
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceArsenic trioxide targets Hsp60, triggering degradation of p53 and survivin†
Xuqiao
Hu‡
a,
Hongyan
Li‡
a,
Tiffany Ka-Yan
Ip
a,
Yam Fung
Cheung
a,
Mohamad
Koohi-Moghadam
ad,
Haibo
Wang
a,
Xinming
Yang
a,
Daniel N.
Tritton
a,
Yuchuan
Wang
a,
Yi
Wang
 a,
Runming
Wang
a,
Kwan-Ming
Ng
ae,
Hua
Naranmandura
b,
Eric Wai-Choi
Tse
c and
Hongzhe
Sun
*a
a,
Runming
Wang
a,
Kwan-Ming
Ng
ae,
Hua
Naranmandura
b,
Eric Wai-Choi
Tse
c and
Hongzhe
Sun
*a
aDepartment of Chemistry and CAS-HKU Joint Laboratory of Metallomics on Health and Environment, The University of Hong Kong, Hong Kong SAR, P. R. China. E-mail: hsun@hku.hk
bDepartment of Toxicology, School of Medicine and Public Health, Zhejiang University, Hangzhou, P.R. China
cDepartment of Medicine, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Queen Mary Hospital, Hong Kong, P. R. China
dDivision of Applied Oral Sciences and Community Dental Care, Faculty of Dentistry, University of Hong Kong, Hong Kong SAR, P. R. China
eDepartment of Chemistry, Shantou University, Shantou, Guangdong 515063, P. R. China
First published on 17th July 2021
Abstract
The mechanisms of action of arsenic trioxide (ATO), a clinically used drug for the treatment of acute promyelocytic leukemia (APL), have been actively studied mainly through characterization of individual putative protein targets. There appear to be no studies at a system level. Herein, we integrate metalloproteomics through a newly developed organoarsenic probe, As-AC (C20H17AsN4O3S2) with quantitative proteomics, allowing 37 arsenic binding and 250 arsenic regulated proteins to be identified in NB4, a human APL cell line. Bioinformatics analysis reveals that ATO disrupts multiple physiological processes, in particular, chaperone-related protein folding and cellular response to stress. Furthermore, we discover heat shock protein 60 (Hsp60) as a vital target of ATO. Through biophysical and cell-based assays, we demonstrate that ATO binds to Hsp60, leading to abolishment of Hsp60 refolding capability. Significantly, the binding of ATO to Hsp60 disrupts the formation of Hsp60-p53 and Hsp60-survivin complexes, resulting in degradation of p53 and survivin. This study provides significant insights into the mechanism of action of ATO at a systemic perspective, and serves as guidance for the rational design of metal-based anticancer drugs.
Introduction
Arsenic trioxide (ATO) is an effective chemotherapeutic drug either used alone or in combination therapy for the treatment of acute promyelocytic leukemia (APL).1–3 It also shows efficacy against other hematologic malignancies and other solid tumors.4,5 It has been demonstrated that arsenic facilitates profound cellular alteration including induction of apoptosis, inhibition of proliferation, stimulation of differentiation and inhibition of angiogenesis.6 The molecular mechanism of action of ATO (i.e., arsenite) for APL has long been attributed to degradation of the disease-causing oncoprotein, PML-RARα by acting on the two fusion partner.7 However, cytotoxicity of ATO was also found for the system that lacks the PML-RARα fusion protein,4 suggesting other biological targets may also be accountable for its efficacies. Given the thiophilic nature of arsenic, ATO binds large numbers of proteins,8 and differentiating key targets from these proteins remains to be challenging. Although enormous efforts have been made towards validation of potential targets of ATO on individual proteins, including hexokinase-2 (HK2),9 peptidyl-prolyl cis–trans isomerase NIMA-interacting 1 (PIN1),10 MTHFD1 (ref. 11) and mutated NPM1,12 such studies are unlikely to provide a systemic view on the mechanism of action of ATO.Proteomics has been well adapted to track proteins regulated by metallodrugs.13 Previously, global profiling of arsenic-binding proteins has been performed using a human proteome microarray, allowing 360 arsenic-binding proteins to be identified.9 However, these proteins may not be authentic arsenic binding proteins in live cells. Owing to the complexity of arsenic–protein interactions in cells, it is a considerable challenge to track arsenic binding proteins particularly in live cells.
Various approaches have been employed to mine arsenic-binding proteins, among which small-molecule-based fluorescent probes have been used in the detection and visualization of proteins owing to their less-obvious steric effects and fast labeling properties.14 For example, biarsenical fluorescent probes e.g. FlAsH-EDT2, which was initially designed to image proteins with genetically fused tetracysteine motifs (CCXXCC) in live cells,15 were also immobilized to beads or resin with some modification for specific capture arsenic-binding proteins.16 However, this type of probe is unable to identify proteins with spatially cysteine-rich regions or with even one critical cysteine.17 For phenylarseneoxide (PAO) based fluorescent probes,18 such as FAsH, although they could image vicinal dithiol rich proteins, downstream identification of these proteins might result in potential dissociation of the probe from proteins of interest. Affinity chromatography-based approaches have also been employed to site-specifically label large libraries of arsenic binding proteins.19
Herein, we design and synthesize a new fluorescent probe AsIII-AC, via conjugation of an arsenic moiety with a fluorophore and an arylazide. The probe can rapidly enter cells to label intracellular arsenic binding proteins. An arylazide was incorporated into the probe, enabling the labelled proteins to be anchored upon photoactivation and subsequently identified through high-throughput proteomics. By integration with quantitative proteomics, we comprehensively identify 37 As-binding and 250 As-regulated proteins in live NB4 cells. Subsequent bioinformatics analysis reveals 10 hub proteins as potential key targets of ATO. We further validate that ATO binds to Hsp60 (HSPD1), abolishing its folding/refolding activity. Significantly, such a binding disrupts the complexes of Hsp60-p53 and Hsp60-survivin, leading to degradation of p53 and survivin, which contributes to ATO-induced apoptosis of leukemia cells. Hsp60 therefore serves as an important target of the anti-leukemia drug.
Results and discussion
Synthesis of an organoarsenic fluorescent probe and systemic identification of AsIII-binding and -regulated proteins in NB4 cells
We design an organoarsenic probe As-AC (1) consisting of an arsenic moiety, coumarin as a fluorophore and arylazide. By taking the advantage of arylazide, the labelled arsenic binding proteins could be subsequently identified via conventional proteomics.20,21 We first synthesized 2-p-aminophenyl-1,3,2-dithiarsenolane (3) from p-arsanilic acid (6) according to a previously reported method.22 The azido compound (2) was synthesized from 7-amino-4-methyl-3-coumarinylacetic acid using the procedure developed previously.21 Finally, (2) and (3) were conjugated together by EDCI (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide) coupling to afford the coumarin azide-based probe As-AC (1) (Fig. 1a and Schemes S1–S3†). The purity of As-AC was confirmed by 1H and 13C NMR (Fig. S1 and S2†) and electrospray ionization mass spectrometry, which gave rise to a peak at m/z of 522.60 assignable to [M + Na]+ (calcd 523.00) (Fig. S3†). As-AC exhibits a maximum absorption at 355 nm and emits at 448 nm (Fig. 1b) with a quantum yield (ΦF) of 0.050 under physiologically relevant conditions. The low quantum yield of As-AC is attributed to the presence of an azide at the seventh position of the coumarin moiety, which quenches the fluorescence of coumarin.23,24 Negligible toxicity of the probe in mammalian cells was noted (Fig. S4†).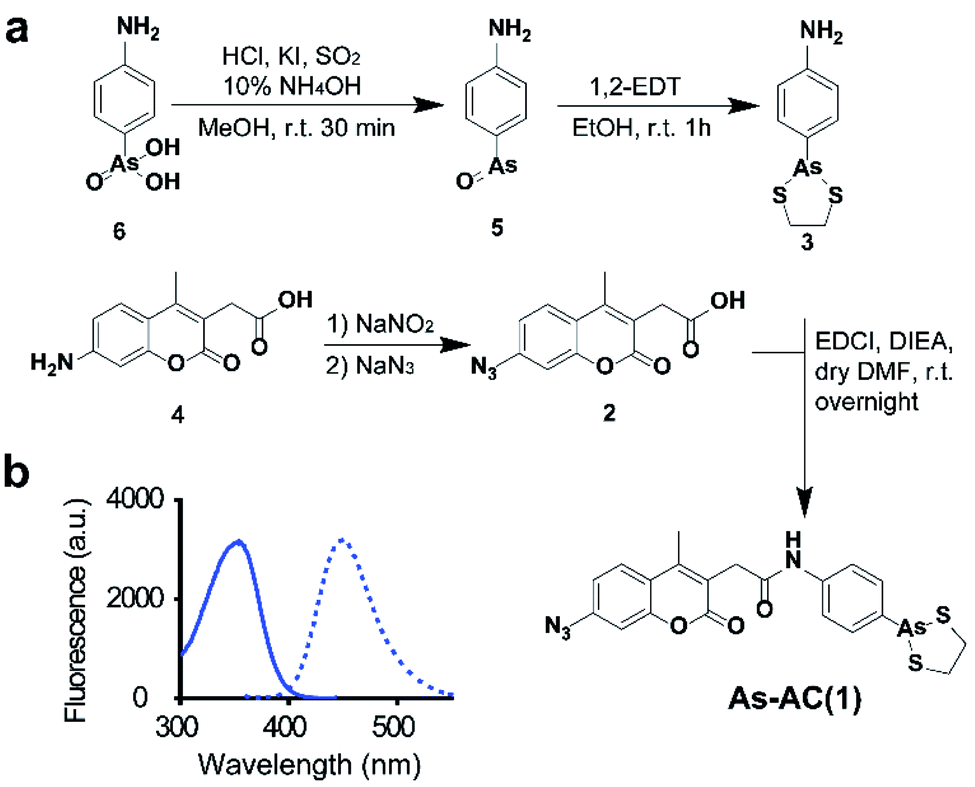 | ||
| Fig. 1 (a) Synthesis of As-AC. (b) Excitation (λex = 355 nm) and emission (λem = 448 nm) spectra of As-AC. | ||
We then examined the feasibility of selective labelling and identification of arsenic-binding proteins by As-AC in vitro. Incubation of 10 molar equivalents of cysteine-containing proteins e.g. BIR3 or SlyD (Fig. S5 and Table S1†) with As-AC (1 μM) led to a time-dependent increase in fluorescence intensity upon photo-activation, with a plateau reached at around 10 min, where ca. 8-fold increase in fluorescence intensity was observed (Fig. 2a and S6†), possibly attributable to the change of the conjugated environment.25 In contrast, nonspecific binding of As-AC to proteins that have no cysteines e.g. SlyDΔC (Fig. S7a†) and ubiquitin (Fig. S7b†) resulted in only 3-fold fluorescence changes. Other thiol-containing small molecules e.g. glutathione (GSH), 1,4-dithiothreitol (DTT), 2-mercaptoethanol (β-ME) or cysteine (Fig. S7c†) also exhibited negligible effects on protein labelling by the probe. The mixtures of As-AC with BIR3 or ubiquitin were irradiated under UV light (365 nm) for ca. 20 min to achieve the maximum fluorescence intensity then subjected to SDS-PAGE analysis and fluorescence imaging. We observed an intense blue fluorescence band at a molecular weight of 10 kDa, corresponding to BIR3 but not ubiquitin in the fluorescence image (Fig. 2b). Binding of BIR3 to AsIII (as ATO or As-AC) was further confirmed by MALDI-TOF mass spectrometry. As shown in Fig. 2c, peaks at m/z of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 987, 11058 and 11365 correspond to the intact BIR3 protein, the protein with one arsenic bound (calcd 11059) and the protein with one probe bound (calcd 11365). Collectively, we demonstrate that the probe selectively targets arsenic binding-proteins, and the arylazide is critical for significant fluorescence turn-on upon UV activation (Fig. S8†).
987, 11058 and 11365 correspond to the intact BIR3 protein, the protein with one arsenic bound (calcd 11059) and the protein with one probe bound (calcd 11365). Collectively, we demonstrate that the probe selectively targets arsenic binding-proteins, and the arylazide is critical for significant fluorescence turn-on upon UV activation (Fig. S8†).
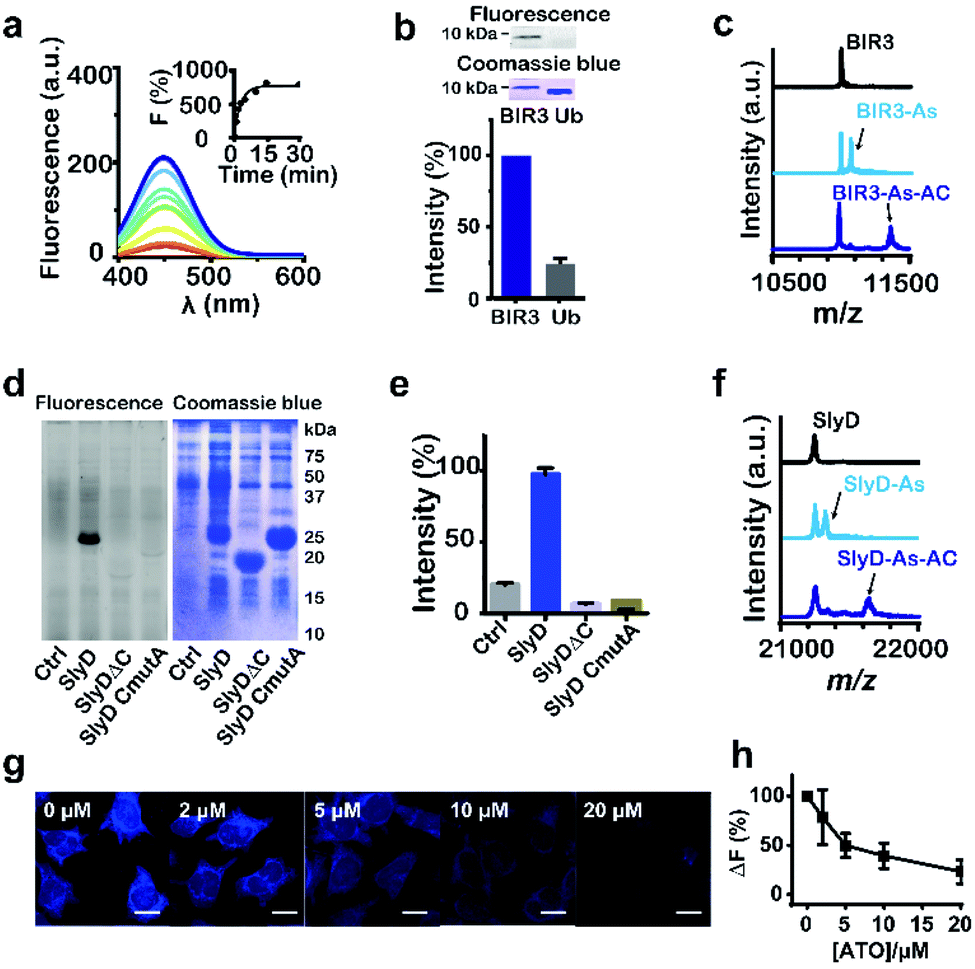 | ||
| Fig. 2 Feasibility of labelling As-binding proteins by As-AC. (a) Fluorescence spectra of As-AC (1 μM) at different time intervals after addition of BIR3 (10 μM). Inset: time-dependent fluorescence changes (λem = 448 nm) of As-AC upon binding to BIR3 protein. (b) Selectivity of As-AC towards BIR3 and ubiquitin (Ub) examined by SDS-PAGE after UV activation. The fluorescence image and SDS-PAGE are also shown. (c) MALDI-TOF-MS spectra of BIR3 protein (30 μM) in the absence and presence of 4 molar equivalents of AsIII (as ATO) and As-AC. (d) and (e) Fluorescence image and SDS-PAGE showing that As-AC selectively labels overexpressed SlyD but not SlyDΔC and SlyD CmutA in E. coli. KML603 cells without (control) and with overexpressed SlyD, SlyDΔC or SlyD CmutA were incubated with As-AC (10 μM) under dark for 30 min at 37 °C, followed by UV activation, lysed, then subjected to 13% SDS-PAGE gel analysis. (f) MALDI-TOF-MS spectra of SlyD (30 μM) in the absence and presence of 4 molar equivalents of AsIII (as ATO) and As-AC. (g) Confocal images of As-AC labelled HeLa cells pre-incubated with different concentrations of AsIII (as ATO). Scale bar: 20 μm. (h) Relative fluorescent intensity plotted against different concentrations of ATO pre-incubated with the cells. | ||
We then evaluated whether the probe could selectively label arsenic binding proteins in live cells. The probe (1 μM) was first incubated with E. coli cells overexpressing either SlyD, SlyDΔC or SlyD CmutA, where five cysteine residues were mutated into alanines, then subjected to UV irradiation prior to SDS-PAGE analysis. An intense blue band was observed on the fluorescence image only for cells overexpressing SlyD, but not SlyDΔC or SlyD CmutA (Fig. 2d and e), confirming that the probe entered bacterial cells and labeled SlyD. Binding of AsIII (as ATO or As-AC) was further confirmed by MALDI-TOF mass spectrometry. As shown in Fig. 2f, the peaks at m/z of 21237, 21312 and 21616, correspond to the intact protein, the protein with one arsenic bound (calcd 21309) and the protein with one probe bound (calcd 21615) respectively. In contrast, negligible fluorescence intensities were observed for the control groups (Fig. 2d and e). Similar experiments were performed with HeLa cells and we observed intense and faint blue fluorescence signals in the cytoplasm and nuclear region of HeLa cells respectively (Fig. S9†). Pretreatment of the cells with increasing amounts of ATO for 1 hour prior to As-AC probe led to dose-dependent decrease in the fluorescence intensity (Fig. 2h), suggesting that ATO competes with As-AC in protein binding.
We finally applied the probe to NB4 cells and another human leukemia cell line HL60 for comparison, to track arsenic binding proteins at a proteome-wide scale. The cells were treated with 10 μM As-AC for 30 min, then subjected to UV irradiation at 365 nm for 20 min prior to lysis, subsequently the labelled proteins were separated by 2D gel electrophoresis and then identified through peptide mass fingerprinting (Fig. S10†). In total, 37 and 31 putative arsenic binding proteins were identified by the As-AC probe from NB4 and HL60 respectively (Tables S2 and S3†), among which 21 mutual proteins were found in both cells. The Gene Ontology (GO) enrichment analysis illustrated different functional and component of categories of the non-mutual proteins (Fig. S11†), demonstrating the utility of the probe in profiling arsenic binding proteins in cells.
Amongst 37 identified As-binding proteins in NB4 cell, ten proteins have also been identified previously by other techniques (Table S2,† highlighted in red).19,26 Further analysis of the structures of newly identified arsenic binding proteins revealed that majority proteins contain Cys–Cys pairs, which likely coordinate to arsenic, including TPI (pdb: 4POC); CLIC1 (pdb: 1K0O); VCP (pdb: 3HU2) etc. However, some proteins e.g. NPM1 (pdb: 5EHD); HSPA9 (pdb: 6P2U) contain no Cys–Cys pairs. Despite of its thiophilic nature, arsenic was also shown previously to coordinate to histidine and serine in the crystal structures.27,28 This suggests conventional “soft-hard’’ theory might not be applicable to proteins.
To verify the identified proteins are indeed authentic As-binding proteins, we overexpressed and purified two proteins i.e., GSTP1 and PPA1 (ESI†) and examined their interaction with ATO by MALDI-TOF mass spectrometry. As shown in Fig. S12,† one and up to six As(III) ions were found to bind GSTP1 and PPA1 respectively. To further validate that these proteins bind to ATO in cellulo, we selected 8 putative AsIII binding proteins, i.e., TPT1, EEF1B2, ENO1, NPM1, VCP, IMPDH2, LDHB and NME1 for verification by cellular thermal shift assay (CETSA), a method that is widely used to investigate target engagement of drug candidates in a cellular context.29 Supplementation of 1 μM ATO to the cell culture medium for 24 h caused thermal melting curves of these proteins shifted evidently (Fig. S13†), indicative of the binding of ATO to these proteins in cellulo.
To profile the intact proteome of leukemia in response to arsenic trioxide, ATO-treated and non-treated NB4 cell extracts were compared in a TMT quantitative experiment. The differences of protein expression between the control and ATO-treated groups were quantified by LC-MS/MS. 870 distinct proteins were identified from biological triplicate runs by at least two high-confidence peptides per protein with a confidence level of 95% and global false discovery rate (FDR) of less than 1%. In total, 250 proteins including 141 up-regulated and 109 down-regulated proteins were identified as significant differentially expressed proteins (p <0.05) with >1.3-fold or <0.77-fold change in abundance compared to the control group (ESI Data 1†).
Bioinformatics analysis of arsenic-associated proteins
We used ClueGO to build gene ontology (GO) analysis network for the 37 AsIII-binding proteins. From the network, we found As-binding proteins were functionally arranged into 6 clusters and the most significantly enriched clusters were proteins related to chaperone cofactor-dependent protein refolding (GO: 005108, p = 6.75 × 10−7) and acute phase response (GO: 000695, p = 1.05 × 10−3) (Fig. 3a and ESI Data 2†). In particular, chaperone proteins, including HSPA5, HSPA8, HSPA9, HSPD1, ST13 that significantly enriched in the former GO term were largely related to the response to stress.30 Then we performed GO enrichment analysis for 250 arsenic-regulated proteins identified by quantitative proteomics. GOPlot was used to perform analysis and those GO terms with p-value <0.05 were selected for further analysis. As shown in Fig. 3b, the proteins that were most enriched in nine GO terms with the best z-scores and most significant p-values are involved in translational initiation, viral transcription, protein folding, ER translocation, nonsense-mediated mRNA decay, rRNA processing, RNA binding, protein binding and unfolded protein binding, indicative of the general functional disruption in NB4 under ATO stress (Tables S4–S6†).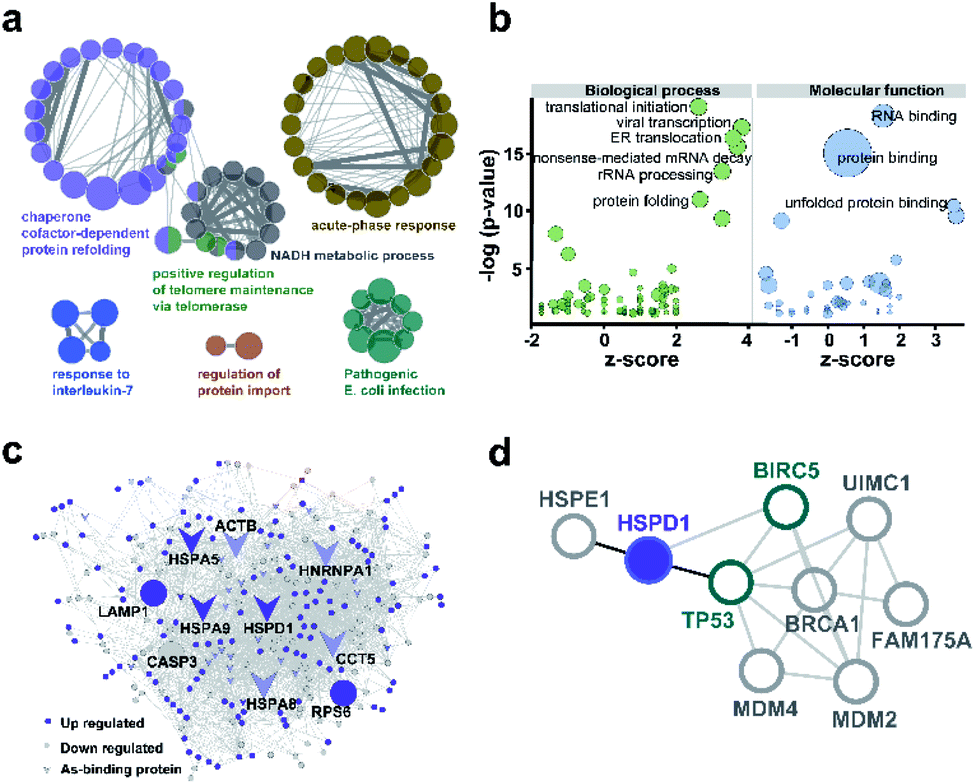 | ||
| Fig. 3 Bioinformatics analysis of arsenic associated proteins. (a) GO enrichment generated by ClueGO for 37 As-binding proteins detected by As-AC probe. Colors represent different GO clusters. Each node represents a GO biological process and the edges show the connectivity between each node based on the connection of the proteins in their biological pathways. The size of the nodes depends on the number of proteins grouped. (b) GO enrichment analysis of proteins differentially regulated by ATO. Genes with p-value <0.05 were selected. −log10 of the enrichment p-values for selected GO categories (biological process, molecular function) are plotted relative to z-scores of differential expression ratios in each category. Those nodes with −log(p-value) >10 are labelled. Circle size denotes the number of genes in each group. (c) As-associated protein–protein interactions (AsPI) network in NB4 cells. Proteins are colored and shaped according to their different properties in the network. The central nodes are represented by those of larger size with high degree and betweenness centrality (BC) values within the top 10% of the total nodes. (d) PPI networks projecting the proteins functionally associated with Hsp60 (HSPD1). | ||
To further functionally categorize the identified arsenic-associated proteins i.e., 37 arsenic-binding and 250 arsenic-regulated proteins, a protein–protein interaction (PPI) network was generated. The top 10 proteins with the highest betweenness centrality scores were selected as hub proteins including HSPD1, HSPA5, HSPA8, HSPA9, LAMP1, HNRNPA1, CASP3, RPS6, ACTB and CCT5 (Fig. 3c), which are likely served as key targets of ATO. Amongst these hub proteins, four of them are referred to heat shock proteins (HSP) superfamily, which were highly expressed in acute myeloid leukemia and have been utilized as targets for the development of therapeutics against AML.31 Ribosomal protein (RPS) is responsible for protein synthesis in living cells.32 Mutations of this ribosomal protein family genes are of high risk in developing leukemia.33 Caspase 3, activated by ATO, plays a central role in the execution-phase of cell apoptosis as well as progression of acute myeloid leukemia.34 Other hub proteins, e.g. ACTB,35 LAMP1,36 HNRNPA1 have been reported to be involved in cancer cell development, invasion, progression and chemoresistance.37 Amongst the 10 hub proteins identified in the AsIII-associated PPI, HSPD1 (Hsp60) appears to be the most up-regulated arsenic binding protein (Fig. S14†). Hsp60 has been previously exploited as a potential target for cancer therapies.38,39 Further analysis of the protein–protein interaction network of Hsp60 (Fig. 3d) revealed that Hsp60 has direct interaction with two important apoptosis related proteins i.e., BIRC5 (survivin) and TP53. We therefore selected Hsp60 for further investigation to see what relevant biological pathways are disrupted by ATO.
ATO binds to Hsp60 and inhibits its activity
Hsp60 protein was overexpressed in E. coli (DE3) and purified with high purity being confirmed by SDS-PAGE (ESI and Fig. S15a†) and size exclusion chromatography (Fig. S15b†). Hsp60 was incubated with six molar equivalents (eq.) of ATO and then subjected to a home-made GE-ICP-MS for simultaneous analysis of metals/metalloids and its bound protein.40127I-labeled proteins were used as an internal standard to calibrate the molecular weight (MW) and Hsp60 without treatment of ATO was used as a control. Incubation of ATO to Hsp60 resulted in appearance of a single 75As peak at the MW of ca. 60 kDa corresponding to a monomer of AsIII-bound Hsp60 (Fig. 4a), suggesting that AsIII binds to Hsp60 in vitro. The stoichiometry of AsIII to Hsp60 was determined to be 1.90 ± 0.11 by ICP-MS through quantification of 34S for protein concentration and 75As in ATO-treated Hsp60, after removal of excess non-bound arsenic ions by size exclusion chromatography (Fig. S16†). Given that arsenic is highly thiophilic, we then examined whether the three Cys residues (Cys213, Cys418 and Cys423) are involved in arsenic binding by the measurement of free thiols of Hsp60 pre-incubated with different molar ratios of ATO using Ellman's assay. As shown in Fig. 4b, about 3 eq. of ATO could completely deplete free thiols, indicating that cysteine residues are involved in arsenic binding. Collectively, we demonstrate that arsenic binds to Hsp60 mainly via cysteine residues. We then examined whether ATO binds to Hsp60 in cells by cellular thermal shift assay (CETSA).29 NB4 cells was exposed to 1 μM ATO (IC50 = 0.9 μM, 24 h) for 24 h, and then subjected to CETSA according to a previous protocol.41 Treatment of ATO led to a shift of melting point from 60.6 °C to 62.6 °C (Fig. 4c), indicative of cellular engagement of AsIII to Hsp60 in NB4 cells.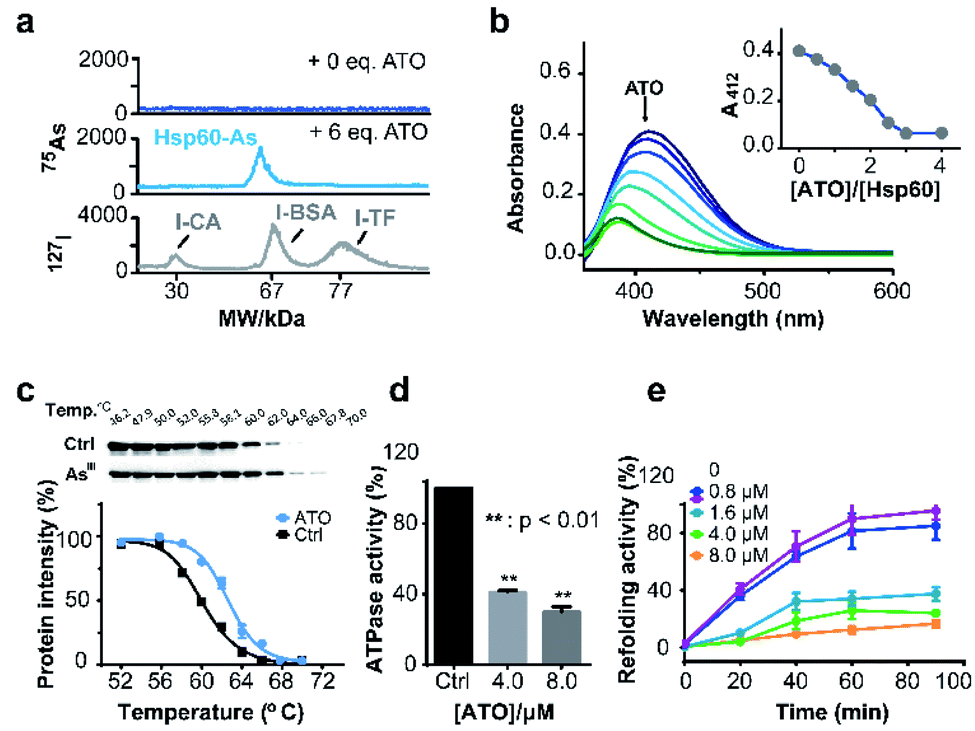 | ||
| Fig. 4 Arsenic binds and functionally disrupts Hsp60. (a) GE-ICP-MS profiles of apo-Hsp60, ATO treated Hsp60, and 127I-labeled internal standards. (b) Free thiol contents in Hsp60 after treatment with different concentrations of ATO measured by Ellman's assay. Inset shows the absorbance at 412 nm vs. amounts of ATO. (c) Thermal melting curves of Hsp60 protein in intact leukemia cells, treated with or without arsenic trioxide. Data are presented as the mean SEM from at least three independent experiments. (d) Effect of ATO on ATPase activity of Hsp60. ATP content was determined by Malachite Green Phosphate Assay Kit (POMG-25H) after incubation of Hsp60 (2 μM) with or without ATO for 30 min at 37 °C. (e) ATO inhibits Hsp60/Hsp10 refolding activity. Time-dependent refolding activity of Hsp60/Hsp10 in the presence of gradient concentrations of ATO. | ||
Hsp60 is an ATP-dependent protein that facilitates the folding of a wide range of substrates to the native state and prevents their aggregation.42 ATPase is essential for Hsp60 to conduct its activity either by recruiting its co-chaperonin Hsp10 or releasing protein after final folding and assembly.43 We first examined whether binding of arsenic inhibited the ATPase activity of Hsp60 by a well-established in vitro assay. As shown in Fig. 4d, ATPase activity of Hsp60 was suppressed by 59.0% at 8 μM ATO. We then examined the refolding activity of Hsp60 using a typical chaperone assay, in which the substrate protein was first denatured chemically. The refolding activity of Hsp60/Hsp10 was then monitored both in the absence and presence of gradient amounts of ATO. In the absence of ATO, the denatured substrate was refolded effectively by Hsp60/Hsp10 in a time-dependent manner, and ca. 80% of the substrate protein was fully-folded in around 60 min. The presence of ATO significantly reduced refolding activity of Hsp60/Hsp10. As shown in Fig. 4e, Hsp60/Hsp10 could only recover 34.3%, 25.8% and 12.3% of the substrate activity in the presence of 1.6, 4.0 and 8.0 μM ATO respectively. Our combined data confirm that binding of ATO to Hsp60 attenuates ATPase activity and abolishes its refolding activity.
Binding of ATO to Hsp60 disrupts the complexes of p53-Hsp60 and survivin-Hsp60, leading to degradation of p53 and survivin
The protein–protein interaction network shows that Hsp60 (HSPD1) functionally associated with BIRC5 (SVV family) and TP53 (P53 family) (Fig. 3d). Hsp60 regulates tumour cell apoptosis by formation of Hsp60-survivin and Hsp60-p53 complexes, which will stabilize survivin levels and restrain the function of p53.44 The wild-type p53 is a tumor suppressor,45 involving in different types of leukemia diagnosis and therapy.46,47 Survivin, belonging to the family of inhibitor of apoptosis protein (IAP), has been considered as an attractive candidate for cancer therapy.48We then asked whether binding of ATO to Hsp60 disrupts the Hsp60-survivin or Hsp60-p53 complexes, resulting in the release of p53 and survivin, which could be degraded rapidly by a protease. We performed co-immunoprecipitation assay for endogenous proteins in NB4 cell lysates. In ATO un-treated control, immunoprecipitated survivin from NB4 cell lysates contained co-associated Hsp60, indicative of formation of a complex between Hsp60 and survivin (Fig. 5a and S17†). Similarly, Hsp60-p53 complex was also detected by co-immunoprecipitation in the cell lysates. However, upon treatment of the cell with increasing amounts of ATO, the co-associated Hsp60 in these protein complexes decreased (Fig. 5b and S17†), confirming that ATO disrupts the ability of Hsp60 to recruit target proteins, i.e., p53 and survivin.
We further examined whether ATO affected the co-localization of Hsp60 with survivin or p53 in NB4 cells by immunofluorescence. In the control group, both Hsp60 and p53, which were immobilized with anti-Hsp60 antibody (green) and anti-p53 antibody (red), were evenly distributed in the cellular compartment with certain co-localization (yellow) of Hsp60 and p53 (Fig. 5c). Upon exposure to ATO, Hsp60 tends to be non-dispersed and concentrated in certain part of the cell, whereas p53 concentrated in different part of the cell and co-localization of the two proteins was hardly observed. Similarly, survivin is evenly distributed inside cells in the control group, whereas co-localization of Hsp60 and survivin was only observed in the control but not ATO-treated cells (Fig. 5d). These data demonstrate that binding of ATO to Hsp60 results in dissociation or disruption of formation of Hsp60-p53 and Hsp60-survivin complexes in cells.
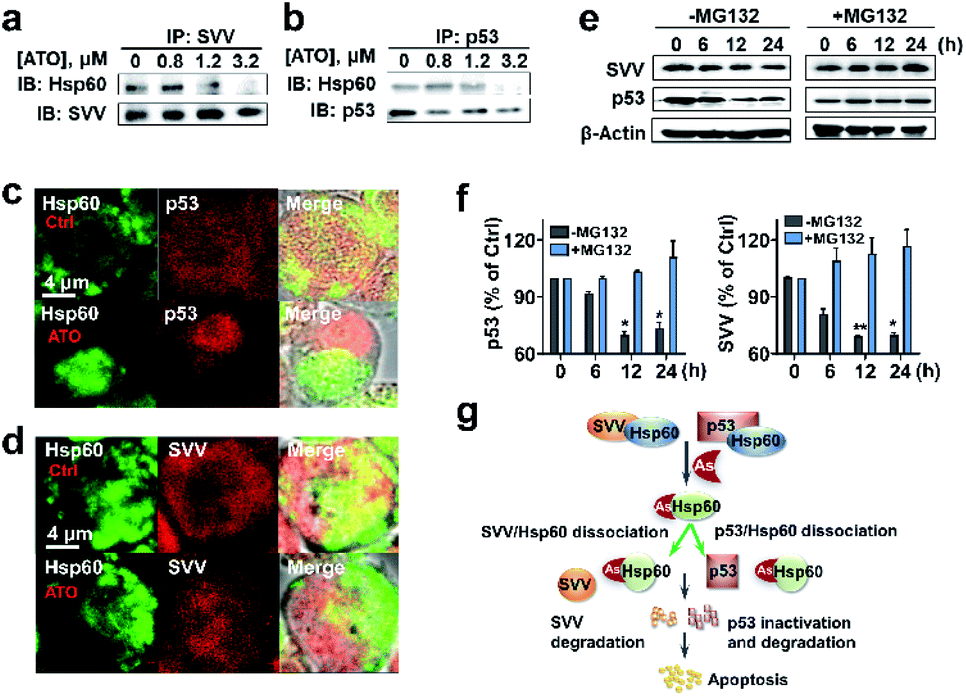 | ||
| Fig. 5 Binding of arsenic to Hsp60 disrupts complexation of Hsp60 with p53 and survivin, leading to degradation of p53 and survivin. (a) and (b) IP-western blots of endogenous Hsp60 associated with survivin or p53 in NB4 cells treated with ATO at different concentrations. (c) and (d) Immunolocalization of endogenous Hsp60 with p53 and survivin. Co-staining was performed using antibodies specific for Hsp60 (green), survivin (red), p53 (red) and a fluorochrome conjugated secondary antibodies against rabbit or mouse IgG. Cells treated with ATO (1.2 μM) or without (as control, upper panel) were analyzed using confocal microscopy (×63 oil lens). The images were visualized with a Zeiss microscope. Scale bars: 4 μm. (e) and (f) Arsenic mediates proteasome degradation of p53 and survivin. Treatment of NB4 cells with MG132, a 26S proteasome inhibitor prevents arsenic induced protein degradation in cells. Western blots were prepared with the extracts of NB4 cells untreated or pretreated with 25 μM MG132 for 4 h in the absence and presence of 0.8 μM As2O3, β-actin was used as control. Protein contents were quantified by densitometric analysis. Means ± SEM; n = 3; *p <0.05, **p <0.01 versus vehicle control. (g) Proposed the mechanism of ATO induced apoptosis through inhibition of key target Hsp60 and degradation of survivin and p53. | ||
We next examined whether the p53 and survivin are degraded via the proteasomal pathway. NB4 cells were pre-treated with or without 25 μM MG132, an inhibitor of 26S proteasome, for 4 h and protein levels of both survivin and p53 in NB4 cells were measured by western blot. As shown in Fig. 5e and f, time-dependent decreases in the levels of both p53 and survivin were observed in ATO-treated NB4 cells in the absence of MG132. However, upon addition of MG132, the protein levels remained almost unchanged. These results indicate that both p53 and survivin undergo proteasomal degradation, in consistence with previous reports.49,50 Taken together, we demonstrate that ATO binds and functionally abolishes Hsp60, leading to dissociation of the complexes of Hsp60-survivin and Hsp60-p53. Both p53 and survivin are subsequently degraded, which may partially contribute to ATO-induced apoptosis (Fig. 5g).
Conclusions
In spite of extensive studies for decades, the mechanism of action of ATO is still not fully understood. Arsenic trioxide was demonstrated to bind multiple proteins or enzymes.8 However, it remains unknown what the primary or key targets are. Conventionally, the putative protein targets of ATO were selected and characterized individually, an approach that does not provide a holistic view on the biological or pathological mechanisms of action of ATO. Given the complexity of arsenic–protein interaction in cells, integration of different approaches is necessary to comprehensively uncover arsenic-associated proteins, which provides rich resources for downstream investigation of biological pathways disrupted by ATO at a system level.We design and synthesize an organo-arsenic fluorescent probe, which could rapidly enter live cells to selectively label AsIII-binding proteins. These proteins were subsequently identified via conventional proteomics approach upon photoactivation of arylazide of the probe. The arylazide not only enabled significant fluorescence turn-on, but also strengthened the binding between the probe and the labelled proteins. We have successfully identified 37 arsenic-binding proteins in NB4 cells. Our approach allows endogenous arsenic binding proteins to be mined at a proteome-wide scale. Moreover, our method enables the weakly, even transiently arsenic bound proteins to be identified. Together with quantitative proteomics, As-binding and As-regulated proteins were tracked at a proteome-wide scale.
Bioinformatics analysis of these arsenic-associated proteins (and proteome) and their PPI networks allowed us to uncover ten central nodes in the AsIII–protein-interaction (API) network with seven of them being As-binding proteins in leukemia cells. We show that Hsp60 binds to ATO both in vitro and in cellulo. Such a binding disrupts its folding/refolding activity together with Hsp10 owing to abolished ATPase activity of Hsp60 by ATO. By using immunoprecipitation and immunofluorescence, we demonstrate that treatment of ATO to NB4 cells resulted in dissociation or disrupting the formation of Hsp60-p53 and Hsp60-survivin complexes, leading to degradation of p53 and survivin. Owing to its high expression levels in tumors, Hsp60 is an ideal target for anticancer therapy due to less toxicity to normal cells/tissues. Moreover, targeting complexes such as survivin-Hsp90,51 Hsp60-p53 and Hsp70/90-p53 have been demonstrated to be effective in anticancer therapies.44,52 Our combined data have demonstrated clearly that Hsp60 serves as an important target of ATO. Other hub proteins identified in this study may warrant further studies in future. The integrative approach we report here also provides a general platform for system-wide understanding therapeutic and toxicology of arsenic (and other metallodrugs).
Author contributions
H. S. and H. L. conceived the project and H. S., H. L., X. H., E. T. and N. H. designed the experiments; X. H., T. K.-Y. I., D. T., H. W. and X. Y. performed the experiments; Y. F. C., M. K. and X. H. performed the MS-based quantitative experiment; K. M., X. H., R. W., and Y. W. performed the bioinformatics; and H. L., X. H. and H. S. principally wrote the manuscript with input from all.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Research Grants Council of Hong Kong (1711618M, 17304614P, 17333616P and 17307017P), the University of Hong Kong (URC), and Norman & Cecilia Yip foundation. We acknowledge Faculty Core Facility of LKS Faculty of Medicine (the University of Hong Kong) for the support on confocal imaging and thank the Center for Genomic Sciences, Li Ka Shing Faculty of Medicine for the mass spectrometry facilities.Notes and references
- H. Sun, in Biological Chemistry of Arsenic, Antimony and Bismuth, John Wiley & Sons, Ltd, Chiester, 2010 Search PubMed.
- E. Lengfelder, F. Lo-Coco, L. Ades, P. Montesinos, D. Grimwade, B. Kishore, S. M. Ramadan, M. Pagoni, M. Breccia, A. J. Huerta, A. M. Nloga, J. D. Gonzalez-Sanmiguel, A. Schmidt, J. F. Lambert, S. Lehmann, E. Di Bona, B. Cassinat, W. K. Hofmann, D. Gorlich, M. C. Sauerland, P. Fenaux, M. Sanz and L. European, Leukemia, 2015, 29, 1084–1091 CrossRef CAS PubMed.
- J.-X. Liu, G.-B. Zhou, S.-J. Chen and Z. Chen, Curr. Opin. Chem. Biol., 2012, 16, 92–98 CrossRef CAS PubMed.
- Z. G. Wang, R. Rivi, L. Delva, A. Konig, D. A. Scheinberg, C. Gambacorti-Passerini, J. L. Gabrilove, R. P. Warrell Jr and P. P. Pandolfi, Blood, 1998, 92, 1497–1504 CrossRef CAS PubMed.
- Y.-H. Ling, J.-D. Jiang, J. F. Holland and R. Perez-Soler, Mol. Pharmacol., 2002, 62, 529–538 CrossRef CAS PubMed.
- Y. Zhou, H. Li and H. Sun, Chem. Commun., 2017, 53, 2970–2973 RSC.
- X.-W. Zhang, X.-J. Yan, Z.-R. Zhou, F.-F. Yang, Z.-Y. Wu, H.-B. Sun, W.-X. Liang, A.-X. Song, V. Lallemand-Breitenbach, M. Jeanne, Q.-Y. Zhang, H.-Y. Yang, Q.-H. Huang, G.-B. Zhou, J.-H. Tong, Y. Zhang, J.-H. Wu, H.-Y. Hu, H. de Thé, S.-J. Chen and Z. Chen, Science, 2010, 328, 240–243 CrossRef CAS PubMed.
- S. Shen, X.-F. Li, W. R. Cullen, M. Weinfeld and X. C. Le, Chem. Rev., 2013, 113, 7769–7792 CrossRef CAS PubMed.
- H.-n. Zhang, L. Yang, J.-y. Ling, D. M. Czajkowsky, J.-F. Wang, X.-W. Zhang, Y.-M. Zhou, F. Ge, M.-k. Yang, Q. Xiong, S.-J. Guo, H.-Y. Le, S.-F. Wu, W. Yan, B. Liu, H. Zhu, Z. Chen and S.-c. Tao, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15084–15089 CrossRef CAS PubMed.
- S. Kozono, Y.-M. Lin, H.-S. Seo, B. Pinch, X. Lian, C. Qiu, M. K. Herbert, C.-H. Chen, L. Tan, Z. J. Gao, W. Massefski, Z. M. Doctor, B. P. Jackson, Y. Chen, S. Dhe-Paganon, K. P. Lu and X. Z. Zhou, Nat. Commun., 2018, 9, 3069 CrossRef PubMed.
- E. Kamynina, E. R. Lachenauer, A. C. DiRisio, R. P. Liebenthal, M. S. Field and P. J. Stover, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E2319–E2326 CrossRef CAS PubMed.
- W. Piao, D. Chau, L. M. Yue, Y. L. Kwong and E. Tse, Leukemia, 2017, 31, 522–526 CrossRef CAS PubMed.
- P. P. Kulkarni, Y. M. She, S. D. Smith, E. A. Roberts and B. Sarkar, Chem.–Eur. J., 2006, 12, 2410–2422 CrossRef CAS PubMed.
- J. Zhang, R. E. Campbell, A. Y. Ting and R. Y. Tsien, Nat. Rev. Mol. Cell Biol., 2002, 3, 906–918 CrossRef CAS PubMed.
- B. Albert Griffin, S. R. Adams, J. Jones and R. Y. Tsien, Methods Enzymol., 2000, 327, 565–578 Search PubMed.
- J. Schulte-Zweckel, F. Rosi, D. Sreenu, H. Schröder, C. Niemeyer and G. Triola, Molecules, 2016, 21, 750 CrossRef PubMed.
- P. Kapahi, T. Takahashi, G. Natoli, S. R. Adams, Y. Chen, R. Y. Tsien and M. Karin, J. Biol. Chem., 2000, 275, 36062–36066 CrossRef CAS PubMed.
- C. Huang, Q. Yin, W. Zhu, Y. Yang, X. Wang, X. Qian and Y. Xu, Angew. Chem., Int. Ed., 2011, 50, 7551–7556 CrossRef CAS PubMed.
- X. Yan, J. Li, Q. Liu, H. Peng, A. Popowich, Z. Wang, X.-F. Li and X. C. Le, Angew. Chem., Int. Ed., 2016, 55, 14051–14056 CrossRef CAS PubMed.
- H. Li, R. Wang and H. Sun, Acc. Chem. Res., 2019, 52, 216–227 CrossRef CAS PubMed.
- Y.-T. Lai, Y.-Y. Chang, L. Hu, Y. Yang, A. Chao, Z.-Y. Du, J. A. Tanner, M.-L. Chye, C. Qian, K.-M. Ng, H. Li and H. Sun, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2948 CrossRef CAS PubMed.
- Y. Liu, D. Duan, J. Yao, B. Zhang, S. Peng, H. Ma, Y. Song and J. Fang, J. Med. Chem., 2014, 57, 5203–5211 CrossRef CAS PubMed.
- B. Chen, W. Li, C. Lv, M. Zhao, H. Jin, H. Jin, J. Du, L. Zhang and X. Tang, Analyst, 2013, 138, 946–951 RSC.
- B. J. M. Thevenin, Z. Shahrokh, R. L. Williard, E. K. Fujimoto, J.-J. Kang, N. Ikemoto and S. B. Shohet, Eur. J. Biochem., 1992, 206, 471–477 CrossRef CAS PubMed.
- W. Tan, K. Wang and T. J. Drake, Curr. Opin. Chem. Biol., 2004, 8, 547–553 CrossRef CAS PubMed.
- T. Zhang, H. Lu, W. Li, R. Hu and Z. Chen, Int. J. Mol. Sci., 2015, 16, 26871–26879 CrossRef CAS PubMed.
- T. Zhou, S. Radaev, B. P. Rosen and D. L. Gatti, J. Biol. Chem., 2001, 276, 30414–30422 CrossRef CAS PubMed.
- T. Zhou, EMBO J., 2000, 19, 4838–4845 CrossRef CAS PubMed.
- D. M. Molina, R. Jafari, M. Ignatushchenko, T. Seki, E. A. Larsson, C. Dan, L. Sreekumar, Y. Cao and P. Nordlund, Science, 2013, 341, 84 CrossRef CAS PubMed.
- P. Walter and D. Ron, Science, 2011, 334, 1081 CrossRef CAS PubMed.
- H. Reikvam, I. Nepstad, A. Sulen, B. T. Gjertsen, K. J. Hatfield and O. Bruserud, Expert Opin. Invest. Drugs, 2013, 22, 551–563 CrossRef CAS PubMed.
- J. Pelletier, G. Thomas and S. Volarević, Nat. Rev. Cancer, 2017, 18, 51 CrossRef PubMed.
- K. M. Goudarzi and M. S. Lindström, Internet J. Oncol., 2016, 48, 1313–1324 CrossRef CAS PubMed.
- N. Man, Y. Tan, X. J. Sun, F. Liu, G. Cheng, S. M. Greenblatt, C. Martinez, D. L. Karl, K. Ando, M. Sun, D. Hou, B. Chen, M. Xu, F. C. Yang, Z. Chen, S. Chen, S. D. Nimer and L. Wang, Blood, 2017, 129, 2782–2792 CrossRef CAS PubMed.
- P. T. Caswell and T. Zech, Trends Cell Biol., 2018, 28, 823–834 CrossRef CAS PubMed.
- E. Machado, S. White-Gilbertson, D. van de Vlekkert, L. Janke, S. Moshiach, Y. Campos, D. Finkelstein, E. Gomero, R. Mosca, X. Qiu, C. L. Morton, I. Annunziata and A. d'Azzo, Sci. Adv., 2015, 1, e1500603 CrossRef PubMed.
- H. Kedzierska and A. Piekielko-Witkowska, Cancer Lett., 2017, 396, 53–65 CrossRef CAS PubMed.
- L. Sedlackova, M. Spacek, E. Holler, Z. Imryskova and I. Hromadnikova, Tumor Biol., 2011, 32, 33–44 CrossRef CAS PubMed.
- S. K. Fung, T. Zou, B. Cao, P.-Y. Lee, Y. M. E. Fung, D. Hu, C.-N. Lok and C.-M. Che, Angew. Chem., Int. Ed., 2017, 56, 3892–3896 CrossRef CAS PubMed.
- L. Hu, T. Cheng, B. He, L. Li, Y. Wang, Y.-T. Lai, G. Jiang and H. Sun, Angew. Chem., Int. Ed., 2013, 52, 4916–4920 CrossRef CAS PubMed.
- R. Jafari, H. Almqvist, H. Axelsson, M. Ignatushchenko, T. Lundbäck, P. Nordlund and D. M. Molina, Nat. Protoc., 2014, 9, 2100–2122 CrossRef CAS PubMed.
- B. Bukau and A. L. Horwich, Cell, 1998, 92, 351–366 CrossRef CAS PubMed.
- H. Saibil, Nat. Rev. Mol. Cell Biol., 2013, 14, 630–642 CrossRef CAS PubMed.
- J. C. Ghosh, T. Dohi, B. H. Kang and D. C. Altieri, J. Biol. Chem., 2008, 283, 5188–5194 CrossRef CAS PubMed.
- B. Vogelstein, D. Lane and A. J. Levine, Nature, 2000, 408, 307–310 CrossRef CAS PubMed.
- A. Quintás-Cardama, C. Hu, A. Qutub, Y. H. Qiu, X. Zhang, S. M. Post, N. Zhang, K. Coombes and S. M. Kornblau, Leukemia, 2017, 31, 1296–1305 CrossRef PubMed.
- M. Prokocimer, A. Molchadsky and V. Rotter, Blood, 2017, 130, 699–712 CrossRef CAS PubMed.
- D. C. Altieri, Nat. Rev. Cancer, 2003, 3, 46–54 CrossRef CAS PubMed.
- W. Yan, Y. Zhang, J. Zhang, S. Liu, S. J. Cho and X. Chen, J. Biol. Chem., 2011, 286, 17478–17486 CrossRef CAS PubMed.
- J. Zhao, T. Tenev, L. M. Martins, J. Downward and N. R. Lemoine, J. Cell Sci., 2000, 113, 4363–4371 CrossRef CAS PubMed.
- J. Plescia, W. Salz, F. Xia, M. Pennati, N. Zaffaroni, M. G. Daidone, M. Meli, T. Dohi, P. Fortugno, Y. Nefedova, D. I. Gabrilovich, G. Colombo and D. C. Altieri, Cancer Cell, 2005, 7, 457–468 CrossRef CAS PubMed.
- S. Yamamoto and T. Iwakuma, Cancers, 2019, 11, 4, DOI:10.3390/cancers11010004.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, supplementary figures, identified proteomes and bioinformatics data. See DOI: 10.1039/d1sc03119h |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |