
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe Pd-catalysed asymmetric allylic alkylation reactions of sulfamidate imines†
Quoc Hoang
Pham
 ,
Andrew J.
Tague
,
Christopher
Richardson
,
Christopher J. T.
Hyland
* and
Stephen G.
Pyne
*
,
Andrew J.
Tague
,
Christopher
Richardson
,
Christopher J. T.
Hyland
* and
Stephen G.
Pyne
*
School of Chemistry and Molecular Bioscience, Molecular Horizons Research Institute, University of Wollongong, Wollongong, New South Wales 2522, Australia. E-mail: chris_hyland@uow.edu.au; spyne@uow.edu.au
First published on 20th August 2021
Abstract
The Pd-catalysed asymmetric allylic alkylation (Pd-AAA) of prochiral enamide anions derived from 5H-oxathiazole 2,2-dioxides has been developed. Various 4,5-disubstituted and 4-substituted cyclic sulfamidate imines have participated in the transformation with a range of allyl carbonates—as well as 2-vinyl oxirane, 2-vinyl-N-tosylaziridine, and 2-vinyl-1,1-cyclopropane dicarboxylate—to furnish the desired C-allylated products in moderate to high yields, with high regioselectivites and generally high enantioselectivities. Conversion between N- and C-allyl products was observed, with the N-allylated products converting to the C-allylated products over time. The resulting high-value allylated heterocyclic products all bear a tetrasubstituted stereogenic centre and can be reduced to an allylated chiral sulfamidate or an amino alcohol.
Introduction
Sulfamidate imines are versatile heterocyclic building blocks that have been utilised in a range of reactions.1 The reactivity of these heterocycles is dominated by nucleophilic attack and reduction of the activated imine portion of the ring to provide sulfamidates – themselves valuable precursors to a myriad of heteroatom containing molecules.2–5 A number of metal-catalysed hydrogenation6–9 and transfer-hydrogenation10–16 reactions disclosed in recent years offer a convenient and efficient pathway to synthetically useful chiral sulfamidates with an α-secondary amine moiety. Similarly, the successful development of various Rh-,17–22 Ir-,23 and Pd-catalysed24 nucleophilic addition reactions allows for the preparation of sulfamidates with an α-tertiary amine. A number of these transformations have been employed in the syntheses of bioactive and medicinally relevant scaffolds such as norephedrine and norpseudoephedrine,25 a medicinally relevant piperazinone derivative,7 a potent β-secretase 1 inhibitor,26 and the potential Alzheimer's medication, verubecestat.24While the electrophilic reactivity of sulfamidate imines is well developed, their reactions as nucleophilic enamide anions Avia deprotonation of the acidic proton(s) adjacent to the imine moiety are not well developed – especially with regard to stereoselective transition metal-catalysed reactions (Fig. 1a). The only reports of such reactions are from the groups of Vicario and Samanta, who have both demonstrated organocatalyzed diastereo- and/or enantio-selective reactions of sulfamidate imines to provide stereodefined scaffolds, such as spiro sulfamidate imine-fused δ-lactone B,27 sulfamidate imine-fused cyclohexanes and trans-decalins C,28,29 and 3,3-disubstituted oxindoles D.30 None of these reactions involve reaction of a 5-substituted cyclic imine to directly form a tetra-substituted stereogenic centre in the product. Further, to the best of the authors' knowledge, there are no transition metal-catalysed processes harnessing enamide anion A. Given the range of nucleophiles deployed in Pd-catalysed asymmetric allylic (Pd-AAA) reactions, it was envisaged that the enamide anion A would be an intriguing and useful prochiral nucleophile in this powerful methodology (Fig. 1b).31–35 While enolates and enamines have been deployed as nucleophiles in Pd-AAA reactions, enamide anions have not been explored.36 The use of A in the Pd-AAA reaction provides the opportunity to introduce a heterocyclic nucleophile with multiple heteroatoms,37–42 and given the availability of 5-substituted sulfamidate imines, Pd-AAA products bearing an enantiodefined tetra-substituted carbon centre could be obtained. Furthermore, the imine moiety would still be present in the allylated products, providing a handle for further synthetic manipulation.
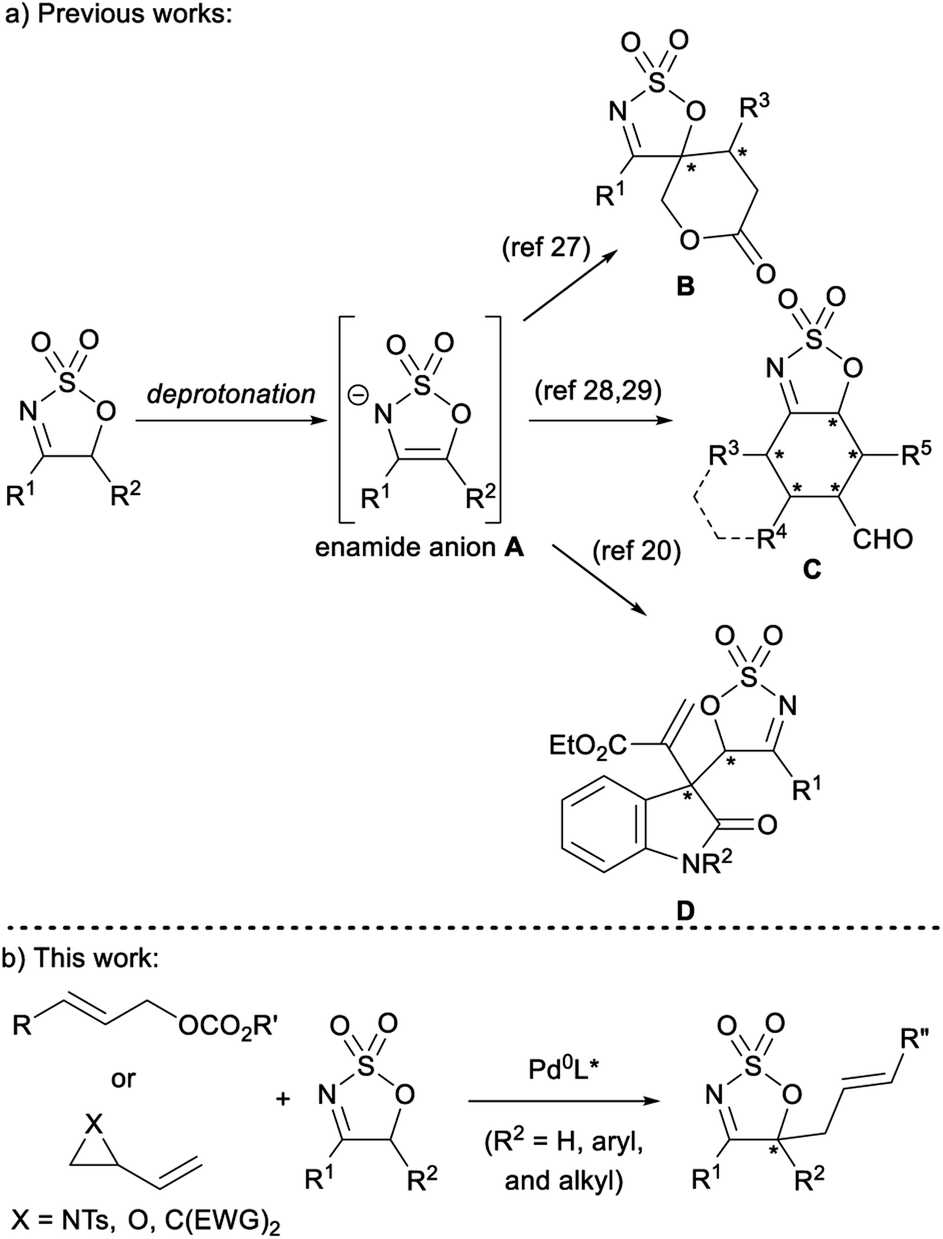 | ||
| Fig. 1 Deployment of cyclic sulfamidate imines as nucleophilic enamide anions. | ||
Herein, we report the successful application of cyclic sulfamidate imines in Pd-catalysed asymmetric allylic alkylation to produce high-value allylated heterocycles.
Results and discussion
Preliminary proof of principle for the proposed enamide anion allylation was carried in a racemic sense using allyl carbonate 1a and 4,5-diphenyl cyclic imine 2a with [Pd(π-C3H5)Cl]2/PPh3 as the catalyst system. The imine substrates 2 are readily prepared from α-hydroxyketones and chlorosulfonyl isocyanate (see ESI for full details†). The reaction proceeded as predicted, however an interesting regioselectivity issue was encountered, with both the desired C-allylated product 3aa and the N-allylated species 4aa being formed (Table 1, entry 1—structures unambiguously assigned by X-ray crystallography, see the ESI†). It was postulated that due to the weaker C–N bond, the N-allylation process should be reversible, and gratifyingly, when re-subjecting the N-allyl product 4aa to the reaction conditions, the thermodynamically more stable C-allyl product 3aa was obtained in high yield (Table 1, entry 2).|
|
|||
|---|---|---|---|
| Entry | Solvent | Yieldb (%) | |
| 3aa | 4aa | ||
| a Reaction conditions: 1a (0.2 mmol, 1.0 equiv.), 2a (0.22 mmol, 1.1 equiv.), [Pd(π-C3H5)Cl]2 (5 mol%), PPh3 (15 mol%), solvent (0.07 M w.r.t. 1a), rt, 21 h. b Yield determined by 1H NMR integration against an internal standard (1,2,3-trimethoxybenzene). c Isolated yield. d 4aa was employed as the SM. e Reaction time: 17 h. | |||
| 1 | MeCN | 49c | 30c |
| 2d,e | MeCN | 77 | — |
| 3 | CH 2 Cl 2 | 70 | — |
| 4 | THF | 89 | — |
| 5 | PhMe | NR | |
| 6 | MeOH | 16 | 17 |
| 7 | DMF | 41 | 22 |
| 8 | DMSO | 61 | 12 |
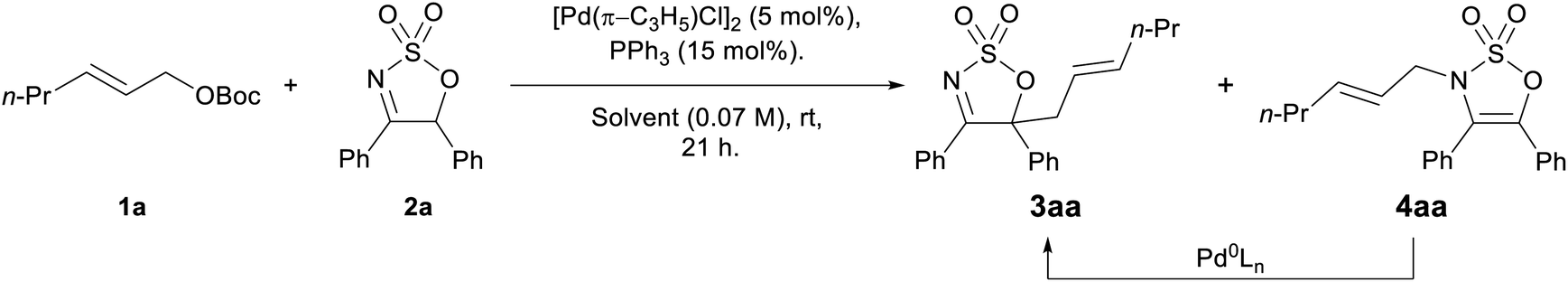
A number of solvents were tested to find the best for generating the desired C-allylated product selectively, and while the reaction progressed well in a range of solvents such as MeCN, DMF, and DMSO—with both C- and N-allylated products being formed in good combined yields (Table 1, entries 1, 5–8)—CH2Cl2 and THF were the only two options allowing the regioselective synthesis of the C-allylated product 3aa in good to high yields (70% and 89%, entries 2 and 3, respectively).
With two optimum solvents for the C-allylated product identified, screening of several chiral ligands was carried out (Table 2). A range of Trost ligands L1–4 was screened, and L1 was found to have excellent performance in both THF and CH2Cl2, affording the desired C-allylated product 3aa in high yields and high enantiomeric ratios (ers) (Table 2, entries 1 and 6). Analysis of the progress of the reaction in this case also showed formation of the N-allyl product 4aa, which slowly disappeared over time, indicating it is a possible reaction intermediate. Trost ligand L4 performed well in both solvents, affording 3aa in moderate to good yields and good ers, although a small amount of 4aa was observed in both cases (entries 4 and 9). (R)-BINAP (L5) performed very well in THF, affording 3aa in excellent yield and high er (entry 5), but was almost completely inactive in CH2Cl2 (entry 10). Trost ligands L2 and L3 were found to be completely inactive in THF (entries 2 and 3, respectively), and performed poorly in CH2Cl2 (entries 7 and 8, respectively).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Solvent | Yieldb (%) | erc | |
| 3aa | 4aa | ||||
| a Reaction conditions: 1a (0.2 mmol, 1.0 equiv.), 2a (0.22 mmol, 1.1 equiv.), [Pd(π-C3H5)Cl]2 (5 mol%), L (15 mol%), solvent (0.07 M w r t. 1a), rt, 21 h. b Yield determined by 1H NMR integration against an internal standard (dimethyl sulfone or trans-stilbene oxide). c Enantiomeric ratio determined by chiral HPLC. d Reaction reached completion after 3 h. e Reaction reached completion after 1 h. f Reversed enantioselectivity compared to entry 5. g [Pd(π-C3H5)Cl]2 (2.5 mol%), L (7.5 mol%). h Reaction concentration halved (0.04 M w. r. t. 1a). i i-1a (1.0 equiv.) used instead of 1a. j Pd2dba3·CHCl3 (5 mol%). k 4aa was employed as SM. | |||||
| 1d | L1 | THF | 90 | — | 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 :7 |
| 2 | L2 | THF | NR | — | |
| 3 | L3 | THF | NR | — | |
| 4 | L4 | THF | 73 | 6 | 83:17 |
| 5 | L5 | THF | 97 | — |
91![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :9 :9
|
| 6 | L1 | CH 2 Cl 2 | 90 | — |
91:9
|
| 7 | L2 | CH2Cl2 | 17 | 10 | 84:16 |
| 8 | L3 | CH2Cl2 | 6 | 9 | 62:38 |
| 9 | L4 | CH2Cl2 | 54 | 6 | 85:15 |
| 10 | L5 | CH2Cl2 | 7 | — | 35:65f |
| 11e,g | L1 | CH2Cl2 | 78 | — | 91:9 |
| 12e,h | L1 | CH2Cl2 | 86 | — | 91:9 |
| 13e,i | L1 | CH2Cl2 | 84 | — | 92:8 |
| 14e,j | L1 | CH2Cl2 | 85 | — | 93:7 |
| 15e,k | L1 | CH2Cl2 | 61 | — | 91:9 |
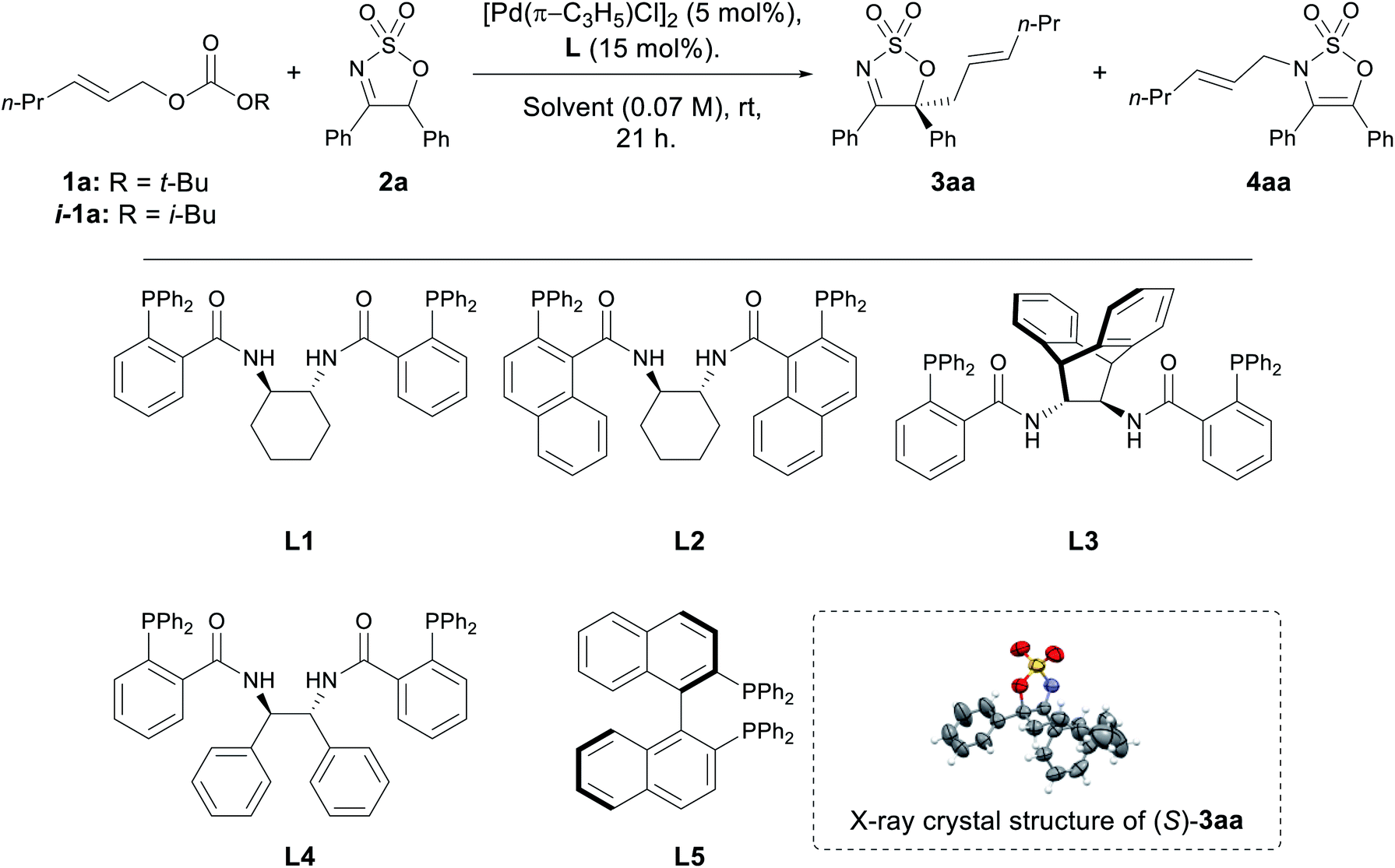
Further optimisation was carried out with L1 in CH2Cl2 due to a faster rate of reaction compared to THF (Table 2). A slight decline in yield was observed when the catalytic load was halved to 2.5 mol% (entry 11), while no noticeable impacts on either yield or enantioselectivity were observed when the reaction was performed at a more dilute concentration (entry 12). The nature of the in situ generated alkoxide seemed to be insignificant, as indicated in the reaction employing i-1a in place of 1a (entry 13). No appreciable changes to the performance of the reaction were observed when employing Pd2dba3·CHCl3 as the Pd(0)-source, suggesting that it can be a viable alternative to the allylpalladium(II) chloride dimer (entry 14).43 Finally, subjecting the isolated N-allyl product 4aa to the optimised reaction conditions furnished the C-allyl product 3aa in moderate yield and with comparable er, confirming that this conversion occurs enantioselectively (entry 15).
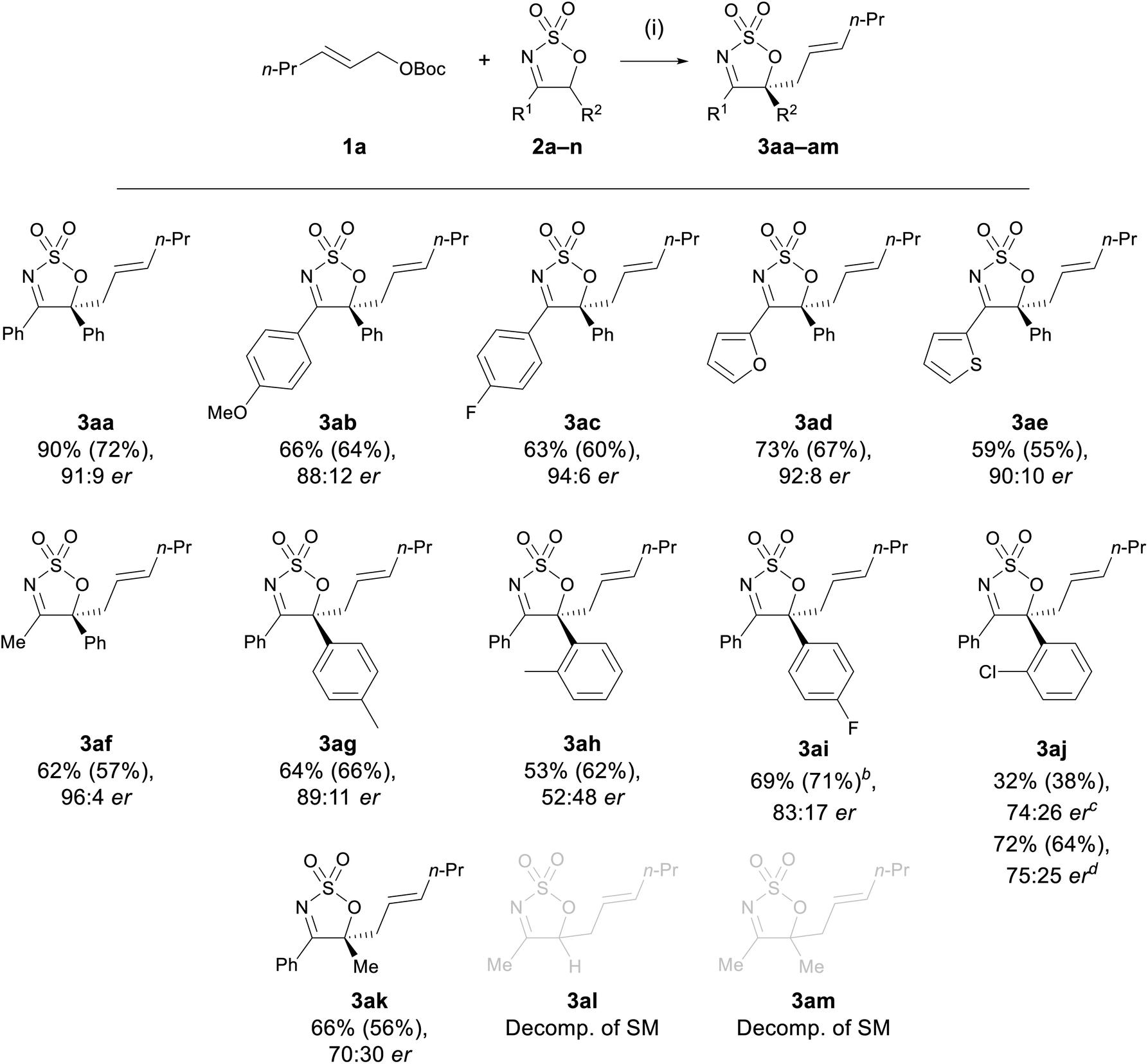 | ||
| Scheme 1 Substrate scope of cyclic sulfamidate imines.a a Reaction conditions: (i) 1a (1.0 equiv.), 2 (1.1 equiv.), [Pd(π-C3H5)Cl]2 (5 mol%), L1 (15 mol%), CH2Cl2, rt, 1 h. Yield determined by 1H NMR integration against an internal standard (dimethyl sulfone or trans-stilbene oxide), isolated yield in parentheses. Enantiomeric ratio determined by chiral HPLC. bCombined yield of a 7.1:1 mixture of 3aj and 3ac (see ESI†). cN-Allylated species 4aj detected in 21% yield by NMR. dModified procedure: 1a (1.1 equiv.), 2 (1.0 equiv.), Pd2dba3·CHCl3 (5 mol%), L1 (15 mol%), CH2Cl2, rt, 24 h. | ||
The absolute (S)-configuration of the C-allylated product was determined via X-ray crystallographic analysis of the enantiopure crystal of 3aa. For all other enantioenriched substrates, the absolute configuration was assigned by analogy.
With the optimised Pd-AAA of 2a established (Table 2, entry 6), the generality of the allylation reaction with other cyclic sulfamidate imines was pursued (Scheme 1). The electronic effect of the substituent at the C4 position of the cyclic imine was first probed. A range of cyclic imines 2a–e bearing different aryl and heteroaryl substituents at the C4 position—either electron rich or poor—reacted efficiently with allyl carbonate 1a to furnish the desired products 3aa–ae in moderate to high yields (59–90%), and in high ers (88:12–94:6 er). Notably, imine 2f bearing a 4-methyl group was found to be well tolerated, selectively producing the allylated product 3af in 62% yield and 96:4 er with no by-products arising from the competitive deprotonation of the C4 methyl group detected, as had been the case in previous methods.44
 | ||
| Scheme 2 Synthesis of 5,5-diallyl cyclic sulfamidate imines. Reaction conditions: (i) 1 (1.0 equiv.), 2 (1.1 equiv.), [Pd(π-C3H5)Cl]2 (5 mol%), L1 (15 mol%), CH2Cl2, rt, 1 h. Yield determined by 1H NMR integration against an internal standard (dimethyl sulfone or trans-stilbene oxide). Enantiomeric ratio determined by chiral HPLC. | ||
Cyclic sulfamidate imines bearing substituents with varying electronic and steric properties at the C5 position (2g–k) were subjected to the reaction conditions to investigate the impact of these two factors on reaction efficacy. All the tested substrates afforded the C-allylated product in moderate to good yields (53–72%), suggesting neither electronic nor steric factors had a significant impact on the reaction yields. No appreciable change in er was observed when varying the electronic properties of the substituent on C5 (3ag and 3ai); however, a significant drop in er was observed with substrates bearing more sterically hindered substituents (3ah, 3aj, and 3ak). In the case of imine 2j, a diminished yield of 3aj was obtained under the optimised conditions and a significant amount of the N-allylated product 4aj (21% by NMR analysis) was detected in the crude reaction mixture. Prolonging the reaction time of 2j to 24 h afforded exclusively 3aj in 72% yield with an er of 75:25. The unchanged er observed for substrate 3aj further confirmed that the conversion from the N- to the C-allyl species is an enantioselective process.
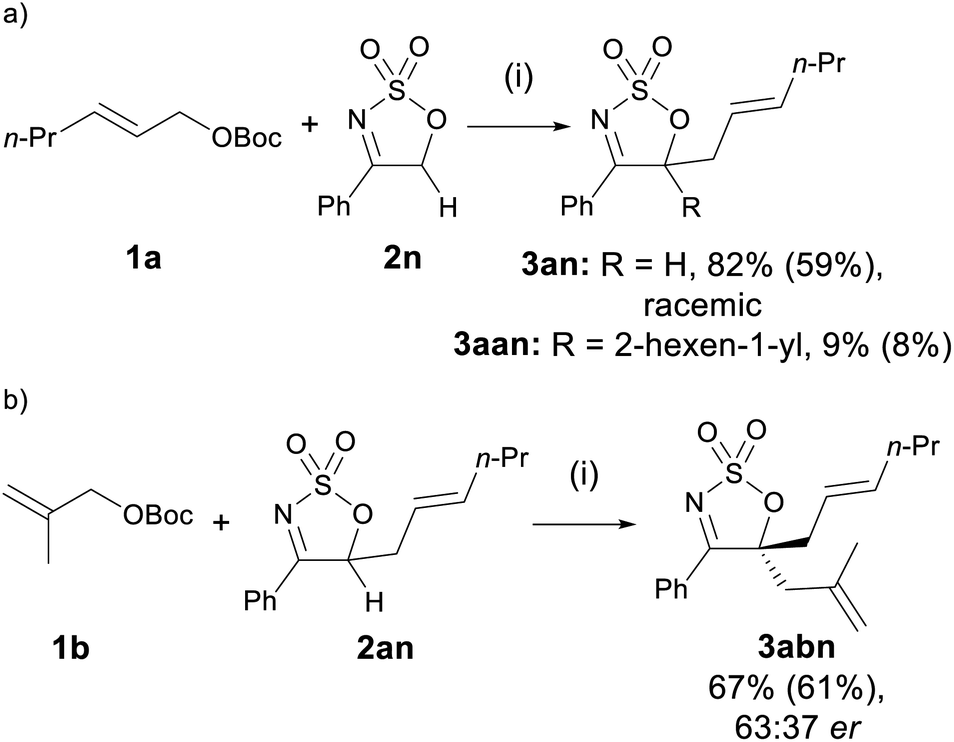
Sulfamidate imine 2n, lacking a substituent at C5, engaged in the allylation reaction to yield the corresponding monoallylated product 3an in 82% yield as a racemate, accompanied by a small amount of di-allylated 3aan (Scheme 2a). The complete lack of enantioselectivity is not surprising, as the presence of an acidic proton at C5 of the mono-allylated product allows rapid racemisation of 3an to occur under the basic reaction conditions. Inspired by the formation of the diallylated product 3aan, product 3an was reacted with allyl carbonate 1b, and the diallylated product 3abn was afforded in good yield and low er (Scheme 2b).
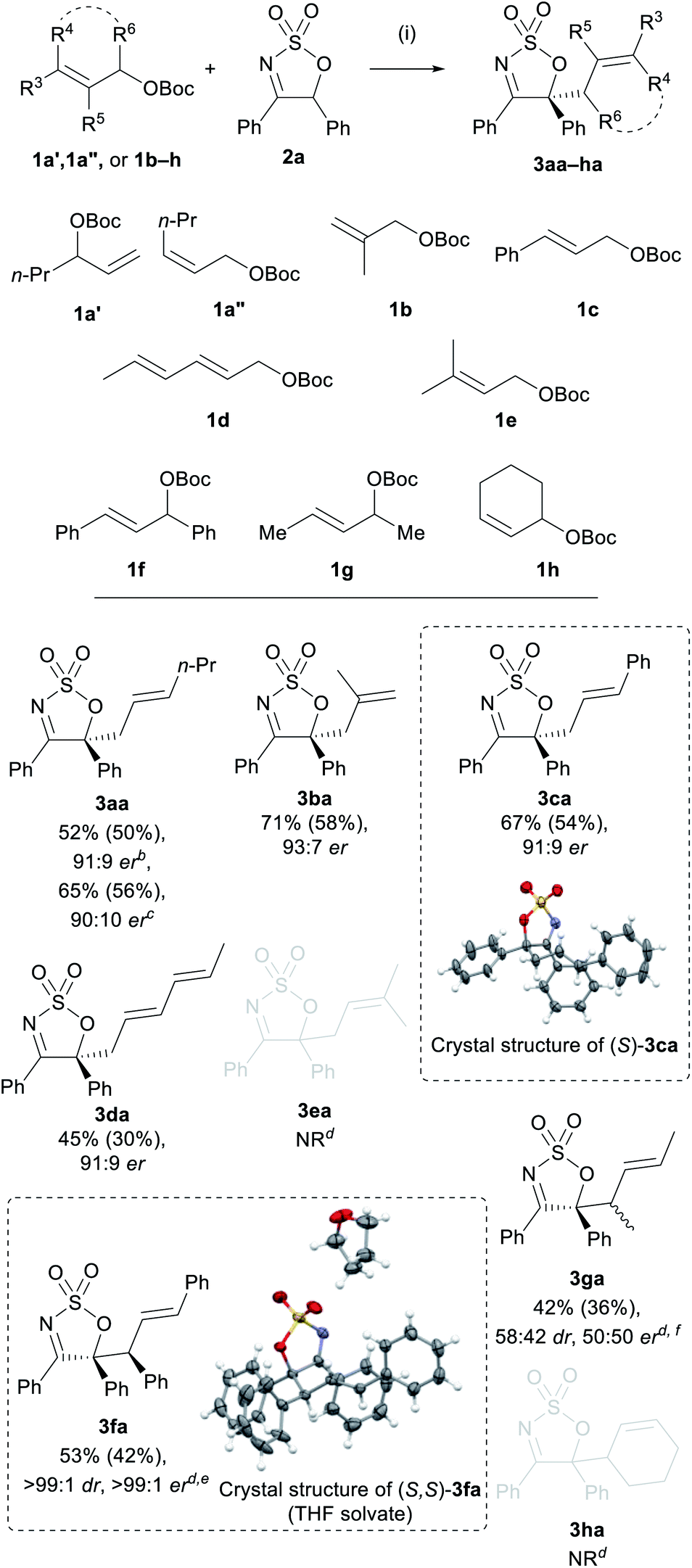
To explore the scope further, a range of monosubstituted allyl carbonates were also examined (Scheme 3). Allyl carbonates 1a′ and 1a′′ were viable substrates, as both furnished imine 3aa in excellent er but in lower yields compared to 1a (52% and 65%, respectively). Carbonates 1b and 1c were also competent substrates, affording the corresponding products 3ba and 3ca in good yields of 71% and 67%, respectively, and very high ers (93:7 and 91:9, respectively). X-ray crystallographic analysis carried out on 3ca confirmed the absolute (S)-configuration of this product. Conjugated diene substrate 1d provided 3da in only 45% yield, but in high enantioselectivity (91:9 er) and without the formation of any other regioisomeric products. No reaction was observed when prenyl carbonate 1e was employed.
 | ||
| Scheme 3 Substrate scope of allyl carbonates.a aReaction conditions: (i) 1 (1.0 equiv.), 2a (1.1 equiv.), [Pd(π-C3H5)Cl]2 (5 mol%), L1 (15 mol%), CH2Cl2, rt, 1 h. Yield determined by 1H NMR integration against an internal standard (dimethyl sulfone or trans-stilbene oxide), isolated yield in parentheses. Enantiomeric ratio determined by chiral HPLC. b1a′ employed as substrate. c1a′′ employed as substrate. dReaction time: 24 h. eModified procedure: 1 (1.0 equiv.), 2a (1.1 equiv.), [Pd(π-C3H5)Cl]2 (5 mol%), (S)-BINAP (15 mol%), THF, rt, 24 h. fIsolated yield, diastereomeric ratio, and enantiomeric ratio determined from the recrystallised material. gDiastereomeric ratio determined from the 1H NMR spectrum of the crude reaction mixture. | ||
A number of disubstituted allyl carbonates (1f–h) were also tested, however, none of these were reactive under the optimised reaction conditions. Allyl carbonates 1f and 1g, however, showed moderate reactivity when the alternate (S)-BINAP/THF (see Table 2, entry 5) conditions were employed, providing 3fa and 3fg. Interestingly, the major diastereoisomer of 3fa was obtained as a single enantiomer following recrystallisation, and X-ray crystallographic analysis of this enantiopure sample confirmed the (S,S)-configuration of the two stereogenic centres. Allyl carbonates 1e and 1h were also subjected to this modified procedure, but no products were obtained in either case. Notably, however, racemic 3ea and 3ha were obtained when Ph3P was employed as the ligand—indicating that the more sterically encumbered chiral bidendate ligands were inhibiting the reaction for these substrates (see the ESI†).
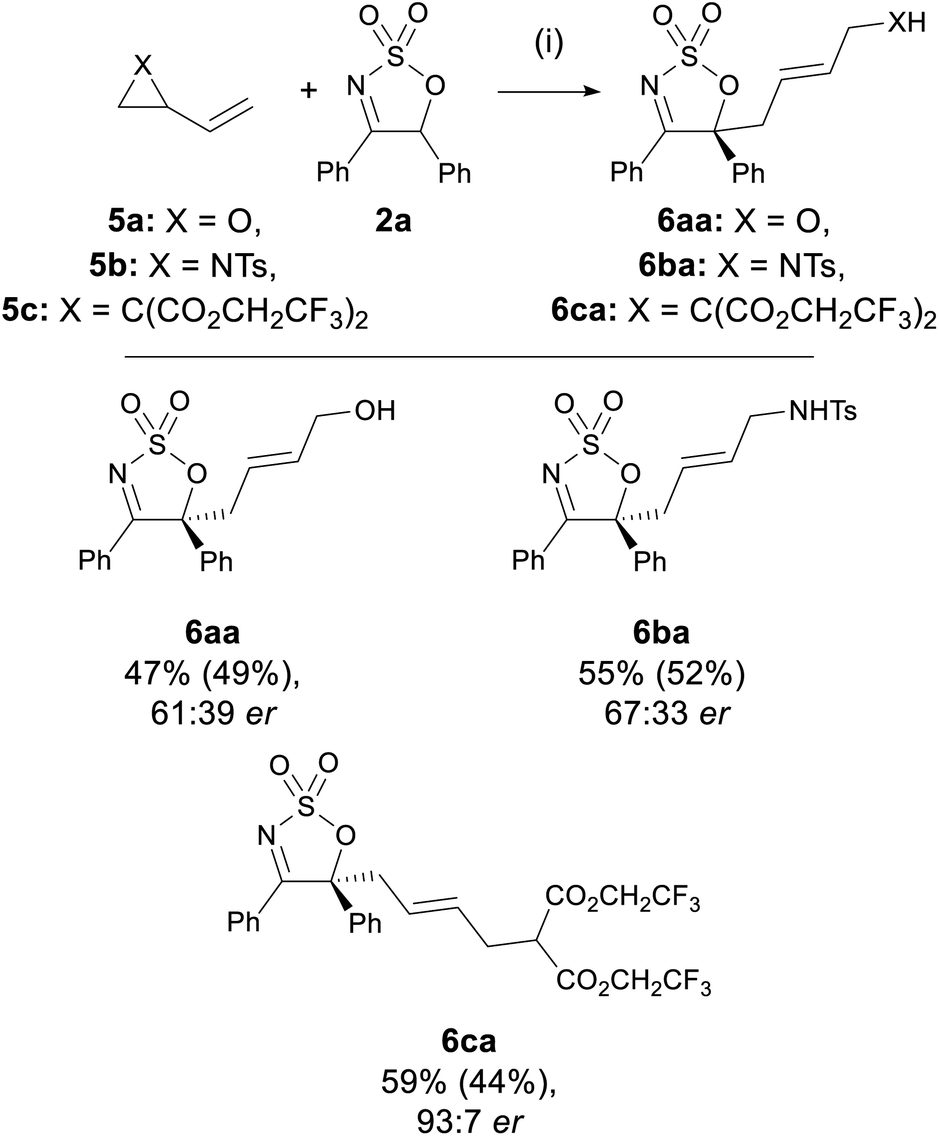
The compatibility of in situ generated Pd-stabilised zwitterionic 1,3-dipoles in the allylation of 3 was also investigated (Scheme 4).45 Under the optimised conditions, dipole precursors 2-vinyl oxirane 5a, N-tosyl-2-vinylaziridine 5b, and 2-vinyl-1,1-cyclopropane dicarboxylate (VCP) 5c all reacted with imine 2a to deliver the corresponding linear products 6aa–ca in moderate yields (44–52%). Interestingly, while 6aa and 6ba were both obtained in modest ers of 61:39–67:33, the VCP-derived product 6ca was formed with a very high er of 93:7, which is comparable to those obtained in previously reported VCP-specific Pd-AAA protocols.38,46 It is also worth noting that there have been few examples on the use of vinyl oxiranes and 2-vinylaziridines as substrates in Pd-AAA with carbon nucleophiles.47–50
 | ||
| Scheme 4 Substrate scope with in situ generated 1,3-dipoles.a aReaction conditions: (i) 4 (1.0 equiv.), 2a (1.1 equiv.), [Pd(π-C3H5)Cl]2 (5 mol%), L1 (15 mol%), CH2Cl2, rt, 24 h. Yield determined by 1H NMR integration against an internal standard (dimethyl sulfone or trans-stilbene oxide), isolated yield in parentheses. Enantiomeric ratio determined by chiral HPLC. | ||
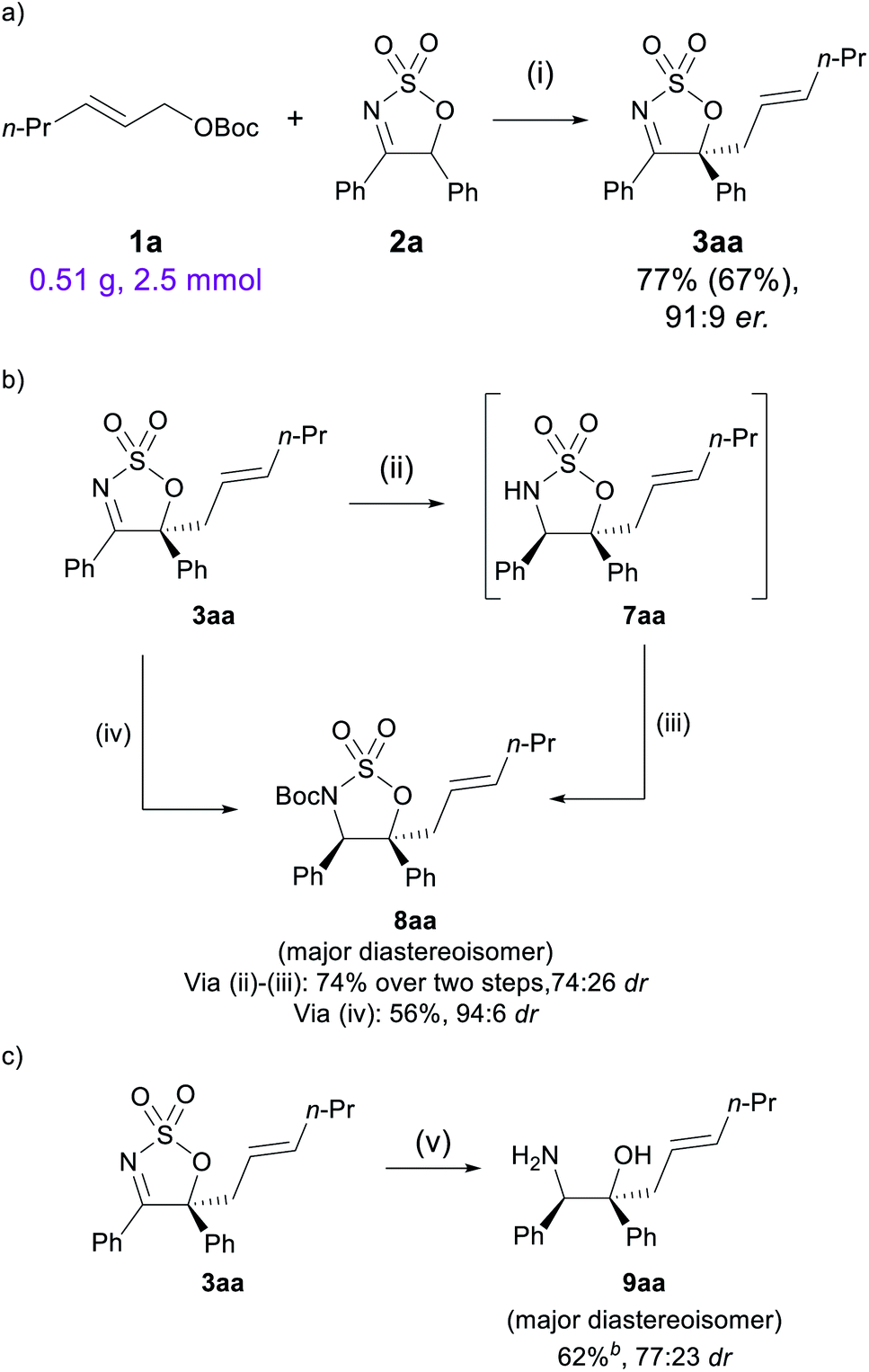
To demonstrate the synthetic practicality of this method, the reaction of 1a with 2a was performed at a 2.5 mmol scale, with a reduced catalytic loading of 2.5 mol% of [Pd(π-C3H5)Cl]2 and 7.5 mol% of L1. Gratifyingly, the allylated product 3aa was obtained in good yield (77% by NMR analysis, 67% isolated) and comparable enantioselectivity (91:9 dr, Scheme 5a). A number of post-synthetic modifications were then attempted to showcase the utility of the sulfamidate imine allylated product. First, 3aa was reduced by NaBH4 to the corresponding sulfamidate 7aa, and following an N-Boc protection step,51 sulfamidate 8aa was obtained in 74% yield and 74:26 dr (Scheme 5b, absolute configuration of 8aa was determined by 1D NOE, see the ESI†). A one-pot reduction/N-Boc protection procedure can be carried out using the more sterically hindered reducing reagent K-Selectride®, in which 8aa was afforded in 56% yield and 94:6 dr. Compared to the hydrogenation products obtained from reduction of the imine, this method can provide more substituted products having two contiguous stereocentres.
 | ||
| Scheme 5 Scale-up reaction and derivatisation of allylated product 3aa.a aReaction conditions: (i) 1 (2.5 mmol, 1.0 equiv.), 2a (2.8 mmol, 1.1 equiv.), [Pd(π-C3H5)Cl]2 (2.5 mol%), L1 (7.5 mol%), CH2Cl2, rt, 1 h. Yield determined by 1H NMR integration against an internal standard (dimethyl sulfone), isolated yield in parentheses. (ii) 3aa (1.0 equiv.), NaBH4 (4.5 equiv.), MeOH, 0 °C, 1 h. (iii) Boc2O (1.3 equiv.), DMAP (0.4 equiv.), CH2Cl2, rt, overnight. (iv) (a) 3aa (1.0 equiv.), K-Selectride® (2.2 equiv.), THF, −10 °C, 1 h. (b) Boc2O (2.3 equiv.), DMAP (14 mol%), −10 °C to rt, overnight. (v) LiAlH4 (3.1 equiv.), THF, reflux, 1.5 h. Isolated yield. Diastereomeric ratio determined from the 1H NMR spectrum of the crude reaction mixture. bMinor diastereoisomer also isolated in 19% yield. | ||
Lastly, treatment of 3aa with LiAlH4 under reflux conditions successfully furnished β-amino alcohol 9aa in 62% yield, which was readily separable by chromatography from its diastereomer, which was isolated in 19% yield (Scheme 5c). This synthetic sequence therefore offers efficient access to highly functionalised allylated β-amino alcohols, which can serve as useful synthetic building blocks.52,53
The stereochemical outcomes of the allylated products can be rationalised using Trost's well established “wall and flap” model (Fig. 2).54,55 Following this predictive tool, ionisation of achiral allyl substrates by the chiral Pd(0)-complex should provide the kinetically favoured π-allyl complex E that is rapidly attacked by the sterically bulky enamide anion of 2 under the flap, rather than under the Pd-allyl (Fig. 2a). The enamide anion is oriented such as to avoid steric interaction between the ligand wall and R1 and R2. A similar stereochemical model has been postulated by Trost and co-workers for the allylation of prochiral tetralones.56,57 The experimentally determined (S)-configuration of the allylated products 3 aligns with that predicted by this model.
In the case of allyl carbonates 1f and 1g—the ionisation of which should provide the pseudo-meso π-allyl complex F—the presence of an additional substituent on the other terminus of the π-allyl complex might have been prohibitive to the approaching sterically hindered enamide nucleophile, therefore explaining their inactivity under the optimised reaction conditions (Fig. 2b). As for the prenyl carbonate 1e and cyclohexenyl carbonate 1h, it is likely that the steric hindrance caused by the anti-substituent(s) on the corresponding π-allyl complexes may have been the cause of their inertness.
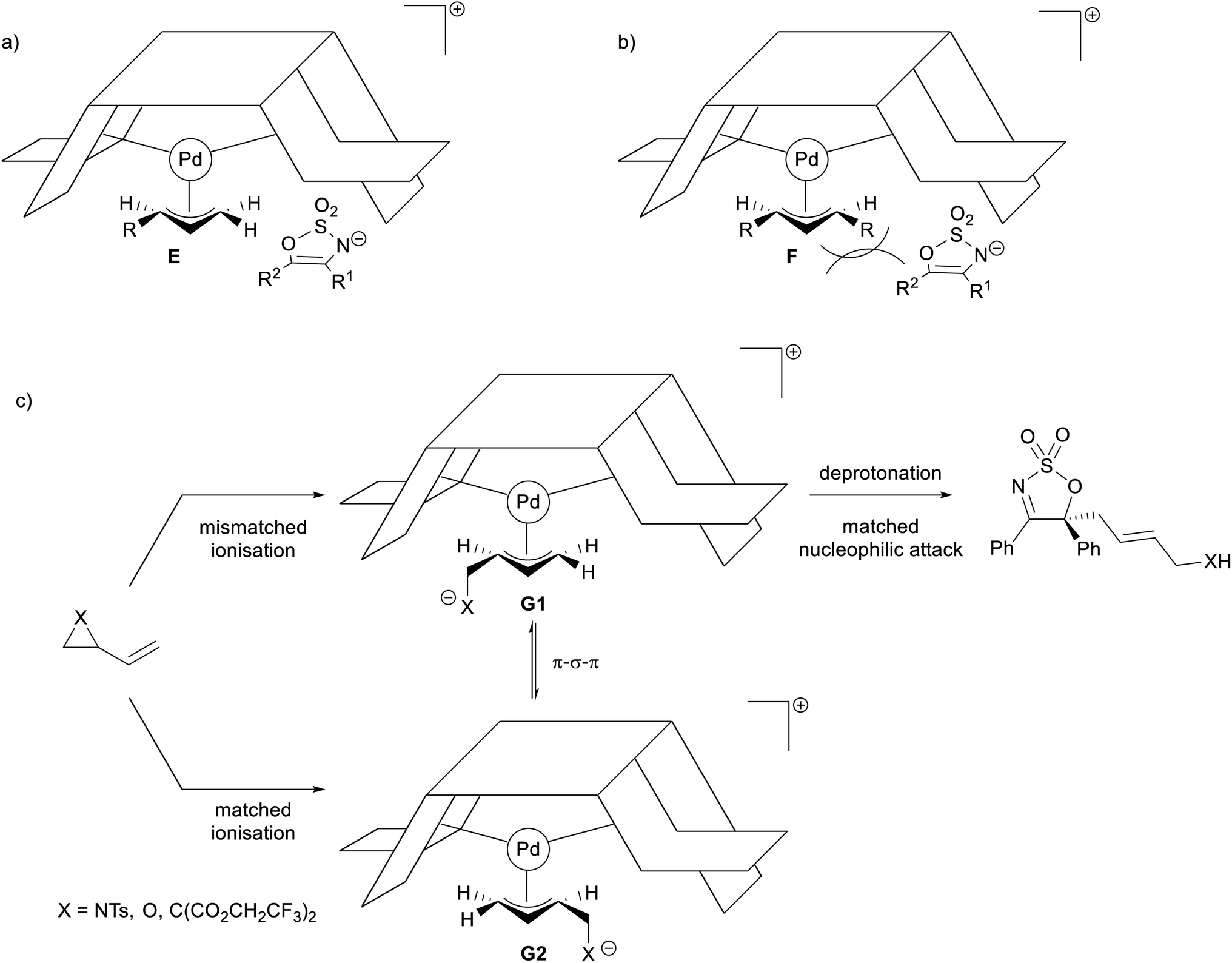 | ||
| Fig. 2 Rationalising the stereochemical outcomes observed for the various allyl substrates. | ||
A clear explanation for the discrepancies in er observed in the reactions employing dipole precursors is unfortunately elusive at this stage, and further experimental and computational investigation is needed. However, it is possible that the varying stability of the in situ generated Pd-stabilised zwitterionic dipoles under the reaction conditions may influence the rate of interconversion between the two diastereomeric π-allyl complexes G1 and G2 (Fig. 2c), with the equilibration rate in the VCP case being more rapid compared to the rate of nucleophilic attack, and therefore furnishing the allylated product in higher er.
Conclusions
In conclusion, the Pd-AAA reactions of 4,5-disubstituted cyclic sulfamidate imines have been successfully developed, which is the first example where enamide anions are utilised in a metal-catalysed reaction. The protocol was found to tolerate a wide range of imines, allyl carbonates, as well as several related vinylic substrates. Under optimised reaction conditions, C-allylated products were exclusively produced in generally high enantiomeric ratios and good yields. Reactions with in situ generated 1,3-dipoles gave exclusively linear products. The allylation reactions can be readily performed efficiently on a 2.5 mmol scale with 5 mol% Pd catalyst without appreciable loss in chemical yield or enantiomeric purity. The allylated products can serve as precursors to chiral 1,2-amino alcohols through reduction reactions, which are useful synthetic building blocks that can be utilised in the preparation of more complex scaffolds containing N- and O-heteroatoms. Other methods are available to prepare trisubstituted β-amino alcohol derivatives similar to 8aa. For example via the addition of aryl or alkyl organometallics to chiral N,N-protected α-amino ketones58 or the aminolysis of trisubstituted epoxides.59 However, the former method is expected to result in racemic products when α-aryl-α-amino ketones are used and the latter method expected to suffer from regiochemical issues when 2-alkyl-2,3-diaryl-epoxides are employed. The new methodology reported here avoids these problems in an enantioselective and efficient manner, and is readily amenable to the synthesis of the enantiomeric series of compounds by using the enantiomeric Trost catalyst. Compound 8aa also comprises a tetrasubstituted carbinol stereocentre – other methods for preparing such alcohols are of recent interest, especially using kinetic resolution.60,61Data availability statement
Data for all compounds in this manuscript are available in the ESI,† which includes experimental details, characterisation and copies of 1H and 13C NMR spectra. Crystallographic data for compounds has been deposited at the CCDC under CCDC 2087721–2087725.Author contributions
CH and SP were involved with conceptualisation of the project, supervision, funding acquisition and writing – reviewing and editing. HP carried out the investigation, formal analysis of data and was involved with writing the original draft of the manuscript. AT was involved in writing – reviewing and editing and formal analysis of data. CR collected the X-ray structures and was involved in writing – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Australian Research Council Discovery grant DP180101332 for funding of this research. QHP is the recipient of a UOW UPA and IPTA PhD scholarship.Notes and references
- Q. H. Pham, C. J. T. Hyland and S. G. Pyne, Org. Biomol. Chem., 2020, 18, 7467–7484 RSC.
- R. E. Meléndez and W. D. Lubell, Tetrahedron, 2003, 59, 2581–2616 CrossRef.
- J. F. Bower, J. Rujirawanich and T. Gallagher, Org. Biomol. Chem., 2010, 8, 1505–1519 RSC.
- A. Megia-fernandez, J. Morales-sanfrutos, F. Hernandez-mateo and F. Santoyo-Gonzalez, Curr. Org. Chem., 2011, 15, 401–432 CrossRef CAS.
- R. Baig, M. Nadagouda and R. Varma, Aldrichimica Acta, 2015, 48, 71–80 Search PubMed.
- Y. Q. Wang, C. Bin Yu, D. W. Wang, X. B. Wang and Y. G. Zhou, Org. Lett., 2008, 10, 2071–2074 CrossRef CAS PubMed.
- M. McLaughlin, K. Belyk, C. Y. Chen, X. Linghu, J. Pan, G. Qian, R. A. Reamer and Y. Xu, Org. Process Res. Dev., 2013, 17, 1052–1060 CrossRef CAS.
- Y. Liu, Y. Huang, Z. Yi, G. Liu, X. Q. Dong and X. Zhang, Adv. Synth. Catal., 2019, 361, 1582–1586 CrossRef CAS.
- Y. Liu, Z. Yi, X. Tan, X. Q. Dong and X. Zhang, iScience, 2019, 19, 63–73 CrossRef CAS PubMed.
- S. Kang, J. Han, E. S. Lee, E. B. Choi and H. K. Lee, Org. Lett., 2010, 12, 4184–4187 CrossRef CAS PubMed.
- J. Han, S. Kang and H. K. Lee, Chem. Commun., 2011, 47, 4004–4006 RSC.
- S. A. Lee, S. H. Kwak and K. I. Lee, Chem. Commun., 2011, 47, 2372–2374 RSC.
- J. A. Kim, Y. J. Seo, S. Kang, J. Han and H. K. Lee, Chem. Commun., 2014, 50, 13706–13709 RSC.
- S. Itsuno, Y. Hashimoto and N. Haraguchi, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 3037–3044 CrossRef CAS.
- Y. J. Seo, J. A. Kim and H. K. Lee, J. Org. Chem., 2015, 80, 8887–8902 CrossRef CAS PubMed.
- H. R. Kim, R. Achary and H. K. Lee, J. Org. Chem., 2018, 83, 11987–11999 CrossRef CAS PubMed.
- H. B. Hepburn, N. Chotsaeng, Y. Luo and H. Lam, Angew. Chem., Int. Ed., 2012, 51, 8309–8313 CrossRef PubMed.
- T. Nishimura, Y. Ebe, H. Fujimoto and T. Hayashi, Chem. Commun., 2013, 49, 5504–5506 RSC.
- Y. J. Chen, Y. H. Chen, C. G. Feng and G. Q. Lin, Org. Lett., 2014, 16, 3400–3403 CrossRef CAS PubMed.
- J. Kong, M. McLaughlin, K. Belyk and R. Mondschein, Org. Lett., 2015, 17, 5520–5523 CrossRef CAS PubMed.
- M.-Q. Liu, T. Jiang, W.-W. Chen and M.-H. Xu, Org. Chem. Front., 2017, 4, 2159–2162 RSC.
- C. Y. Wu, Y. F. Zhang and M. H. Xu, Org. Lett., 2018, 20, 1789–1793 CrossRef CAS PubMed.
- Y. Ebe, M. Hatano and T. Nishimura, Adv. Synth. Catal., 2015, 357, 1425–1436 CrossRef CAS.
- W. Chen, D. Meng, B. N'Zemba and W. J. Morris, Org. Lett., 2018, 20, 1265–1268 CrossRef CAS PubMed.
- H.-K. Lee, K. Soyeong and E. B. Choi, J. Org. Chem., 2012, 77, 5454–5460 CrossRef CAS PubMed.
- B. D. Allison and N. S. Mani, ACS Omega, 2017, 2, 397–408 CrossRef CAS PubMed.
- D. Majee, S. Biswas, S. M. Mobin and S. Samanta, Tetrahedron Lett., 2014, 55, 4553–4558 CrossRef CAS.
- I. Riaño, U. Uria, L. Carrillo, E. Reyes and J. L. Vicario, Org. Chem. Front., 2015, 2, 206–210 RSC.
- I. Riano, U. Uria, E. Reyes, L. Carrillo and J. L. Vicario, J. Org. Chem., 2018, 83, 4180–4189 CrossRef CAS PubMed.
- S. K. Arupula, S. Guin, A. Yadav, S. M. Mobin and S. Samanta, J. Org. Chem., 2018, 83, 2660–2675 CrossRef CAS PubMed.
- B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2944 CrossRef CAS PubMed.
- B. M. Trost, Tetrahedron, 2015, 71, 5708–5733 CrossRef CAS PubMed.
- B. M. Trost and J. E. Schultz, Synthesis, 2019, 51, 1–30 CrossRef CAS.
- S. Parisotto and A. Deagostino, Synthesis, 2019, 51, 1892–1912 CrossRef CAS.
- D. Haas, J. M. Hammann, R. Greiner and P. Knochel, ACS Catal., 2016, 6, 1540–1552 CrossRef CAS.
- O. Pàmies, J. Margalef, S. Cañellas, J. James, E. Judge, P. J. Guiry, C. Moberg, J. E. Bäckvall, A. Pfaltz, M. A. Pericàs and M. Diéguez, Chem. Rev., 2021, 121, 4373 CrossRef PubMed.
- H. Zhou, H. Yang, M. Liu, C. Xia and G. Jiang, Org. Lett., 2014, 16, 5350–5353 CrossRef CAS PubMed.
- Z. Liu, X. Feng, J. Xu, X. Jiang and X. Cai, Tetrahedron Lett., 2020, 61, 151694 CrossRef CAS.
- M. Serra, E. Bernardi, G. Marrubini, E. De Lorenzi and L. Colombo, Eur. J. Org. Chem., 2019, 2019, 732–741 CrossRef CAS.
- B. M. Trost, P. J. Morris and S. J. Sprague, J. Am. Chem. Soc., 2012, 134, 17823–17831 CrossRef CAS PubMed.
- Y. N. Wang, Q. Xiong, L. Q. Lu, Q. L. Zhang, Y. Wang, Y. Lan and W. J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 11013–11017 CrossRef CAS PubMed.
- M. Nascimento de Oliveira, S. Arseniyadis and J. Cossy, Chem.–Eur. J., 2018, 24, 4810–4814 CrossRef CAS PubMed.
- [Pd(π-C3H5)Cl]2 was chosen over Pd2dba3·CHCl3 due to the more consistent quality of the commercially available reagents.
- D. Majee, A. Srivastava, S. M. Mobin and S. Samanta, RSC Adv., 2013, 3, 11502–11506 RSC.
- B. D. W. Allen, C. P. Lakeland and J. P. A. Harrity, Chem.–Eur. J., 2017, 23, 13830–13857 CrossRef CAS PubMed.
- B. M. Trost, W. J. Bai, C. Hohn, Y. Bai and J. J. Cregg, J. Am. Chem. Soc., 2018, 140, 6710–6717 CrossRef CAS PubMed.
- B. M. Trost and C. Jiang, J. Am. Chem. Soc., 2001, 123, 12907–12908 CrossRef CAS PubMed.
- B. M. Trost and C. Jiang, Org. Lett., 2003, 5, 1563–1565 CrossRef CAS PubMed.
- C. Du, L. Li, Y. Li and Z. Xie, Angew. Chem., Int. Ed., 2009, 48, 7853–7856 CrossRef CAS PubMed.
- Y. Wang, J. Chai, C. You, J. Zhang, X. Mi, L. Zhang and S. Luo, J. Am. Chem. Soc., 2020, 142, 3184–3195 CrossRef CAS PubMed.
- Imine 7aa is unstable on silica gel.
- H. Nakano, I. A. Owolabi, M. Chennapuram, Y. Okuyama, E. Kwon, C. Seki, M. Tokiwa and M. Takeshita, Heterocycles, 2018, 97, 647–667 CrossRef CAS.
- F. D. Klingler, Acc. Chem. Res., 2007, 40, 1367–1376 CrossRef CAS PubMed.
- B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 1999, 121, 4545–4554 CrossRef CAS.
- B. M. Trost, M. R. Machacek and A. Aponick, Acc. Chem. Res., 2006, 39, 747–760 CrossRef CAS PubMed.
- B. M. Trost and G. M. Schroeder, J. Am. Chem. Soc., 1999, 121, 6759–6760 CrossRef CAS.
- For a related argument, see: B. M. Trost, J. Xie and J. D. Sieber, J. Am. Chem. Soc., 2011, 133, 20611–20622 CrossRef CAS PubMed.
- T. R. Manfred and A. Schmitzb, Tetrahedron Lett., 1999, 40, 2737–2740 CrossRef.
- K. S. Reddy, L. Solà, A. Moyano, M. A. Pericàs and A. Riera, J. Org. Chem., 1999, 64, 3969–3974 CrossRef CAS.
- S. Rajkumar, S. He and X. Yang, Angew. Chem., Int. Ed., 2019, 58, 10315–10319 CrossRef CAS PubMed.
- C.-H. Zhang, Q. Gao, M. Li, J.-F. Wang, C.-M. Yu and B. Mao, Org. Lett., 2021, 23, 3949–3954 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2087721–2087725. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03268b |
| This journal is © The Royal Society of Chemistry 2021 |