
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanochemistry unveils stress transfer during sacrificial bond fracture of tough multiple network elastomers†
Yinjun
Chen
,
Gabriel
Sanoja
and
Costantino
Creton
 *
*
Laboratoire Sciences et Ingénierie de la Matière Molle, ESPCI Paris, PSL University, Sorbonne Université, CNRS, F-75005 Paris, France. E-mail: costantino.creton@espci.psl.eu
First published on 23rd July 2021
Abstract
The molecular level transfer of stress from a stiff percolating filler to a stretchable matrix is a crucial and generic mechanism of toughening in soft materials. Yet the molecular details of how this transfer occurs have so far been experimentally unreachable. Model multiple network elastomers containing spiropyran (SP) force sensors incorporated into the stiff filler network or into the stretchable matrix network are used here to detect and investigate the mechanism of stress transfer between distinct populations of polymer strands. We find that as the filler network progressively breaks by random bond scission, there is a critical stress where cooperative bond scission occurs and the macroscopic stretch increases discontinuously by necking. Surprisingly, SP molecules reveal that even in the necked region both filler and matrix chains share the load, with roughly 90% of the SPs force-activated in the filler chains before necking still being loaded in the necked region where significant activation of the SP incorporated into the matrix chains occurs. This result, where both networks remain loaded upon necking, is qualitatively consistent with the model proposed by Brown, where holes or microcracks are formed in the stiff regions and are bridged by stretched matrix chains. Detection of merocyanine (i.e. activated SP) fluorescence by confocal microscopy shows that such microcrack formation is also active at the crack tip even for materials that do not exhibit macroscopic necking. Additionally, we demonstrate that when the ethyl acrylate monomer is replaced by hexyl methacrylate in the first network, preventing molecular connections between the two networks, the stress transmission is less efficient. This study outlines the different roles played by these multiple networks in the onset of fracture and provides molecular insights for the construction of molecular models of fracture of elastomers.
1. Introduction
Elastomers are widely used in industrial applications because of their unique combination of fully reversible deformation and high fracture toughness. The classic strategy to increase their stiffness and improve their fracture toughness while maintaining a high reversible elasticity involves incorporating nanofillers,1–5 mainly carbon black6,7 and nanosilica8,9 in industry and more recently graphene10–12 and carbon nanotubes10 at the academic level. Yet, these nanofillers aggregate and scatter light, making the preparation of tough, transparent, and highly elastic elastomers (with the notable exception of strain-crystallizing natural rubber) still challenging. In the last 20 years new materials have been designed that might just fill that gap,13–16 and some generic multiscale toughening strategies have been proposed.1,17 These strategies involve the incorporation of sacrificial bonds, covalent14,15,18,19 or more commonly non-covalent,16,20–24 to increase the energy needed to grow a crack,25 as well as the extensibility and fracture toughness. Microscopic26,27 and continuum mechanical models28,29 based on interpenetrated networks have been proposed to describe the toughening mechanism but while these models are qualitatively consistent with the observed behavior, they are based on untested molecular hypotheses.The key question can be appreciated in the following way: for the toughening of interpenetrated networks to be effective there needs to be a mechanism to transfer the load from a stiff to a stretchable structure capable of sustaining the load without catastrophic failure. Intuitively, the more delocalized and random the process of failure of the stiff structure is, the less likely is the creation of large weak spots that eventually nucleate cracks. Yet the molecular requirements to obtain that random weakening without creating macroscopic cracks remain unknown for lack of suitable experimental techniques and model systems.
The design of tough unfilled elastomers based on multiple networks,19,30–32 synthesized via an approach pioneered in hydrogels by Gong and coworkers,14,33–35 bears some clear resemblance to the design of nanofilled elastomers (where the role of the stiff network is played by the percolating network of nanofillers2) and may be a good model system to investigate their toughening at the molecular level. In multiple network elastomers, the first network is highly crosslinked and acts as a percolating filler (i.e., stiff structure) into a loosely crosslinked matrix (i.e., stretchable structure). Thus, the covalent bonds in the first network serve as sacrificial bonds that dissipate energy when ruptured during loading. In a particularly insightful model,26 Brown proposed the following idea to explain why such a molecular design would lead to toughening: at a critical “yield stress” internal cracks nucleate in the stiff structure creating damaged zones but these zones do not coalesce, like crazes in glassy polymers.36 At the critical point, final failure occurs because the damage zone becomes sufficiently wide to create a stress concentration at the damage zone tip and grow a crack in the damaged region. Such a model is based on the growth of cracks into a craze (a plasticity driven damage zone) into a polymer glass37,38 and is qualitatively consistent with the observed toughening but details remain difficult to verify experimentally.
Thanks to the development of mechanochemistry as an effective tool to detect the force and scission of polymer chains, we can revisit the question of molecular fracture in elastomers. The molecules used in mechanochemistry, called mechanophores, are force-sensitive and can convert mechanical stimuli into observables like luminescence,39–41 fluorescence,42 and color.43 Spiropyran (SP), a classic mechanochromic mechanophore, has in particular attracted lots of attention due to its distinct change in color and fluorescence upon activation and has been used as a molecular probe to sense stress and show damage inside polymeric materials by many groups, including those of Moore and Sottos,43–45 Craig,46,47 Weng48–51 and Qiao.52
When SP is incorporated into a polymer chain and elongated using single molecule force spectroscopy (SMFS) (tip velocity of 300 nm s−1), it turns into merocyanine (MC) at forces above 240 pN.53 Although in our acrylate material the activation force may be a bit different, we did not observe any strain rate dependence of the activation (see Fig. S5†) and will consider that this is a reasonable approximation. In addition to the examples given above, the mechanosensitivity of SP and of MC has also been used to map the stress history of elastomers,32 as these turn from colourless to blue upon loading and from blue to purple upon unloading. While the colorless-to-blue transition is clearly due to the formation of MC,43,53,54 the molecular origin of the blue-to-purple transition upon unloading (blue in the loaded state and purple in the unloaded state) has been suggested to be the isomerization of MC because of its low activation energy32,46,55 but direct spectroscopic proof has not been reported.
In a previous report,15 the luminescence of a dioxetane mechanophore used as a crosslinker in multiple network elastomers showed conclusively the occurrence of bond scission during uniaxial extension and crack propagation. This molecular evidence was instrumental in proving that the progressive increase in size of the region around the crack tip where bond scission (and hence energy dissipation) occurs was an effective mechanism to increase the fracture energy of elastomers. Yet the limited spatial resolution of the experiments precluded more advanced interpretations on how the load redistributes among the undamaged regions upon bond scission.
The question of how the load redistributes upon damage of the first network was first experimentally addressed in analogous double network (DN) hydrogels by Gong and coworkers.33 They observed that some DN hydrogels exhibited a stable necking process in uniaxial extension33 and upon unloading and reloading, the necked region significantly softens and has an elastic modulus and tensile behavior almost identical to that of the second network alone. Based on this evidence, they proposed a mechanism of transfer of load from the progressively broken stiff filler network to the stretchable matrix.33,35 Intrigued by this unusual behavior, they investigated the homogeneity of the structure in the neck by Small Angle Neutron Scattering (SANS) by deuterating the matrix network.56 A pronounced scattering peak revealed the existence of matrix concentration inhomogeneities along the tensile direction spaced by a characteristic length scale around 1.5 μm. Yet the applied strain was only 50% and in a pure shear geometry making it difficult to ascertain that this SANS length scale was the same as that potentially formed in the necked region in uniaxial tension at strains of more than 500%. Nevertheless based on the mechanical macroscopic evidence, Gong and coworkers suggested a plausible mechanism of DN fracture where the stiff filler network breaks in separate islands that are physically connected by the second network as schematically illustrated in Fig. 1.33,35,57,58 Such hypothesis, also based on the very heterogeneous structure of the gel filler network of poly(2-acrylamido-2-methyl-1-propanesulfonic acid) as detected by light scattering,59 implies that most of the filler network chains in the island should be unloaded after necking.
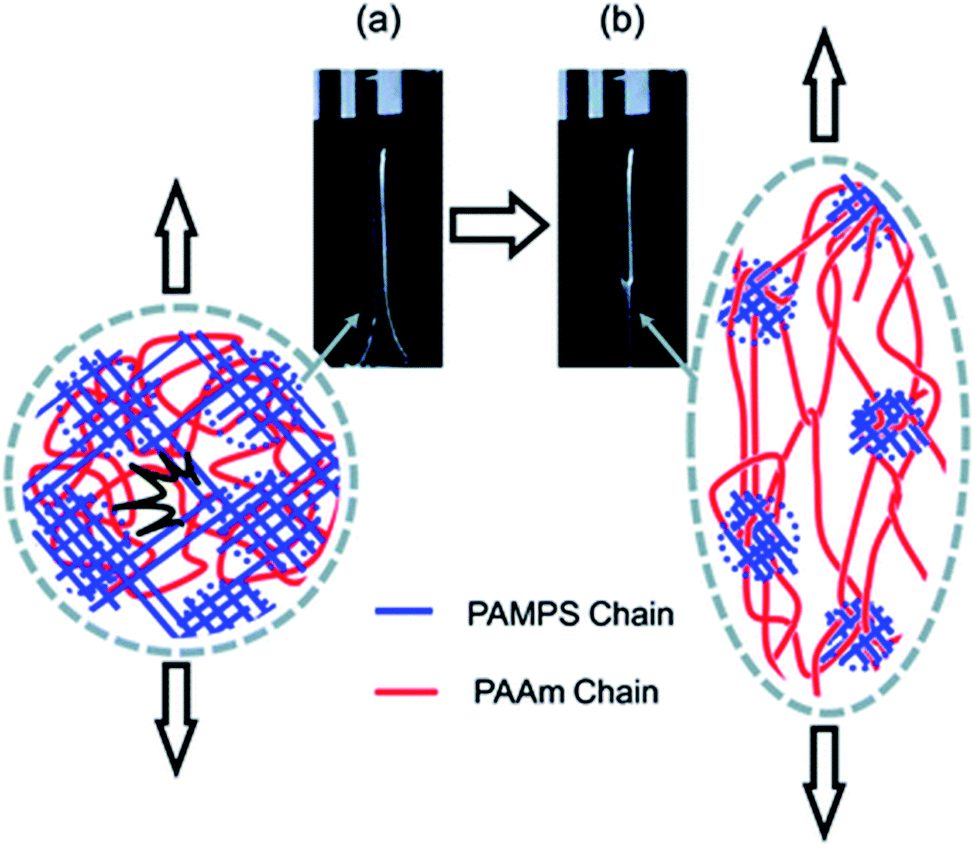 | ||
| Fig. 1 Scheme of the network structure of the DN gel before (a) and after (b) necking as seen by Gong. Above a critical stress, the filler network fractures into separate islands and the DN gel becomes much softer after the necking. Reprinted with permission from ref. 35. | ||
Our own group discovered that some multiple network elastomers also undergo elastic necking in uniaxial tension19 and addressed the question of bond scission, and indirectly load transfer. In contrast to the case of DN gels, we found that the necked region in the elastomers was softer than the original undamaged material but still stiffer than the matrix network alone. Additionally using the time resolved capability of the dioxetane mechanoluminescent crosslinkers we quantitatively compared the sacrificial bond scission events of the first network occurring up to the necking point, and during the propagation of the necking front. To our surprise as many sacrificial bonds per unit volume appeared to break before the necking point (in a regime where the material does not soften much) than during the propagation of the necking front (where the material softens and increases its extensibility dramatically). These observations both suggest that the picture of breaking into well-separated islands proposed by Gong for gels may not be general or be too simplistic.
More recently, we labeled the filler network of multiple network elastomers with SP and quantified the stress ahead of the crack tip before propagation using the color change from (i) colorless to blue upon loading,31 and (ii) blue to purple upon unloading.32 We found that the shade of purple depended both on the concentration of MC formed upon loading and on the average force on the chains. Note that such a connection between the network architecture and macroscopic activation stress was also carried out in similar materials by Qiao et al. to demonstrate that an increasing prestretch of the filler network leads to an activation of SP into MC at a lower stretch level.52
Here we use SP mechanophores to gain direct insight into the average force experienced by the crosslinkers of multiple network elastomers. The novelty of the present work is that we relate the force-induced molecular activation of SP to the load sustained by the crosslinkers of the filler and matrix networks in uniaxial extension and ahead of the crack tip prior to crack propagation. This information is unprecedented and instrumental to build a molecular model of stress transfer in tough elastomers.
2. Results
2.1 Synthesis and mechanical properties of multiple network elastomers
Multiple network elastomers were synthesized from the same filler network, a densely crosslinked single network composed of 99.5 mol% ethyl acrylate (EA) monomer and 0.5 mol% 1,4-butanediol diacrylate (BDA) crosslinker (family 1 in Table 1), that was subsequently swollen to polymerize loosely crosslinked networks composed of 99.99 mol% EA and 0.01 mol% BDA. Details of the synthesis have been previously reported,15,19,31,32,60 and are summarized in the ESI.† These multiple networks consist of a filler network (the first network) and a matrix network (the second/third/fourth networks). The filler network acts as a sacrificial network and is damaged by covalent bond scission as the material is stretched. This damage leads to the mechanical reinforcement seen in the stress–strain curves shown in Fig. 2 and measured in uniaxial extension. The single network (SN) EA(1) breaks at a very low stress level and before any strain stiffening is observed, while as the number of polymer networks is increased so does the Young's modulus and stress at break. Double networks (DN) EA(2), triple networks (TN) EA(3) and quadruple networks (QN) EA(4) all display a strain stiffening at large strains as previously described.19,31| Family | SN | DN | TN | QN |
|---|---|---|---|---|
| 1 | EA(1) | EA(2) | EA(3) | EA(4) |
| 2 | EA(1)-1 | EA(2)-1 | EA(3)-1 | EA(4)-1 |
| 3 | EA(1) | EA(2)-2 | EA(3)-2 | EA(4)-2 |
| 4 | EA(1) | EA(2) | EA(3)-3 | EA(4)-3 |
| 5 | HMA(1) | HMA(2)-2 | HMA(3)-2 | HMA(4)-2 |
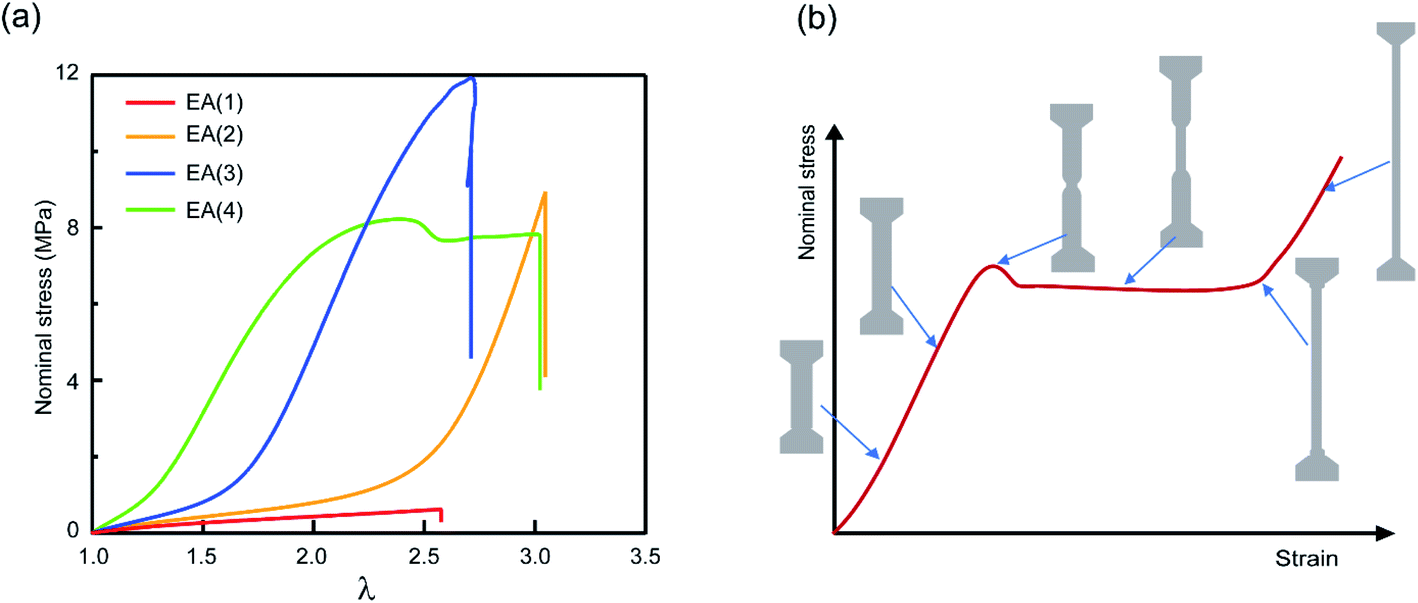 | ||
| Fig. 2 Mechanical properties of multiple network elastomers. (a) Stress–strain curves of multiple network elastomers (family 1 in Table 1); (b) scheme of stress–strain curve for an elastomer displaying a necking phenomenon and the corresponding representations of the dogbone specimen at different levels of stretch. If the neck extends throughout the specimen, the elastomer displays a second strain stiffening in the stress–strain curve. | ||
The striking feature of the data shown in Fig. 2a is that while the extension at break does not change much with the network architecture, the stress at break increases dramatically. As discussed by Millereau et al.19 this effect of network architecture on the stress at break also causes the dramatic increase in fracture energy Γ reported in Table S1.† Interestingly, Fig. 2a reveals that the EA(4) specimen presents a necking phenomenon described by Millereau19 and Gong et al.18,33 and shown schematically in Fig. 2b. As discussed in the introduction this necking is accompanied by an extensive level of bond scission in the filler network followed by a stable load transfer (i.e., stress transmission) between the filler and the matrix.
2.2 Labelling multiple network elastomers with a force-activated mechanophore
To study the stress transmission, SP, a force sensitive molecule, was separately incorporated into the first, second, and third networks of multiple network elastomers having the same mechanical properties (families 2–4 in Table 1). Details of the synthesis of the labelled elastomers have been previously reported15,19,31,60 and are summarized in the ESI.† The compositions of the materials are shown in Table 1, where samples are labeled as A(x)-y, with ‘A’ as the monomer of the first network, ‘x’ the total number of sequential polymerization steps used, and ‘y’ the number of the network in which SP was incorporated. For example, if EA is the monomer used to prepare the filler and matrix networks, a TN elastomer with SP incorporated into the second network will be labeled as EA(3)-2.Fig. 3a shows the color change of EA(4)-1 in uniaxial extension before and after yielding. As discussed in previous work, if the SP is subjected to a sufficient force, it can ring-open and become merocyanine (MC) (see Fig. 3b). This rearrangement is accompanied in an acrylate environment by a change in absorption in the visible range and the appearance of a deep blue color. A similar color change is observed when loading EA(3)-1 (Fig. 3c), though the intensity is somewhat lower. It is noteworthy that a second color change can be seen during unloading of EA(3)-1. The material gradually turns from deep blue into purple, which is even observed in the completely unloaded state of EA(3)-1 (Fig. 3c).
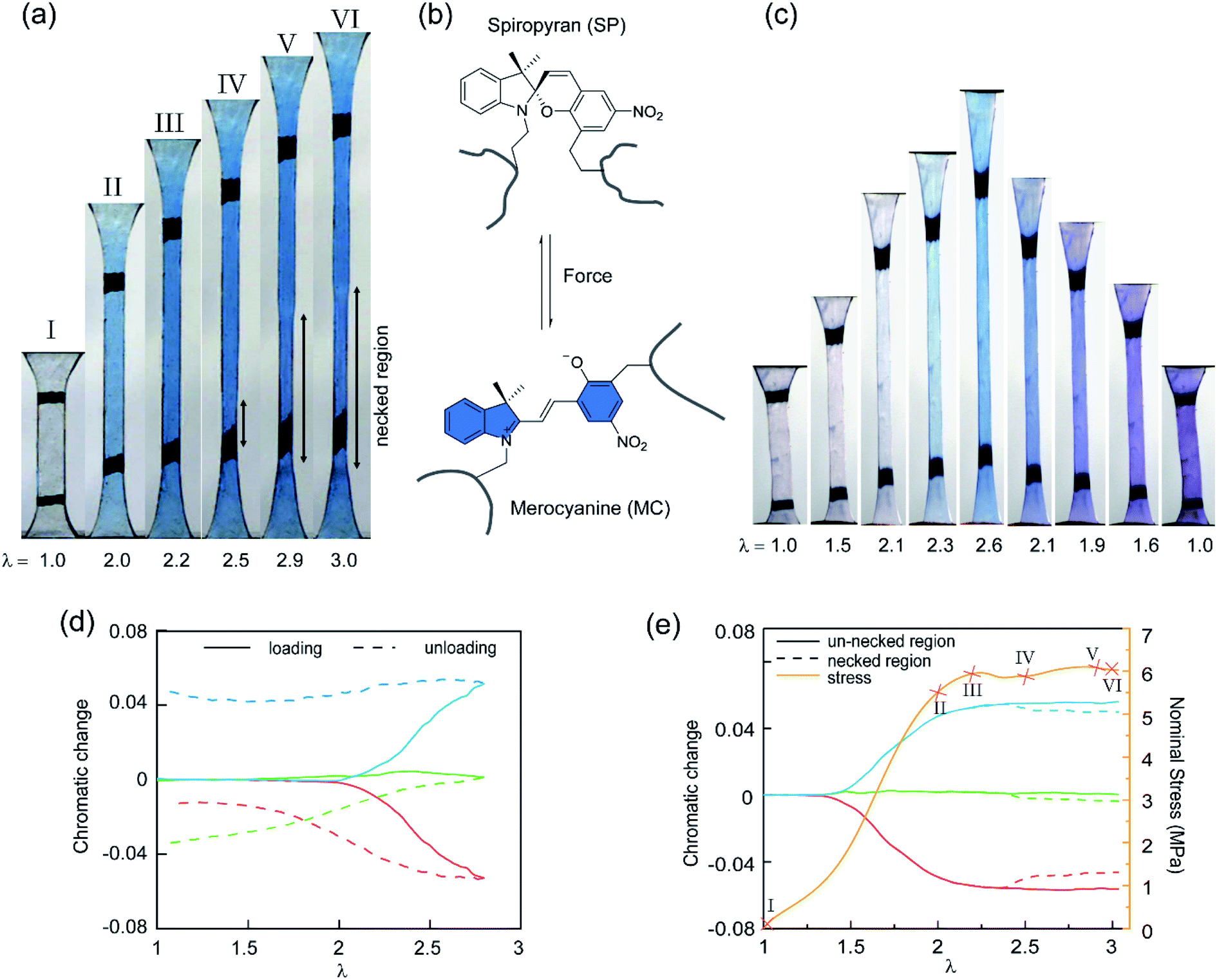 | ||
| Fig. 3 (a) Images of the EA(4)-1 sample showing the color change in uniaxial extension. (b) Scheme of SP transformed into MC under the stimulus of force. (c) Images of the EA(3)-1 showing two different color changes during loading and unloading, respectively. (d) Chromatic changes, ΔRratio, ΔBratio and ΔGratio, quantified with the variation of the chromatic ratios with respect to those in the original (i.e., pristine) unloaded state. It is noteworthy that the onset of chromatic changes, ΔRratio (red), ΔBratio (blue), and ΔGratio (green), of EA(3)-1 in (c) occurs at a stretch λ > 2.0, upon loading (solid lines), and there is an important change in ΔGratio and ΔRratio upon unloading (dashed lines). (e) Stress–stretch curve (yellow) and chromatic change of an EA(4)-1 in the necked (dashed lines) and un-necked (solid lines) regions during uniaxial extension. Images of the EA(4)-1 in (a) are mapped in the stress–stretch curve. | ||
Although the incorporation of SP into multiple network elastomers results in a stress-induced color change, it is challenging to probe differences among the loaded deep blue and unloaded purple regions. Close inspection of Fig. 3a reveals that the necked region of EA(4)-1 shifts from a deep blue to a slightly more purple color (see Video S1†), but the change is not easily distinguishable. To quantify the extent of color change and relate it to the load transfer mechanism, a Red, Green and Blue analysis (RGB analysis) was performed with a methodology previously described in detail.31,32 The basic idea is to define the chromatic change, ΔSratio, for each color channel (i.e., S = R, G, or B) as the variation of the chromatic ratio of the channel intensity, Sratio = S/∑S, to that of the original unloaded state, Sratio,0 = S0/∑S0. As illustrated in Fig. 3d, the blue chromatic change, ΔBratio, is directly proportional to the increasing concentration of MC during loading.31 However, during unloading the MC concentration is assumed to remain constant so that the relative proportion of the MC isomers can be detected from the green chromatic change, ΔGratio.32 Thus, the chromatic change exhibits a completely different behavior between loading (transition from SP to MC) and unloading (presumed isomerization of MC), which can be related to the average load in the elastomer. Note that the transition from colorless to blue is irreversible within the time scale of the experiment while the transition from blue to purple is reversible and detectable within the 40 ms separating two video frames.32
Fig. 3e shows the difference in chromaticity between the necked and unnecked regions in EA(4)-1. It is noteworthy that the chromatic change due to the variation in thickness with uniaxial extension can be neglected based on previous results.31,32 The slight color change from deep blue to purple occurs instantaneously upon appearance of the necked region and is consistent with a decrease in concentration of the loaded MC.32,46 This change in chromaticity likely occurs because some of the polymer strands covalently bonded with MC are unloaded during the necking process and the filler network is in a partially unloaded state. The sensitivity of the green chromatic change ΔGratio to unloading can be further used to estimate the fraction of unloaded polymer strands in the filler network. Interestingly, a comparison of the ΔGratio in the necked region with that in the unloaded and unnecked specimen after cyclic loading reveals that only about 9% of the filler network strands that were still loaded at the onset of the necking have been unloaded by bond scission in the necked region. Thus, most polymer chains in the filler network are still loaded in the neck. In our opinion this observation is not very consistent with the schematic picture proposed in Fig. 1 for DN gels,35 where the filler network breaks into separated clusters in the necked area so that filler network strands would be mostly unloaded and the matrix network would sustain most of the stress. The damage process inside the neck of multiple network elastomers more likely follows the pathway shown in Fig. 4: some damage clearly occurs in the filler network with a local decrease in the crosslinking density. However, the fact that the majority of the MC remains in the loaded state suggests that the filler network, while damaged, remains percolated and capable of sustaining an important fraction of the load.
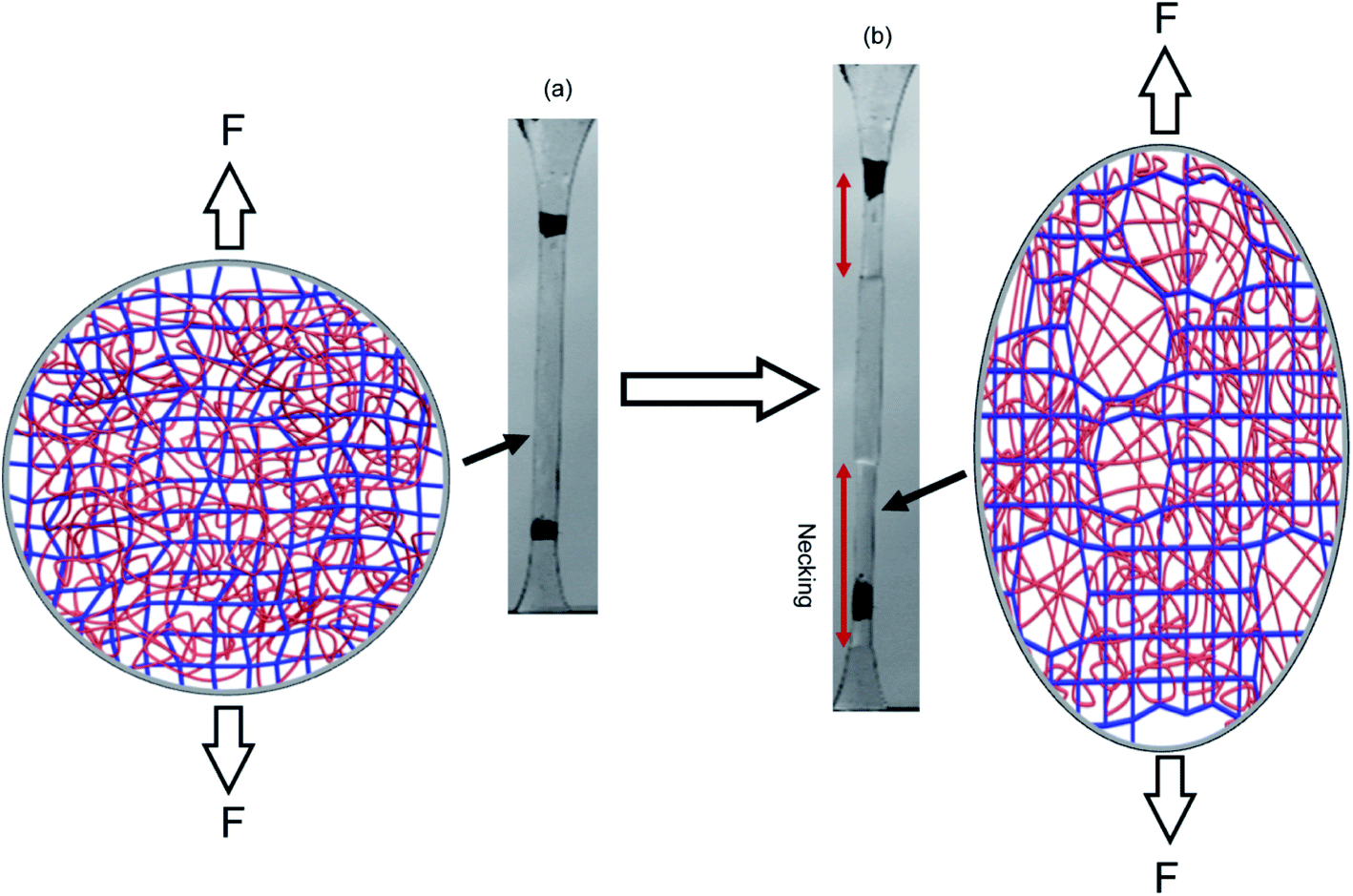 | ||
| Fig. 4 Schematic illustration of the network structure of multiple network elastomers (a) before and (b) after necking. Above a critical stress, the filler network fractures and decreases its crosslinking density but remains connected and capable of sustaining an important fraction of the load. | ||
To explore further the mechanism of stress transmission between the filler and matrix networks, we also synthesized and uniaxially deformed elastomers labeled with SP in the matrix: EA(4)-2 and EA(4)-3. Color changes and mechanical properties are shown in Fig. 5a–d. Note that the necking stress in Fig. 5d scales directly with the areal density of filler network strands along the tensile direction (i.e. the degree of prestretch of the filler network), which is slightly different between the three materials. Renormalized data are available in Fig. S8,† and are in agreement with recent reports from Qiao and co-workers on analogous multiple network elastomers.52 The images extracted from Videos S2 and S3† (Fig. 5a and b) show that the necked region of EA(4)-2 exhibits a distinctive blue color, while that of EA(4)-3 remains clear. More quantitative information can be obtained with an RGB analysis on the three QN elastomers labeled with SP either in the first, second, or third networks. A volume element inside the necked region was chosen to perform the RGB analysis. Results are summarized in Fig. 5c and reveal that the chromatic change of EA(4)-1 occurs at low strain (λ < 1.5), that of EA(4)-2 occurs at the point where the necked region nucleates (i.e., the yield point), and that of EA(4)-3 is negligible. These observations shed some light on how the molecules break and redistribute forces inside multiple network elastomers subject to uniaxial extension.
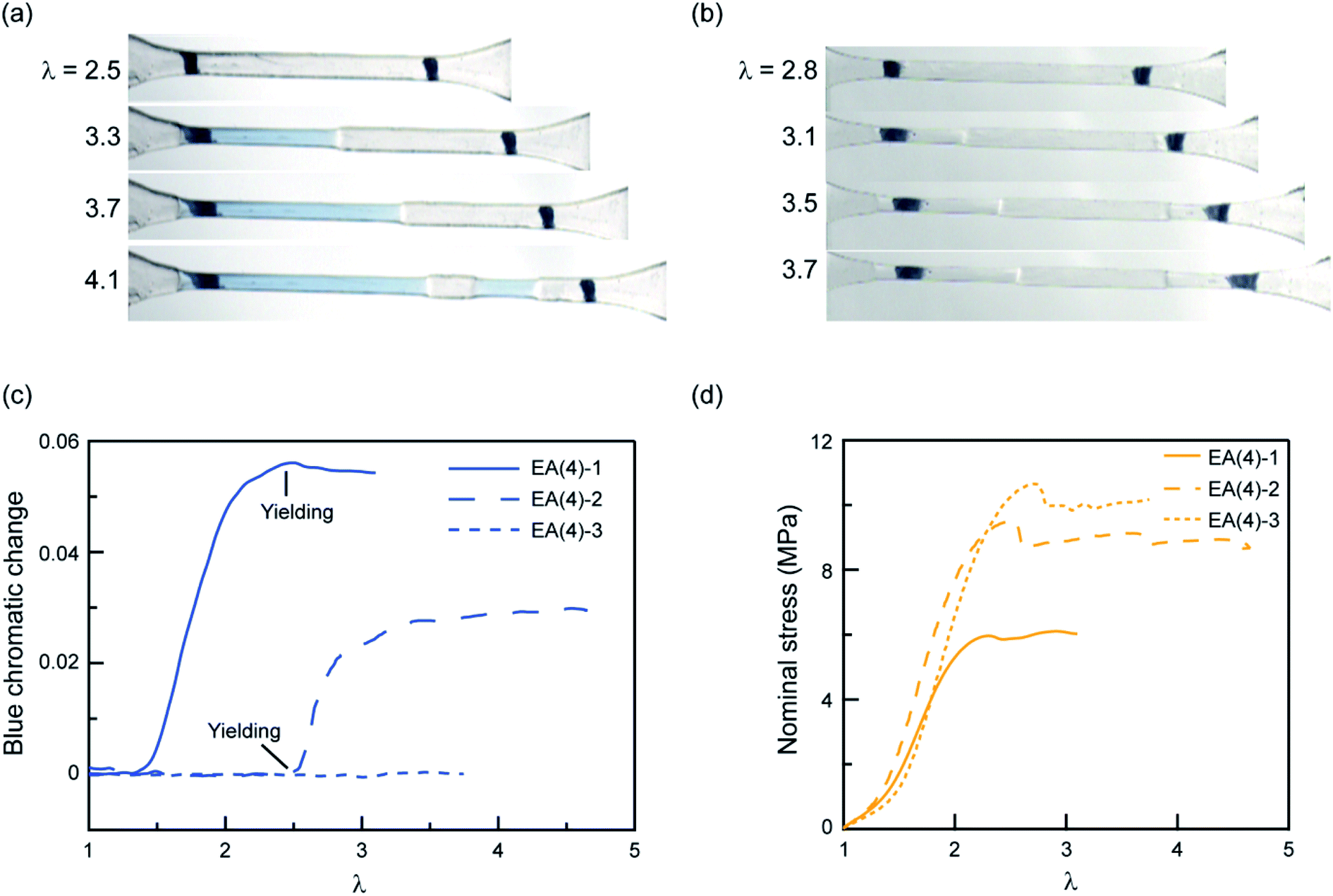 | ||
| Fig. 5 Chromatic change and stress–strain curves for QN elastomers with the SP cross-linker incorporated into the first, second, or third network. Images for (a) EA(4)-2 and (b) EA(4)-3 illustrating color changes during uniaxial tensile tests; (c) chromatic change and (d) stress–strain curves for the QN elastomers EA(4)-1, EA(4)-2 and EA(4)-3. | ||
The uniaxial tension tests carried out above on QN elastomers show that during the necking process the breakup of the stiff, filler network results in a transfer of load mainly to the softer, matrix, second network (i.e., polymerized during the second step). However, this transfer of load does not significantly reduce the fraction of first network strands that are under load, suggesting the creation of open holes or microcracks in a percolated first network (i.e., damaged fish net) as opposed to isolated islands. How these microcracks subsequently open in the first network in the absence of solvent and without changing the material density is an interesting open question.
2.3 The effect of connecting covalent bonds between the first and the second network
The observed color change of the necked region of EA(4)-2 confirms that unloaded (or weakly loaded) second network chains suddenly carry a load as the first network extensively breaks at the yield point and the material necks.As previously pointed out, there are likely some covalent bonds connecting the filler and the matrix networks due to chain transfer reactions61 during the free radical polymerization of EA. The hydrogen in the alpha position of acrylate monomers in the filler network (Fig. 6a) can react with a propagating radical during the polymerization of the matrix networks. In DN hydrogels, Gong and coworkers showed that the presence of this internetwork connection has a significant effect on the mechanical properties.62 In our system, we have the possibility to avoid this effect by synthesizing a filler network based on hexyl methacrylate in place of ethyl acrylate (Fig. 6a).
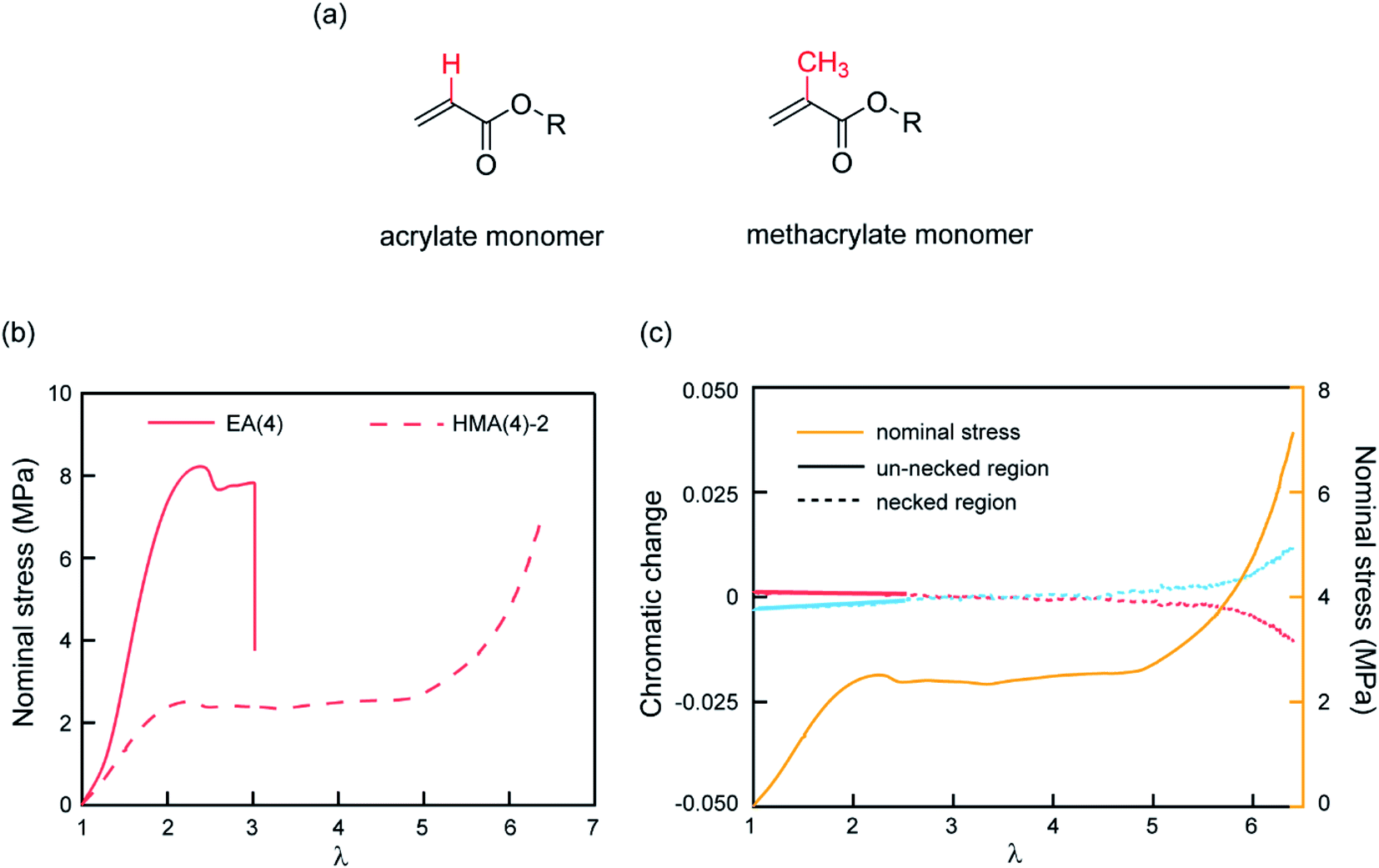 | ||
| Fig. 6 (a) Molecular structure of acrylate and methacrylate monomers; (b) stress–stretch curves of EA(4) and HMA(4)-2; (c) chromatic change curves and stress–stretch curve of HMA(4)-2. After yielding the necked region was used to do RGB analysis and the chromatic change is shown as a dashed line (red and blue). A slight chromatic change, ΔRratio (red) and ΔBratio (blue), is only observed after necking has taken place over the whole gauge length of the dogbone sample (i.e. at the onset of the second strain hardening). | ||
The influence of the presence or absence of internetwork connections on the stress transmission was investigated by comparing EA(4) and HMA(4)-2. Fig. 6b shows that the stress–strain curves of these materials look qualitatively similar with a strain hardening and yielding, but with clear quantitative differences in yield stress and stress at break. In addition, HMA(4)-2 exhibits a second strain hardening at high elongations.
In terms of SP activation, the chromatic change shown in Fig. 6c shows the color change of a sample of HMA(4)-2. A necked region in uniaxial extension is clearly visible and extends to the whole sample before the sample breaks (Fig. S6a†). Interestingly, a slight color change appears only after the necking area extends to the whole sample where a second stress hardening is present, and not during the neck propagation process as in sample EA(4)-2. That point was verified in several independent tensile tests of HMA(4)-2 specimens where materials fail at different degrees of stretching (for details see Fig. S6†).
Comparing the results of EA(4)-2 with those of HMA(4)-2, it is clear that the process of stress transfer between the multiple networks is affected by the presence of internetwork covalent bonds. The difference between the two cases is evident in the necked region: in the case of HMA(4)-2, the second network is not loaded significantly in the necked region, but only when the necked region itself experiences a second strain hardening at λ = 6 (Fig. 6b and c). Although this result may appear puzzling compared to the result obtained for the EA networks one should keep in mind that the onset of the color reports the concentration of the SP that has been exposed to a sufficient force to open the SP cycle. The absence of covalent internetwork connections may favor a relative slippage of the entangled second network through the crosslinked and broken filler network. This partial slip may have reduced the overall stress necessary for necking and the average force seen by the matrix strands below the activation threshold.
2.4 Load transfer mechanisms during the crack propagation process
A central hypothesis of the molecular model proposed by Brown26 is the creation of micro-cracks in the filler network at the crack tip. However, while we have shown in Section 2.2 that for highly prestretched and diluted filler networks this process of micro-crack formation is plausible and load transfer occurs, we have not demonstrated yet whether it occurs more generally at the crack tip of multiple network elastomers that are toughened relative to single networks, but too brittle to display macroscopic necking.In a previous publication we showed that when the SP is incorporated into the filler network, it can be used to map and quantify the nominal stress (in 2D and integrated over the specimen thickness).31 When a prenotched specimen is uniaxially loaded, the stress concentration in front of the crack tip can be computed theoretically (with a material model)63 and visualized experimentally.31,32 The question is then whether this molecular stress transfer process, that in principle should be detectable with the same tools, is indeed relevant for the reinforcement of multiple network elastomers that do not exhibit necking in uniaxial tension.
To explore this stress transmission mechanism in a spatially resolved way in a damaged zone around the crack tip, we carried out fracture tests on prenotched rectangular shape EA(4)-2. As a reminder, these specimens result from four polymerizations (composition in Table 1) and contain the SP mechanophore in the second network.
As we recorded the specimen during loading with a RGB color camera, no obvious color change was observed in front of the crack tip prior to crack propagation (Fig. S4†). A similar result was obtained when observing the fracture surface after crack propagation, in stark contrast to the case when SP is incorporated into the filler network.32 These observations suggest that if the stress transfer mechanism between the filler and matrix network exists, it is very localized (i.e., over regions smaller than the spatial resolution of the RGB camera). The lack of detection could be due to the low concentration of MC (i.e. activated SP) which makes it difficult to probe it with a method based on absorption.
A more sensitive method to detect MC is based on fluorescence. It has been shown that the activation of SP into MC not only results in a change in the absorption but also in the emission spectrum. To probe the potential activation of SP into MC around the crack tip, we uniaxially loaded notched specimens of EA(4)-2, EA(4)-3 and EA(3)-2 (with a stretch rate of ∼10−3 s−1) under a laser scanning confocal microscope (with excitation at λex = 561 nm and data collection in a window centered at λ = 615 nm) (Fig. 7a–c). Details of the experiment are given in the ESI.† It is noteworthy that EA(4)-2 and EA(4)-3 exhibited necking in uniaxial extension but EA(3)-2 did not (Fig. 7d).
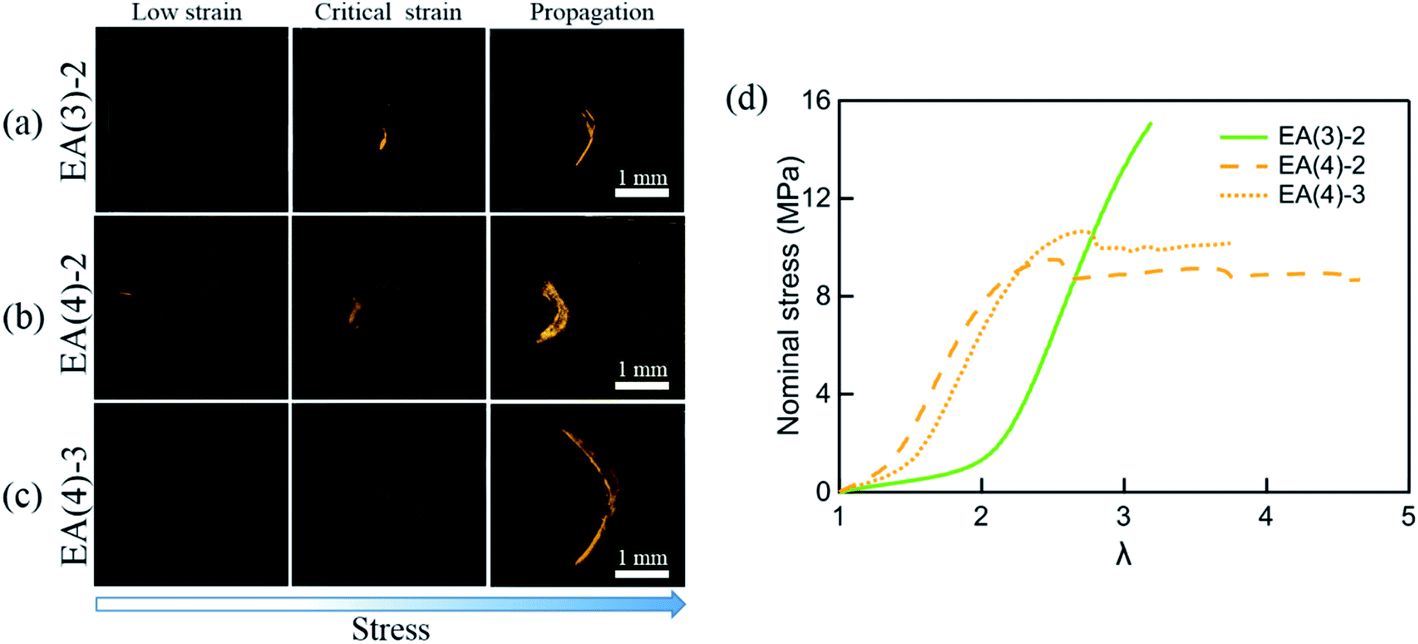 | ||
| Fig. 7 Single-edge notched samples of (a) EA(3)-2, (b) EA(4)-2 and (c) EA(4)-3 are made to detect the activation of SP in the matrix network around the crack tip with a Deben tensile stage. The fluorescent signal of MC around the crack tip is detected by confocal microscopy. The 3D fluorescence images of samples (top view) at low strain, critical strain, and after crack propagation are presented in the left column, middle column, and right column, respectively. Details of the experiment are given in the ESI.† (d) Stress–strain curves of EA(3)-2, EA(4)-2 and EA(4)-3. Note that EA(3)-2 does not present a necking phenomenon in uniaxial tension. | ||
Interestingly, EA(3)-2 and EA(4)-2 signal and quantify the extent of SP activation in front of the crack tip at the critical strain (i.e., before crack propagation), while EA(4)-3 only fluoresces after crack propagation (Fig. 7c). Although we did not attempt to calibrate the extent of stress transfer around the crack, activation of SP embedded in the second network does occur over a region ∼100 μm thick ahead of the crack tip. In addition, activation of SP embedded in the third network is only observed after the crack propagates, consistent with the chromatic change and stress–strain curve in Fig. 5. Some SP activation also occurs in EA(3)-2 that does not neck in uniaxial extension (Fig. 1), demonstrating that stress transfer does occur over a localized region in front of the crack tip as hypothesized by Brown and Tanaka.26,27
If the internetwork connectivity is mitigated by replacing ethyl acrylate with hexyl methacrylate in the filler network, a weak fluorescent signal can still be observed in HMA(4)-2 near the crack tip before propagation (Fig. S9†). This observation illustrates that the second network contributes to the stress-carrying ability of the elastomer despite the lack of crosslinking between the first and the second networks. The entanglements between networks may equalize the load in the second network and reduce or suppress the concentration of MC upon local necking. In other words the load is distributed over more of the second network chain at a lower force per chain.
More generally, these fluorescence images indicate that localized damage exists at the crack tip, similar to crazing in glassy materials.64,65 At high extensions, the filler network first breaks randomly19 and then in a more correlated way (maybe forming local weak spots or microcracks), resulting in significant changes in extensibility as the load is transferred from the stiff filler to the more extensible matrix. The matrix network can then stretch up to its own strain hardening causing activation of SP in labeled specimens. One can imagine that at this final state microcracks are open and bridged by matrix network strands that must break to let the crack propagate. An interesting general question concerning the origin of the high toughness of multiple network elastomers is whether the very specific network architecture mainly dissipates energy during steady-state propagation (and hence slows the crack growth) or mainly delays nucleation of the crack.
3. Conclusion
The incorporation of a force-sensitive probe into multiple network elastomers has been used to shed light on the toughening mechanisms of double network gels and multiple network elastomers.Four groups of multiple network elastomers that exhibit yielding in uniaxial extension have been synthesized by incorporating SP into the first, second, and third networks, respectively. The color change of SP observed under strain confirms that the filler network first sustains the full load and only transfers into the second network at the point where macroscopic necking is observed. However, this stress transfer is only partial as roughly 9% of the filler network chains are broken at the yield point despite the large jump in stretch, i.e. the first network still holds the main load. In quadruple networks, a high level of stress in the third network is not observed. This result points at a scenario of microcracks as proposed by Brown, rather than a scenario of islands as proposed by Gong for double network gels. It should be pointed out, however, that these details may be dependent on the network architecture.
Moreover, when the interconnection between the first and the second networks owing to the chain transfer reaction is altered, by replacing ethyl acrylate with hexyl methacrylate, the stress transmission is delayed and the second network does not activate the SP until the whole specimen is necked.
Finally, since MC is fluorescent, the activation of SP into MC can also be detected by confocal microscopy revealing that the stress transfer does occur over a localized region in front of the crack tip even in materials that do not undergo macroscopic necking in uniaxial extension. This novel picture of fracture of tough elastomers, revealing the existence of a damage zone at the crack tip with alternating soft and stiff regions, may be much more general than for our model system and may also provide some helpful ideas for more conventional elastomers filled with percolating nanofillers.
Author contributions
The project was planned by C. C. and Y. C. All experiments were carried out by Y. C. The data were analyzed by all authors, and the paper was written mainly by C. C. and Y. C. with input from GES.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge helpful discussions with Prof. Hugh R. Brown, Dr Artem Kovalenko, Dr Jean Comtet and Dr Huan Zhang. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program under grant agreement AdG No. 695351. Dr Yinjun Chen has benefitted from a scholarship from the Chinese Scholarship Council. All data needed to evaluate the conclusions in the paper are present in the paper and/or the ESI.† Additional data are available from the authors upon request.References
- C. Creton, 50th Anniversary Perspective: Networks and Gels: Soft but Dynamic and Tough, Macromolecules, 2017, 50(21), 8297–8316 CrossRef CAS.
- G. Heinrich and M. Kluppel, Recent advances in the theory of filler networking in elastomers, Adv. Polym. Sci., 2002, 160, 1–44 CrossRef CAS.
- G. Heinrich, M. Kluppel and T. A. Vilgis, Reinforcement of elastomers, Curr. Opin. Solid State Mater. Sci., 2002, 6(3), 195–203 CrossRef CAS.
- A. N. Gent, Engineering with Rubber, Hanser, Munich, 2001, vol. 1, p. 365 Search PubMed.
- S. K. Kumar, B. C. Benicewicz, R. A. Vaia and K. I. Winey, 50th Anniversary Perspective: Are Polymer Nanocomposites Practical for Applications?, Macromolecules, 2017, 50(3), 714–731 CrossRef CAS.
- L. Karásek and M. Sumita, Characterization of dispersion state of filler and polymer-filler interactions in rubber-carbon black composites, J. Mater. Sci., 1996, 31(2), 281–289 CrossRef.
- Z. Rigbi, Reinforcement of Rubber by Carbon-Black, Adv. Polym. Sci., 1980, 36, 21–68 CrossRef CAS.
- F. Yatsuyanagi, N. Suzuki, M. Ito and H. Kaidou, Effects of secondary structure of fillers on the mechanical properties of silica filled rubber systems, Polymer, 2001, 42(23), 9523–9529 CrossRef CAS.
- D. R. Paul and J. E. Mark, Fillers for polysiloxane (silicone) elastomers, Prog. Polym. Sci., 2010, 35(7), 893–901 CrossRef CAS.
- H. Li, L. Yang, G. Weng, W. Xing, J. Wu and G. Huang, Toughening rubbers with a hybrid filler network of graphene and carbon nanotubes, J. Mater. Chem. A, 2015, 3(44), 22385–22392 RSC.
- C. Li and G. Shi, Functional Gels Based on Chemically Modified Graphenes, Adv. Mater., 2014, 26(24), 3992–4012 CrossRef CAS PubMed.
- S. Wan and Q. Cheng, Fatigue-Resistant Bioinspired Graphene-Based Nanocomposites, Adv. Funct. Mater., 2017, 27(43), 1703459 CrossRef.
- J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Highly stretchable and tough hydrogels, Nature, 2012, 489(7414), 133–136 CrossRef CAS PubMed.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Double-network hydrogels with extremely high mechanical strength, Adv. Mater., 2003, 15(14), 1155–1158 CrossRef CAS.
- E. Ducrot, Y. Chen, M. Bulters, R. P. Sijbesma and C. Creton, Toughening Elastomers with Sacrificial Bonds and Watching them Break, Science, 2014, 344(6180), 186–189 CrossRef CAS PubMed.
- E. Filippidi, T. R. Cristiani, C. D. Eisenbach, J. Herbert Waite, J. N. Israelachvili, B. Kollbe Ahn and M. T. Valentine, Toughening elastomers using mussel-inspired iron-catechol complexes, Science, 2017, 358(6362), 502–505 CrossRef CAS PubMed.
- X. Zhao, Multi-scale multi-mechanism design of tough hydrogels: building dissipation into stretchy networks, Soft Matter, 2014, 10(5), 672–687 RSC.
- T. Matsuda, T. Nakajima, Y. Fukuda, W. Hong, T. Sakai, T. Kurokawa, U.-i. Chung and J. P. Gong, Yielding Criteria of Double Network Hydrogels, Macromolecules, 2016, 49(5), 1865–1872 CrossRef CAS.
- P. Millereau, E. Ducrot, J. M. Clough, M. E. Wiseman, H. R. Brown, R. P. Sijbesma and C. Creton, Mechanics of elastomeric molecular composites, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(37), 9110–9115 CrossRef CAS PubMed.
- J. Liu, C. S. Y. Tan, Z. Y. Yu, N. Li, C. Abell and O. A. Scherman, Tough Supramolecular Polymer Networks with Extreme Stretchability and Fast Room-Temperature Self-Healing, Adv. Mater., 2017, 29, 1605325 CrossRef PubMed.
- S. Burattini, B. W. Greenland, W. Hayes, M. E. Mackay, S. J. Rowan and H. M. Colquhoun, A Supramolecular Polymer Based on Tweezer-Type π–π Stacking Interactions: Molecular Design for Healability and Enhanced Toughness, Chem. Mater., 2011, 23(1), 6–8 CrossRef CAS.
- Y. Chen, Z. Tang, Y. Liu, S. Wu and B. Guo, Mechanically Robust, Self-Healable, and Reprocessable Elastomers Enabled by Dynamic Dual Cross-Links, Macromolecules, 2019, 52(10), 3805–3812 CrossRef CAS.
- Z. Tang, J. Huang, B. Guo, L. Zhang and F. Liu, Bioinspired Engineering of Sacrificial Metal–Ligand Bonds into Elastomers with Supramechanical Performance and Adaptive Recovery, Macromolecules, 2016, 49(5), 1781–1789 CrossRef CAS.
- M. Vatankhah-Varnosfaderani, A. N. Keith, Y. Cong, H. Liang, M. Rosenthal, M. Sztucki, C. Clair, S. Magonov, D. A. Ivanov, A. V. Dobrynin and S. S. Sheiko, Chameleon-like elastomers with molecularly encoded strain-adaptive stiffening and coloration, Science, 2018, 359(6383), 1509–1513 CrossRef CAS PubMed.
- T. Zhang, S. Lin, H. Yuk and X. Zhao, Predicting fracture energies and crack-tip fields of soft tough materials, Extreme Mech. Lett., 2015, 4, 1–8 CrossRef.
- H. R. Brown, A model of the fracture of double network gels, Macromolecules, 2007, 40(10), 3815–3818 CrossRef CAS.
- Y. Tanaka, A local damage model for anomalous high toughness of double-network gels, Europhys. Lett., 2007, 78(5), 56005 CrossRef.
- X. Wang and W. Hong, Pseudo-elasticity of a double network gel, Soft Matter, 2011, 7(18), 8576–8581 RSC.
- S. R. Lavoie, P. Millereau, C. Creton, R. Long and T. Tang, A continuum model for progressive damage in tough multinetwork elastomers, J. Mech. Phys. Solids, 2019, 125, 523–549 CrossRef CAS.
- E. Ducrot, H. Montes and C. Creton, Structure of tough multiple network elastomers by Small Angle Neutron Scattering, Macromolecules, 2015, 48(21), 7945–7952 CrossRef CAS.
- Y. Chen, C. J. Yeh, Y. Qi, R. Long and C. Creton, From Force Responsive Molecules to Quantifying and Mapping Stresses in Soft Materials, Sci. Adv., 2020, 6(20), eaaz5093 CrossRef CAS PubMed.
- Y. Chen, C. J. Yeh, Q. Guo, Y. Qi, R. Long and C. Creton, Fast reversible isomerization of merocyanine as a tool to quantify stress history in elastomers, Chem. Sci., 2021, 12, 1693–1701 RSC.
- Y. H. Na, Y. Tanaka, Y. Kawauchi, H. Furukawa, T. Sumiyoshi, J. P. Gong and Y. Osada, Necking phenomenon of double-network gels, Macromolecules, 2006, 39(14), 4641–4645 CrossRef CAS.
- Y. Tanaka, Y. Kawauchi, T. Kurokawa, H. Furukawa, T. Okajima and J. P. Gong, Localized Yielding Around Crack Tips of Double-Network Gels, Macromol. Rapid Commun., 2008, 29(18), 1514–1520 CrossRef CAS.
- J. P. Gong, Why are double network hydrogels so tough?, Soft Matter, 2010, 6(12), 2583–2590 RSC.
- E. J. Kramer, Microscopic and Molecular fundamentals of crazing, Adv. Polym. Sci., 1983, 52/53, 1–56 CrossRef.
- H. R. Brown, A Molecular Interpretation of the toughness of glassy polymers, Macromolecules, 1991, 24, 2752–2756 CrossRef CAS.
- P. G. de Gennes, Soft Interfaces, Cambridge University Press, Cambridge, 1st edn, 1997, p. 117 Search PubMed.
- Y. Chen, A. J. H. Spiering, S. Karthikeyan, G. W. M. Peters, E. W. Meijer and R. P. Sijbesma, Mechanically induced chemiluminescence from polymers incorporating a 1,2-dioxetane unit in the main chain, Nat. Chem., 2012, 4(7), 559–562 CrossRef CAS PubMed.
- J. M. Clough, A. Balan, T. L. J. van Daal and R. P. Sijbesma, Probing Force with Mechanobase-Induced Chemiluminescence, Angew. Chem., Int. Ed., 2016, 55(4), 1445–1449 CrossRef CAS PubMed.
- J. M. Clough, C. Creton, S. L. Craig and R. P. Sijbesma, Covalent Bond Scission in the Mullins Effect of a Filled Elastomer: Real-Time Visualization with Mechanoluminescence, Adv. Funct. Mater., 2016, 26(48), 9063–9074 CrossRef CAS.
- R. Gostl and R. P. Sijbesma, [small pi]-extended anthracenes as sensitive probes for mechanical stress, Chem. Sci., 2016, 7(1), 370–375 RSC.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martinez, S. R. White, J. S. Moore and N. R. Sottos, Force-induced activation of covalent bonds in mechanoresponsive polymeric materials, Nature, 2009, 459(7243), 68–72 CrossRef CAS PubMed.
- B. A. Beiermann, D. A. Davis, S. L. B. Kramer, J. S. Moore, N. R. Sottos and S. R. White, Environmental effects on mechanochemical activation of spiropyran in linear PMMA, J. Mater. Chem., 2011, 21(23), 8443–8447 RSC.
- M. E. Grady, B. A. Beiermann, J. S. Moore and N. R. Sottos, Shockwave Loading of Mechanochemically Active Polymer Coatings, ACS Appl. Mater. Interfaces, 2014, 6(8), 5350–5355 CrossRef CAS PubMed.
- G. R. Gossweiler, G. B. Hewage, G. Soriano, Q. Wang, G. W. Welshofer, X. Zhao and S. L. Craig, Mechanochemical Activation of Covalent Bonds in Polymers with Full and Repeatable Macroscopic Shape Recovery, ACS Macro Lett., 2014, 3(3), 216–219 CrossRef CAS.
- G. R. Gossweiler, C. L. Brown, G. B. Hewage, E. Sapiro-Gheiler, W. J. Trautman, G. W. Welshofer and S. L. Craig, Mechanochemically Active Soft Robots, ACS Appl. Mater. Interfaces, 2015, 7(40), 22431–22435 CrossRef CAS PubMed.
- X. Fang, H. Zhang, Y. Chen, Y. Lin, Y. Xu and W. Weng, Biomimetic Modular Polymer with Tough and Stress Sensing Properties, Macromolecules, 2013, 46(16), 6566–6574 CrossRef CAS.
- S. Jiang, L. Zhang, T. Xie, Y. Lin, H. Zhang, Y. Xu, W. Weng and L. Dai, Mechanoresponsive PS-PnBA-PS Triblock Copolymers via Covalently Embedding Mechanophore, ACS Macro Lett., 2013, 2(8), 705–709 CrossRef CAS.
- Y. Chen, H. Zhang, X. Fang, Y. Lin, Y. Xu and W. Weng, Mechanical Activation of Mechanophore Enhanced by Strong Hydrogen Bonding Interactions, ACS Macro Lett., 2014, 3(2), 141–145 CrossRef CAS.
- H. Zhang, Y. J. Chen, Y. J. Lin, X. L. Fang, Y. Z. Xu, Y. H. Ruan and W. G. Weng, Spiropyran as a Mechanochromic Probe in Dual Cross-Linked Elastomers, Macromolecules, 2014, 47(19), 6783–6790 CrossRef CAS.
- W. Qiu, P. A. Gurr and G. G. Qiao, Regulating Color Activation Energy of Mechanophore-Linked Multinetwork Elastomers, Macromolecules, 2020, 53(10), 4090–4098 CrossRef CAS.
- G. R. Gossweiler, T. B. Kouznetsova and S. L. Craig, Force-Rate Characterization of Two Spiropyran-Based Molecular Force Probes, J. Am. Chem. Soc., 2015, 137(19), 6148–6151 CrossRef CAS PubMed.
- M. Li, Q. Zhang, Y.-N. Zhou and S. Zhu, Let spiropyran help polymers feel force!, Prog. Polym. Sci., 2018, 79, 26–39 CrossRef CAS.
- J. Hobley and V. Malatesta, Energy barrier to TTC-TTT isomerisation for the merocyanine of a photochromic spiropyran, Phys. Chem. Chem. Phys., 2000, 2(1), 57–59 RSC.
- T. Tominaga, V. R. Tirumala, E. K. Lin, J. P. Gong, H. Furukawa, Y. Osada and W.-l. Wu, The molecular origin of enhanced toughness in double-network hydrogels: A neutron scattering study, Polymer, 2007, 48(26), 7449–7454 CrossRef CAS.
- M. Huang, H. Furukawa, Y. Tanaka, T. Nakajima, Y. Osada and J. P. Gong, Importance of Entanglement between First and Second Components in High-Strength Double Network Gels, Macromolecules, 2007, 40(18), 6658–6664 CrossRef CAS.
- K. Fukao, T. Nakajima, T. Nonoyama, T. Kurokawa, T. Kawai and J. P. Gong, Effect of Relative Strength of Two Networks on the Internal Fracture Process of Double Network Hydrogels As Revealed by in Situ Small-Angle X-ray Scattering, Macromolecules, 2020, 53(4), 1154–1163 CrossRef CAS.
- Y. H. Na, T. Kurokawa, Y. Katsuyama, H. Tsukeshiba, J. P. Gong, Y. Osada, S. Okabe, T. Karino and M. Shibayama, Structural characteristics of double network gels with extremely high mechanical strength, Macromolecules, 2004, 37(14), 5370–5374 CrossRef CAS.
- E. Ducrot and C. Creton, Characterizing Large Strain Elasticity of Brittle Elastomeric Networks by Embedding Them in a Soft Extensible Matrix, Adv. Funct. Mater., 2016, 26(15), 2482–2492 CrossRef CAS.
- N. M. Ahmad, F. Heatley, D. Britton and P. A. Lovell, Chain transfer to polymer in emulsion polymerization, Macromol. Symp., 1999, 143(1), 231–241 CrossRef CAS.
- T. Nakajima, H. Furukawa, Y. Tanaka, T. Kurokawa, Y. Osada and J. P. Gong, True Chemical Structure of Double Network Hydrogels, Macromolecules, 2009, 42(6), 2184–2189 CrossRef CAS.
- Y. Qi, Z. Zou, J. Xiao and R. Long, Mapping the nonlinear crack tip deformation field in soft elastomer with a particle tracking method, J. Mech. Phys. Solids, 2019, 125, 326–346 CrossRef CAS.
- W. Döll, Optical interference measurements and fracture mechanics analysis of crack tip craze zones, Adv. Polym. Sci., 1983, 52/53, 105–168 CrossRef.
- M. George, Y. Nziakou, S. Goerke, A. C. Genix, B. Bresson, S. Roux, H. Delacroix, J. L. Halary and M. Ciccotti, In situ AFM investigation of slow crack propagation mechanisms in a glassy polymer, J. Mech. Phys. Solids, 2018, 112, 109–125 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03352b |
| This journal is © The Royal Society of Chemistry 2021 |