reactant) (Fig. 4c and d), which were identified from the spectra of the isolated HMPPA, HIBA, and pentylamine (Fig. S41 and S42 in ESI†). The observation of HMPPA supports the proposed amine-assisted degradation scheme (Scheme S2 in ESI†). The degradation was carried out without removing moisture. HIBA (Scheme 4) was generated probably because of the presence of moisture, which might provide hydroxide in the presence of the amine. However, similar to the NaOH-assisted degradation, no other products than HMPPA and HIBA were observed in the 1H and 13C NMR spectra (Fig. 4c and d) for an unclear reason.
Although the degradation mechanism is yet to be definitive, this experiment demonstrated that PDMDL degraded even with a milder base (amine). The facile operation (no need to remove moisture) and mild temperature (65 °C) are attractive.
Degradation of PPEGMA-r-PDMDL
Besides the PDMDL homopolymer, we studied the degradation of a PPEGMA-r-PDMDL random copolymer, where PPEGMA is poly(ethylene glycol) methyl ether methacrylate. This random copolymer is hydrophilic and soluble in water. We dissolved a PPEGMA-r-PDMDL (Mn = 39000, Đ = 2.22, and FDMDL = 17%) (0.06 g, 1 wt%, 1 equiv. of the DMDL monomer unit) in a 0.9 M KOH aqueous solution (6.0 g, 99 wt%, 67 equiv. of KOH) and monitored the degradation at room temperature for 23 h. Fig. 5a and b show the GPC chromatograms and the plot of Mnvs. time. The original Mn (= 39000) value of the copolymer became approximately a half (Mn = 21000) for 1 h, approximately 1/3 (Mn = 13000) for 8 h, and approximately a quarter (Mn = 9300) for 23 h. To confirm that the observed decrease in the Mn value was not ascribed to the pendant chain cleavage of the PEGMA units, we studied the degradation of a PPEGMA homopolymer (Mn = 72000 and Đ = 2.82) in the same condition. After 24 h, the Mn value decreased from 72000 to only 70000 (Fig. S43 in ESI†), confirming no significant pendant chain cleavage. Thus, the observed degradation (significant decrease in the Mn value) of the PPEGMA-r-PDMDL is ascribed to the main-chain cleavage of the DMDL units in the polymer. The FDMDL value of the PPEGMA-r-PDMDL was 17%, meaning that a DMDL unit was incorporated in every approximately 6 PEGMA units on average. Upon the full main-chain cleavage of the DMDL units, the Mn value may decrease to 1800 (6 units of PEGMA). The observed Mn value (=9400) after 23 h of the reaction was larger than 1800, possibly because of a compositional drift during the polymerization (smaller DMDL compositions in the polymers generated at an early stage of polymerization and larger DMDL compositions in those generated at a later stage of polymerization).46
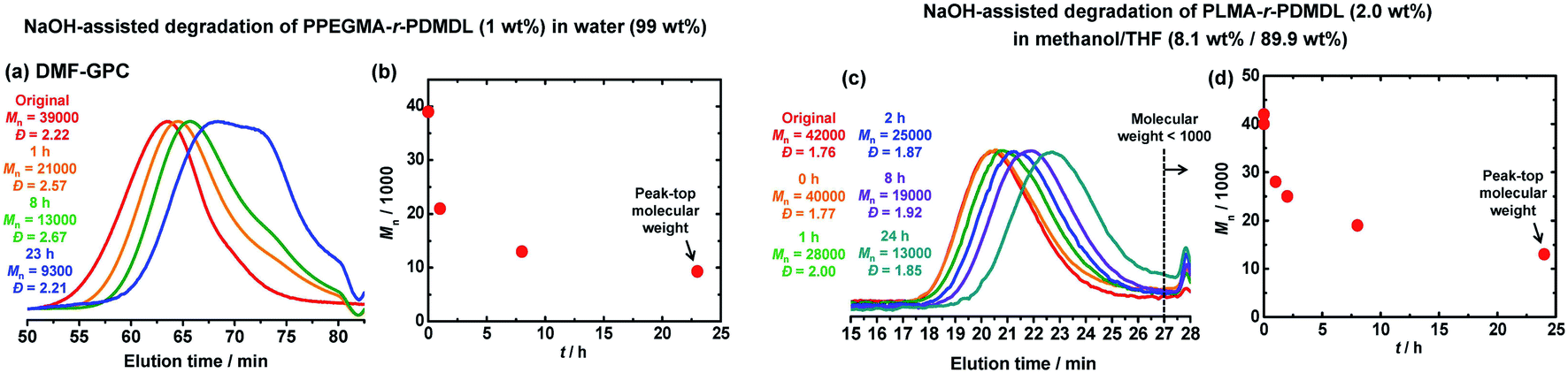 | ||
| Fig. 5 Degradation of PPEGMA-r-PDMDL (Mn = 39000, Đ = 2.22, and FDMDL = 17%) (1 wt%) using 67 equiv. of KOH in water (99 wt%) at room temperature: (a) GPC chromatograms and (b) plot of Mnvs. time. Degradation of PLMA-r-PDMDL (Mn = 42000, Đ = 1.76, and FDMDL = 16%) (1.8 wt%) with 3.5 equiv. of KOH in a mixture of methanol (8.1 wt%) and THF (89.9 wt%) at room temperature; (c) GPC chromatograms and (d) plot of Mnvs. time. | ||
Degradation of PLMA-r-PDMDL
We also studied the degradation of a PLMA-r-PDMDL random copolymer, where PLMA is poly(lauryl methacrylate). This random copolymer is hydrophobic. We used a mixture of methanol and THF to dissolve this hydrophobic copolymer. We dissolved a PLMA-r-PDMDL (Mn = 42000, Đ = 1.76, and FDMDL = 16%) (0.04 g, 2.0 wt%, 1 equiv. of the DMDL monomer unit) in a mixture of a 0.9 M KOH methanol solution (0.16 g, 8.1 wt%, 3.5 equiv. of KOH) and THF (1.78 g, 89.9 wt%) and monitored the degradation at room temperature for 24 h (Fig. 5c and d). The original Mn (= 42000) value of the copolymer became approximately its 60% (Mn = 25000) for 2 h, approximately its 45% (Mn = 19000) for 8 h, and approximately its 30% (Mn = 13000) for 24 h. Thus, the degradation of the PLMA-r-PDMDL successfully occurred.
Recovery of HIBA via NaOH-assisted degradation of PDMDL
HIBA is the starting material (compound 1 (R1 = CH3) in Scheme 3) of the DMDL monomer. Hence, we attempted to recover HIBA after the NaOH-assisted degradation of PDMDL, use the recovered HIBA to synthesize DMDL monomer, and use this monomer to synthesize PDMDL for demonstrating the concept of the recycling of PDMDL (Scheme 3). As mentioned, four of the six carbons of the DMDL monomer are recycled via the recovery of HIBA.A PDMDL (Mn = 56000 and Đ = 2.47) (1.0 g, 1 equiv. of the DMDL monomer unit) was dissolved in a mixture of a 0.9 M KOH methanol solution (28 g, 4 equiv. of KOH) and THF (300 g) and was stirred at room temperature for 1 h. The mixture of methanol and THF was used instead of a mixture of DMSO and water (Fig. 3), because DMSO is difficult to evaporate (in the evaporation process below). KOH was used instead of NaOH (Fig. 3), because KOH is soluble in the mixture of methanol and THF. After the reaction, acidic water and diethyl ether were added to the reaction mixture. HIBA was more soluble in the organic phase than in the aqueous phase in an acidic condition. After the extraction with diethyl ether three times, the collected organic layer was evaporated. The 1H and 13C NMR spectra (Fig. 6) show that the compound extracted in the organic layer was nearly pure HIBA (we observed only HIBA). We collected 0.39 g of HIBA. The theoretically recoverable maximum amount of HIBA from 1 g of PDMDL is 0.81 g. Thus, the recovery yield was 48% (= (0.39 g)/(0.81 g)).
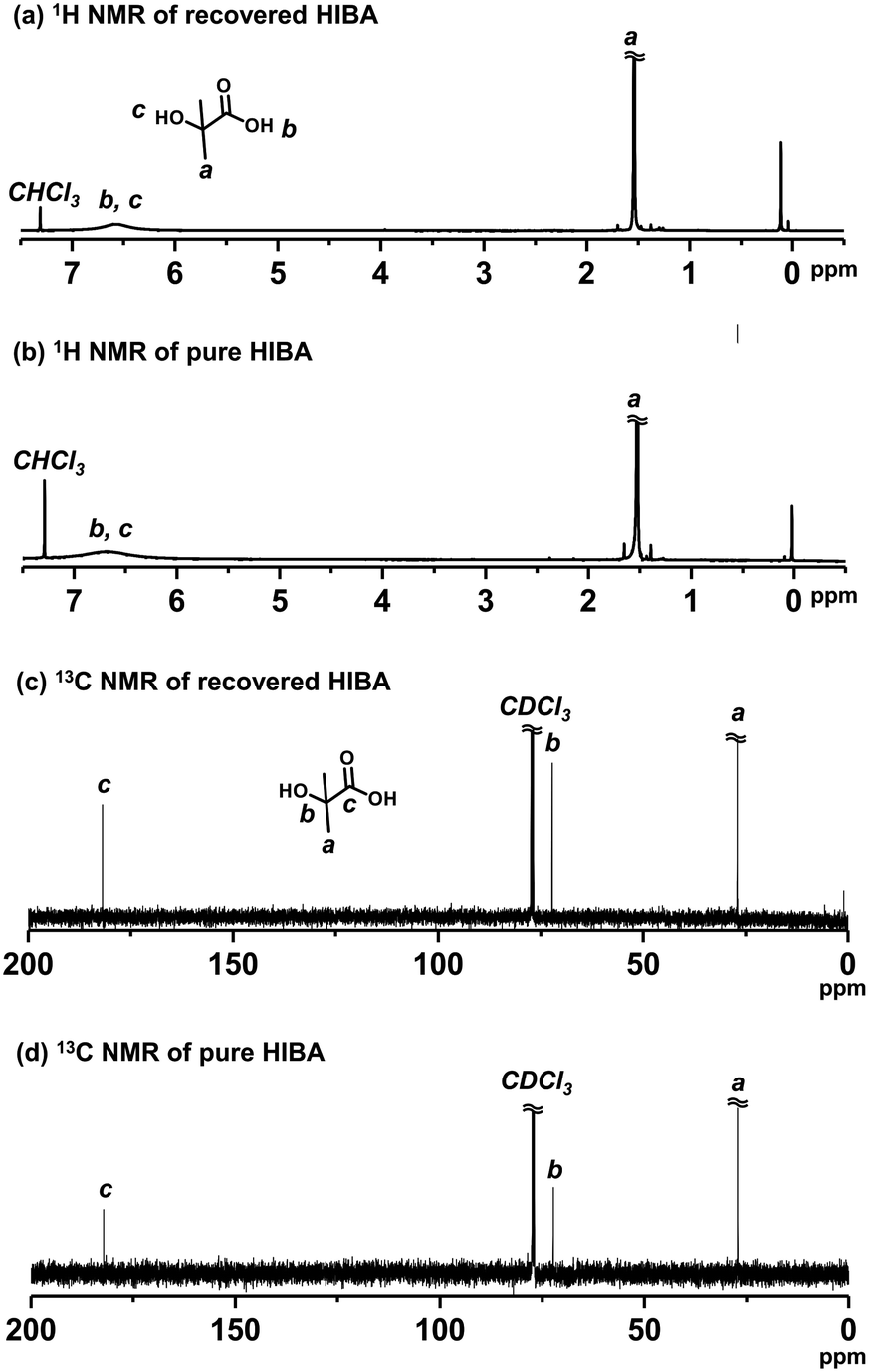 | ||
| Fig. 6 1H NMR spectra of (a) recovered HIBA and (b) pure HIBA. 13C NMR spectra of (c) recovered HIBA and (d) pure HIBA. (400 MHz for 1H NMR (100 MHz for 13C NMR) in CDCl3). | ||
We used the recovered HIBA to synthesize DMDL (Fig. S45–49 in ESI†). We carried out a polymerization of this DMDL (100 equiv.) with AIBN (0.5 equiv.) at 70 °C for 6 h (in the same condition as Table 1, entry 2) and obtained a PDMDL with Mn = 52000 and Đ = 2.32 with an 80% monomer conversion. Thus, we successfully experimentally demonstrated the concept of the chemical recycling of PDMDL.
Conclusions
We successfully synthesized polymers using two new CKHE monomers (DMDL and PhDL). The two monomers were synthesized in high yields (80–90%), which is an attractive feature. DMDL generated homopolymers, and DMDL and PhDL were copolymerized with important families of vinyl monomers, i.e., methacrylates, acrylates, styrene, acrylonitrile, vinyl pyrrolidinone, and acrylamide, and various functional methacrylates and acrylate, exhibiting a wide scope of the accessible polymers. The obtained homopolymers and random copolymers of DMDL degraded in the presence of a hydroxide (NaOH or KOH) and an amine (pentylamine) at mild temperatures (room temperature to 65 °C). The degradation of the PDMDL homopolymer generated HIBA. We recovered HIBA, used the recovered HIBA to synthesize DMDL monomer, and used this monomer to synthesize PDMDL, demonstrating the concept of the chemical recycling of PDMDL. HIBA is not only an ingredient to regenerate DMDL but also a useful ingredient of other important chemicals such as methacrylic acid, tetramethylglycolide, and chloro- and amino-derivatives of HIBA47,48 and can serve for a range of chemical recycling. The degradability and chemical recyclability of PDMDL may be useful for circular materials economy.Data availability
All supporting data have been included in the ESI.†Author contributions
X. Y. Oh performed all experiments. A. Goto and X. Y. Oh contributed to the conception of the experiments, discussion of the results and preparation of the manuscript. Y. Ge assisted in monomer synthetic work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by National Research Foundation (NRF) Investigatorship in Singapore (NRF-NRFI05-2019-0001) and Research, Innovation and Enterprise 2020 Advanced Manufacturing and Engineering Industry Alignment Fund-Pre-Positioning Programme (RIE2020-AME-IAF-PP) of Agency for Science, Technology and Research (A*STAR) (A1786a0029).References
- N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Chem. Rev., 2016, 116, 2602–2663 CrossRef CAS PubMed.
- M. Hong and E. Y. X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- E. Guegain, J. Tran, Q. Deguettes and J. Nicolas, Chem. Sci., 2018, 9, 8291–8306 RSC.
- T. P. Haider, C. Volker, J. Kramm, K. Lanfester and F. R. Wurm, Angew. Chem., Int. Ed., 2019, 58, 50–62 CrossRef CAS PubMed.
- K. H. Paek and S. G. Im, Green Chem., 2020, 22, 4570–4580 RSC.
- K. W. Meereboer, M. Misra and A. K. Mohanty, Green Chem., 2020, 22, 5519–5558 RSC.
- A. Barron and T. D. Sparks, iScience, 2020, 23, 101353 CrossRef CAS PubMed.
- Y. Xu, S. Sen, Q. Wu, X. Zhong, R. H. Ewoldt and S. C. Zimmerman, Chem. Sci., 2020, 11, 3326 RSC.
- J. Yang and Y. Xia, Chem. Sci., 2021, 12, 4389 RSC.
- A. Tardy, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, Chem. Rev., 2017, 117, 1319–1406 CrossRef CAS PubMed.
- A. W. Jackson, Polym. Chem., 2020, 11, 3525–3545 RSC.
- J. Folini, W. Murad, F. Mehner, W. Meier and J. Gaitzsch, Eur. Polym. J., 2020, 134, 109851 CrossRef CAS.
- T. Pesenti and J. Nicolas, ACS Macro Lett., 2020, 9, 1812–1835 CrossRef CAS.
- J. Folini, C.-H. Huang, J. C. Anderson, W. P. Meier and J. Gaitzsch, Polym. Chem., 2019, 10, 5285 RSC.
- W. Bailey, Z. Ni and S.-R. Wu, J. Polym. Sci., Polym. Chem. Ed., 1982, 20, 3021–3030 CrossRef CAS.
- A. Tardy, J.-C. Honoré, D. Siri, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, Polym. Chem., 2017, 8, 5139–5147 RSC.
- G. G. Hedir, M. C. Arno, M. Langlais, J. T. Husband, R. K. O'Reilly and A. P. Dove, Angew. Chem., Int. Ed., 2017, 56, 9178–9182 CrossRef CAS PubMed.
- J. Tran, T. Pesenti, J. Cressonnier, C. Lefay, D. Gigmes, Y. Guillaneuf and J. Nicolas, Biomacromolecules, 2019, 20, 305–317 CrossRef CAS PubMed.
- J.-B. Lena, A. W. Jackson, L. R. Chennamaneni, C. T. Wong, Y. Andriani, P. Thaniyot and A. M. V. Herk, Macromolecules, 2020, 53, 3994–4011 CrossRef CAS.
- A. W. Jackson, L. R. Chennamaneni and P. Thoniyot, Eur. Polym. J., 2020, 122, 109391 CrossRef CAS.
- M. C. D. Carter, A. Hejl, S. Woodfin, B. Einsla, M. Janco, J. DeFelippis, R. J. Cooper and R. C. Even, ACS Macro Lett., 2021, 10, 591–597 CrossRef CAS.
- T.-Y. Zeng, L. Xia, Z. Zhang, C.-Y. Hong and Y.-Z. You, Polym. Chem., 2021, 12, 165 RSC.
- S. Komatsu, T. Sato and A. Kikuchi, Polym. J., 2021, 53, 731–739 CrossRef CAS.
- W. Bailey, Z. Ni and S.-R. Wu, Macromolecules, 1982, 15, 711–714 CrossRef CAS.
- F. Xie, X. Deng, D. Kratzer, K. C. K. Cheng, C. Friedmann, S. Qi, L. Solorio and J. Lahann, Angew. Chem., Int. Ed., 2017, 56, 203–207 CrossRef CAS PubMed.
- C. Zhu and J. Nicolas, Polym. Chem., 2021, 12, 594–607 RSC.
- W. Bailey, S.-R. Wu and Z. Ni, Makromol. Chem., 1982, 183, 1913–1920 CrossRef CAS.
- M. R. Hill, E. Guégain, J. Tran, C. A. Figg, A. C. Turner, J. Nicolas and B. S. Sumerlin, ACS Macro Lett., 2017, 6, 1071–1077 CrossRef CAS.
- J. Tran, E. Guégain, N. Ibrahim, S. Harrisson and J. Nicolas, Polym. Chem., 2016, 7, 4427–4435 RSC.
- E. Guegain, C. Zhu, E. Giovanardi and J. Nicolas, Macromolecules, 2019, 52, 3612–3624 CrossRef CAS.
- A. Kazama and Y. Kohsaka, Polym. Chem., 2019, 10, 2764–2768 RSC.
- R. Friary, J. Heterocycl. Chem., 1978, 15, 63–64 CrossRef CAS.
- O. Igglessi-Markopoulou, G. Athanasellis, A. Detsi, K. Prousis and J. Markopoulos, Synthesis, 2003, 2015–2022 CrossRef.
- L. Zheng, D. Zheng, Y. Wang, C. Yu, K. Zhang and H. Jiang, Org. Biomol. Chem., 2019, 17, 9573–9577 RSC.
- P. J. Ma, F. Tang, Y. Yao and C. D. Lu, Org. Lett., 2019, 21, 4671–4675 CrossRef CAS PubMed.
- M. Haim-Zada, A. Basu, T. Hagigit, R. Schlinger, M. Grishko, A. Kraminsky, E. Hanuka and A. J. Domb, Biomacromolecules, 2016, 17, 2253–2259 CrossRef CAS PubMed.
- A. L. J. Beckwith, S. Brumby and C. L. L. Chai, J. Chem. Soc., Perkin Trans. 2, 1992, 2, 2117–2121 RSC.
- A. Rudin and P. Choi, in The Elements of Polymer Science & Engineering, ed. A. Rudin and P. Choi, Academic Press, 3rd edn, 2013, ch. 9, pp. 402–406 Search PubMed.
- S. Agarwal, Polym. J., 2007, 39, 163–174 CrossRef CAS.
- J. Undin, T. Illanes, A. Finne-Wistrand and A.-C. Albertsson, Polym. Chem., 2012, 3, 1260–1266 RSC.
- H. Wickel, S. Agarwal and A. Greiner, Macromolecules, 2003, 36, 2397–2403 CrossRef CAS.
- J. Huang, R. Gil and K. Matyjaszewski, Polymer, 2005, 46, 11698–11706 CrossRef CAS.
- H. Wickel and S. Agarwal, Macromolecules, 2003, 36, 6152–6159 CrossRef CAS.
- Polymer Handbook Fourth Edition, ed. J. Brandrup, E. H. Immergut and E. A. Grulke, Interscience Publisher, New York, 1999 Search PubMed.
- Y. Inokuma, T. Yoneda, Y. Ide and S. Yoshioka, Chem. Commun., 2020, 56, 9079–9093 RSC.
- D. Gigmes, P. H. M. Van Steenberge, D. Siri, D. R. D'hooge, Y. Guillaneuf and C. Lefay, Macromol. Rapid Commun., 2018, 1800193 CrossRef PubMed.
- T. Rohwerder and R. H. Müller, Microb. Cell Fact., 2010, 9, 13 CrossRef PubMed.
- M. Pirmoradi and J. R. Kastner, ACS Sustainable Chem. Eng., 2016, 5, 1517–1527 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03560f |
| This journal is © The Royal Society of Chemistry 2021 |